
Superior Frontal Gyrus TOMM40-APOE Locus DNA Methylation in Alzheimer’s Disease
Abstract
Background:
The APOE ɛ4 allele is the strongest known genetic risk factor for sporadic Alzheimer’s disease (AD). The neighboring TOMM40 gene has also been implicated in AD due to its close proximity to APOE.
Objective:
Here we tested whether methylation of the TOMM40-APOE locus may influence ApoE protein levels and AD pathology.
Methods:
DNA methylation levels across the TOMM40-APOE locus and ApoE levels were measured in superior frontal gyrus tissues of 62 human brains genotyped for APOE and scored for AD neuropathology.
Results:
Methylation levels within the TOMM40 CpG island in the promoter or APOE CpG island in Exon 4 did not differ between APOE ɛ4 carriers versus non-carriers. However, APOE ɛ4 carriers had significantly higher methylation the APOE promoter compared with non-carriers. Although DNA methylation at TOMM40, APOE promoter region, or APOE did not differ between AD pathological groups, there was a negative association between TOMM40 methylation and CERAD scores. ApoE protein concentrations did not significantly different between APOE ɛ4 carriers and non-carriers, or between AD pathological groups. Finally, there was no correlation between ApoE protein concentrations and DNA methylation levels.
Conclusion:
APOE gene methylation may not be affected by genotype, relate to AD pathology or ApoE protein levels in the superior frontal gyrus, though, DNA methylation at the ApoE promoter differed between genotype. DNA methylation at TOMM40 associated with amyloid-β plaques and longitudinal fluid intelligence. In sum, these results suggest a complicated regulation of the TOMM40-APOE locus in the brain in controlling ApoE protein levels and AD neuropathology.
INTRODUCTION
Apolipoprotein E, encoded by the APOE gene, is an important lipid transport protein in the central nervous system. There are three major isoforms (ɛ2, ɛ3, and ɛ4) in humans. The presence of the ɛ4 allele is the strongest known genetic risk factor for late-onset Alzheimer’s disease (AD); carrying one ɛ4 allele increases the risk of AD by 2- to 3-fold, while the presence of two ɛ4 alleles increases the risk by approximately 12-fold compared to individuals without any ɛ4 alleles. Carriers of APOE ɛ2 allele, on the other hand, are protected against AD [1]. However, not all ɛ4 carriers develop AD and therefore it may be possible that other ɛ4-allele-associated interacting factors, such as epigenetic mechanisms, could contribute towards the complex disease etiology [2].
Exon 4 of APOE contains a CpG island, and interestingly the two C/T single nucleotide polymorphisms (SNPs; rs429358 and rs7412) that determine the different ɛ2, ɛ3, and ɛ4 alleles, both of which can alter CpG dinucleotides. The ɛ4 allele introduces two additional CpG dinucleotides in the CpG island at rs429358 and rs7412, while ɛ3 introduces a CpG at rs7412 only and the ɛ2 allele lacks both the two CpG sites present in the ɛ4 allele [3]. It is hypothesized that such allelic differences in the CpG island could lead to allele-specific epigenetic changes that may contribute toward AD risk.
TOMM40, coding for the gene translocase of outer mitochondrial membrane 40, is in strong linkage disequilibrium with APOE. Genome-wide studies have identified variants within TOMM40 that associate with risk of AD [4, 5], while a further study showed blood TOMM40 expression was downregulated in subjects with AD compared with controls and the expression also correlated with cognitive decline [6]. The superior frontal gyrus, known to be heavily involved in a variety of cognitive tasks [7], is among the regions that show the greatest age-related surface area reductions [8], which further predicts risk of cognitive decline and dementia [9]. Furthermore, it has been shown that relative to ɛ3/3 genotypes, brains of ɛ3/4 genotypes show a significantly thinner cortex within the superior frontal gyrus [10].
Here we investigate whether the TOMM40-APOE locus is differentially methylated in AD brains and controls. We hypothesized that TOMM40 methylation is increased in AD brains and correlated with cognitive decline. We further considered if APOE methylation is also increased in AD brains, leading to reduced levels of ApoE protein by regulating ApoE protein expression, and that the methylation levels might associate with AD pathology. Finally, we tested whether the APOE alleles affect DNA methylation and could differentially regulate AD pathology.
METHODS
Study population
Fresh, frozen tissue was taken from the superior frontal gyrus (Brodmann area 8). Samples were acquired from donors through the Manchester Brain Bank. Ethical approval was granted from the Man-chester Brain Bank Committee. Donors were participants of a large prospective cognitive ageing cohort known as The University of Manchester Age and Cognitive Performance Research Cohort [11, 12] and included all those with available brain tissue and neuropathological data. The participants included 46 females and 21 males of Caucasian population from Manchester and Newcastle. The study took place over four waves between 1985 and 2002. Cognitive phenotypes (g factors) for vocabulary ability, memory, processing speed, and fluid intelligence for both cross-sectional (baseline scores) and longitudinal (baseline and subsequent scores) data were generated using Stata (StataCorp., College Station, TX: StataCorp LP). Empirical Bayes’ estimates for each individual were obtained from a random effects model fitted by maximum likelihood to the standardized age-regressed residuals obtained for each sex from the cognitive test scores. All g factors were corrected for age and the longitudinal data corrected for practice effects. Standardized residuals were used for analyses. For longitudinal g factors, missing data points were imputed by sampling the posterior distribution of factor scores using Mplus. The vocabulary ability (crystallized intelligence) factor was generated using tests that comprised the Mill Hill and Wais vocabulary tests [13, 14]. Cognitive tests used for fluid factors were the two parts of the Alice Heim test 4 [15] and the four subtests of the Culture Fair Test [16]. Speed factors were derived from the Alphabet Coding Task and the Random Letters test [17]. Factors for memory were generated from immediate and delayed recall, propositions, and spatial memory tests. These tests ceased in 2004 due to the increasing frailty of the volunteers (mean age, 78.9 years; 2004–2006 n = 729). The method of the cognitive assessment was previously described [11].
