
Aluminum and Tau in Neurofibrillary Tangles in Familial Alzheimer’s Disease
Abstract
Background:
Familial Alzheimer’s disease (fAD) is driven by genetic predispositions affecting the expression and metabolism of the amyloid-β protein precursor. Aluminum is a non-essential yet biologically-reactive metal implicated in the etiology of AD. Recent research has identified aluminum intricately and unequivocally associated with amyloid-β in senile plaques and, more tentatively, co-deposited with neuropil-like threads in the brains of a Colombian cohort of donors with fAD.
Objective:
Herein, we have assessed the co-localization of aluminum to immunolabelled phosphorylated tau to probe the potential preferential binding of aluminum to senile plaques or neurofibrillary tangles in the same Colombian kindred.
Methods:
Herein, we have performed phosphorylated tau-specific immunolabelling followed by aluminum-specific fluorescence microscopy of the identical brain tissue sections via a sequential labelling method.
Results:
Aluminum was co-localized with immunoreactive phosphorylated tau in the brains of donors with fAD. While aluminum was predominantly co-located to neurofibrillary tangles in the temporal cortex, aluminum was more frequently co-deposited with cortical senile plaques.
Conclusion:
These data suggest that the co-deposition of aluminum with amyloid-β precedes that with neurofibrillary tangles. Extracellularly deposited amyloid-β may also be more immediately available to bind aluminum versus intracellular aggregates of tau. Therapeutic approaches to reduce tau have demonstrated the amelioration of its synergistic interactions with amyloid-β, ultimately reducing tau pathology and reducing neuronal loss. These data support the intricate associations of aluminum in the neuropathology of fAD, of which its subsequent reduction may further therapeutic benefits observed in ongoing clinical trials in vivo.
INTRODUCTION
The microtubule-associated protein tau in its normal physiological form supports the intracellular assembly of microtubules, stabilizing the neuronal cytoskeleton [1, 2]. Human tau is a natively unfolded protein with little secondary structure [3]. The six isoforms of human tau arise through alternative RNA splicing, which is defined by either three (3R) or four (4R) microtubule-binding domain repeats [3, 4]. Tau is predominantly found in neurons in the brain, though it is also expressed at lower glia levels [1, 2]. Tau binds to microtubules through its C-terminal domain with the N-terminus binding to cytosolic components, including the membrane-associated protein actin, the latter defining a role for tau in the organization of cytoskeletal networks [2, 3].
Post-translational modifications of tau and its phosphorylation by protein kinases, including glycogen synthase kinase 3β (GSK3β), have been shown to act as a critical mediator influencing both the function and stability of tau [1–3]. Under physiological conditions, the phosphorylation of tau modulates its binding to microtubules in competition with kinesin and dynein motors, thereby regulating the anterograde and retrograde transport of cellular cargoes, respectively [2, 5]. In healthy neuronal cells, tau is typically found along a concentration gradient, increasing toward the axonal terminals of the cells as maintained by the tau diffusion barrier of the axon initial segment (AIS) that acts to modulate against the retrograde transport of tau towards neuronal dendrites and cell soma [3, 6]. The low levels of tau found in axons are suggested to promote long-term potentiation via the strengthening of synaptic terminals [3, 7].
The excessive or hyperphosphorylation of tau under pathophysiological conditions have defined the tauopathies broadly encompassing neurodegenerative conditions, including Alzheimer’s disease (AD) [1]. The subsequent breakdown of the AIS that follows allows for the redistribution of tau from axons allowing its retrograde diffusion into the somatodendritic compartment of neurons [3, 6]. In combination with tau detachment from microtubules upon hyperphosphorylation, increasing intraneuronal concentrations of the protein propagate from the cell soma to dendrites causing the concomitant loss of dendritic spines and neurite outgrowth [1, 3]. The increased accumulation and hyperphosphorylation of tau lead to the formation of neurofibrillary tangles (NFTs) of the insoluble aggregated protein [1]. In concert with synaptic dysfunction and loss, the depolymerization of microtubules disrupts the cytoskeletal network, thus acting to drive the progressive and mechanistic loss of affected neurons [1, 5].
In addition to the large scale loss of neurons in the frontal cortex and astrogliosis, intracellular NFTs of tau protein and extracellular senile plaques of the amyloidogenic amyloid-β (Aβ) peptide constitute the principal neuropathological hallmarks of AD [8–10]. While sporadic forms of AD typically occur in patients aged 65 and over, familial Alzheimer’s disease (fAD) occurs at an earlier age of onset, driven by autosomal dominant mutations in the presenilin 1 (PSEN1) and 2 (PSEN2) proteins and the transmembrane amyloid-β protein precursor (AβPP) [11–13]. Aβ42 is the principal pathogenic peptide located in senile plaque cores [15, 16]. Mutations in PSENs are known to increase the production of the Aβ42 peptide via the enhanced and sequential proteolytic cleavage of AβPP via β-site APP cleaving enzyme 1 (BACE1) and γ-secretases [10, 12, 13].
Insoluble and aggregated forms of Aβ in senile plaques and hyperphosphorylated tau in NFTs dominate the pathological landscape of familial and sporadic AD brains. However, oligomeric forms of amyloidogenic proteins are thought to underlie neurotoxicity in vivo [17]. Therefore, such has continued to lend support to an updated and genetically driven view of the amyloid cascade hypothesis of AD, in which the deleterious effects observed in brain parenchyma have been cemented in the studies of fAD [11–13, 18]. In more recent studies, Aβ has been shown to modulate AIS function through its ability to activate its tethered GSK3β protein kinase [3, 6]. The downstream phosphorylation of non-tau targets that follows has been implicated in increased concentrations of endogenous and exogenous tau in the somatodendritic compartment of neurons due to impaired microtubule dynamics in the AIS [6, 19].
Furthermore, the ability of Aβ to synergistically activate cofilin, an enzyme that acts to depolymerize actin filaments (F-actin), results in the remodeling of cytoskeletal networks, causing the collapse of the AIS [6]. Such demonstrates the ability of Aβ and tau to synergistically drive the structural disintegration of neurons in the AD brain [1–3, 5, 6]. Interestingly, cross-seeding between Aβ and tau promotes aggregation of amyloidogenic β-pleated sheets and NFTs, forming mutated and stable complexes rich in aggregation-prone hydrophobic interactions [20, 21]. Those complexes formed are suggested to be aberrant and hence contributory to AD pathology [20].
Aluminum is non-essential yet in its free solvated and trivalent form, Al3 + (aq) , is biologically reactive [22]. Our previous study of the brains of fAD donors carrying a PSEN1-E280A (Glu280Ala) mutation found aluminum co-localized with fibrillar Aβ in senile plaques [22, 23]. Donors in this kindred are unique in that this mutation is found in large numbers in the Antioquia region of Colombia [24]. Individuals with fAD carrying this mutation develop elevated cortical Aβ at an early age of onset of <50 years. The high brain aluminum content of fAD donors versus statistically lower amounts found in non-neurologically impaired individuals has highlighted the potential for aluminum to act as a nidus to overcome the precipitation of Aβ [23, 25].
In addition to a role for aluminum in senile plaque formation in fAD, we have investigated the potential for aluminum to bind to NFTs of hyperphosphorylated tau protein in the same kindred [26]. The large number of phosphate groups available on the surface of NFTs would be expected to bind intracellular aluminum with high avidity [27]. Herein, we have performed phosphorylated (Ser202 and Thr205) tau-specific immunolabelling to unequivocally identify NFTs in the brain tissue of Colombian fAD donors, carrying a PSEN1-E280A mutation [28]. The unequivocal identification of NFTs has been followed by aluminum-specific fluorescence microscopy to probe the potential preferential binding of aluminum to senile plaques or NFTs in fAD brain tissue.
