
Robust Pharmacodynamic Effect of LY3202626, a Central Nervous System Penetrant, Low Dose BACE1 Inhibitor, in Humans and Nonclinical Species
Abstract
Background:
The development of beta-site amyloid-beta precursor protein cleaving enzyme (BACE) 1 inhibitors for the treatment of Alzheimer’s disease requires optimization of inhibitor potency, selectivity, and brain penetration. Moreover, there is a need for low-dose compounds since liver toxicity was found with some BACE inhibitors.
Objective:
To determine whether the high in vitro potency and robust pharmacodynamic effect of the BACE inhibitor LY3202626 observed in nonclinical species translated to humans.
Methods:
The effect of LY3202626 versus vehicle on amyloid-β (Aβ) levels was evaluated in a series of in vitro assays, as well as in in vivo and multi-part clinical pharmacology studies. Aβ levels were measured using analytical biochemistry assays in brain, plasma, and cerebrospinal fluid (CSF) of mice, dogs and humans. Nonclinical data were analyzed using an ANOVA followed by Tukey’s post hoc test and clinical data used summary statistics.
Results:
LY3202626 exhibited significant human BACE1 inhibition, with an IC50 of 0.615±0.101 nM in a fluorescence resonance energy transfer assay and an EC50 of 0.275±0.176 nM for lowering Aβ1–40 and 0.228±0.244 nM for Aβ1–42 in PDAPP neuronal cultures. In dogs, CSF Aβ1hboxx concentrations were significantly reduced by ∼80% at 9 hours following a 1.5 mg/kg dose. In humans, CSF Aβ1–42 was reduced by 73.1±7.96 % following administration of 6 mg QD. LY3202626 was found to freely cross the blood-brain barrier in dogs and humans.
Conclusion:
LY3202626 is a potent BACE1 inhibitor with high blood-brain barrier permeability. The favorable safety and pharmacokinetic/pharmacodynamic profile of LY3202626 supports further clinical development.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by clinical progressive cognitive decline and loss of function. Pathologically, accumulation of proteins in the brain, specifically extracellular plaques comprised of the amyloid-β (Aβ) peptide and intracellular neurofibrillary tangles of hyperphosphorylated tau, is believed to be linked to neuronal atrophy in specific brain regions, resulting in diminished memory, learning, and cognition. Several forms of Aβ result from amyloid precursor protein processing, with the most prevalent being Aβ1–42 and Aβ1–40 [1]. The beta-site amyloid-beta precursor protein cleaving enzyme (BACE) 1 enzyme plays a critical role in the production of these Aβ peptides, and, consequently, BACE1 inhibitors have been extensively investigated as potential AD therapies [2]. Given the strong genetic evidence supporting Aβ as a pathologic driver of AD, BACE1 inhibitors have been an active area of clinical development and a number of phase 2/3 clinical trials have been launched [3]. A concern that has arisen in the AD research field is that decreased enzymatic processing of other BACE1 substrates may be at least partly responsible for the lack of efficacy observed with BACE1 inhibitors in these late phase trials [4]. Alternatively, given that Aβ plaques build up years before disease symptoms emerge, therapies targeting the slow, protracted production of Aβ might only be effective if patients are treated in the presymptomatic stage of the disease [5]. These potential therapies would require a highly favorable safety profile in order to be administered to an asymptomatic population. We have previously reported the discovery and clinical development of LY2811376 and LY2886721, the latter of which advanced to Phase II clinical trials in which development was stopped because liver enzymes were abnormally elevated [6, 7]. This finding was not observed in nonclinical toxicology studies, nor was it suggested based upon the chemical structure of the compound (internal data). Based on this safety finding and the lack of any obvious predictor of liver injury for LY2886721, we sought to develop a more potent BACE inhibitor that could be administered at lower doses. This strategy was based upon the reports that idiosyncratic drug-induced liver injury may be associated with overall dose load [19, 20]. Herein, we report the discovery and Phase I clinical development of LY3202626, a highly potent, central nervous system (CNS)-penetrant, and low-dose BACE1 inhibitor.
MATERIALS AND METHODS
Preparation of LY3202626
N-[3-(4aR,7aS)-2-Amino-6-(5-fluoropyrimidin-2-yl)-4,4a,5,7-tetrahydropyrrolo3,4-d][1,3]thiazin-7a-yl]-4-fluoro-phenyl]-5-methoxy-pyrazine-2-carboxamide (LY3202626) can be prepared as described in US Patent 8,841,293 [8]. Nonclinical studies were conducted with the hydrochloride salt of LY3202626 or with the free-base form of LY3202626. All clinical studies were conducted using the free-base form of LY3202626.
Nonclinical characterization of LY3202626
In vitro potency and selectivity of LY3202626
BACE1 and related aspartyl protease inhibition assays. To determine the in vitro potency of inhibition, LY3202626 was tested against purified recombinant human BACE1 or BACE2 as an enzyme source and a synthetic Fluorescence Resonance Energy Transfer (FRET) peptide (sequence: methylcourmarine (MCA)-S-E-V-N-L-D-A-E-F-R-K(dinitrophenol)-R-R-R-R-NH2) as a substrate. Human BACE1 and BACE2 enzyme assays were conducted as previously described [6, 9].
Cathepsin D, pepsin, and renin assays were conducted with commercially available reagents using the protocols described by May et al. [6]. Human liver cathepsin D was purchased from Calbiochem (San Diego, CA, USA; Catalog #219401). Porcine gastric mucosa pepsin was purchased from Sigma-Aldrich (St. Louis, MO, USA; Catalog #P6887). Renin recombinant human enzyme was purchased from AnaSpec, Inc. (Fremont, CA, USA; Catalog #AS-72041). For all enzyme assays, a 10-point inhibition curve was plotted and fitted with a 4-parameter logistic equation to obtain the IC50 values.
Mouse primary neuronal cultures. Primary cortical neuronal cultures from embryonic PDAPP mice—containing a PDGF-driven human APP minigene with the V717F (Indiana) mutation—provided a second cellular model to assess BACE inhibition in vitro. Primary cortical neurons were prepared as described by May et al. [6] from PDAPP embryos (gestational day 16) and cultured in 96-well plates (15×104 cells/well) in DMEM/F12 (1:1) containing B27 supplement. Neurons were incubated at 37°C for 24 h in the presence/absence of inhibitors at the desired concentration. Assays were run in triplicate. At the end of the incubation period, conditioned media were analyzed for Aβ peptides using a sandwich Enzyme-Linked Immunosorbent Assay (ELISA) assay using monoclonal 2G3 as a capture antibody for Aβ1–40 and monoclonal 21F12 as a capture antibody for Aβ1–42. Both ELISAs used biotinylated 3D6 as the reporting antibody. Treated PDAPP neuronal cultures from the conditioned medium were diluted 1:5 for Aβ1–42 and 1:10 for Aβ1–40 in buffer (PBS, 0.25% Casein, 0.05% Tween20, pH7.4) and 100μL aliquots assayed in triplicate.
Compound cytotoxicity was assessed by evaluating cellular viability following LY3202626 treatment using the MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]) assay [10]. The plates were read on a spectrophotometer at 595 nm with absorbance proportional to cell viability.
In vivo pharmacology of LY3202626 in PDAPP mice
The in vivo pharmacologic properties of LY3202626 were initially tested in young (2-3 months old) female PDAPP mice from Taconic Biosciences, Inc. (Rensselaer, NY, USA; private line 6042T; [11]) to evaluate in vivo pharmacologic effects of LY3202626 on Aβ processing (as described in May et al. [7]). Mice received single doses of vehicle, 0.3, 1.0, or 3.0 mg/kg LY3202626 delivered by oral gavage in a 7% Pharmasolve vehicle at 10 mL/kg. Three hours after dosing, mice were anesthetized with tribromoethanol (Avertin™) and blood, cerebrospinal fluid (CSF), and brain tissue were collected.
For brain tissue collection, bilateral hippocampal and cortical brain regions were rapidly microdissected, frozen on dry ice, and stored at –80°C until analysis. The remaining brain tissue was flash-frozen and used for compound exposure assessment. Brain samples were homogenized in 5.5 M guanidine-HCl buffer and extracts were collected, diluted, and filtered. Then, sandwich ELISAs were used to measure Aβ1hboxx, C99, soluble amyloid-β protein precursor beta (sAβPPβ), and alpha (sAβPPα) levels in the CSF. To detect Aβ1hboxx levels in the brain and CSF, m266.2 was used as the capture antibody and biotinylated 3D6 was used as the reporter antibody. For determination of C99 protein levels in the brain, an anti-APP C-terminal antibody (rabbit polyclonal antibody; LLY-57clone 2, Epitomics International Inc, Burlingame, CA, USA) was used for capture and biotinylated 3D6 antibody was used to detect the amino-terminus of C99. To determine sAβPPβ levels in brain homogenates, 8E5 was used for capture, the anti-neo-epitope antibody 16-7-7 rMAb (Epitomics International Inc.) was used to bind the C-terminus and an anti-rabbit-HRP (Horseradish Peroxidase) conjugate used for colorometric detection. CSF sAβPPβ detection similarly used 8E5 for capture, but used an end-specific reporter antibody (Sigma-Genosys, St. Louis, MO, USA) designed internally as the reporter antibody followed by detection with a goat anti-rabbit-HRP conjugate. CSF levels of sAβPPα were determined using 8E5 for capture and biotinylated 2H3 as the reporter. Eli Lilly is an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited institution and animal use in these studies conformed to Institutional Animal Care and Use Committee (IACUC) and OLAW guidelines as defined by the Guide for the Care and Use of Laboratory Animals (NRC 2011, 8th Edition).
In vivo central pharmacology of LY3202626 in beagle dogs
Young beagle dogs were used to determine pharmacokinetic (PK) and pharmacodynamic (PD) effects of LY3202626. In this model established at MPI Research (Mattawan, MI, USA), beagle dogs were implanted with a cannula in the lumbar spine region that was threaded up towards the cervical region, allowing for multiple CSF collections throughout a single, 48 h study period. Following a 1.5 mg/kg oral dose of LY3202626 in 1% hydroxyethylcellulose (HEC), plasma samples from 6 male dogs were obtained before dosing and at 0.5, 1, 2, 3, 6, 9, 12, 24, and 48 h later. To assess central pharmacology of LY3202626, CSF was also collected 0.5 h before dosing to establish a baseline and then again at 3, 6, 9, 24, and 48 h after dosing. Aβ1 - x levels were measured in CSF and plasma using a standard sandwich ELISA protocol (m266.2 and biotinylated 3D6 as the capture and reporter antibodies, respectively), averaged across all dogs, and then plotted as a function of time relative to baseline. All dog studies were conducted at MPI Research, which is a registered facility under the Animal Welfare Act by the USDA, has an approved Assurance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and is accredited by the AAALAC International.