Thal phase [18], Braak stage [19], and CERAD score [20] were investigated as well as stratification into groups (None, Low, Intermediate, and High) based on the ‘ABC score’ recommended by National Institute on Aging–Alzheimer’s Association [21], which incorporates histopathological assessments for amyloid-β deposits (A), staging of neurofibrillary tangles (B), and scoring of neuritic plaques (C) to define 4 stages: Not, Low, Intermediate and High. Raw scores between two values were rounded up.
DNA methylation analysis
The TOMM40 promoter, APOE exon 4, and APOE promoter region were identified using previous publications [3, 22, 23] and the University of California Santa Cruz genome browser. Figure 1 shows the approximate location of the investigated CpGs in the TOMM40-APOE locus.
Fig. 1
Schematic representation of TOMM40-APOE locus showing approximate locations of the regions investigated in this study. Black bars represent CpG islands. Grey bars represent gene exons. Transcription of the genes occurs from left to right as shown by arrows.
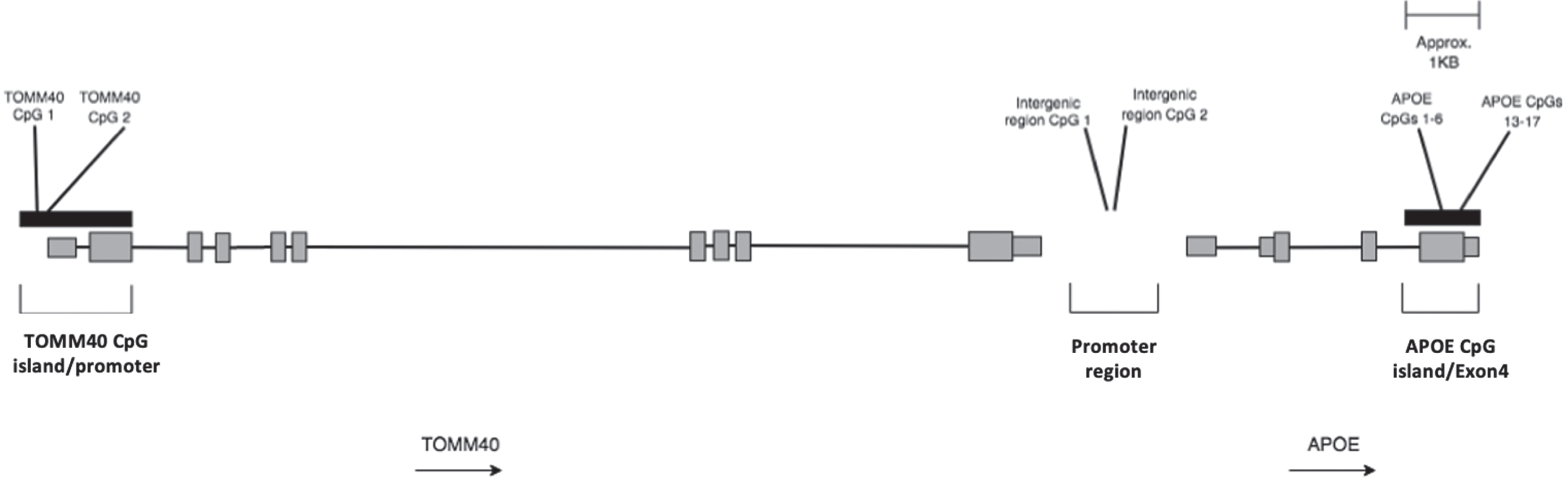
Table 2
Sequences in TOMM40 promoter CpG island APOE promoter region and APOE exon 4 CpG island analyzed in this study and primers used for their amplification and sequencing
| Gene location | Primers; Forward, reverse, sequencing * Biotin tagged | Sequence analyzed (CpGs numbered) 5′-3′ |
| APOE exon 4 | F: ATGAAGGAGTTGAAGGTTTATAAAT | 1.GTTGTAGGYGGYGTAGGTTYGGTTGGGYGYGGATATGGAGGAYGTGYGYGGTYGTTTGGTGTAGTATYGYGGYGAG |
| R: AACCCCCACCTAATACACTACCAA* | 2.YGTAAGTTGYGTAAGYGGTTTTTTYGYGATGTYGATGATTTGTAGAAGYGTTTGGTAGTGTATTAGGTYG | |
| S1: GGGTAAGGTTGTTTAAGGA | ||
| S2: GGGTGAGTTTAGTTTTTTATTTG | ||
| TOMM40 | F: GTTTTAGGGTAGAAGGATAGGTAAAGG | YGATTTAAAATYGGAAGTGTTGGTTTTTTTTAGAGTGTTT |
| R: AACACTCTAAAAAAAACCAACACTT* | ||
| S: TTTGGTTAGGAGTAGT | ||
| APOE promoter region | F: TTGGGATTTATAATAGGGTTTAGGAAAGTG | GYGTTTGAGYGTTTATTGTGGTTTGTTTATTGTTAGTTTTAATATAGGATYGTTGTGTGTTAGGGTTGTTTTTTATGTTTAATATAYGTTAGTTTGTTATTAAATATATTYGTGTYGTTGTTTTTTTAGTTTGATGAGTAAAGGAATTTGATGTTTAGAGAGGATAAGTTATTTGTTTAAGGTTATATAGTTGGTAATTGGTAGAGTTAG |
| R: CCTCTCTAAACATCAAATTCCTTTACT* | ||
| S: AGGGTTTAGGAAAGTGATA |
Genomic DNA was extracted using the Isolate II Genomic DNA kit (Bioline, London, UK) and 500 ng bisulfite-converted using the EpiMark Bisulfite Conversion Kit (New England Biolabs, Hitchen, UK) as previously described [24]. Primers used for DNA amplification and pyrosequencing were designed using Pyromark Assay Design SW 2.0 (Qiagen, Hilden, Germany) and manufactured by In vitro gen (Thermo Fisher Scientific, Cambridge, UK). PCRs were performed using 2μl of the bisulphite converted template DNA in MyTaq HS mix PCR reagents (Bioline, London, UK) including 0.2μM each of the forward and reverse (biotinylated) primers with the following conditions for all primer sets: initial denaturation of 5 min 95°C; 49 cycles of 95°C for 30 s, 58°C for 30 s, 72°C for 30 s; final extension of 72°C for 5 