MATERIALS AND METHODS
Human brain tissue
Human brain tissue consisting of formalin-fixed paraffin-embedded (FFPE) blocks were obtained following ethical approval from Universidad de Antioquia, Medellin, Colombia. Donor brain tissue was received from anonymized donors with fAD, all carrying the PSEN1-E280A mutation and research was performed following ethical approval obtained from Keele University, UK (ERP 2391). FFPE brain tissue blocks of a 45-year-old female, 57-year-old female, and 60-year-old male, all carrying a PSEN1 E280A mutation, were selected for imaging, based upon representative depictions of aluminum and NFT co-localization across frontal, parietal, temporal and occipital lobes.
Microtomy
Human brain tissue blocks were cooled on wet ice for 10 min. Ribbons of adjacent tissue sections were prepared at a thickness of 6μm by use of a Leica RM2025 rotary microtome fitted with Surgipath® DB80 LX low-profile stainless-steel blades (Leica Microsystems, UK). Sections were floated onto ultrapure water (conductivity < 0.067μS cm–1) at 40°C and transferred onto Polysine adhesion slides (Thermo Scientific, UK). Sections were dried overnight at ambient temperature. Excess wax was removed from tissue sections by heating at 62°C for 20 min. Sections were allowed to cool fully before dewaxing and rehydration procedures were performed.
Dewaxing and rehydration of human brain tissue sections
Human brain tissue sections were dewaxed with gentle agitation in Histo-Clear (National Diagnostics, US) for 3 min. Removal of any remaining wax was performed via a second wash in fresh Histo-Clear for 1 min. Sections were transferred into pure (HPLC grade) ethanol (Fisher Scientific, UK) for 2 min. Rehydration of tissue sections was achieved by transfer through an ethanol gradient (95, 70, 50, and 30%v/v) for 1 minute in each solvent. Sections were twice washed in ultrapure water for 30 s to remove the remaining ethanol before staining.
Lumogallion staining
Rehydrated tissue sections were placed in Copl-in jars and fully immersed in 1 mM lumogallion (4-chloro-3-(2,4-dihydroxyphenylazo)-2-hydroxyb-enzene-1-sulphonic acid, TCI Europe N.V., Belgium) in 50 mM PIPES, pH 7.4. For the assessment of autofluorescence, the adjacent serial section was immersed in 50 mM PIPES buffer, pH 7.4 only. Sections were incubated for 6 h at ambient temperature away from light. Sections were subsequently washed six times in fresh PIPES buffer, rinsed for 30 s in ultrapure water and mounted under glass coverslips with the aqueous mounting medium, Fluoromount™ (Sigma Aldrich, UK). Lumogallion staining, ThS counter-staining (ca 0.075%w/v ThS in 50%v/v ethanol) [23, 26], and AT8 immunolabelling were performed and analyzed in sequence, following each labelling method. Every such staining method implemented was performed on the identical tissue section.
Immunohistochemistry
All reagents for immunohistochemistry (IHC) were purchased from Abcam, UK, unless otherwise stated. Following analysis of lumogallion-stained tissue sections, coverslips were removed via immersion in ultrapure water with gentle agitation overnight. Sections were rinsed thoroughly with ultrapure water, outlined with a hydrophobic PAP pen and transferred into a humidity chamber for all subsequent staining procedures. Thereafter, IHC was performed according to the manufacturer’s instructions. Briefly, sections were immunolabelled with an AT8 recombinant primary anti-tau monoclonal antibody (rabbit) against human tau phosphorylated on Ser202 and Thr205 (0.472 mg/mL: Abcam, UK: ab210703).
Antigen retrieval was performed by adding Proteinase K antigen retrieval solution to sections, foll-owed by incubation at ambient temperature for 7 min. Sections were rinsed in 1 X Tris-buffered saline (TBS), followed by further rinses in TBS plus 0.025%v/v Triton X-100. Blocking was achieved by incubating sections in 10%v/v normal goat serum with 1%w/v bovine serum albumin (BSA) in TBS for 2 h at ambient temperature. Pre-optimized dilutions (1:1,000) of the unlabeled primary AT8 antibody in TBS, including 1%w/v BSA, were applied to sections and incubated overnight at 4°C to ensure complete saturation of antibody binding.
All staining procedures were performed at ambient temperature. Following AT8 labelling, sections were rinsed with TBS, including 0.025%v/v Triton X-100. Endogenous tissue peroxidase activity was blocked by incubation in 0.3%v/v H2O2 in TBS for 15 min. Sections were subsequently stained using a rabbit-specific horseradish peroxidase (HRP) 3,3′-diaminobenzidine (DAB) ABC IHC staining kit. A biotinylated goat anti-polyvalent secondary antibody was added to sections and incubated for 10 min. Sections were washed in TBS, and HRP-labelled streptavidin was applied for 10 min. After washing with TBS, DAB chromogen (30μL DAB chromogen added to 1.5 mL DAB substrate) was applied to sections and developed by incubation for 10 min. Sections were washed in TBS and finally mounted under glass coverslips using the aqueous mounting medium, Faramount™ (Agilent Dako, UK).
Microscopy
Brightfield and fluorescence micrographs were acquired using an Olympus BX50 fluorescence mic-roscope (mercury source) equipped with a vertical illuminator and BX-FLA reflected light attachment. A U-MNIB3 fluorescence filter cube (bandpassλex: 470–495 nm, dichromatic mirror: 505 nm, longp-assλem: 510 nm) was used to acquire lumogallion fluorescence (Olympus, UK). Images were captured using an Olympus DP74 (CMOS processor) color camera and the OLYMPUS cellSens (Standard 3.1) software suite. Optimal exposure times for collecting lumogallion fluorescence were set automatically by the superfluorescence (SFL) mode of the Olympus DP74 camera. The same optimized exposure settings were then subsequently fixed for each respective and corresponding autofluorescence micrograph. Merging of fluorescence and brightfield channels was achieved using Photoshop (Adobe Systems Inc., US).
RESULTS
Aluminum and neurofibrillary tangles are found co-localized in familial Alzheimer’s disease
IHC labelling using an AT8 primary antibody demonstrated the presence of flame-like NFTs in predominantly cortical brain regions of Colombian fAD donors. Optimization of the IHC staining method using progressive serial dilutions of the AT8 primary antibody improved the signal to noise ratio sequentially. IHC of the parietal cortex of a 60-year-old male Colombian fAD donor using serial dilutions of the primary AT8 antibody of 1:40, 1:80, 1:200, and 1:1,000 identified NFTs via dark brown DAB staining at every antibody dilution tested. IHC performed in the absence of the AT8 primary antibody produced no detectable DAB staining (see Supplementary Figure 1).