Nonclinical bioanalytical methods for LY3202626
Brain samples were weighed and a 3-fold volume of water/methanol (4:1, v/v) was added before homogenization with an ultrasonic tissue disrupter. Aliquots of homogenized brain, plasma, or CSF samples and appropriate calibration standards were mixed with methanol/acetonitrile (1:1, v/v) containing internal standard to precipitate proteins, then centrifuged to pellet the precipitant. A volume of each sample’s supernatant was transferred to a new 96-well plate and diluted 2-fold with water/methanol (1:1, v/v) then analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS) using a BETACIL™ C18 (Thermo Fisher Scientific, Waltham, MA, USA) 20 mm×2.1 mm×5μm Javelin column and a gradient mobile phase system with A: Water/1M NH4HCO3 (2000:10, v/v) and B: MeOH/1M NH4HCO3 (2000:10, v/v) delivered at 1.5 mL/min. Selected Reaction Monitoring (SRM) transitions (positive ion mode) with (m/z 499.1 > 423.1) and internal standard (m/z 263.1 > 148.1) were acquired with an API4000 (Applied Biosystems/MDS Sciex, Foster City, CA, USA) using Analyst software (version 1.4.2). LY3202626 plasma (mouse, dog, human) and brain (mouse) protein-binding was determined in vitro using equilibrium dialysis, as described previously elsewhere [12]. Unbound brain and plasma concentrations were derived by multiplying the total measured concentrations by the fraction unbound (free) determined in vitro.
Statistical methods used for nonclinical evaluations
Statistical significance was determined by ANOVA using Graphpad Prism software (version 6). If significant, a Tukey’s post hoc analysis was conducted to determine cross-group differences in the mouse PDAPP and dog PK/PD data. Significance was set as a p-value of < 0.05.
Clinical characterization of LY3202626
A clinical study (NCT02323334) of LY3202626 in healthy subjects was conducted in 4 parts: part A explored single-dose safety, tolerability, plasma pharmacokinetics/pharmacodynamics (PK/PD); part B examined single-dose CSF PK/PD; part C examined safety and tolerability following 14 days of daily dosing, as well as plasma and CSF PK/PD. Part D was intended to explore multiple daily doses in an AD population; but was not completed due to difficulty enrolling patients. Each part of the trial was randomized, placebo-controlled, subject- and investigator-blinded, and performed at a single site. Key inclusion criteria were that subjects be either healthy males or females of nonchildbearing potential, at least 20 years at screening with clinical laboratory test results within the normal range, BMI between 18.0 and 32.0 kg/m2, and no evidence of neuropsychiatric disease. In addition, for Parts B and C, patients were to have no abnormalities of the spine or medical history that would prevent them from receiving a lumbar puncture Additionally, part A included pilot evaluations of food effect on the PK of LY3202626 and the potential for CYP3A-mediated drug-drug interactions. The results from the food effect and drug-drug interactions, as well as the results from part D, were considered preliminary assessments and are not reported here. The dose range in Part A was selected to encompass the predicted exposures of LY3202626, based on PK/PD modelling, required to replicate the reductions in CSF Aβ observed in the dog studies. Because the intention of this study was to provide a first assessment of human PK, PD, and tolerability, a young and healthy subject population, typically used in Phase I drug development studies, was judged to be appropriate to achieve the study aims. All trials were conducted at PAREXEL Early Phase LA (Glendale, CA, USA), in compliance with the Declaration of Helsinki and International Conference on Harmonisation/Good Clinical Practice guidelines. Ethical review board approval was given by Aspire Institutional Review Board (Santee, CA, USA).
Single-dose escalations
Doses for the single-dose escalation portion of the study were selected using nonclinical data. Projected human clearance was estimated using the unbound fraction corrected intercept method of allometric scaling [13]. Dose calculations assumed that free plasma concentrations could be estimated using human plasma protein binding (measured in vitro) and that CSF:free plasma ratios would be similar between dogs and humans. The maximum dose in the Part A was chosen to ensure that safety and tolerability was determined at plasma exposures expected to produce human CSF exposures exceeding that associated with significant BACE1 inhibition in dog pharmacology experiments. In single-dose escalations, plasma PK and PD effects on plasma Aβ1–40 and Aβ1–42 concentrations were evaluated following single doses of placebo or 0.1 (n = 3 per group), 0.4, 1.6, 5, 15, or 45 mg LY3202626 (n = 8 per group). Plasma samples were obtained immediately before dosing and up to 120 h after dosing. For the 0.1 and 0.4 mg doses, plasma LY3202626 concentrations were generally below the limit of quantification. Accordingly, meaningful estimates of apparent terminal half-life (t1/2), area under the concentration curve (AUC) from 0 to infinity (AUC0∞) and associated PK parameters could not be obtained at these doses. Because LY3202626 concentrations at the 15 mg dose level were significantly lower than concentrations anticipated based on the otherwise linear dose by exposure relationship observed at other dose levels, the 15 mg dose was re-administered in the same cohort of patients.
The PK/PD of LY3202626 were subsequently assessed in CSF and plasma after administration of single doses of 1.6, 10, and 26 mg LY3202626 or placebo. Three subjects were assigned to receive placebo; 5 subjects were assigned to each group receiving LY3202626. Because of difficulties associated with CSF collection in the 10 mg group, an additional 2 subjects were included, for a total of 7 subjects. CSF sampling was conducted as previously described [6]. Serial CSF samples were collected through an indwelling subarachnoid lumbar catheter from 4 h before dosing up to 36 h after dosing. Any CSF sample that showed visual or laboratory evidence of blood contamination was excluded from the PK/PD and statistical analyses.
Multiple-dose assessments
In multiple-dose assessments, subjects (n = 12 per group) received daily administration of LY3202626 (1, 6, or 26 mg) or placebo for 14 days. Plasma samples were obtained following the first dose for up to 24 h after dose, before the last dose (day 14), and for up to 120 h after dosing on day 14. Additionally, every 2 days, plasma samples were collected before dosing to assess the time to reach steady-state. CSF was sampled by a single, lumbar puncture performed before dosing and again approximately 24 h after administration of the last dose of LY3202626 or placebo on day 14. At each lumbar puncture, a maximum of 25 mL of CSF was collected to determine Aβ1–40, Aβ1–42, sAβPPα, sAβPPβ, and LY3202626 concentrations.
Analysis of clinical samples
Plasma Aβ1 – 40 and Aβ1 – 42 were simultaneously measured using a validated modification of the INNO-BIA plasma Aβ forms assay run (Fujirebio, Ghent, Belgium) on a xMAP platform (Luminex, Austin, TX, USA), as previously reported [14]. CSF Aβ1 – 40 and Aβ1 – 42 were analyzed using validated INNOTEST® β-AMYLOID (1 –40) and INNOTEST® β-AMYLOID (1 –42) ELISA kits (Fujirebio, Ghent, Belgium), respectively, as previously reported [15]. CSF sAβPPα and sAβPPβ were measured using a validated immunoassay by Meso Scale Discovery (Rockville, MD, USA), as previously reported [6]. Human plasma and CSF samples were analyzed for LY3202626 by using validated LC-MS/MS detection methods by Covance Laboratories (Indianapolis, IN, USA).
Pharmacokinetic analysis. PK parameter estimates for LY3202626 were calculated by standard noncompartmental methods of analysis using Phoenix WinNonlin (version 6.4). Clearance and apparent volume of distribution were reported for all subjects. Time of maximum concentration (tmax) and apparent terminal half-life (t1/2) were also reported. The accumulation ratio at steady state was calculated by dividing the AUC-versus-time period over a single dosing interval (AUCτ) at steady state (AUCτ,ss) by AUCτ after the initial dose for each subject.
Pharmacodynamic analysis. For each subject with serially determined concentrations of plasma or CSF Aβ1–40 and Aβ1–42, the time of the lowest observed concentration (Cnadir) was reported, along with change from baseline at Cnadir and the 24 h time-averaged change from baseline following dosing. Steady-state CSF Aβ1–40, Aβ1–42, sAβPPα, and sAβPPβ concentrations were expressed as a percentage change from baseline. Summary statistics were calculated for each PD parameter.
Safety analysis
For all studies, safety was assessed by recording adverse events, physical, neurologic, and ophthalmologic examinations, vital signs, electrocardiograms, and results of clinical safety laboratory tests.
Statistical analysis
PK/PD analyses were conducted on the full analysis set. This set includes all data from all randomized subjects receiving at least 1 dose of the investigational product according to the treatment the subjects actually received. Safety analyses were conducted for all enrolled subjects, whether or not they completed all protocol requirements.
Summary statistics were performed for all PK/PD parameters. No formal statistical comparisons were conducted for PK/PD parameters between dose groups. All tests of treatment effects in the safety analyses were conducted at a 2-sided alpha level of 0.10 (0.05 each side), unless otherwise stated.
RESULTS
Nonclinical characterization of LY3202626
In vitro potency and selectivity of LY3202626
BACE1 and related aspartyl protease inhibition. LY3202626 was evaluated for in vitro potency against recombinant human BACE1 using a small synthetic (methylcourmarine; mca) FRET substrate (Table 1). LY3202626 exhibited potent human BACE1 inhibition with an IC50 of 0.615 nM (SD 0.101 nM; n = 5). Against human BACE2, LY3202626 demonstrated an IC50 of 0.871 nM (SD 0.241 nM; n = 6). In a broader assessment of activity against other related aspartyl proteases, such as cathepsin D, pepsin, and renin, LY3202626 did not appreciably inhibit these proteases (IC50 > 14,000 nM). Taken together, these data indicate that LY3202626 is a potent BACE1 and BACE2 inhibitor with selectivity against cathepsin D, pepsin, and renin.
Table 1
Potency of LY2886721 in BACE1 and other Aspartyl Proteases and potency of LY3202626 in cellular assays
| Aspartyl Protease Assays Absolute IC50, nM (SD) | ||||||
| hBACE1 mca | hBACE2 mca | Cathepsin D | Pepsin FRET | Renin FRET | ||
| FRET Assay | FRET Assay | FRET Assay | FRET | Assay | ||
| n = 5 | n = 6 | n = 4 | n = 2 | n = 1/2 | ||
| 0.615 (0.101) | 0.871 (0.241) | 64,500 (12,800) | 77,400 (37,600) | 14,100 (ND) | ||
| Cellular Assays Relative IC50, nM (SD) | ||||||
| PDAPP 1° | PDAPP 1° | PDAPP 1° | ||||
| Neuron | Neuron | Neuron | ||||
| Aβ1–40 | Aβ1–42 | Cytotox | ||||
| n = 4 | n = 3/6 | n = 1/7 | ||||
| 0.275 (0.176) | 0.228 (0.244) | > 100,000 | ||||
FRET, Fluorescence Resonance Energy Transfer; hBACE1, Human recombinant Beta-APP Cleaving Enzyme1; hBACE2, Human recombinant Beta-APP Cleaving Enzyme 2; HEK293Swe, Human embryonic kidney cell line transfected with APP encoding a Swedish mutation; IC50, Concentration at which enzyme activity is inhibited by 50%; mca, methylcoumarin; ND, not determined; SD, standard deviation.
Mouse primary neuronal cultures. In primary cortical neuronal cultures from embryonic PDAPP mice (containing the Indiana mutation APPV717F), a 24 h exposure to LY3202626 produced a concentration-dependent decrease in Aβ secretion with an EC50 of around 0.275 nM (SD 0.176 nM) for Aβ1–40 and 0.228 nM (SD 0.244 nM) for Aβ1–42. Assessment of PDAPP neuronal cytotoxicity indicated that the LY3202626 EC50 for inducing cytotoxicity is > 100,000 nM. Results used the geometric mean and were based upon at least 3 independent studies. These data indicate potent inhibition of native murine BACE1 within the context of an intact cellular system.