min. For the TOMM40 (CpG island and promoter) we assessed 2 CpG sites using a forward and biotinylated reverse primer, for the APOE promoter region 2 CpG sites were also assesses while 11 CpG sites were assessed across the APOE exon 4 regions (see Table 1 for all primer sequences). Following PCR, single-stranded biotinylated product was purified by mixing 10μl of the amplification mixture, 2μl of streptavidin sepharose HP (Amersham Biosciences, UK), and 40μl of binding buffer. The sepharose beads containing the immobilized biotinylated product were purified, washed, and denatured in 0.2 mol/l NaOH and washed again using the Pyrosequencing Vacuum Prep Tool (Qiagen). The biotinylated DNA was resuspended in 12μl of annealing buffer containing 0.3μmol/l pyrosequencing primer (see Table 1 for all pyrosequencing primer sequences). Two sequencing primers covering neighboring regions of the APOE exon 4 region were used and the sequence to analyze altered to allow for the polymorphic sites in the different genotypes. DNA methylation of CpG residues within the different regions were quantified by pyrosequencing using the PyroMark Q24 Advanced system with the PyroGold SQA reagent kit (Qiagen, Manchester, UK). The percentage methylation for each of the CpG sites was calculated using Pyro Q-CpG software (Qiagen). All assays performed in duplicate and final values represent an average of this.
Table 1
Cohort characteristics stratified by AD pathological change group
| AD pathological change group | |||||
| Variable | Not (n = 16) | Low (n = 19) | Intermediate (n = 25) | High (n = 6) | p (test statistic) |
| Age at death, y (SD) | 86.5 (6.4) | 87.6 (5.1) | 87.7 (6.3) | 88.5 (8.5) | 0.895 (F = 0.202) |
| Females, n (%) | 9 (56) | 15 (79) | 17 (68) | 5 (83) | 0.440 (χ2 = 2.702) |
| Postmortem delay, h (SD)a | 76.6 (56.6) | 72.1 (44.0) | 79.8 (38.9) | 67.2 (29.2) | 0.922 (F = 0.161) |
| Whole brain weight, g (SD)b | 1217 (156.6) | 1167 (118.7) | 1227 (149.2) | 1312 (31.8) | 0.445 (F = 0.910) |
| APOE ɛ4 carrier, n (%) | 1 (6) | 6 (32) | 12 (48) | 3 (50) | 0.037 (χ2 = 8.478) |
aData available in: Not n = 14, Low n = 19, Intermediate n = 21, High n = 5 bData available in: Not n = 8, Low n = 14, Intermediate n = 18, High n = 2.
ApoE protein quantification
The lysate used for the ApoE protein quantification was extracted from the brain tissue using RIPA buffer (Sigma, UK) and 1x protease inhibitor cocktail (Sigma, UK), as described previously [25]. The concentration of ApoE protein in tissue samples were measured using the Apolipoprotein E Human ELISA kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions and as previously described in brain lysates [26]. Absorbances at 450 and 570 nm were measured using Synergy HT microplate reader (BioTek Instruments, Inc., VT, US) and a 7-point standard curve generated in GraphPad. Sample ApoE concentrations were then interpolated and concentrations were standardized to account for the total protein input.