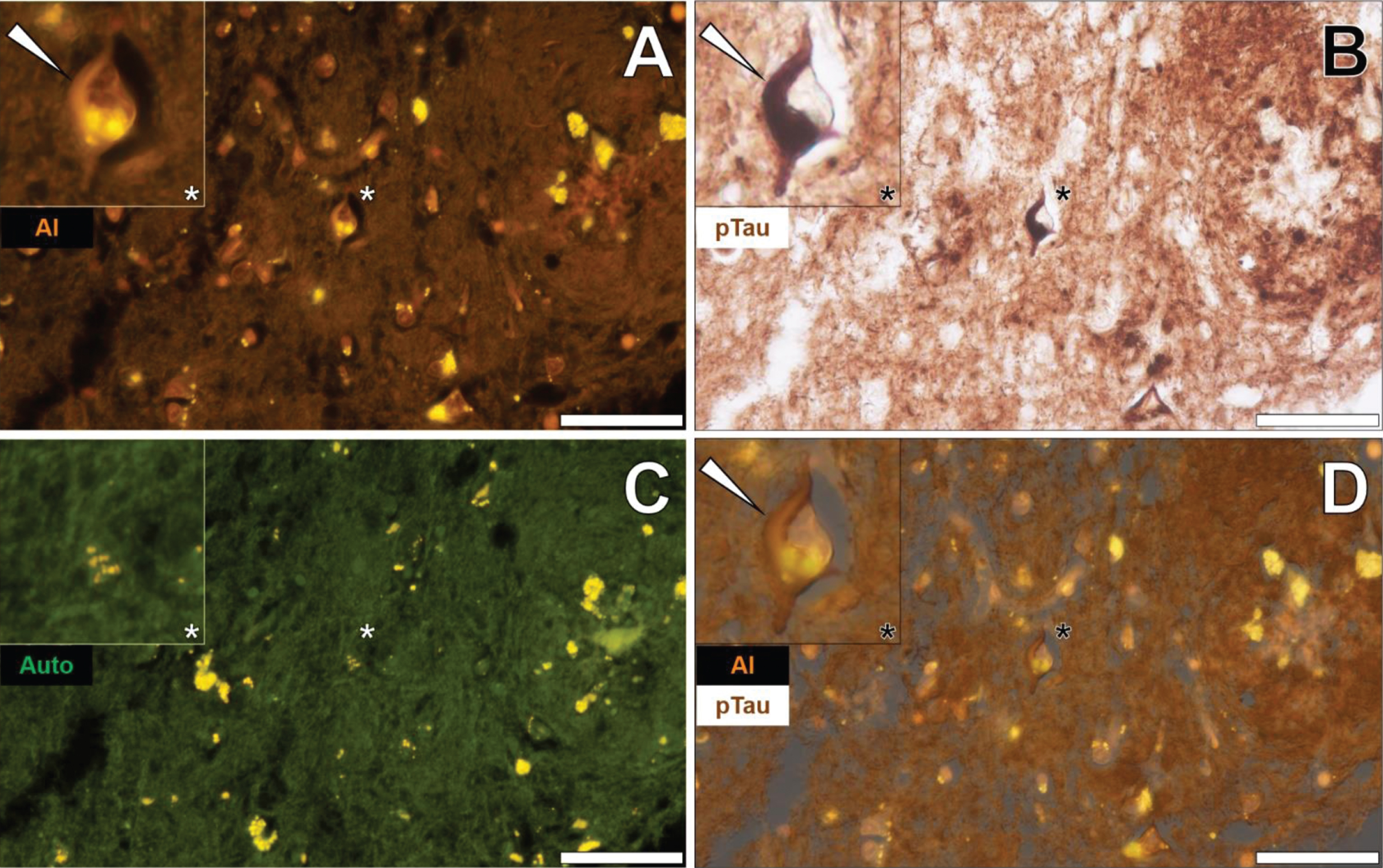
In the temporal cortex of a 45-year-old female Colombian fAD donor, aluminum was found in neuronal cells close to extracellular deposits of aluminum as identified by an intense orange fluorescence emission (Fig. 1A) [26]. Punctate fluorescence was especially noted in the cell soma of neuronal cells and was readily differentiated from the yellow/green fluorescence of cytosolic lipofuscin deposits in neighboring cells (Fig. 1A) [26]. AT8 immunolabelling for phosphorylated tau revealed the dense and intracellular immunoprecipitation of DAB via dark brown staining (Fig. 1B).
Fig. 1
Neuronal cells loaded with aluminum in close proximity to extracellular aluminum in the temporal cortex of a 45-year-old female Colombian donor with familial Alzheimer’s disease. A) Intracellular aluminum (Al) in close proximity to aluminum-rich cellular debris (orange). B) AT8 immunoreactive phosphorylated tau (pTau) in neurons (arrows) located via DAB staining in the identical section (brown). C) Merging of lumogallion and brightfield channels depicting aluminum and pTau in the identical neuron. D) Overlay of thioflavin S fluorescence (green) highlighting NFTs of pTau. Asterisks denote magnified inserts. Figure adapted from [26] under CC license. Magnification: X 400, scale bars: 50μm.
![Neuronal cells loaded with aluminum in close proximity to extracellular aluminum in the temporal cortex of a 45-year-old female Colombian donor with familial Alzheimer’s disease. A) Intracellular aluminum (Al) in close proximity to aluminum-rich cellular debris (orange). B) AT8 immunoreactive phosphorylated tau (pTau) in neurons (arrows) located via DAB staining in the identical section (brown). C) Merging of lumogallion and brightfield channels depicting aluminum and pTau in the identical neuron. D) Overlay of thioflavin S fluorescence (green) highlighting NFTs of pTau. Asterisks denote magnified inserts. Figure adapted from [26] under CC license. Magnification: X 400, scale bars: 50μm.](https://content.iospress.com:443/media/adr/2021/5-1/adr-5-1-adr210011/adr-5-adr210011-g001.jpg)
Merging of the fluorescence (Fig. 1A) and brightfield (Fig. 1B) channels revealed the presence of intraneuronal aluminum and NFTs in a single neuron (Fig. 1C). Subsequent merging of the original ThS fluorescence channel [26] demonstrated the presence of a peripherally located intraneuronal green fluorescence emission, overlaying DAB immunostaining of phosphorylated tau. Higher magnifications demonstrated co-localization of intracellular particulate aluminum deposits and AT8-reactive phosphorylated tau (Fig. 1D). Lumogallion-reactive aluminum was also observed in neuronal cells within pia mater of the same donor that produced an orange fluorescence emission versus green autofluorescence of the non-stained section (see Supplementary Figure 2). Similarly, the merging of fluorescence and brightfield channels identified aluminum co-loca-lized with AT8-reactive DAB staining in the same neuron (see Supplementary Figure 2).
Co-localization of aluminum and phosphorylated tau predominates in the temporal cortex
Lumogallion staining of the temporal cortex of a 57-year-old female Colombian fAD donor revealed lumogallion-reactive aluminum in pyramidal neurons, as highlighted via an intracellular orange fluorescence emission (Fig. 2A). Intracellular lipofuscin was frequently observed as punctate yellow fluorescence in the cytoplasm of neuronal cells in both lumogallion-stained and non-stained tissue sections for autofluorescence. The yellow fluorescence emission of intracellular deposits of lipofuscin was readily distinguished from the uniform green autofluorescence of the adjacent serial section (Fig. 2C). Subsequent AT8 immunolabelling of the identical lumogallion-stained tissue section revealed dark brown DAB precipitation appearing as a classical flame-like NFT in a single pyramidal neuron (Fig. 2B). Merging of lumogallion fluorescence (Fig. 2A) and the brightfield channel (Fig. 2B) revealed that intracellular aluminum was occasionally found co-localized with AT8-reactive tau in the cell soma of pyramidal neurons (Fig. 2D).