In vivo pharmacology of LY3202626 in PDAPP mice
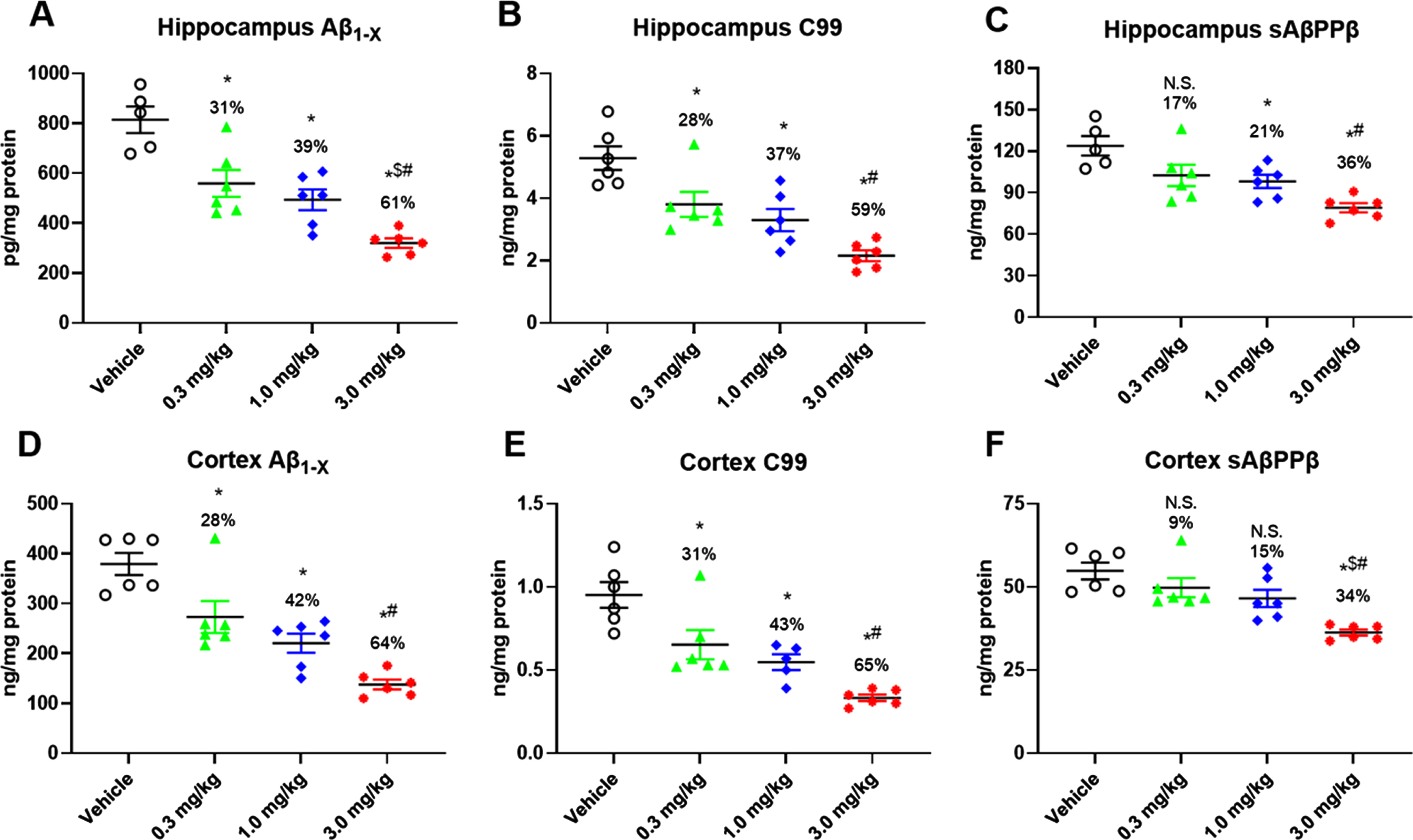
The in vivo PD effects of LY3202626 were assessed 3 h following a single oral administration of vehicle (7% Pharmasolve) or 0.3, 1.0, or 3.0 mg/kg LY3202626 (n = 6–8 per group). All 3 doses of LY3202626 significantly reduced Aβ1hboxx in a dose-dependent manner (Fig. 1A, D). These PD effects occurred at relatively low brain exposures; measured concentrations in brain homogenate were 28.1, 38.1, and 110.2 nM at 0.3, 1.0, and 3.0 mg/kg doses, respectively. Based on a free fraction of 0.026 (determined in brain homogenate), these concentrations equated to approximately 0.7 to 3.0 nM free LY3202626 in brain tissue. These free brain exposures exceeded the in vitro human BACE1 IC50 (0.615 nM) by 1.2–to 4.6-fold. Consistent with downstream effects of BACE1 inhibition, C99 (Fig. 1B, E) and sAβPPβ (Fig. 1C, F) levels in the brain were reduced by LY3202626 treatment. Cortical C99 levels were significantly reduced by all LY3202626 doses, with a maximum decrease of 65% at the 3.0 mg/kg dose (Fig. 1E), whereas decreases in sAβPPβ levels were less pronounced, with a 34% decrease produced at the 3.0 mg/kg dose (Fig. 1F).
Fig. 1
Pharmacologic effects in vivo of oral administration of LY3202626. Young PDAPP mice (n = 6–8 per group) were treated orally by gavage with increasing doses of LY3202626 or vehicle, and hippocampal Aβ1hboxx (A), C99 (B), and sAβPPβ (C) as well as cortical Aβ1hboxx (D), C99 (E), and sAβPPβ (F) levels were determined from brain extracts obtained 3 h after dosing. LY2886721 produced dose-dependent decreases in all amyloid-β protein precursor-related pharmacodynamic markers of BACE1 inhibition in PDAPP mice. All ANOVA p-values were < 0.0001; Tukey post hoc analysis, *p < 0.05 versus vehicle control, $p < 0.05 versus 0.3 mg/kg, #p < 0.05 versus 1.0 mg/kg. p < 0.01 versus vehicle control, ANOVA/Dunnett’s post hoc analysis. Aβ, amyloid-β peptide; sAβPPβ, secreted amyloid-β protein precursor beta.
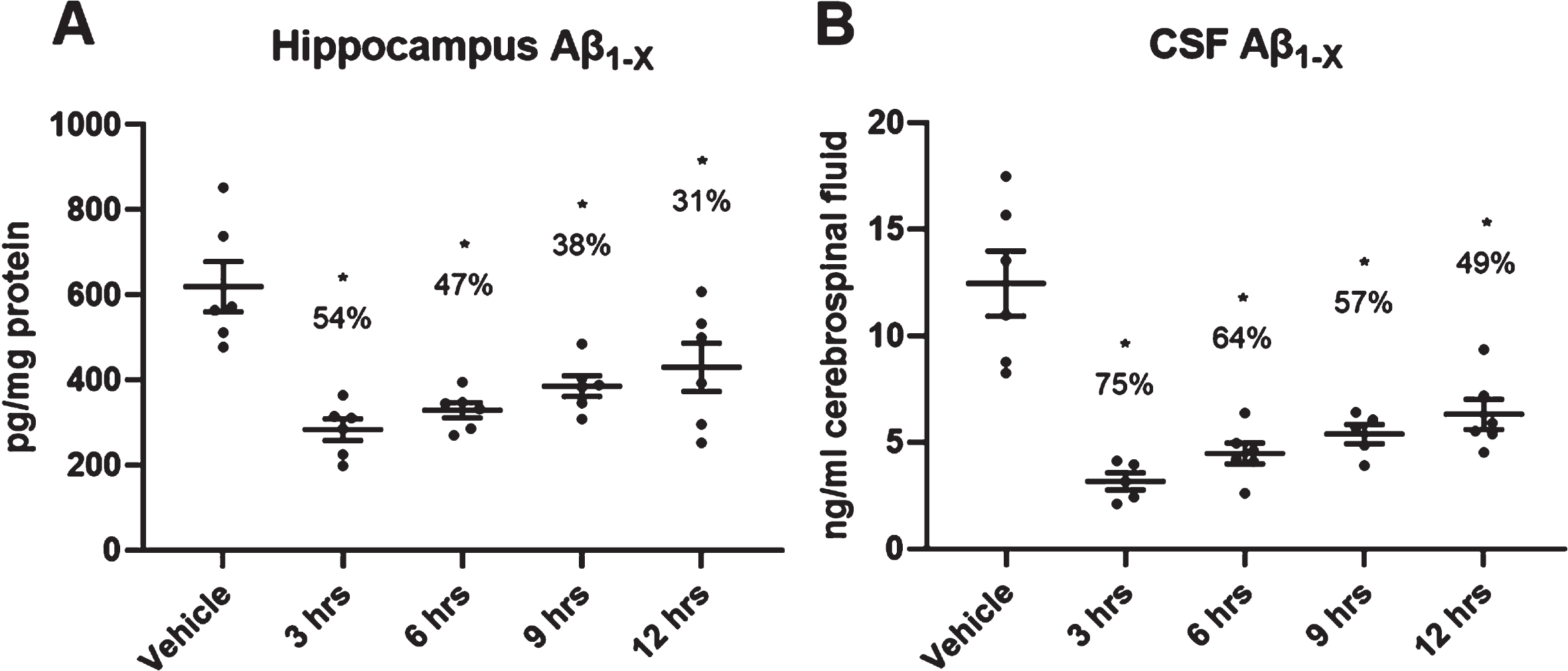
In a time-course experiment following a single oral dose of 3.0 mg/kg LY3202626, cortical and hippocampal Aβ1hboxx levels were significantly suppressed from 3 to 12 h post-dose (Fig. 2A). Moreover, CSF levels of Aβ1hboxx were reduced by 75% at 3 h post-dose and remained significantly lower even at 12 h post-dose relative to the 3 h vehicle control (Fig. 2B).
Fig. 2
Duration of pharmacodynamic effects of LY3202626·HCl in the hippocampus of PDAPP mice. PDAPP mice (n = 6 per group) were treated orally by gavage with LY3202626 (3.0 mg/kg) or vehicle, and hippocampal (A) or cerobrospinal fluid (CSF) (B) Aβ1hboxx levels were measured 3, 6, 9, and 12 h after treatment. Aβ1hboxx levels in the hippocampus and CSF were significantly suppressed from 3 to 12 h post-dose. Hippocampus: F(4,25)=10.13, p < 0.0001 (ANOVA); CSF: F(4,23)=17.24, p < 0.0001 (ANOVA). Tukey post hoc analysis, *p < 0.05 versus vehicle control; time points did not differ from one another. Aβ1hboxx, amyloid-β peptide 1-x.