Genotyping
Genotyping of the APOE rs7412 and rs429358 polymorphisms to enable APOE allele status was previously performed using the Sequenom MassARRAY iPLEX platform [27]. APOE alleles were defined as: ɛ2 = T (rs429358)/T (rs7412); ɛ3 = T (rs429358) / C (rs7412); ɛ4 = C (rs429358) / C (rs7412).
Statistical analysis
All statistical analyses were performed in GraphPad Prism (version 9). To assess differences between continuous patient characteristic variables between groups, we used either a one-way ANOVA or unpaired t-test. For categorical patient characteristic variables, differences between groups were assessed with either a Pearson chi-squared or Fisher’s exact test. DNA methylation of individual CpG sites with each region of interest were averaged using the geometric mean and log2-transformed prior to statistical testing. ApoE protein concentrations were square-root transformed prior to statistical testing. Group differences were assessed using either a one-way ANOVA or unpaired t-test. Spearman rank correlation was used to assess correlations between variables. Linear regression, using SPSS version 24, was used to levels of TOMM40 methylation with longitudinal and cross-sectional measures of cognition.
RESULTS
Cohort characteristics
Clinical and pathological characteristics of the study sample stratified by AD pathological groups can be found in Table 1. All groups were well matched for donor age at death, sex, postmortem delay time, and whole brain weight. Cohort characteristics stratified by APOE ɛ4 status can be found Supplementary Table 1. The frequency of APOE ɛ4 carriers increased with more severe AD pathological changes.
TOMM40-APOE locus DNA methylation
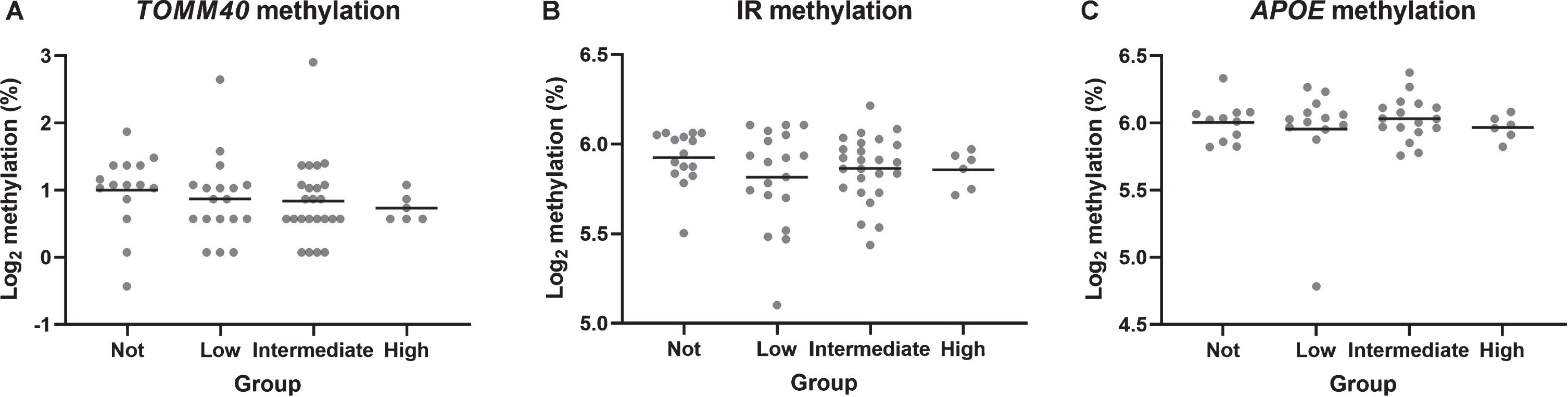
Levels of DNA methylation at the TOMM40 CpG island (F = 0.386, p = 0.764), APOE promoter region (F = 0.806, p = 0.495), and APOE CpG island (F = 0.306, p = 0.821) did not significantly differ be-tween AD pathology groups (Fig. 2) or between gender (all p > 0.05). TOMM40 methylation was negatively association with CERAD severity scores (rho = –0.254, p = 0.045; Supplementary Figure 1) though, this is a nominal p-value and not corrected multiple testing. When testing TOMM40 methyla-tion with cognitive measures, regression analyses revealed significant negative correlations with both cross-sectional fluid intelligence (B = –0.655, p < 0.001) and longitudinal fluid intelligence (B =–0.368, p = 0.013), but not with vocabulary (cross-sectional, B = –0.493, p = 0.24; longitudinal, B =0.537, p = 0.201) or memory (cross-sectional, B =0.134, p = 0.469; longitudinal, B = –0.082, p = 0.658).
Fig. 2
TOMM40 CpG island (A), APOE promoter (B), and APOE CpG island (C) methylation differences between groups. Horizontal lines represent mean group values. TOMM40 n = 67; APOE promoter n = 66, APOE CpG island n = 64.