Fig. 2
Intracellular aluminum in pyramidal neurons in the temporal cortex of a 57-year-old female Colombian donor with familial Alzheimer’s disease. A) Intracellular aluminum (Al) in pyramidal neurons (orange). B) AT8 immunoreactive phosphorylated tau (pTau) in a single neuron (black arrow) located via DAB staining (brown) and the absence of staining in an adjacent neuron (white arrow). C). Autofluorescence (green) of the non-stained adjacent section highlighting deposits of intracellular lipofuscin (yellow). D) Merging of lumogallion and brightfield channels depicting aluminum and pTau in an identical neuron (black arrow) and the absence of DAB staining in an adjacent neuron (white arrow). Asterisks denote magnified inserts. Magnification: X 400, scale bars: 50μm.
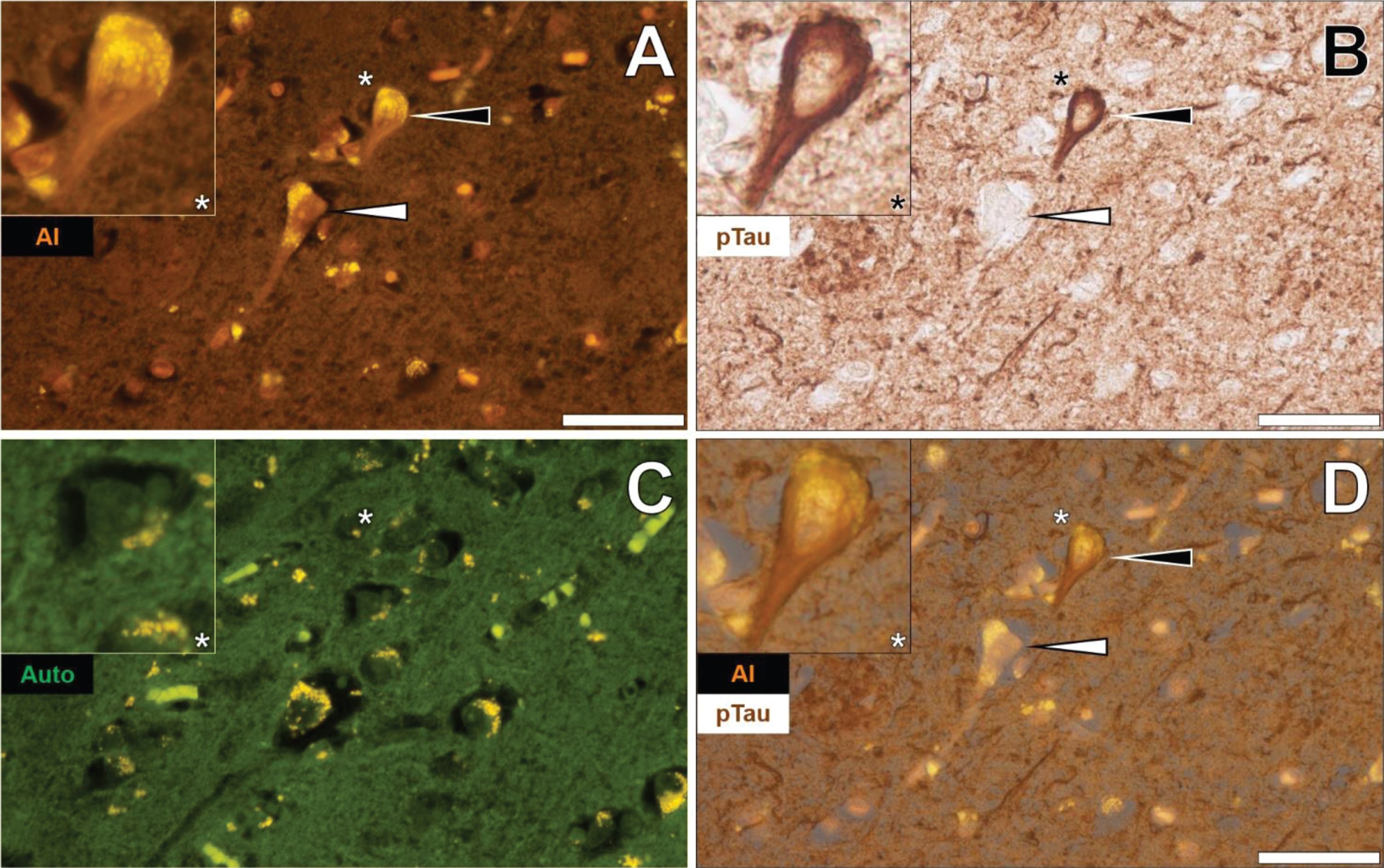
Similarly, lumogallion-reactive aluminum was detected in neuronal cells in pia mater in the temporal cortex of the same donor, as highlighted via an intense orange fluorescence emission (Fig. 3A). AT8 counter-immunolabelling of the identical tissue section produced brown intraneuronal DAB precipitation of an NFT-like morphology (Fig. 3B). The adjacent non-stained serial section yielded a green autofluorescence emission with yellow punctate lipofuscin deposits identified intracellularly (Fig. 3C). Merging of the lumogallion and brightfield channel demonstrated the co-localization of aluminum and AT8-reactive tau in the same neuron (Fig. 3D).
Fig. 3
Intraneuronal aluminum in the temporal cortex of a 57-year-old female Colombian donor with familial Alzheimer’s disease. A) Intracellular aluminum (Al) in a neuron (orange). B) AT8 immunoreactive phosphorylated tau (pTau) located via DAB staining (brown). C). Autofluorescence (green) of the non-stained adjacent section highlighting occasional deposits of intracellular lipofuscin (yellow). D) Merging of lumogallion and brightfield channels depicting aluminum and pTau in the identical neuron. Asterisks denote magnified inserts. Magnification: X 400, scale bars: 50μm.