In vivo central pharmacology of LY3202626 in beagle dogs
Plasma exposure, CSF exposure, and central PD effects of LY3202626 were evaluated in cannulated beagle dogs (n = 6). LY3202626 (1.5 mg/kg) was dosed orally, and plasma and CSF samples were collected over a 48 h period. Robust suppression of plasma Aβ1hboxx levels (up to 80%) occurred by 2 h after dosing, and Aβ1hboxx was still reduced by approximately 40% at the 48 h time point (Fig. 3). Reduction of Aβ1hboxx levels was delayed in CSF relative to plasma, with maximal reductions of Aβ1hboxx (approximately 80%) occurring at the 9 h time point; CSF Aβ1hboxx remained significantly suppressed for 24 h after dosing. Following the 1.5 mg/kg oral dose, the mean±SD plasma AUC(0-∞) was 10812±5195 nM·h and the mean±SD maximum plasma concentration (Cmax) was 2989±750 nM. Given a measured dog plasma free fraction of 0.009 (determined by equilibrium dialysis of dog plasma), free plasma concentrations of LY3202626 exceeded the BACE1 IC50 (0.615 nM) for 12 h after dosing. LY3202626 concentrations were quantifiable in CSF at the 3, 6, and 9 h timepoints. The mean CSF AUC (3–9 h) was 25 nM*h and the mean free plasma AUC (3–9 h) was 28 nM*h, suggesting free penetration of LY3202626 across the blood-brain barrier.
Fig. 3
Peripheral and central pharmacodynamic effects of LY3202626 in beagle dogs after a single 1.5 mg/kg dose. Baseline plasma and CSF samples were collected from cannulated beagle dogs (n = 6) before and after dosing with 1.5 mg/kg LY3202626 at various times and stored for analysis; Aβ1hboxx levels are measured in plasma and CSF and averaged across dogs. Baseline plasma Aβ1hboxx was 248±17 pg/mL and baseline CSF Aβ1hboxx was 6.6±1.3 ng/mL (Mean±SEM). LY3202626 produced robust and time-dependent decreases in Aβ1hboxx in both plasma and CSF of dog relative to baseline. Plasma: F(2.40,11.76)=58.02, p < 0.0001. CSF: F(2.38,10.93)=11.79, p = 0.001. Tukey post hoc comparisons: *p < 0.05 versus Time 0 (baseline), #p < 0.05 versus 48 h time point, $ p < 0.05 versus all time points except baseline. Notes: 1) Because time points were not uniform between CSF and plasma, separate one-way repeated-measures ANOVAs were conducted, 2) analysis was conducted on raw data, but to compare Aβ1hboxx levels between CSF and plasma, the figure reflects change from baseline, and 3) two collection time points were missing in the CSF data set.
Clinical characterization of LY3202626 in healthy volunteers
A total of 94 subjects were enrolled in the study: 36 subjects in part A, 20 subjects in part B, and 36 subjects in part C (2 additional subjects were recruited for Part D – their data are not reported here). Subject characteristics for each part are shown in Table 2.
Table 2
Subject characteristics by study
| Part A (n = 36) | Part B (n = 20) | Part C (n = 36) | |
| Age, mean y (min-max) | 36.3 (21–61) | 38.7 (23–65) | 39.5 (24–65) |
| Sex, % male | 83.3% | 100.0% | 94.4% |
| Weight, mean kg, (SD) | 74.3 (14.8) | 80.1 (9.6) | 81.5 (14.3) |
| Race (%) | |||
| Asian (Japanese) | 44.4% | 0% | 33.3% |
| Black | 19.4% | 20.0% | 19.4% |
| White | 33.3% | 80.0% | 44.4% |
| Mixed | 2.8% | 0% | 2.8% |
Subjects in Part A were divided into 3 subcohorts of 12 subjects each, with 2 of these subcohorts participating in the dose escalation portion of Part A. The third subcohort participated in the food-effect portion of the trial, which is not reported here. Part B was divided into 3 subcohorts (numbers in each subcohort are not equal due to the need for replacement subjects). Part C was divided into 3 subcohorts of 12 subjects each. MAD, multiple ascending dose; SAD, single ascending dose.
Pharmacokinetics
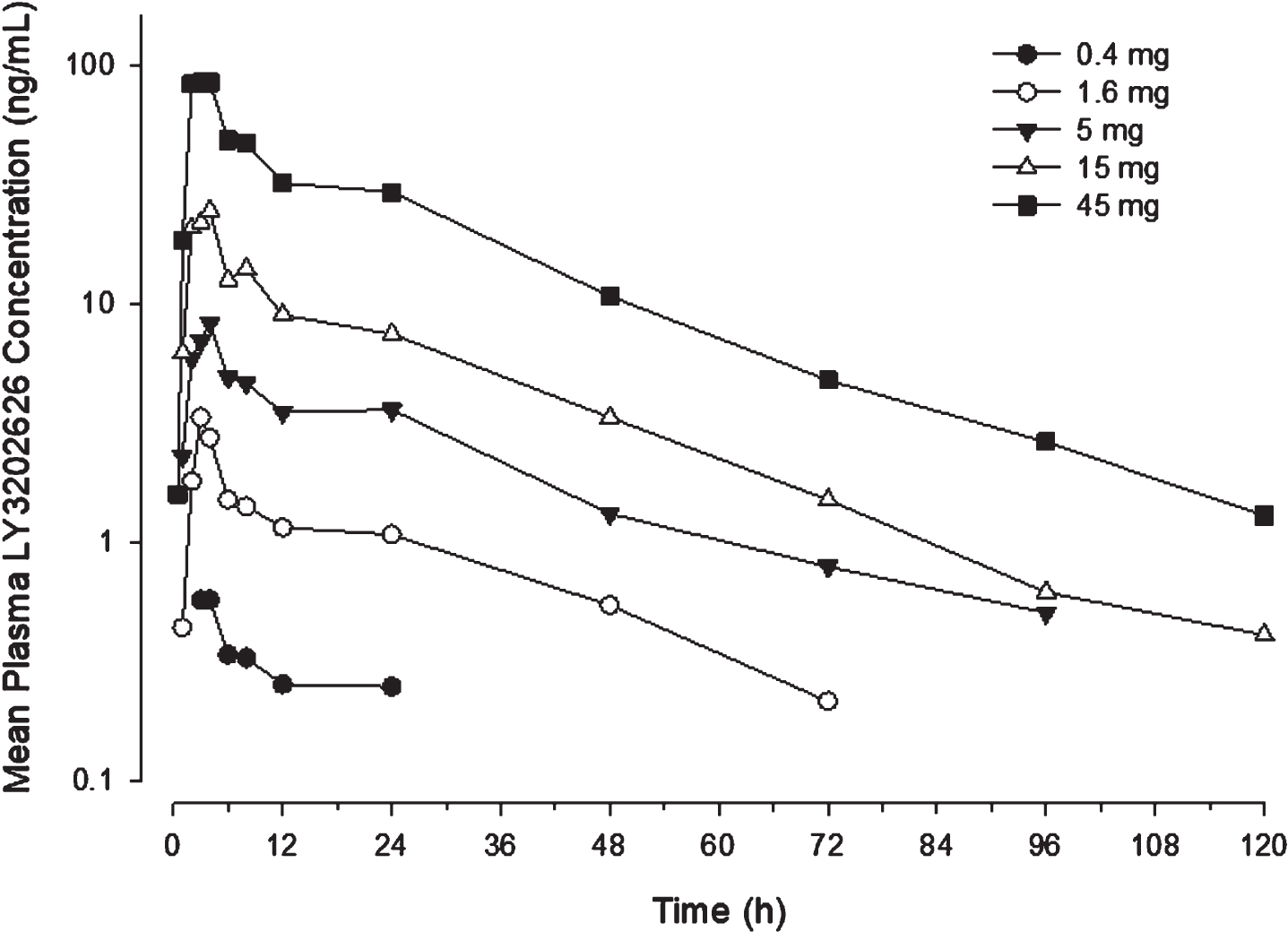
Single-dose pharmacokinetics. Following oral administration, LY3202626 concentrations rose until reaching Cmax approximately 2.5 to 4 h after dosing. After Cmax, LY3202626 concentrations fell in a generally bi-exponential manner, with a terminal t1/2 of approximately 20 h. Notably, there was substantial variability in PK parameters, with the coefficients of variation (CV) in Cmax and AUC ranging up to 84% at those dose levels where exposure parameters could be reliably quantified (Table 3). Renal clearance was approximately 1 L/h across dose levels. Apparent oral clearance was approximately 26 L/h across dose levels (with the exception of the 15 mg dose level), suggesting that the PK of LY3202626 were dose proportional.
Table 3
Pharmacokinetic parameters of LY3202626 following single or multiple doses assessed in the plasma
| LY3202626 Geometric Mean (CV%) | |||||||||
| Single Dose (Part A) | Multiple Dose (Part C)a | ||||||||
| 0.1 mg (n = 3) | 0.4 mg (n = 8) | 1.6 mg (n = 8) | 5 mg (n = 8) | 15 mg (n = 8) | 45 mg (n = 8) | 1 mg (n = 8) | 6 mg (n = 9) | 26 mg (n = 9) | |
| Cmax (ng/mL) | 0.169 (50) | 0.486 (81) | 2.89 (70) | 7.91 (56) | 21.3 (77) | 92.5 (47) | 2.57 (25) | 11.2 (71) | 72.8 (61) |
| tmax (h)b | 3.08 (3.00–6.00) | 3.55 (3.00–8.00) | 3.00 (3.00–4.00) | 4.03 (2.90–24.12) | 3.01 (1.98–4.00) | 2.54 (2.00–4.07) | 4.04 (3.98–4.23) | 3.87 (1.95–4.00) | 4.03 (2.00–5.78) |
| t1/2 (h) | NC | NC | 23.0 (21) | 19.8 (42) | 19.5 (25) | 21.5 (19) | 33.1 (38) | 38.1 (29) | 46.3 (25) |
| AUC (0∞) c (ng·h/mL) | NC | NC | 60.6 (49) | 197 (38) | 393 (84) | 1680 (34) | 38.6 (25) | 151 (54) | 1020 (50) |
| CLR, L/h | NA | NA | NA | 1.09 (64) | 0.829 (37) | 1.03 (53) | 1.61 (46) | 1.09 (86) | 1.29 (39) |
aParameters from part C are associated with steady state (observed on day 14), bMedian (range), cAUCτ,ss is reported for the Part C doses. Pharmacokinetic parameters from each dose level in Parts A and C of the clinical study are provided. While single doses of 15 mg were administered on 2 different occasions due to lower-than-anticipated exposures on the first occasion, only the first occasion is shown here, as there were no meaningful differences in exposure on the two occasions. AUC (0 - ∞) , area under the concentration-time profile, extrapolated to infinity; AUCτ,ss, AUC during 1 dosing interval at steady state; Cmax, maximum observed drug concentration; CLR, renal clearance; CV, coefficient of variation; N, number of subjects; NA, not applicable; NC, not calculated, as there were not enough data to characterize the elimination phase; t1/2, half-life associated with the terminal rate constant in noncompartmental analysis; tmax, time of Cmax.