APOE DNA methylation, protein concentrations and APOE ɛ4 carrier status
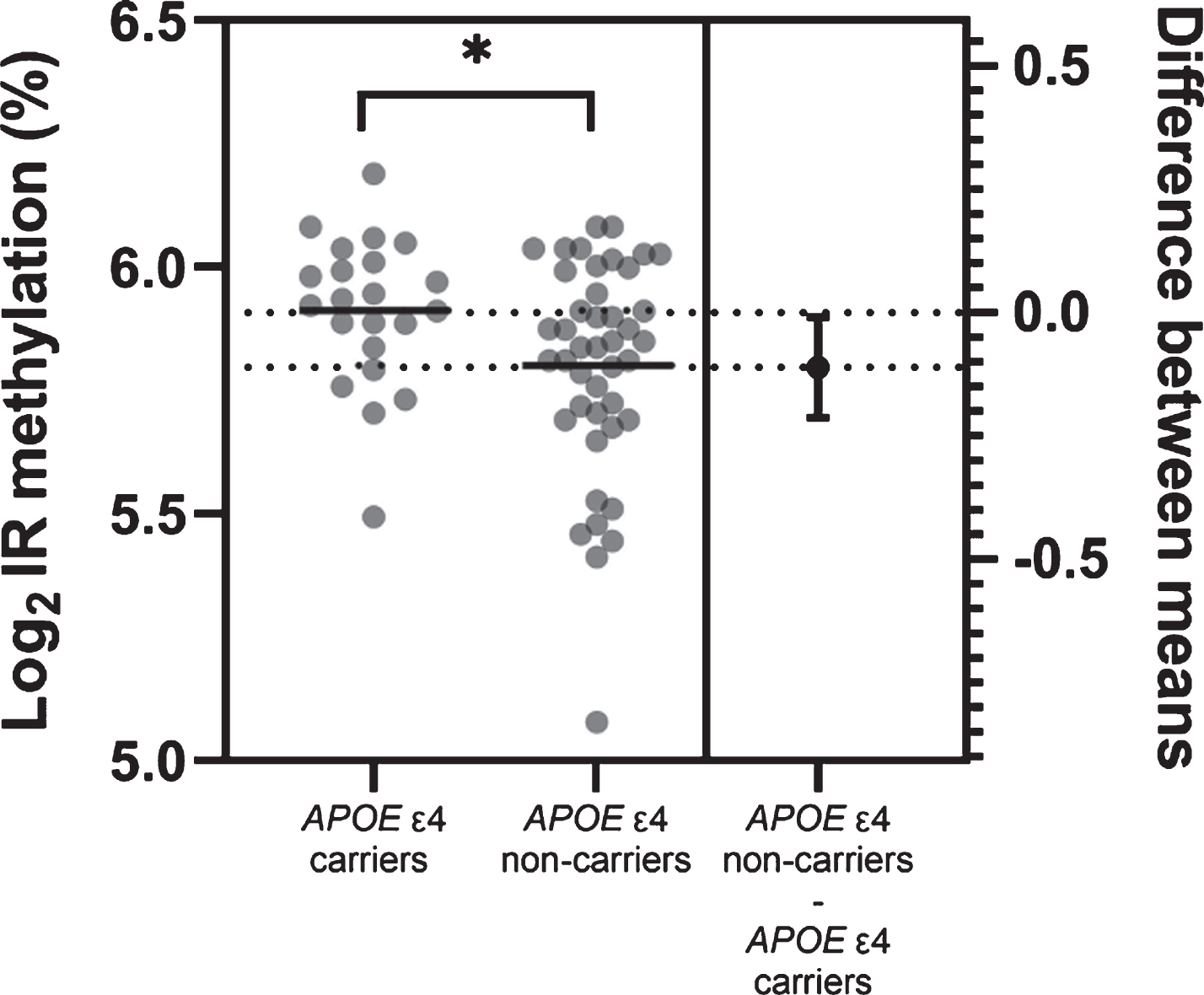
Next, we grouped samples according to APOE ɛ4 status. There was no difference in methylation levels for the TOMM40 (t = 0.755, p = 0.456) or APOE (t = 0.334, p = 0.742) regions between APOE ɛ4 carriers (homozygous/heterozygous ɛ4) and non-carriers. However, APOE ɛ4 carriers had significantly higher methylation levels at the APOE promoter region, compared with non-carriers (t = 2.158, p = 0.035; Fig. 3).
Fig. 3
Estimation plot for APOE promoter region methylation differences between APOE ɛ4 carriers and non-carriers. Horizontal lines represent mean group values. Difference between group means with 95%confidence intervals is also plotted. n = 66. *p < 0.05.
ApoE protein levels did not significantly different between APOE ɛ4 carriers and non-carriers (t = 1.624, p = 0.110), or between AD pathological groups (F = 1.069, p = 0.370). Further, when analyzing all samples collectively, there was no correlation between ApoE protein levels and DNA methylation levels (rho = –0.041, p = 0.793; Supplementary Figure 1).
DISCUSSION
The results of this study show that APOE ɛ4 allele status did not affect APOE methylation but was associated with a difference in methylation levels within the APOE promoter region. Though DNA methylation at TOMM40 or APOE did not differ between AD pathological groups, TOMM40 methylation was associated with lower CERAD scores and reduced measures of cross-sectional and longitudinal fluid intelligence. ApoE protein concentrations did not significantly differ with APOE ɛ4 allele status or between AD pathological groups, and there were no correlations between ApoE protein concentrations and DNA methylation levels at the APOE promoter region or Exon 4 CpG island.