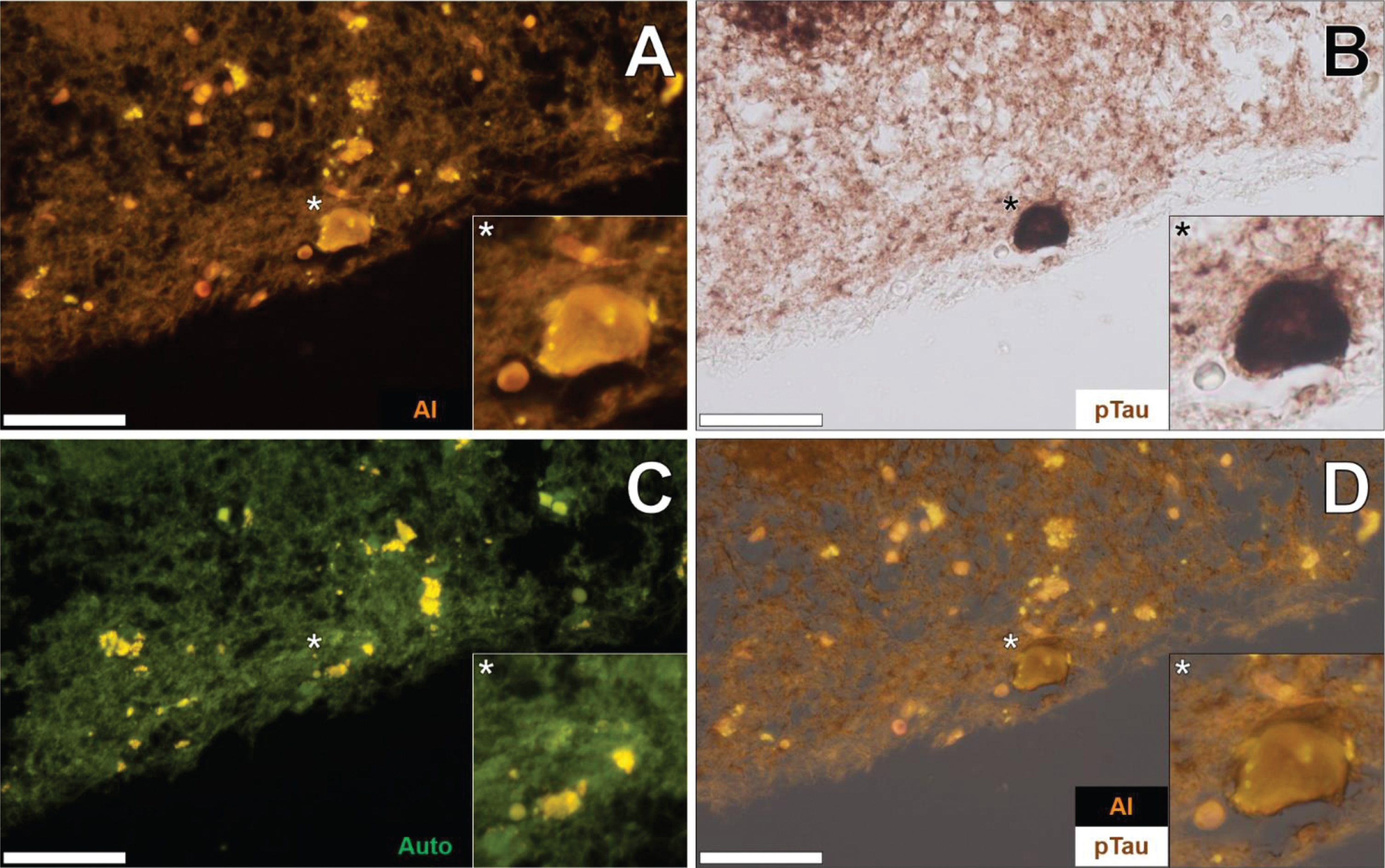
The co-localization of aluminum and immunoreactive tau was not restricted to the temporal lobe. Such deposition was also noted in the parietal (see Supplementary Figure 3) and frontal cortex (see Supplementary Figure 4) of the same 57-year-old female Colombian fAD donor. In both cases, aluminum was found intracellularly in neurons close to the pia mater. The orange fluorescence intensity produced was generally weaker in neurons in the frontal lobe, though markedly brighter than green autofluorescence typically noted for non-stained sections. AT8 immunolabelling produced dark brown staining, resembling intracellular NFTs. In the identical neurons, aluminum was found co-localized with AT8-labelled DAB-reactive tau deposits. In the occipital lobe, few neurons produced a positive lumogallion fluorescence signal for aluminum above background autofluorescence.
Additional brain tissue sections of the temporal lobe of fAD donors were investigated to support observations of aluminum-rich NFTs. In the temporal cortex of a 60-year-old male Colombian fAD donor, intracellular lumogallion-reactive aluminum produced an orange fluorescence emission that exhibited a flame-like morphology (Fig. 4A). IHC for phosphorylated tau revealed intracellular dark brown DAB staining in the same neuron, appearing as a characteristic flame-like NFT (Fig. 4B). Similar lipofuscin loading was noted in the same cell via punctate yellow autofluorescence of the non-stained adjacent tissue section (Fig. 4C). Merging of lumogallion fluorescence and the AT8 brightfield channel confirmed that both aluminum and immunolabelled tau were intraneuronal and co-localized, of which both adopted a flame-like structure, similar to that of NFTs (Fig. 4D). Such confirmed similar observations made of aluminum co-localized with DAB-reactive NFTs, in different cortical regions of the same lobe (see Supplementary Figure 5).
Fig. 4
Intraneuronal aluminum in the temporal cortex of a 60-year-old male Colombian donor with familial Alzheimer’s disease. A) Intracellular aluminum (Al) in a neuron (orange). B) AT8 immunoreactive phosphorylated tau (pTau) located via DAB staining (brown) appearing as flame-like NFT. C). Autofluorescence (green) of the non-stained adjacent section highlighting occasional deposits of intracellular lipofuscin (yellow). D) Merging of lumogallion and brightfield channels depicting aluminum and pTau co-localized in the identical neuron. Asterisks denote magnified inserts. Magnification: X 400, scale bars: 50μm.
Phosphorylated tau immunostaining and aluminum deposition in senile plaques
Senile plaques were frequently found to produce a bright orange fluorescence emission when stained with lumogallion across all lobes investig-ated (frontal, parietal, occipital, and temporal cortex) from each Colombian fAD donor. For example, in the temporal cortex of a 60-year-old male Colombian fAD donor, a bright orange fluorescence emission was noted in a senile plaque, in which yellow punctate fluorescence of lipofuscin was found at its periphery (Fig. 5A). Upon counter-staining against phosphorylated tau using an AT8 primary antibody, DAB-reactive staining was found in neuronal-like bodies that, similarly to lipofuscin, were also located at the periphery of the identical senile plaque (Fig. 5B).
Fig. 5
Aluminum in a senile plaque in the temporal cortex of a 60-year-old male Colombian donor with familial Alzheimer’s disease. A) Aluminum (Al) staining of a senile plaque (orange). B) AT8 immunoreactive phosphorylated tau (pTau) located via DAB staining (brown) in dystrophic neurites (arrows) at the periphery of the identical senile plaque C). Autofluorescence (green) of the non-stained adjacent section highlighting occasional deposits of intracellular lipofuscin (yellow). D) Merging of lumogallion and brightfield channels depicting aluminum and pTau (arrows) in the identical senile plaque. Asterisks denote magnified inserts. Magnification: X 400, scale bars: 50μm.