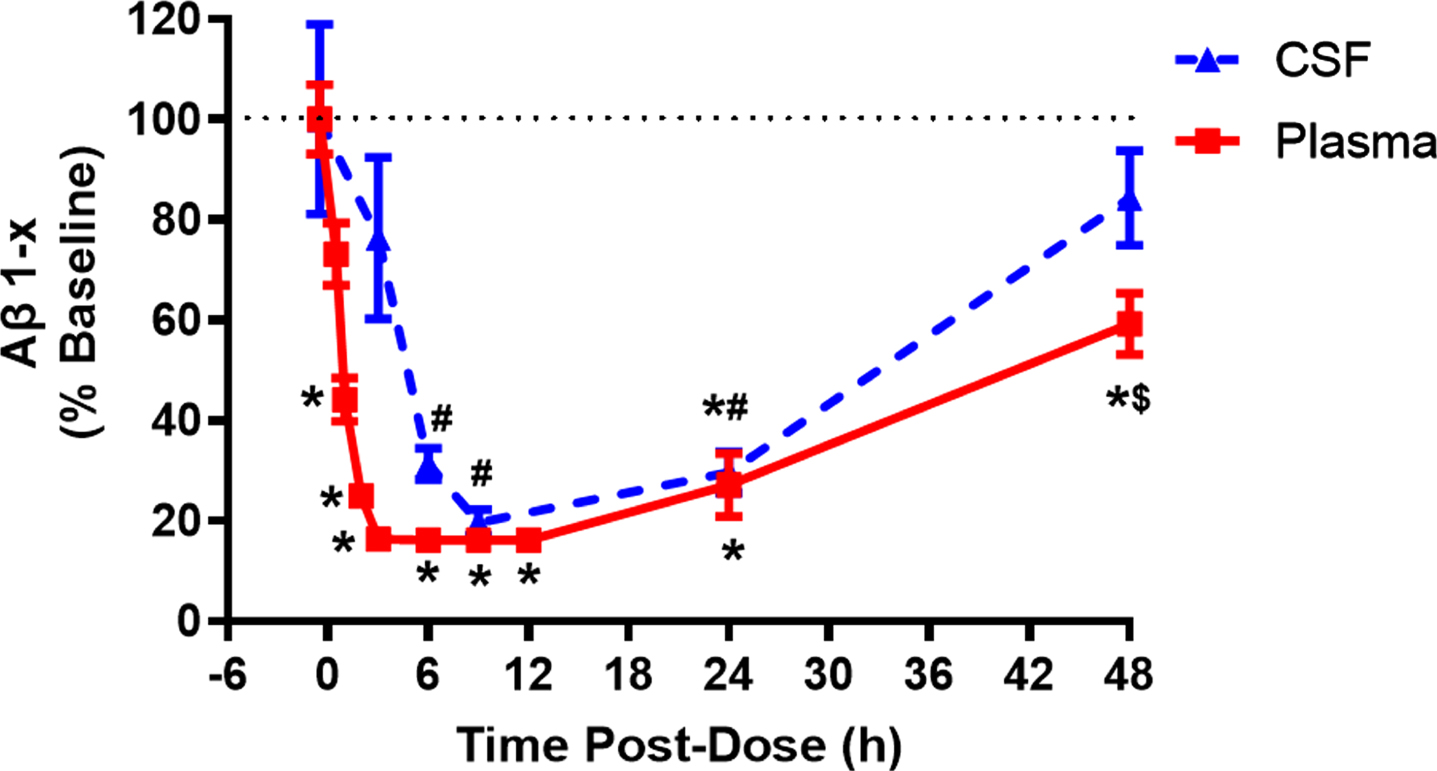
Two notable observations during the single-dose portion of the trial were lower-than-expected exposures following administration of the 15 mg dose and increases in plasma concentrations at 24 h following dosing (not consistent with the overall PK profile, and exceeding concentrations at the 12 h time point in some patients) at all dose levels (Fig. 4). When the 15 mg dose level was repeated in the same cohort of subjects, exposures remained low relative to the otherwise linear exposures observed at other doses. To explore the possibility that the concentration elevations at 24 h were associated with enterohepatic recirculation, additional PK samples were collected before and after the 24 h time point for the cohort receiving the repeated dose. However, the results of the additional PK sampling were inconclusive, with the increase in LY3202626 concentrations not having any apparent correlation with time of breakfast (i.e., the meal closest to the 24 h time point).
Fig. 4
Plasma LY3202626 concentrations following single doses of 0.4 mg to 45 mg of LY3202626 in healthy subjects. Subjects received a single dose of 0.4, 1.6, 5, 15, or 45 mg LY3202626 (n = 8 per dose). Samples were drawn at times up to 120 h after dosing. The time of maximum concentration generally occurred 2.5 to 4 h after dosing. The shape of the concentration-time profile following absorption was generally bi-exponential, with a secondary peak occurring in 2–5 patients per dose group at approximately 24 h after dosing.
In CSF, maximum concentrations were achieved approximately 6 h after dosing. Due to the relatively short sampling window, a t1/2 in CSF could not be determined. Comparison of CSF:plasma AUC0–24 values afforded ratios ranging between 0.0278 and 0.0428, which were similar to the plasma-free fraction and therefore suggestive of free penetration of LY3202626 across the blood-brain barrier (Supplementary Table 1).
Multiple-dose pharmacokinetics. Steady state was achieved approximately 4 to 8 days after the initiation of daily dosing, based on a comparison of trough concentrations. On day 14, median Cmax was achieved at approximately the same time as on day 1. Variability in exposure estimates was high, and similar to what was observed after single doses. The t1/2 was higher at steady state than following single doses, ranging from 33.1 to 46.3 h across dose levels. The accumulation ratio, based on AUC, was approximately 2.8. There was no apparent difference in clearance or volume of distribution between Japanese and non-Japanese subjects. The ratio of CSF-to-plasma LY3202626 concentrations were generally consistent with the values anticipated from the single-dose data, with mean values ranging from 0.0400 to 0.0514, further indicating free penetration of LY3202626 across the blood-brain barrier.
Pharmacodynamics
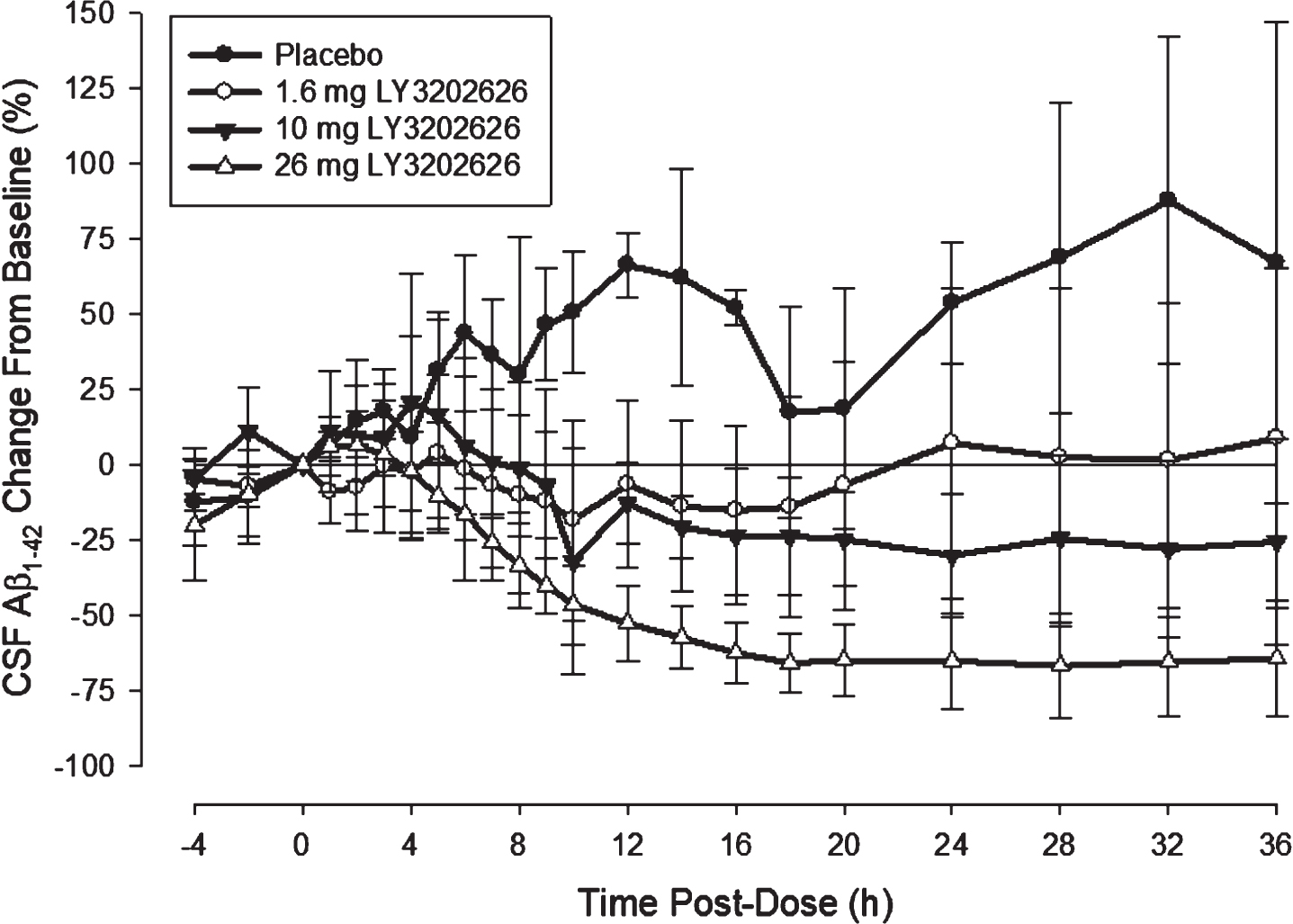
Single-dose CSF A β. Two subjects were excluded from the analysis due to contamination of CSF samples with blood or inability to collect samples. Following administration of LY3202626, Aβ concentrations tended to increase for approximately 2 to 4 h in a fashion similar to subjects administered placebo, at which point both Aβ1–40 and Aβ1–42 isoforms began to decline (Fig. 5). Reductions in CSF Aβ became more substantial with increasing doses of LY3202626. At the 26 mg dose level, neither Aβ isoform had returned to baseline within the 36 h observation period. The time at which nadir concentrations were observed tended to increase with higher doses, ranging from approximately 8 h at 1.6 mg to approximately 28 h at 26 mg (Fig. 5).
Fig. 5
CSF levels of Aβ1–42 after a single doses of LY3202626 in healthy subjects. CSF concentrations of Aβ1–42 decreased in a dose-dependent manner after a single dose administration of LY3202626 in healthy subjects (n = 5 per LY3202626-treated group, n = 3 for placebo). The mean nadir increased with increasing dose, occurring at approximately 10 h for the 1.6 mg dose, and at approximately 28 h for the 26 mg dose. At nadir, the mean reduction from baseline was –33.4 (standard deviation = 13.4%) at the 1.6 mg dose level and –71.6 (10.4%) at the 26 mg dose level. At the 10 mg and 26 mg dose levels, concentrations did not return to baseline over the 36 h observation period. Aβ1–42, amyloid-β 1–42.