It was hypothesized that the APOE exon 4 region methylation level may differ between the genotypes, especially as the rs7412 (C/T) removes a CpG site, and the rs429358 (T/C) creates an extra CpG site. This study, however, did not find any significant difference between the methylation levels of the APOE exon 4 CpG island. One previous study, has shown higher APOE exon 4 CpG island methylation in the frontal lobe among carriers of ɛ3/ɛ4 genotypes compared to the ɛ3/ɛ3 carriers [3]. However, the effect was only seen in the controls but not in AD cases and the sample size was small (n = 25). Other studies that investigated the APOE locus methylation did not observe any genotype effect on the methylation levels [23], and they did not investigate the difference in methylation between different genotypes [2, 22].
Interestingly, there were lower levels of methylation in the promoter region of APOE ɛ4 carriers. Two studies investigating blood DNA, reported differential methylation at CpGs in the APOE promoter (cg04406254, cg01032398) with AD and cognition, suggesting a possible regulatory role [23]. Both these CpGs were in close proximity to the CpGs we found associated in this study. A twin study found that methylation in the promoter, but not the exon 4 CpG island, increased the risk of AD; however, they found no association with APOE ɛ4 and methylation and stratification on genotype suggesting the association between promoter methylation and AD was not genotype-specific. Another study found methylation at the promoter was significantly negatively correlated with cognitive function in a study of 289 African Americans with a mean age of 67 [28].
DNA methylation at TOMM40 or APOE did not differ between AD neuropathological groups when stratified using the ABC score. However, DNA methylation at the TOMM40 promoter negatively associated with CERAD scores supporting that its regulation might relate to amyloid-β plaque amyloid deposition [29]. That APOE methylation did not differ with AD neuropathology appears contrary to some previously published studies showing differential methylation of APOE Exon 4 CpG island in AD brains compared with controls, in an APOE-genotype-specific manner [3], and lower methylation in frontal lobe, but not cerebellar, tissue from AD brains compared with controls, suggesting tissue-specific dysregulations [2]. Shao et al. [22] found CpG methylation across the locus significantly differed between AD and cognitively normal controls in a small number of 12 cerebellum and hippocampal samples, while methylation did not different between AD and cognitively normal controls in 67 blood samples. Perhaps, the results in the superior frontal gyrus might suggest a different mechanism.
TOMM40 polymorphisms have been reported to increase susceptibility to AD [30]. TOMM40 rs10524523 variable poly-T repeats were found to be associated with AD risk and age at onset [31], while rs157581-G conferred an increased risk for developing AD and mild cognitive impairment (MCI) in patients [32]. In the previously mentioned study by Liu et al., a further CpG site in TOMM40 was found to show a significant inverse association between methylation level and delayed recall [28]. A further study on the University of Manchester Age and Cognitive Performance Research Cohort found that poly-T repeat polymorphism, which associates with reduced gene expression, associated with reduced cognition in the same cases studied here [33]. It is therefore interesting that the present study suggests that methylation at the TOMM40 promoter negatively associated with a measure of cognition.
Here we found that ApoE protein levels in the brains did not differ with APOE ɛ4 allele status, between AD pathological groups, or correlate with DNA methylation levels at the APOE promoter region or Exon 4 CpG island suggesting that APOE genotypes may modulate AD risk independently of total ApoE protein production. That ApoE protein levels did not differ between APOE ɛ4 carriers and non carriers would suggest that APOE genotype does not affect protein expression in the superior frontal gyrus. These findings differ from some previous publications in which APOE ɛ4 allele has been associated with decreased ApoE protein levels in in vitro studies [34], mouse brains and human frontotemporal lobe [35]. However, the absolute levels of the ApoE ɛ3 protein were similar between carriers of ɛ3/ɛ3 and ɛ3/ɛ4 genotype, suggesting that the overall decrease in the ApoE protein levels could be due to the reduction in the ɛ4 isoform. Here we only measured the total ApoE levels and not the levels of the specific APOE isoforms. This could mean that the overall ApoE levels do not significantly affect AD pathology development and the effect is isoform-specific, though the few findings from studies on this are inconsistent [36]. According to Riddell et al., APOE ɛ4 has a shorter half-life and is degraded at a higher rate than APOE ɛ3, which might be the cause for an overall decreased ApoE protein levels in the APOE ɛ4 carriers [34]. An isoform-specific ApoE protein quantification would allow us to assess the effect of the individual genotypes more accurately.
There are several limitations to this study. With multifactorial diseases such as dementia and AD, individual SNPs or gene-specific methylation, may confer only a modest influence on the pathology and the diseases risk, and thus may require larger sample sizes to provide the necessary statistical power to detect small effects. As a result of a large postmortem delay in some samples, the levels of mRNA were of poor quality with low RIN values. Therefore, we were unable to measure ApoE and TOMM40 mRNA levels which may provide an insight into how APOE methylation directly affects the level of mRNA transcripts and which CpG sites may have a significant effect. Another limitation are the unknown potential effects that the AD pathological changes could have on cell fractions, for example reduced neurons and increased glial cells and how these changes may influence the results. Finally, we should also consider the possible role of sex-differences. Though methylation did not differ between genders, this study was not large enough to test for sex-differences in gene-specific regulation in AD neuropathology.