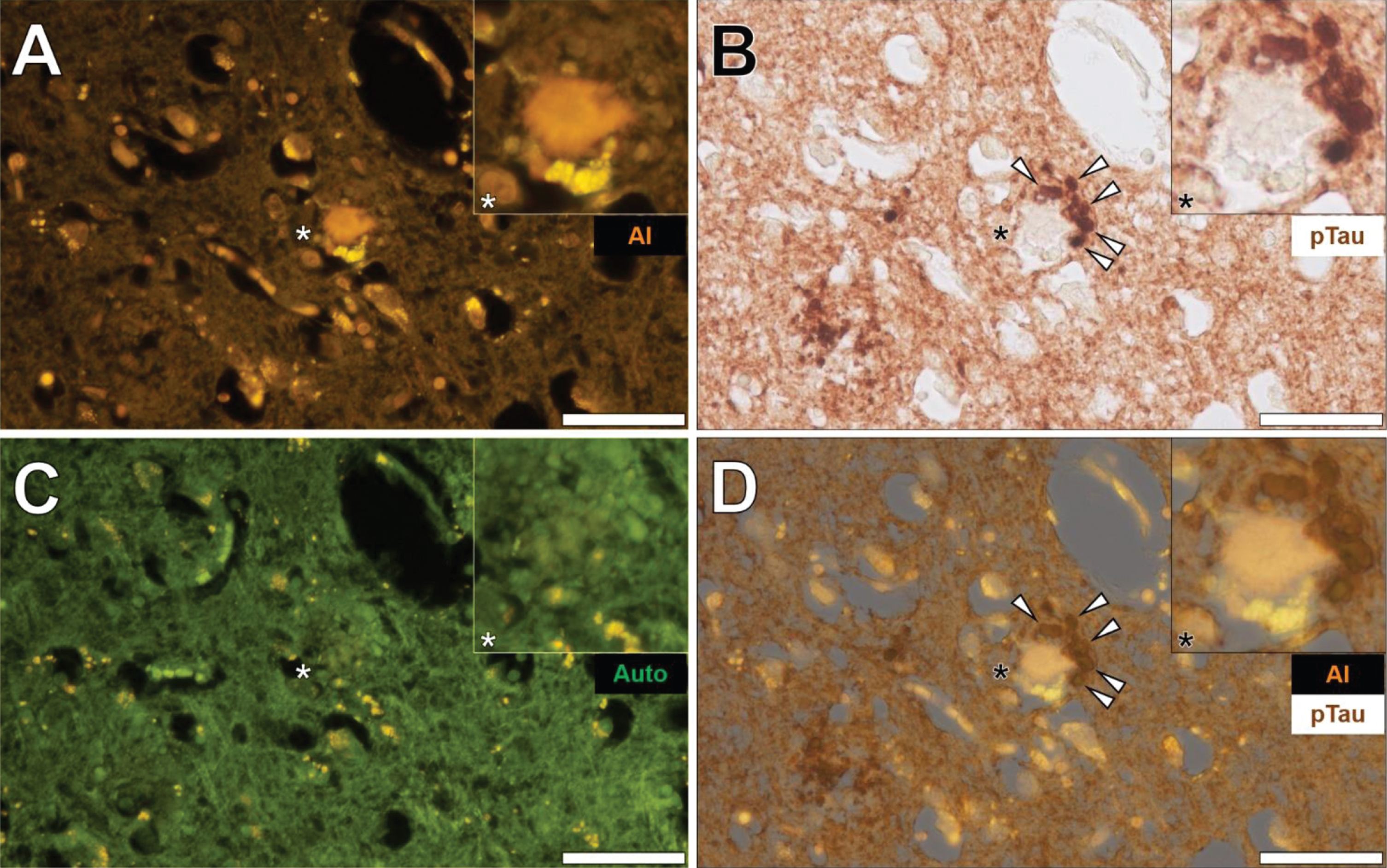
A weak green autofluorescence emission was noted in the immediate vicinity of the same senile plaque, with discreet particulate-like deposits of lipofuscin located at its periphery (Fig. 5C). Merging of the lumogallion fluorescence and the brightfield channel highlighted the presence of aluminum in the senile plaque core with no clear co-localization to DAB-reactive neuronal bodies at its periphery (Fig. 5D). Similar observations were made in larger senile plaques (>50μm), in which a dark brown AT8-reactive DAB staining was noted in peripheral dystrophic-like neurites and discreet neuronal bodies, depositing away from the aluminum-rich core regions of senile plaques (see Supplementary Figure 6).
DISCUSSION
We have demonstrated the neuronal co-localiz-ation of aluminum and phosphorylated tau in NFTs in fAD donor brain tissue, carrying a PSEN1-E280A mutation. Aluminum-specific fluorescence microscopy utilizing the fluorophore lumogallion revealed the presence of aluminum in neurons co-deposited with neuropil-like threads [26]. The latter was previously detected by the use of the benzothiazole-based dye, thioflavin S (ThS), which binds to β-pleated sheet-like moieties, including mature NFTs and Aβ fibrils in senile plaques [29]. While a key benefit of ThS labelling is its ability to produce fluorescence only with mature and pathologically-relevant misfolded amyloidogenic proteins, such is not specific to hyperphosphorylated tau in NFTs [29].
AT8 is an anti-tau primary antibody that recognizes the well-characterized Ser202 and Thr205 epitopes of phosphorylated tau [28]. Owing to the clear DAB labelling above background at 1:1,000 dilutions of the primary AT8 antibody, this concentration was used for all subsequent IHC experiments identifying NFTs. In cortical brain regions, AT8 revealed intracellular pathological NFT inclusions. As the most prominent of the AT-series monoclonal antibodies directed against phosphatase-sensitive epitopes, AT8 labels both intracellular and extracellular NFTs as distinguished by the presence of collapsed dendrites [30]. These late-stage or ghost-like tangles that form extracellularly following apoptosis of the affected neuronal cell show the distinctive absence of a nucleus with no discernable cytoplasm [29–31]. Late-stage ghost NFTs frequently possess a central void. Their characteristic presence adopts tangles of a paired helical filament morphology, located in the somatodendritic compartments of the ruptured cells [3, 30, 31].
Herein, we identified AT8-immunoreactive ghost NFTs co-localized with aluminum, confirming our previous observations of ThS-reactive tangles in identical neurons in the temporal cortex of a 45-year-old female fAD donor [26]. As expected, the use of IHC to detect phosphorylated tau was more sensitive than fluorescent staining using ThS and showed improvements in visualizing the various stages of tau tangle formation [29, 32]. Aluminum was observed co-located with NFTs across multiple brain regions, including the frontal, temporal, and parietal cortex, of which fewer positive staining examples were found in the occipital cortex.
Of all lobes investigated, aluminum was found most commonly co-located in NFTs in the temporal cortex. The temporal cortex is of the first brain regions in which tau is known to propagate as NFTs. The expansion of NFT pathology in the brain is graded via the Braak stages of severity, of which 0-IV ascribe the confinement of NFT pathology to the medial temporal lobe [33, 34]. At stage I, NFT pathology is confined to the transentorhinal region, which migrates to deeper entorhinal regions and into the hippocampus at stage II. Widespread hippocampal lesions are observed at stage III, spreading to the medial temporal gyrus at stage IV [34]. Later stages (V and VI) of disease progression are marked by NFT deposition in the striate and parastriate regions of the occipital cortex [34].
In living fAD donors carrying a PSEN1-E280A mutation, positron emission tomography has revealed that NFT formation ensues ten to fifteen years after marked increases of soluble tau are typically detected in the cerebrospinal fluid [35, 36]. Subsequent onset to severe dementia arises approximately seven years later, marking tau pathology as an essential biomarker of early disease progression. As a consequence of the late onset of tau pathology in the PSEN1-E280A brain, such may account for the early and widespread distribution of NFTs in the temporal cortex and perhaps their association with aluminum [35, 36].
Elevated soluble cortical Aβ in the same kindred is mirrored by widespread senile plaque deposition, preceding NFT formation in vivo [24, 35, 36]. Therefore, the abundance of extracellular Aβ depositing as β-pleated sheets in senile plaques would provide numerous binding sites for aluminum before potential competitive interactions with the intracellular phosphorylated binding sites available in NFTs [27]. Such a mechanism may account for the high levels of aluminum observed in the brain tissue of fAD donors carrying a PSEN1-E280A mutation versus donors with neither a clinical nor pathological diagnosis of neurodegenerative disease [23, 25, 37].
Interestingly, while numerous ghost NFTs were present in the cerebral cortex of fAD donors that produced positive DAB staining for phosphorylated tau, their co-localization with aluminum was detected less frequently. While immunoreactive phosphorylated tau was found co-located with aluminum in cortical neurons, they were fewer in incidence than co-deposits of aluminum in the core of extra-cellular senile plaques. Therefore, AT8 immunolabelling confirmed the preferential binding of aluminum exhibited in senile plaques versus NFTs [23, 26]. These data suggest that the co-deposition of aluminum with Aβ precedes that with NFTs. Extracellularly deposited Aβ may also be more immediately available to bind aluminum versus intracellular aggregates of tau. AT8 immunoreactive tau was found deposited at the periphery of senile plaques in both intracellular neuronal bodies and extracellular ghost NFTs. Lumogallion-reactive aluminum was frequently observed in the cores of neuritic plaques in which its deposition was less evident within surrounding tau-reactive neurons.