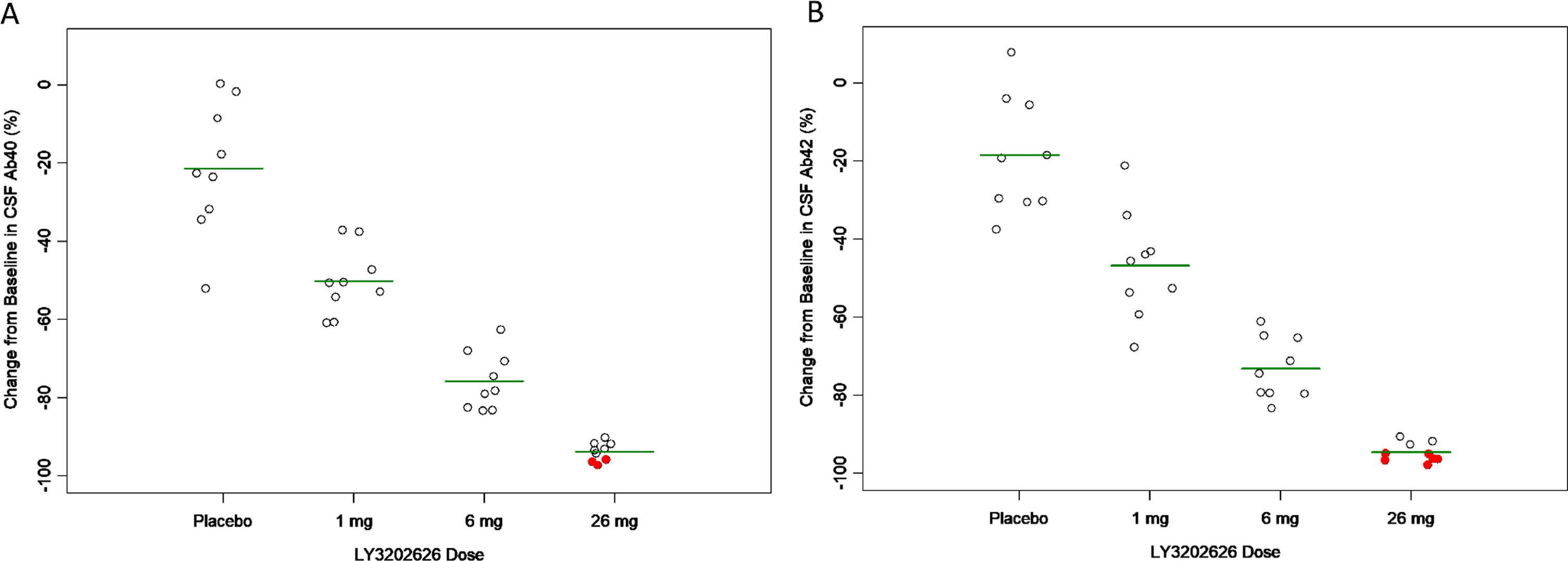
Multiple-dose CSF A β. Following 14 days of daily dosing of LY3202626, CSF concentrations of Aβ1–40 and Aβ1–42 were decreased relative to placebo in a dose-dependent manner (Fig. 6A, B). Concentrations of Aβ1–40 were reduced by 75.7% (7.38%) at the 6 mg dose level, while Aβ1–42 was reduced by 73.1% (7.96%). At the 26 mg dose level, 3 Aβ1–40 samples (33%) and 6 Aβ1–42 samples (67%) were below the lower limit of quantification (LLOQ) (7.81 pg/mL for Aβ1–40, 29.3 pg/mL for Aβ1–42). Assigning a value of one-half the LLOQ to those samples below the assay limits, the reduction in Aβ1–40 and Aβ1–42 was estimated to be approximately 94% at 26 mg.
Fig. 6
Change from baseline in CSF Aβ1–40 (A) and Aβ1–42 (B) observed 24 h following final dose in clinical study Part C. Subjects (n = 9 per group) received daily doses of LY3202626 for 14 days. On day 15, 24 h ± 4 h after the last dose of LY3202626 on day 14, CSF samples were collected for measurement of Aβ1–40 and Aβ1–42. Values are expressed as the percentage change from baseline levels. Horizontal lines indicate the mean reduction at each dose level, while red (solid) circles indicate that the concentration on day 15 was below the limit of quantification (a value of half the lower-limit of quantification was assigned to calculate change from baseline in these cases). As the dose increased, increasing reductions in CSF Aβ1–40 and Aβ1–42 were observed. At the 1 mg dose, the mean level of decrease in CSF Aβ1–40 and Aβ1–42, was approximately 50% of baseline levels. At the 6 mg dose, the decrease was approximately 74% (all p < 0.001, as determined by ANCOVA analysis). Aβ1–40, amyloid-β peptide 1–40; Aβ1–42, amyloid-β peptide 1–42.
Multiple-dose CSF sA βPP α and sA βPP β. To examine LY3202626 target engagement and the indirect and direct impact of BACE inhibition on AβPP processing, concentrations of CSF sAβPPα and sAβPPβ were measured after repeated daily dosing for 14 days. There was a clear dose-response relationship from placebo to 26 mg daily dosing for the mean change from baseline in CSF concentrations of sAβPPα and sAβPPβ. At the highest dose administered, sAβPPα increased by 111% (SD = 53.0%), while sAβPPβ was reduced by 92.7% (SD = 2.26%).
Safety
There were no serious adverse events reported for LY3202626 administered up to a single dose of 45 mg or multiple doses of 26 mg. In addition, no subject withdrew from the study because of an adverse event. Overall, the most common adverse events after administration of LY3202626 were post-lumbar puncture syndrome, discolored feces, headache, and dizziness. The adverse event profile after administration of multiple doses of LY3202626 is shown in Table 4. The post-lumbar puncture syndrome adverse events were procedural adverse events related to the CSF sampling procedure in parts B, C, and D of the study. The discolored feces adverse events were attributable to the color of the excipients in the formulation of LY3202626 that was administered in the study. There were two adverse events deemed treatment-related. One subject experienced orthostatic hypotension approximately 8 h after receiving a single 10 mg LY3202626 dose in part A of the study. The orthostatic hypotension was asymptomatic, lasted approximately 15 min, and did not occur at other time points. Another subject experienced postural orthostatic tachycardia syndrome approximately 6 h after receiving the first 26 mg LY3202626 dose in a multiple-dosing cohort in part C. The adverse event lasted approximately 2 h and the subject did not experience another episode of postural orthostatic tachycardia syndrome after administration of 26 mg LY3202626 for the remaining duration of the multiple-dosing period. Overall, there was no dose-dependence in severity or incidence of adverse events, with most adverse events being mild in severity. There were no clinically significant findings in clinical laboratory tests, including liver functions tests (alanine aminotransferase, aspartate aminotransferase, bilirubin, Table 5). There were also no clinically significant changes in electrocardiograms (including QTc interval), ophthalmologic examinations, or assessments of alertness and attention. Any changes in vital signs were unremarkable, aside from the orthostatic-related cases described above. LY3202626 was well tolerated in the AD subjects in part D of the study, with only 1 adverse event of post-lumbar puncture syndrome reported. There were no apparent differences in the safety profile of LY3202626 between Japanese and non-Japanese subjects.
Table 4
Adverse event profile after administration of multiple doses of LY3202626
| Number of Adverse Events (number of subjects with adverse event) | |||||
| Adverse Event | Placebo (N = 9) | 1 mg (N = 9) | 6 mg (N = 9) | 26 mg (n = 9) | Overall (N = 36) |
| Feces discoloured | 5 (5) | 0 (0) | 2 (2) | 9 (9) | 16 (16) |
| Dizziness | 0 (0) | 0 (0) | 0 (0) | 2 (2) | 2 (2) |
| Postural orthostatic tachycardia syndrome | 0 (0) | 0 (0) | 0 (0) | 1 (1) | 1 (1) |
| Constipation | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 1 (1) |
| Post-lumbar puncture syndrome | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 1 (1) |
| Sunburn | 1 (1) | 0 (0) | 0 (0) | 0 (0) | 1 (1) |
| Burning sensation | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 1 (1) |
| Dysgeusia | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 1 (1) |
| Headache | 1 (1) | 0 (0) | 0 (0) | 0 (0) | 1 (1) |
| Memory impairment | 0 (0) | 0 (0) | 0 (0) | 1 (1) | 1 (1) |
| Abnormal dreams | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 1 (1) |
| Nightmare | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 1 (1) |
Adverse event, treatment-emergent adverse events related to study drug, by treatment, in order of frequency.
Table 5
Liver safety data over time following treatment with LY3202626 or placebo
| Day 11 | Day 2 | Day 3 | Day 4 | Day 6 | Day 8 | Day 10 | Day 12 | Day 14 | Day 15 | Day 17 | ||
| Placebo | ALT, U/L | 23. 7 (13.4) | 22.9 (9.7) | 22.2 (8.6) | 24.1 (9.2) | 23.0 (9.0) | 21.3 (7.3) | 20.6 (8.3) | 19.7 (8.7) | 19.8 (7.1) | 20.2 (6.5) | 24.0 (10.3) |
| AST, U/L | 18.8 (4.2) | 18.9 (4.9) | 17.6 (4.3) | 19.2 (6.2) | 19.2 (6.5) | 18.9 (6.0) | 20.2 (11.4) | 20.3 (10.6) | 16.9 (3.3) | 18.0 (5.3) | 19.6 (6.2) | |
| ALP, U/L | 61.7 (15.9) | 63.3 (13.9) | 62.4 (15.6) | 63.2 (14.8) | 61.0 (13.7) | 61.4 (12.7) | 60.7 (14.6) | 61.6 (12.9) | 57.9 (16.3) | 57.4 (17.1) | 61.8 (15.1) | |
| GGT, U/L | 21.3 (9.2) | 21.9 (7.9) | 21.4 (8.1) | 21.8 (9.0) | 20.1 (8.5) | 20.3 (7.5) | 19.2 (7.5) | 19.7 (6.6) | 18.7 (6.7) | 19.0 (6.1) | 21.0 (7.6) | |
| Total Bilirubin, umol/L | 9.3 (2.8) | 10.4 (4.6) | 8.4 (2.9) | 7.8 (5.0) | 9.1 (7.8) | 6.6 (2.8) | 7.6 (2.4) | 6.7 (2.6) | 8.2 (2.9) | 8.1 (2.6) | 8.2 (3.8) | |
| Albumin, g/L | 44.2 (3.1) | 45.4 (2.5) | 45.2 (2.3) | 46.1 (2.1) | 44.4 (2.9) | 45.9 (2.0) | 44.7 (2.0) | 45.4 (2.8) | 43.4 (2.7) | 43.8 (2.5) | 46.8 (1.9) | |
| LY3202626, 1 mg | ALT, U/L | 19.0 (11.8) | 18.9 (11.8) | 18.7 (10.6) | 20.4 (10.1) | 18.9 (7.0) | 20.8 (8.4) | 24.6 (19.6) | 24.2 (21.2) | 22.8 (17.2) | 20.8 (15.5) | 23.2 (12.8) |
| AST, U/L | 18.4 (6.7) | 18.4 (6.5) | 16.1 (4.7) | 19.6 (5.5) | 17.7 (4.8) | 21.3 (13.2) | 30.2 (39.0) | 26.2 (29.7) | 20.2 (13.1) | 18.3 (9.7) | 19.3 (6.0) | |
| ALP, U/L | 62.8 (7.6) | 62.2 (8.7) | 62.3 (8.4) | 64.7 (10.7) | 61.0 (10.9) | 62.4 (11.3) | 62.6 (10.0) | 62.1 (9.7) | 61.3 (10.6) | 59.6 (9.6) | 63.9 (10.6) | |
| GGT, U/L | 22.3 (14.8) | 21.8 (15.6) | 20.9 (13.9) | 21.4 (14.5) | 19.4 (11.8) | 18.8 (11.5) | 18.7 (11.3) | 18.0 (9.9) | 18.0 (10.1) | 18.2 (11.0) | 20.1 (11.8) | |
| Total Bilirubin, umol/L | 10.7 (2.8) | 10.7 (2.6) | 8.9 (2.3) | 7.4 (4.2) | 8.5 (4.5) | 7.9 (5.0) | 7.2 (2.1) | 8.0 (2.8) | 9.1 (2.7) | 9.2 (2.5) | 8.2 (4.0) | |
| Albumin, g/L | 43.9 (1.5) | 43.8 (3.0) | 43.1 (2.4) | 44.0 (2.4) | 43.1 (2.4) | 44.1 (2.0) | 43.3 (2.2) | 43.2 (1.5) | 43.6 (2.6) | 42.6 (2.2) | 46.0 (2.8) | |
| LY3202626, 6 mg | ALT, U/L | 17.6 (5.5) | 17.8 (3.8) | 18.9 (4.7) | 20.9 (5.2) | 18.9 (4.4) | 18.6 (2.5) | 17.9 (2.3) | 16.9 (2.5) | 15.2 (2.5) | 16.1 (2.9) | 19.8 (4.3) |
| AST, U/L | 18.9 (4.0) | 18.0 (3.0) | 17.8 (2.9) | 19.8 (4.7) | 19.7 (4.9) | 20.8 (6.2) | 19.6 (6.0) | 17.2 (3.8) | 15.3 (4.3) | 15.9 (3.9) | 18.9 (3.5) | |
| ALP, U/L | 64.6 (16.9) | 66.4 (20.2) | 65.8 (18.6) | 67.8 (18.4) | 63.9 (17.8) | 64.6 (17.6) | 62.9 (15.4) | 63.6 (17.2) | 62.2 (16.0) | 58.9 (14.3) | 65.0 (18.7) | |
| GGT, U/L | 21.6 (10.7) | 21.7 (9.5) | 22.3 (11.2) | 24.0 (11.0) | 22.0 (10.8) | 21.7 (10.8) | 20.6 (8.7) | 20.3 (8.2) | 19.3 (10.3) | 18.6 (8.6) | 20.9 (9.7) | |
| Total Bilirubin, umol/L | 8.9 (3.6) | 9.8 (4.5) | 7.4 (3.1) | 6.9 (2.4) | 8.4 (4.0) | 8.1 (2.8) | 8.2 (2.7) | 7.4 (2.4) | 8.1 (5.0) | 7.3 (3.0) | 6.3 (4.7) | |
| Albumin, g/L | 45.0 (1.7) | 45.8 (2.5) | 46.3 (2.1) | 47.0 (1.9) | 45.3 (2.7) | 46.3 (1.6) | 46.0 (1.7) | 46.7 (1.6) | 44.4 (1.8) | 43.8 (2.1) | 46.0 (1.7) | |