Sporadic AD is a complex disorder, displaying heterogeneous pathogenesis, and numerous genetic variants together with environmental factors may interact to influence the diseases risk. TOMM40-APOE locus methylation might influence the risk of AD by modulating ApoE protein expression directly in the brain. This is the first study to investigate APOE methylation with protein levels in the brain suggesting there is no association, or we lack the power to detect it. The association between APOE genotype/APOE promoter methylation and the TOMM40 promoter methylation, suggests possible regulatory mechanisms that may or may not interact. More work is needed to understand these molecular mechanisms within the brain and AD neuropathology.
CONFLICT OF INTEREST
The authors have no conflict of interests to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-201000.
REFERENCES
[1] | Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[2] | Tulloch J , Leong L , Thomson Z , Chen S , Lee EG , Keene CD , Millard SP , Yu CE ((2018) ) Glia-specific APOE epigenetic changes in the Alzheimer’s disease brain. Brain Res 1698: , 179–186. |
[3] | Foraker J , Millard SP , Leong L , Thomson Z , Chen S , Keene CD , Bekris LM , Yu CE , Fischer A ((2015) ) The APOE gene is differentially methylated in Alzheimer’s disease. J Alzheimers Dis 48: , 745–755. |
[4] | Omoumi A , Fok A , Greenwood T , Sadovnick AD , Feldman HH , Hsiung GYR ((2014) ) Evaluation of late-onset Alzheimer disease genetic susceptibility risks in a Canadian population. Neurobiol Aging 35: , 936.e5–936.e12. |
[5] | Roses AD , Lutz MW , Amrine-Madsen H , Saunders AM , Crenshaw DG , Sundseth SS , Huentelman MJ , Welsh-Bohmer KA , Reiman EM ((2010) ) A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 10: , 375–384. |
[6] | Mise A , Yoshino Y , Yamazaki K , Ozaki Y , Sao T , Yoshida T , Mori T , Mori Y , Ochi S , Iga JI , Ueno SI ((2017) ) TOMM40 and APOE gene expression and cognitive decline in Japanese Alzheimer’s disease subjects. J Alzheimers Dis 60: , 1107–1117. |
[7] | Li W , Qin W , Liu H , Fan L , Wang J , Jiang T , Yu C ((2013) ) Subregions of the human superior frontal gyrus and their connections. Neuroimage 78: , 46–58. |
[8] | Lemaitre H , Goldman AL , Sambataro F , Verchinski BA , Meyer-Lindenberg A , Weinberger DR , Mattay VS ((2012) ) Normal age-related brain morphometric changes: Nonuniformity across cortical thickness, surface area and gray matter volume? Neurobiol Aging 33: , 617.e1–617.e9. |
[9] | Gourley D , Pasha EP , Kaur SS , Haley AP , Tanaka H ((2020) ) Association of dementia and vascular risk scores with cortical thickness and cognition in low-risk middle-aged adults. Alzheimer Dis Assoc Disord 34: , 313–317. |
[10] | Fennema-Notestine C , Panizzon MS , Thompson WR , Chen CH , Eyler LT , Fischl B , Franz CE , Grant MD , Jak AJ , Jernigan TL , Lyons MJ , Neale MC , Seidman LJ , Tsuang MT , Xian H , Dale AM , Kremen WS ((2011) ) Presence of ApoE ɛ4 allele associated with thinner frontal cortex in middle age. J Alzheimers Dis 26: , 49–60. |
[11] | Rabbitt PMA , McInnes L , Diggle P , Holland F , Bent N , Abson V , Pendleton N , Horan M ((2004) ) The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age, 1983 through 2003. Aging Neuropsychol Cogn 11: , 245–279. |
[12] | Robinson AC , Davidson YS , Horan MA , Pendleton N , Mann DMA ((2018) ) Pathological correlates of cognitive impairment in The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age. J Alzheimers Dis 64: , 483–496. |
[13] | Raven J ((1965) ) The Mill Hill Vocabulary Scale, HK Lewis, London. |
[14] | Weshler D ((1986) ) The Wechsler Adult Intelligence Test-Revised. New York. |
[15] | Heim A (1970) AH4 Group Test of General Intelligence. |
[16] | Catell RB , Cattell A (1960) The individual or group Culture Fair intelligence test, I.P.A.T. Champaign, Illinois. |
[17] | Savage R (1984) Alphabet Coding Task-15, Murdock University, Perth, W. Australia. |
[18] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[19] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[20] | Mirra SS , Heyman A , McKeel D , Sumi SM , Crain BJ , Brownlee LM , Vogel FS , Hughes JP , van Belle G , Berg L , Ball MJ , Bierer LM , Claasen D , Hansen LR , Hart M , Hedreen J , Baltimore B , Hen Derson V , Hyman BT , Joachim C , Mark-Esbery W , Mar Tinez AJ , McKee A , Miller C , Moossy J , Nochlin D , Perl D , Petito C , Rao GR , Schelper RL , Slager U , Terry RD ((1991) ) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41: , 479–486. |