In addition to AT8-labelled dystrophic neurites, lipofuscin was also observed deposited at the periphery of senile plaques. As a marker of lysosomal degradation, autofluorescent lipid and misfolded protein-rich deposits are known to accumulate in the cytoplasm of neurons [38]. Aggregates of lipofuscin have previously been suggested to play an active role in neurodegeneration [38, 39]. As a marker of impeded lysosomal function and proteolytic activity, the release of lipofuscin into the extracellular milieu following neuronal death has been implicated in the formation of senile plaques [39].
Failure in the misfolded protein response promotes the formation of lipofuscin and its subsequent release from dying cells, and hence, the presence of lipofuscin in brain tissue sections increases upon ageing [38]. Such may explain the intimate associations observed between Aβ and coronal lipofuscin in senile plaques [37, 38, 40, 41]. In concert with Aβ, co-aggregated lipofuscin deposits bind aluminum that may act further to exacerbate the accumulation of aluminum in senile plaque cores, as has been previously reported in AD brain tissue [23, 37, 42]. Conversely, intracellular aluminum found in neurons heavily laden with lipofuscin was not always found co-located with NFTs. Therefore, intracellular lipofuscin presence may not necessarily dictate aluminum binding as was observed herein in pyramidal neurons in the temporal cortex of a 57-year-old female Colombian fAD donor.
AT8-immunoreactive phosphorylated tau protein and its silver-impregnated derivatives have previously been reported at the periphery of cored neuritic plaques in the hippocampus of AD donors and within dystrophic neurites surrounding senile plaques in the neighboring entorhinal cortex [43–45]. Early observations of Braak staging of NFT pathology in AD highlighted the presence of AT8-reactive NFTs in the amygdala of elderly (aged≥70 years) AD donors [33]. The appearance of tau depositing as NFTs has been predominantly reported in pyramidal neurons upon labelling for phosphorylated tau. Therein, hyperphosphorylated tau filaments were observed near Aβ in approximately a third of AT8-immunolabelled senile plaques [43]. Furthermore, the co-localization of tau in senile plaques was not only restricted to AT8-reactive tau, as additional immunoreactivity to the phosphorylated-tau Ser396 isoform was always detected [43]. Therefore, our observations of coronal tau surrounding senile plaques would be expected to originate from phosphorylated forms of the protein in either dystrophic neurites or depositing as NFTs.
In summary, we have demonstrated the unequivocal association of aluminum and phosphorylated tau in NFTs in fAD. While aluminum binding to Aβ in senile plaques is expected in early disease progression concomitant with elevated amyloidogenic protein levels, an association with tau may occur at later disease stages [23, 24, 26, 35, 36]. Senile plaques containing dystrophic neurites are associated with neuronal death in AD, of which the ensuing neurotoxicity may be synergistically mediated by the co-existence of Aβ and aluminum in these neuropathological hallmarks [46–49]. Indeed, the latter potentiates the redox cycling of iron in the presence of Aβ42, generating reactive oxygen species, including the superoxide radical anion [50]. The subsequent extracellular release of NFTs from dying neurons may allow for a mechanism of aluminum binding in senile plaques to phosphorylated tau, exposed in filamentous regions of the misfolded protein [42]. Therapeutic approaches to reduce tau have been suggested to ameliorate its synergistic interactions with Aβ, reducing tau pathology and subsequent neuronal loss [51]. These data support the intricate associations of aluminum in the neuropathology of fAD, of which its subsequent reduction may further therapeutic benefits observed in ongoing clinical trials, in vivo [52].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
MM is a Children’s Medical Safety Research Institute (CMSRI: a charity based in Washington DC, USA) Research Fellow. We thank the families of all donors who donated tissues to the brain bank of the Universidad de Antioquia, Medellin, Colombia. Dr Johana Gómez-Ramírez and Dr Andrés Villegas-Lanau are thanked for tissue acquisition and processing for the delivery of FFPE brain tissue blocks to Keele University.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-210011.
REFERENCES
[1] | Hanger DP , Lau DHW , Phillips EC , Bondulich MK , Guo T , Woodward BW , Pooler AM , Noble W ((2014) ) Intracellular and extracellular roles for tau in neurodegenerative disease. J Alzheimers Dis 40: , S37–S45. |
[2] | Wang Y , Mandelkow E ((2016) ) Tau in physiology and pathology. Nat Rev Neurosci 17: , 22–35. |
[3] | Teravskis PJ , Ashe KH , Liao D ((2020) ) The accumulation of tau in postsynaptic structures: A common feature in multiple neurodegenerative diseases? Neuroscientist 26: , 503–520. |
[4] | Andreadis A ((2005) ) Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 1739: , 91–103. |
[5] | Bodea LG , Eckert A , Ittner LM , Piguet O , Götz J ((2016) ) Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J Neurochem 138: , 71–94. |
[6] | Zempel H , Dennissen FJA , Kumar Y , Luedtke J , Biernat J , Mandelkow EM , Mandelkow E ((2017) ) Axodendritic sorting and pathological missorting of tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem 292: , 12192–12207. |
[7] | Babür E , Tan B , DelibaŞ S , Yousef M , Dursun N , Süer C ((2019) ) Depotentiation of long-term potentiation is associated with epitope-specific tau hyper-/hypophosphorylation in the hippocampus of adult rats. J Mol Neurosci 67: , 193–203. |
[8] | Shastry BS ((2003) ) Neurodegenerative disorders of protein aggregation. Neurochem Int 43: , 1–7. |
[9] | Andrade-Moraes CH , Oliveira-Pinto AV , Castro-Fonseca E , da Silva CG , Guimarães DM , Szczupak D , Parente-Bruno DR , Carvalho LRB , Polichiso L , Gomes BV , Oliveira LM , Rodriguez RD , Leite REP , Ferretti-Rebustini REL , Jacob-Filho W , Pasqualucci CA , Grinberg LT , Lent R ((2013) ) Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 136: , 3738–3752. |
[10] | Karran E , Mercken M , De Strooper B ((2011) ) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10: , 698–712. |
[11] | Zhu XC , Tan L , Wang HF , Jiang T , Cao L , Wang C , Wang J , Tan CC , Meng XF , Yu JT ((2015) ) Rate of early onset Alzheimer’s disease: A systemic review and meta-analysis. Ann Transl Med 3: , 38. |
[12] | Goate A , Chartierharlin MC , Mullan M , Brown J , Crawford F ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: , 704–706. |
[13] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[14] | Exley C ((2006) ) Aluminium and iron, but neither copper nor zinc, are key to the precipitation of β-sheets Aβ42 in senile plaque cores in Alzheimer’s disease. J Alzheimers Dis 10: , 173–177. |
[15] | Dong J , Atwood CS , Anderson VE , Siedlak SL , Smith MA , Perry G , Carey PR ((2003) ) Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42: , 2768–2773. |