| LY3202626, 26 mg | ALT, U/L | 16.9 (6.3) | 15.6 (6.2) | 16.2 (6.4) | 16.9 (5.6) | 16.2 (5.2) | 16.8 (4.5) | 15.8 (5.5) | 16.9 (5.6) | 17.1 (7.4) | 16.8 (7.8) | 20.0 (9.8) |
| AST, U/L | 15.2 (2.5) | 14.6 (2.7) | 14.9 (3.6) | 16.8 (3.8) | 17.8 (3.2) | 16.4 (3.0) | 16.1 (2.7) | 18.3 (3.5) | 16.3 (4.0) | 16.2 (4.6) | 18.6 (6.2) | |
| ALP, U/L | 56.9 (12.0) | 57.9 (11.3) | 57.2 (11.1) | 58.6 (10.8) | 57.6 (12.0) | 59.3 (10.8) | 60.7 (11.4) | 60.1 (9.6) | 59.3 (10.2) | 58.6 (9.6) | 65.7 (11.5) | |
| GGT, U/L | 18.3 (8.2) | 18.9 (8.5) | 18.6 (8.8) | 19.0 (9.6) | 18.2 (8.5) | 18.6 (7.2) | 17.9 (7.4) | 17.7 (7.4) | 18.1 (8.8) | 17.6 (8.8) | 20.7 (11.1) | |
| Total Bilirubin, umol/L | 9.0 (2.6) | 8.6 (2.6) | 7.1 (2.3) | 6.7 (1.4) | 8.6 (3.0) | 8.6 (4.6) | 8.7 (4.2) | 8.3 (4.5) | 5.8 (2.4) | 7.2 (1.9) | 7.3 (4.6) | |
| Albumin, g/L | 46.3 (1.9) | 46.2 (1.9) | 45.0 (2.7) | 46.0 (1.9) | 44.9 (2.0) | 46.3 (2.9) | 45.8 (2.0) | 44.9 (2.3) | 43.7 (2.2) | 44.0 (2.6) | 45.9 (1.3) |
1Predose. ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; ALP, Alkaline phosphatase; GGT, Gamma glutamyl-transferase. Values are expressed as mean (standard deviation). N = 9 per group.
DISCUSSION
For many years, inhibition of BACE1 represented an extraordinarily promising mechanism for potential treatment of AD, largely due to early reports suggesting that no negative phenotypic changes were associated with BACE1 knockout in mice [16]. These findings implied that BACE1 inhibition could test the amyloid hypothesis by halting the production of Aβ with a low-likelihood of on-target side effects [16]. Despite this initial optimism, various adverse findings were subsequently observed in both nonclinical and clinical studies. The first BACE inhibitor (LY2811376) entering clinical testing was halted due to abnormalities in the retinal epithelium that were observed in toxicological studies [6]. This finding was subsequently attributed to inhibition of cathepsin D [17]. The development of other BACE inhibitors, including LY2886721, was stopped due to hepatotoxicity observed in clinical studies, the mechanism of which is unknown [7, 18]. LY3202626 was predicted to have reduced risk of these toxicities due to high selectivity against cathepsin D and a low daily dose (which is known to be associated with a lower rate of idiosyncratic drug-induced liver injury) [19, 20].
To mitigate peripheral effects for tolerability and safety, discovery efforts focused on developing a molecule with high potency against BACE1 and maximizing penetration across the blood-brain barrier [21]. The IC50 for LY3202626 in a FRET assay measuring BACE1 activity was 0.615 nM, markedly lower than the value for LY2886721 (20.3 nM) or LY2811376 (239 nM) [6, 7]. This potency was supported by studies in PDAPP mice and beagle dogs, each demonstrating substantial reductions in plasma and CSF Aβ associated with low plasma and CNS LY3202626 exposures. The ratio of CSF to plasma exposures in nonclinical species were similar to the ex vivo plasma free fraction measured for those species, suggesting free penetration into the CNS. Together, these data indicated that LY3202626 had the potential to lower CSF Aβ by at least 70% using a dose less than 10 mg. In the clinical multiple-dose study, concentrations of Aβ1–40, Aβ1–42, and the proximal biomarker sAβPPβ all decreased by more than 70% following multiple doses of 6 mg, confirming projections from the nonclinical studies. Modeling of the clinical data suggested that a dose of 12 mg QD was sufficient to lower CSF Aβ species by at least 90%. These results suggest that LY3202626 is substantially more potent than other BACE1 inhibitors for which clinical data have been reported [22, 23].
LY3202626 was generally well tolerated in the Phase 1 study. Excluding adverse events associated with CSF sampling, the most commonly reported adverse events (headache and dizziness) were all considered mild. While hepatoxicity has been observed in clinical studies with several BACE inhibitors, the doses of LY3202626 necessary to substantially reduce Aβ were deemed sufficiently low to mitigate the risk of hepatotoxicity. Consistent with this, safety data from this study showed no meaningful alterations in liver enzymes that would suggest hepatic injury due to LY3202626 administration [24].
The PK of LY3202626 were marked by relatively high intersubject and intrasubject variability. This could be due in part to variable absorption from the gastrointestinal tract. Notwithstanding the observed variability, the PK were generally linear with respect to time and dose, with a t1/2 supporting once-daily dosing. The PK of LY3202626 supported continued development of the compound.
The overall profile of LY3202626 rendered it suitable for testing in patients with mild AD in a Phase II proof-of-concept study. A dose of 12 mg QD was chosen as the high dose in the study, which was anticipated to produce 90% reduction in CSF Aβ. Subsequently and during further clinical development, the Phase II study of LY3202626 (NCT02791191), a proof-of-principle study powered to detect change on flortaucipir PET tracer signal, was ultimately terminated following an interim analysis of safety and efficacy data [25]. The decision to terminate the study was not due to safety concerns, rather due to a low statistical probability to detect a significant effect on clinical outcomes (either cognitive or functional) observed from the interim data. This interim analysis occurred after approximately 140 patients were randomized and observed for at least 24 weeks. Around the time of this interim analysis, there were discouraging Phase III data from other BACE inhibitors [26–28]. These other studies failed to demonstrate a slowing of AD progression at relatively high levels of BACE1 inhibition, and some studies suggested there was cognitive worsening soon after initiating treatment with these BACE inhibitors. In this interim analysis, no clinically meaningful separation was observed between either the 3 mg or 12 mg dose arms and placebo in the analysis of measures of cognition, function, and composite measures of cognition and function (ADAS-Cog13, ADCS-iADL, and iADRS, respectively) following 52 weeks of treatment [25]. Consistent with the results of the Phase I study, no evidence of hepatotoxicity was observed in this study, no elevations of liver function tests meeting the definition of Hy’s law occurred, and no statistical differences were observed in any liver function test between patients treated with LY3202626 or placebo. Nevertheless, the efficacy-to-risk benefit suggested that further investigation of LY3202626 in this clinical study was not warranted.
In evaluating the clinical efficacy of BACE inhibitors, the general strategy taken by sponsors was to prioritize doses that were anticipated to lead to significant suppression of Aβ production [26, 28–30]. While this approach failed to achieve a beneficial impact for patients with AD, several avenues remain for continued investigation of this class of compounds. In a study of Icelandic adults, it was determined that the APP A673T mutation (the Icelandic mutation), which interferes with cleavage of AβPP by BACE, was associated with reduced AD rates [31]. A subsequent study indicates that carriers of the A673T mutation have mildly reduced (approximately 28%) plasma Aβ as compared to noncarriers [32]. The BACE inhibitors that have been tested in Phase III studies all explored substantially more profound Aβ reductions (e.g., approximately 85% Aβ reduction for verubecestat). It has been hypothesized that the apparent changes in brain volume and cognition are due to the inhibition of BACE1 processing of substrates other than AβPP. For instance, the BACE1 enzyme interacts potently with other important proteins implicated in neuronal function and plasticity (e.g., SEZ6, NRG1, CHL1, JAG1) [33]. Thus, it remains possible, although an untested hypothesis to date, that an asymptomatic populations can be protected from developing AD by a low degree of BACE inhibition, one that that does not incure the negative effects observed with high degrees of BACE inhibition [29, 30, 34]. Recent results from monoclonal antibodies targeting deposited amyloid plaque has led to renewed interest in the amyloid hypothesis [35, 36], and therefore agents that target this pathology. However, it remains to be seen if this interest will translate to further study of BACE inhibitors in asymptomatic populations.
In conclusion, we found that the high in vitro potency and robust pharmacodynamic effect of the BACE inhibitor LY3202626 observed in nonclinical species translated to humans, even at low dose. This highly selective BACE1 inhibitor displayed high blood-brain barrier permeability. The favorable clinical safety and pharmacokinetic/pharmacodynamic profile of LY3202626 supports further clinical development.