[21] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters H V , Montine TJ ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[22] | Shao Y , Shaw M , Todd K , Khrestian M , D’Aleo G , Barnard PJ , Zahratka J , Pillai J , Yu CE , Keene CD , Leverenz JB , Bekris LM ((2018) ) DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J Hum Genet 63: , 459–471. |
[23] | Karlsson IK , Ploner A , Wang Y , Gatz M , Pedersen NL , Hägg S ((2018) ) Apolipoprotein E DNA methylation and late-life disease. Int J Epidemiol 47: , 899–907. |
[24] | Aarons T , Bradburn S , Robinson A , Payton A , Pendleton N , Murgatroyd C ((2019) ) Dysregulation of BDNF in prefrontal cortex in Alzheimer’s disease. J Alzheimers Dis 69: , 1089–1097. |
[25] | Bradburn S , McPhee J , Bagley L , Carroll M , Slevin M , Al-Shanti N , Barnouin Y , Hogrel J-Y , Pääsuke M , Gapeyeva H , Maier A , Sipilä S , Narici M , Robinson A , Mann D , Payton A , Pendleton N , Butler-Browne G , Murgatroyd C ((2018) ) Dysregulation of C-X-C motif ligand 10 during aging and association with cognitive performance. Neurobiol Aging 63: , 54–64. |
[26] | Cao J , Gaamouch F El , Meabon JS , Meeker KD , Zhu L , Zhong MB , Bendik J , Elder G , Jing P , Xia J , Luo W , Cook DG , Cai D ((2017) ) ApoE4-associated phospholipid dysregulation contributes to development of Tau hyper-phosphorylation after traumatic brain injury. Sci Rep 7: , 11372. |
[27] | Davies G , Harris SE , Reynolds CA , Payton A , Knight HM , Liewald DC , Lopez LM , Luciano M , Gow AJ , Corley J , Henderson R , Murray C , Pattie A , Fox HC , Redmond P , Lutz MW , Chiba-Falek O , Linnertz C , Saith S , Haggarty P , McNeill G , Ke X , Ollier W , Horan M , Roses AD , Ponting CP , Porteous DJ , Tenesa A , Pickles A , Starr JM , Whalley LJ , Pedersen NL , Pendleton N , Visscher PM , Deary IJ ((2014) ) A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol Psychiatry 19: , 76–87. |
[28] | Liu J , Zhao W , Ware EB , Turner ST , Mosley TH , Smith JA ((2018) ) DNA methylation in the APOE genomic region is associated with cognitive function in African Americans. BMC Med Genomics 11: , 43. |
[29] | Valant V , Keenan BT , Anderson CD , Shulman JM , Devan WJ , Ayres AM , Schwab K , Goldstein JN , Viswanathan A , Greenberg SM , Bennett DA , de Jager PL , Rosand J , Biffi A ((2012) ) TOMM40 in cerebral amyloid angiopathy related intracerebral hemorrhage: Comparative genetic analysis with Alzheimer’s disease. Transl Stroke Res 3: , 102–112. |
[30] | Jiao B , Liu X , Zhou L , Wang MH , Zhou Y , Xiao T , Zhang W , Sun R , Waye MMY , Tang B , Shen L ((2015) ) Polygenic analysis of late-onset Alzheimer’s disease from mainland China. PLoS One 10: , e0144898. |
[31] | Roses AD , Lutz MW , Crenshaw DG , Grossman I , Saunders AM , Gottschalk WK ((2013) ) TOMM40 and APOE: Requirements for replication studies of association with age of disease onset and enrichment of a clinical trial. Alzheimers Dement 9: , 132–136. |
[32] | Bagnoli S , Piaceri I , Tedde A , Bessi V , Bracco L , Sorbi S , Nacmias B ((2013) ) Tomm40 polymorphisms in Italian Alzheimer’s disease and frontotemporal dementia patients. Neurol Sci 34: , 995–998. |
[33] | Payton A , Sindrewicz P , Pessoa V , Platt H , Horan M , Ollier W , Bubb VJ , Pendleton N , Quinn JP ((2016) ) A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in nonpathologic aging. Neurobiol Aging 39: , 217.e1–217.e7. |
[34] | Riddell DR , Zhou H , Atchison K , Warwick HK , Atkinson PJ , Jefferson J , Xu L , Aschmies S , Kirksey Y , Hu Y , Wagner E , Parratt A , Xu J , Li Z , Zaleska MM , Jacobsen JS , Pangalos MN , Reinhart PH ((2008) ) Impact of Apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci 28: , 11445–11453. |
[35] | Bray NJ , Jehu L , Moskvina V , Buxbaum JD , Dracheva S , Haroutunian V , Williams J , Buckland PR , Owen MJ , O’Donovan MC ((2004) ) Allelic expression of APOE in human brain: Effects of epsilon status and promoter haplotypes. Hum Mol Genet 13: , 2885–2892. |
[36] | Minta K , Brinkmalm G , Janelidze S , Sjödin S , Portelius E , Stomrud E , Zetterberg H , Blennow K , Hansson O , Andreasson U ((2020) ) Quantification of total apolipoprotein e and its isoforms in cerebrospinal fluid from patients with neurodegenerative diseases. Alzheimers Res Ther 12: , 19. |