[16] | Iwatsubo T , Odaka A , Suzuki N , Mizusawa H , Nukina N , Ihara Y ((1994) ) Visualisation of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13: , 45–53. |
[17] | Amar F , Sherman MA , Rush T , Larson M , Boyle G , Chang L , Götz J , Buisson A , Lesné SE ((2017) ) The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal 10: , eaa12021. |
[18] | Selkoe DJ , Hardy J ((2016) ) The amyloid cascade hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[19] | Wong DY , Sept D ((2011) ) The interaction of cofilin with the actin filament. J Mol Biol 413: , 97–105. |
[20] | Guo JP , Arai T , Miklossy J , McGeer PL ((2006) ) Aβ and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 1953–1958. |
[21] | Atsmon-Raz Y , Miller Y ((2015) ) Insight into atomic resolution of the cross-seeding between tau/mutated tau and amyloid-β in neurodegenerative diseases. Isr J Chem 55: , 628–636. |
[22] | Exley C , Mold M ((2015) ) The binding, transport and fate of aluminium in biological cells. J Trace Elem Med Biol 30: , 90–95. |
[23] | Mold M , Linhart C , Gómez-Ramírez J , Villegas-Lanau A , Exley C ((2020) ) Aluminum and amyloid-β in familial Alzheimer’s disease. J Alzheimers Dis 73: , 1627–1635. |
[24] | Lopera F , Ardilla A , Martínez A , Madrigal L , Arango-Viana JC , Lemere CA , Arango-Lasprilla JC , Hincapíe L , Arcos-Burgos M , Ossa JE , Behrens IM , Norton J , Lendon C , Goate AM , Ruiz-Linares A , Rosselli M , Kosik KS ((1997) ) Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 277: , 793–799. |
[25] | Exley C , Clarkson E ((2020) ) Aluminium in human brain tissue from donors without neurodegenerative disease: A comparison with Alzheimer’s disease, multiple sclerosis and autism. Sci Rep 10: , 7770. |
[26] | Mold MJ , O’Farrell A , Morris B , Exley C ((2020) ) Aluminum and neurofibrillary tangle co-localization in familial Alzheimer’s disease and related neurological disorders. J Alzheimers Dis 78: , 139–149. |
[27] | Luque NB , Mujika JI , Rezabal E , Ugalde JM , Lopez X ((2014) ) Mapping the affinity of aluminium(III) for biophosphates: interaction mode and binding affinity in 1:1 complexes. Phys Chem Chem Phys 16: , 20107–20119. |
[28] | Gourmaud S , Shou H , Irwin DJ , Sansalone K , Jacobs LM , Lucas TH , Marsh ED , Davis KA , Jensen FE , Talos DM ((2020) ) Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 143: , 191–209. |
[29] | Guntern R , Bouras C , Hof PR , Vallet PG ((1992) ) An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer’s disease. Experientia 48: , 8–10. |
[30] | Augustinack JC , Schneider A , Mandelkow EM , Hyman BT ((2002) ) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103: , 26–35. |
[31] | Mercken M , Vandermeeren M , Lubke U , Six J , Boons J , Van de Voorde A , Martin JJ , Gheuens J ((1992) ) Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol 84: , 265–272. |
[32] | Bussière T , Bard F , Barbour R , Grajeda H , Guido T , Khan K , Schenk D , Games D , Seubert P , Buttini M ((2004) ) Morphological characterisation of thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Aβ immunotherapy on their clearance. Am J Pathol 165: , 987–995. |
[33] | Nelson PT , Abner EL , Schmitt FA , Kryscio RJ , Jicha GA , Santacruz K , Smith CD , Patel E , Markesbery WR ((2009) ) Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 68: , 774–784. |
[34] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Tredici KD ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[35] | Quiroz YT , Sperling RA , Norton DJ , Baena A , Arboleda-Velasquez JF , Cosio D , Schultz A , Lapoint M , Guzman-Velez E , Miller JB , Kim LA , Chen K , Tariot PN , Lopera F , Reiman EM , Johnson KA ((2018) ) Association between amyloid and tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol 75: , 548–556. |
[36] | McDade E , Randall DO , Bateman RJ ((2018) ) Tau positron emission tomography in autosomal dominant Alzheimer disease: Small windows, big picture. JAMA Neurol 75: , 536–538. |
[37] | Mirza A , King A , Troakes C , Exley C ((2016) ) The identification of aluminum in human brain tissue using lumogallion and fluorescence microscopy. J Alzheimers Dis 54: , 1333–1338. |
[38] | Moreno-García A , Kun A , Calero O , Medina M , Calero M ((2018) ) An overview of the role of lipofuscin in age-related neurodegeneration. Front Neurosci 12: , 464. |
[39] | Giaccone G , Orsi L , Cupidi C , Tagliavini F ((2011) ) Lipofuscin hypothesis of Alzheimer’s disease. Dement Geriatr Cogn Disord Extra 1: , 292–296. |
[40] | Riga D , Riga S , Halalau F , Schneider F ((2006) ) Brain lipopigment accumulation in normal and pathological aging. Ann N Y Acad Sci 1067: , 158–163. |
[41] | Michael R , Lenferink A , Vrensen GFJM , Barraquer EGRI , Otto C ((2017) ) Hyperspectral Raman imaging of neuritic plaques and neurofibrillary tangles in brain tissue from Alzheimer’s disease patients. Sci Rep 7: , 15603. |
[42] | Tokutake S , Nagase H , Morisaki S , Oyanagi S ((1995) ) Aluminium detected in senile plaques and neurofibrillary tangles is contained in lipofuscin granules with silicon, probably as aluminosilicate. Neurosci Lett 185: , 99–102. |
[43] | Furcila D , DeFelipe J , Alonso-Nanclares L ((2018) ) A study of amyloid-β and phosphotau in plaques and neurons in the hippocampus of Alzheimer’s disease patients. J Alzheimers Dis 64: , 417–435. |
[44] | Schmidt ML , Lee VM , Trojanowski JQ ((1991) ) Comparative epitope analysis of neuronal cytoskeletal proteins in Alzheimer’s disease senile plaque, neurites and neuropil threads. Lab Invest 64: , 352–35. |
[45] | Nelson PT , Jicha GA , Schmitt FA , Liu H , Davis DG , Mendiondo MS , Abner EL , Markesbery WR ((2007) ) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: Neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66: , 1136–1146. |
[46] | Niikura T , Tajima H , Kita Y ((2006) ) Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr Neuropharmacol 4: , 139–147. |
[47] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[48] | Malek-Ahmadi M , Perez SE , Chen K , Mufson EJ ((2016) ) Neuritic and diffuse plaque associations with memory in non-cognitively impaired elderly. J Alzheimers Dis 53: , 1641–1652. |
[49] | Knowles RB , Wyart C , Buldyrev SV , Cruz L , Urbanc B , Hasselmo ME , Stanley HE , Hyman BT ((1999) ) Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc Natl Acad Sci U S A 96: , 5274–5279. |
[50] | Khan A , Dobson JP , Exley C ((2006) ) Redox cycling of iron by Aβ42. Free Radical Biol Med 40: , 557–569. |
[51] | DeVos SL , Corjuc BT , Commins C , Dujardin S , Bannon RN , Corjuc D , Moore BD , Bennett RE , Jorfi M , Gonzales JA , Dooley PM , Roe AD , Pitstick R , Irimia D , Frosch MP , Carlson GA , Hyman BT ((2018) ) Tau reduction in the presence of amyloid-β prevents tau pathology and neuronal death in vivo. Brain 141: , 2194–2212. |
[52] | Tolar M , Abushakra S , Hey JA , Porsteinsson A , Sabbagh M ((2020) ) Aducanumab, gantenerumab, BAN2401 and ALZ-801 –the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther 12: , 95. |