ACKNOWLEDGMENTS
Formerly of Eli Lilly and Company, authors Robert A. Dean and David L. McKinzie are both now affiliated with Indiana University School of Medicine, and Leonard N. Boggs, Patrick J. Cocke, and Patrick C. May are now retired.
FUNDING
This study was funded by Eli Lilly and Company.
CONFLICT OF INTEREST
BAW, SLL, SAM, PJC, CDA, LNB, ARB, RAB, RAD, SJG, DEJ, QL, ACL, PCM, BMW, LLW, ZY, JAZ, DLM, DJM are all employed by and minor stockholders in Eli Lilly and Company or were at the time these studies were conducted.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-210037.
REFERENCES
[1] | Gregory GC , Halliday GM ((2005) ) What is the dominant Abeta species in human brain tissue? A review. Neurotox Res 7: , 29–41. |
[2] | Hampel H , Vassar R , De Strooper B , Hardy J , Willem M , Singh N , Zhou J , Yan R , Vanmechelen E , De Vos A , Nistico R , Corbo M , Imbimbo BP , Streffer J , Voytyuk I , Timmers M , Tahami Monfared AA , Irizarry M , Albala B , Koyama A , Watanabe N , Kimura T , Yarenis L , Lista S , Kramer L , Vergallo A ((2021) ) The beta-secretase BACE1 in Alzheimer’s disease. Biol Psychiatry 89: , 745–756. |
[3] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[4] | Munro KM , Nash A , Pigoni M , Lichtenthaler SF , Gunnersen JM ((2016) ) Functions of the Alzheimer’s disease protease BACE1 at the synapse in the central nervous system. J Mol Neurosci 60: , 305–315. |
[5] | Selkoe DJ ((2013) ) The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann Neurol 74: , 328–336. |
[6] | May PC , Dean RA , Lowe SL , Martenyi F , Sheehan SM , Boggs LN , Monk SA , Mathes BM , Mergott DJ , Watson BM , Stout SL , Timm DE , Smith Labell E , Gonzales CR , Nakano M , Jhee SS , Yen M , Ereshefsky L , Lindstrom TD , Calligaro DO , Cocke PJ , Greg Hall D , Friedrich S , Citron M , Audia JE ((2011) ) Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci 31: , 16507–16516. |
[7] | May PC , Willis BA , Lowe SL , Dean RA , Monk SA , Cocke PJ , Audia JE , Boggs LN , Borders AR , Brier RA , Calligaro DO , Day TA , Ereshefsky L , Erickson JA , Gevorkyan H , Gonzales CR , James DE , Jhee SS , Komjathy SF , Li L , Lindstrom TD , Mathes BM , Martenyi F , Sheehan SM , Stout SL , Timm DE , Vaught GM , Watson BM , Winneroski LL , Yang Z , Mergott DJ ((2015) ) The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J Neurosci 35: , 1199–1210. |
[8] | Green SJ , Mergott DJ , Watson BM , Winneroski LL Tetrahydropyrrolothiazine compounds. Eli Lilly and Company (Indianapolis, IN, US), United States. US Patent Classification: 514/224.2. Application Number: 8841293. Patent Number: 8841293. Issue Date: September 23, 2014. |
[9] | Yang HC , Chai X , Mosior M , Kohn W , Boggs LN , Erickson JA , McClure DB , Yeh WK , Zhang L , Gonzalez-DeWhitt P , Mayer JP , Martin JA , Hu J , Chen SH , Bueno AB , Little SP , McCarthy JR , May PC ((2004) ) Biochemical and kinetic characterization of BACE1: Investigation into the putative species-specificity for beta- and beta’-cleavage sites by human and murine BACE1. J Neurochem 91: , 1249–1259. |
[10] | Mosmann T ((1983) ) Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 65: , 55–63. |
[11] | Games D , Adams D , Alessandrini R , Barbour R , Berthelette P , Blackwell C , Carr T , Clemens J , Donaldson T , Gillespie F , et al. ((1995) ) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373: , 523–527. |
[12] | Zamek-Gliszczynski MJ , Ruterbories KJ , Ajamie RT , Wickremsinhe ER , Pothuri L , Rao MV , Basavanakatti VN , Pinjari J , Ramanathan VK , Chaudhary AK ((2011) ) Validation of 96-well equilibrium dialysis with non-radiolabeled drug for definitive measurement of protein binding and application to clinical development of highly-bound drugs. J Pharm Sci 100: , 2498–2507. |
[13] | Tang H , Mayersohn M ((2005) ) A novel model for prediction of human drug clearance by allometric scaling. Drug Metab Dispos 33: , 1297–1303. |
[14] | Lachno DR , Emerson JK , Vanderstichele H , Gonzales C , Martenyi F , Konrad RJ , Talbot JA , Lowe SL , Oefinger PE , Dean RA ((2012) ) Validation of a multiplex assay for simultaneous quantification of amyloid-beta peptide species in human plasma with utility for measurements in studies of Alzheimer’s disease therapeutics. J Alzheimers Dis 32: , 905–918. |
[15] | Lachno DR , Evert BA , Maloney K , Willis BA , Talbot JA , Vandijck M , Dean RA ((2015) ) Validation and clinical utility of ELISA methods for quantification of amyloid-beta of peptides in cerebrospinal fluid specimens from Alzheimer’s disease studies. J Alzheimers Dis 45: , 527–542. |
[16] | Vassar R , Kuhn PH , Haass C , Kennedy ME , Rajendran L , Wong PC , Lichtenthaler SF ((2014) ) Function, therapeutic potential and cell biology of BACE proteases: Current status and future prospects. J Neurochem 130: , 4–28. |
[17] | Zuhl AM , Nolan CE , Brodney MA , Niessen S , Atchison K , Houle C , Karanian DA , Ambroise C , Brulet JW , Beck EM , Doran SD , O’Neill BT , Am Ende CW , Chang C , Geoghegan KF , West GM , Judkins JC , Hou X , Riddell DR , Johnson DS ((2016) ) Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of beta-secretase inhibitors. Nat Commun 7: , 13042. |
[18] | Cummings J , Lee G , Ritter A , Sabbagh M , Zhong K ((2019) ) Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y) 5: , 272–293. |
[19] | Lammert C , Einarsson S , Saha C , Niklasson A , Bjornsson E , Chalasani N ((2008) ) Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 47: , 2003–2009. |
[20] | Lammert C , Bjornsson E , Niklasson A , Chalasani N ((2010) ) Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology 51: , 615–620. |
[21] | McKinzie DL , Winneroski LL , Green SJ , Hembre EJ , Erickson JA , Willis BA , Monk SA , Aluise CD , Baker TK , Lopez JE , Hendle J , Beck JP , Brier RA , Boggs LN , Borders AR , Cocke PJ , Garcia-Losada P , Lowe SL , Mathes BM , May PC , Porter WJ , Stout SL , Timm DE , Watson BM , Yang Z , Mergott DJ ((2021) ) Discovery and early clinical development of LY3202626, a low-dose, CNS-penetrant BACE inhibitor. J Med Chem 64: , 8076–8100. |
[22] | Imbimbo BP , Watling M ((2019) ) Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs 28: , 967–975. |
[23] | Lopez Lopez C , Tariot PN , Caputo A , Langbaum JB , Liu F , Riviere ME , Langlois C , Rouzade-Dominguez ML , Zalesak M , Hendrix S , Thomas RG , Viglietta V , Lenz R , Ryan JM , Graf A , Reiman EM ((2019) ) The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimers Dement (N Y) 5: , 216–227. |
[24] | Lo AC , Evans CD , Mancini M , Wang H , Shcherbinin S , Lu M , Natanegara F , Willis BA ((2021) ) Phase II (NAVIGATE-AD study) results of LY3202626 effects on patients with mild Alzheimer’s disease dementia. J Alzheimers Dis Rep 5: , 321–336. |
[25] | Lo AC , Evans CD , Mancini M , Lin Q , Wang H , Shcherbinin S , Lu M , Natanegara F , Willis BA ((2021) ) Phase 2 (NAVIGATE-AD study) results of LY3202626 effects on patients with mild Alzheimer’s disease dementia. J Alzheimers Dis Rep 5: , 321–336. |
[26] | Egan MF , Kost J , Tariot PN , Aisen PS , Cummings JL , Vellas B , Sur C , Mukai Y , Voss T , Furtek C , Mahoney E , Harper Mozley L , Vandenberghe R , Mo Y , Michelson D ((2018) ) Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 378: , 1691–1703. |
[27] | Egan MF , Kost J , Voss T , Mukai Y , Aisen PS , Cummings JL , Tariot PN , Vellas B , van Dyck CH , Boada M , Zhang Y , Li W , Furtek C , Mahoney E , Harper Mozley L , Mo Y , Sur C , Michelson D ((2019) ) Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med 380: , 1408–1420. |
[28] | Henley D , Raghavan N , Sperling R , Aisen P , Raman R , Romano G ((2019) ) Preliminary results of a trial of atabecestat in preclinical Alzheimer’s disease. N Engl J Med 380: , 1483–1485. |
[29] | Sims JR , Selzler KJ , Downing AM , Willis BA , Aluise CD , Zimmer J , Bragg S , Andersen S , Ayan-Oshodi M , Liffick E , Eads J , Wessels AM , Monk S , Schumi J , Mullen J ((2017) ) Development review of the BACE1 inhibitor lanabecestat (AZD3293/LY3314814). J Prev Alzheimers Dis 4: , 247–254. |
[30] | Timmers M , Streffer JR , Russu A , Tominaga Y , Shimizu H , Shiraishi A , Tatikola K , Smekens P , Borjesson-Hanson A , Andreasen N , Matias-Guiu J , Baquero M , Boada M , Tesseur I , Tritsmans L , Van Nueten L , Engelborghs S ((2018) ) Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimers Res Ther 10: , 85. |
[31] | Jonsson T , Atwal JK , Steinberg S , Snaedal J , Jonsson PV , Bjornsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jonsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488: , 96–99. |
[32] | Martiskainen H , Herukka SK , Stancakova A , Paananen J , Soininen H , Kuusisto J , Laakso M , Hiltunen M ((2017) ) Decreased plasma beta-amyloid in the Alzheimer’s disease APP A673T variant carriers. Ann Neurol 82: , 128–132. |
[33] | Barao S , Moechars D , Lichtenthaler SF , De Strooper B ((2016) ) BACE1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci 39: , 158–169. |
[34] | Kennedy ME , Stamford AW , Chen X , Cox K , Cumming JN , Dockendorf MF , Egan M , Ereshefsky L , Hodgson RA , Hyde LA , Jhee S , Kleijn HJ , Kuvelkar R , Li W , Mattson BA , Mei H , Palcza J , Scott JD , Tanen M , Troyer MD , Tseng JL , Stone JA , Parker EM , Forman MS ((2016) ) The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 8: , 363ra150. |
[35] | Mintun MA , Lo AC , Duggan Evans C , Wessels AM , Ardayfio PA , Andersen SW , Shcherbinin S , Sparks J , Sims JR , Brys M , Apostolova LG , Salloway SP , Skovronsky DM ((2021) ) Donanemab in early Alzheimer’s disease. N Engl J Med 384: , 1691–1704. |
[36] | Mullard A ((2021) ) FDA approval for Biogen’s aducanumab sparks Alzheimer disease firestorm. Nat Rev Drug Discov 20: , 496. |


