
In-Frame and Frameshift Mutations in Zebrafish Presenilin 2 Affect Different Cellular Functions in Young Adult Brains
Abstract
Background:
Mutations in PRESENILIN 2 (PSEN2) cause early onset familial Alzheimer’s disease (EOfAD) but their mode of action remains elusive. One consistent observation for all PRESENILIN gene mutations causing EOfAD is that a transcript is produced with a reading frame terminated by the normal stop codon—the “reading frame preservation rule”. Mutations that do not obey this rule do not cause the disease. The reasons for this are debated.
Objective:
To predict cellular functions affected by heterozygosity for a frameshift, or a reading frame-preserving mutation in zebrafish psen2 using bioinformatic techniques.
Methods:
A frameshift mutation (psen2N140fs) and a reading frame-preserving (in-frame) mutation (psen2T141 _ L142delinsMISLISV) were previously isolated during genome editing directed at the N140 codon of zebrafish psen2 (equivalent to N141 of human PSEN2). We mated a pair of fish heterozygous for each mutation to generate a family of siblings including wild type and heterozygous mutant genotypes. Transcriptomes from young adult (6 months) brains of these genotypes were analyzed.
Results:
The in-frame mutation uniquely caused subtle, but statistically significant, changes to expression of genes involved in oxidative phosphorylation, long-term potentiation and the cell cycle. The frameshift mutation uniquely affected genes involved in Notch and MAPK signaling, extracellular matrix receptor interactions and focal adhesion. Both mutations affected ribosomal protein gene expression but in opposite directions.
Conclusion:
A frameshift and an in-frame mutation at the same position in zebrafish psen2 cause discrete effects. Changes in oxidative phosphorylation, long-term potentiation and the cell cycle may promote EOfAD pathogenesis in humans.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder which develops silently over decades [1]. The pathological hallmarks of AD include the presence of extracellular senile neuritic plaques consisting, primarily, of amyloid-β (Aβ) peptides, intracellular neurofibrillary tangles (primarily consisting of hyperphosphorylated tau proteins), and progressive neuronal loss (reviewed in [2]). The majority of therapeutics for AD are aimed at reducing Aβ levels (reviewed in [3]). However, all compounds to date show little or no effect on cognitive symptoms (reviewed in [4, 5]). This likely reflects our ignorance of the pathogenic mechanism underlying AD. Also, once cognitive changes occur, damage to the brain is considerable and may be irreversible. A comprehensive understanding of the early molecular changes/stresses occurring many years before disease onset is required to develop preventative treatments to reduce the prevalence of AD.
The majority of AD cases arise sporadically with an age of onset after 65 years (late onset AD, LOAD). Genome-wide association studies (GWAS) have identified at least 20 genes associated with increased risk for LOAD, with the most influential being the APOLIPOPROTEIN E (APOE) locus [6, 7]. Rare, familial forms of AD also exist. Early onset familial AD (EOfAD) cases have an age of onset before 65 years and arise due to mutations in the PRESENILIN genes (PSEN1 and PSEN2), and the genes AMYLOID β A4 PRECURSOR PROTEIN (AβPP) and SORTILIN-RELATED RECEPTOR 1 (SORL1) (reviewed in [8–10]).
PSEN2 is the less commonly mutated PSEN gene in EOfAD. To date, only 13 pathogenic mutations have been described in PSEN2 compared to over 185 pathogenic mutations in the homologous gene PSEN1 [11]. The first PSEN2 EOfAD mutation characterized was N141I, found in Volga German families and affecting the second transmembrane domain of the PSEN2 protein [12]. We previously attempted to introduce this mutation into zebrafish by genome editing. While we were not successful at introducing an exact equivalent of the human N141I mutation into zebrafish, we did isolate an in-frame mutation at this position: psen2T141 _ L142delinsMISLISV [13]. This in-frame mutation alters two codons and inserts five additional novel codons and was predicted to largely preserve the transmembrane structure of the resultant protein. We also isolated a frameshift mutation at the same position: psen2N140fs. This is a deletion of 7 nucleotides resulting in a premature stop codon at novel codon position 142 [13]. Since all EOfAD mutations in the PSENs follow a “reading frame preservation rule” [14], psen2T141 _ L142delinsMISLISV models such a mutation in PSEN2. In contrast, the psen2N140fs allele is predicted to express a truncated protein, is not EOfAD-like, and so can act as a negative control to investigate the differences between in-frame and frameshift mutations of PSEN2.
The overall goal of the Alzheimer’s Disease Genetics Laboratory has been to identify the cellular processes affected in common by EOfAD mutations in different genes in young adult, heterozygous mutant brains (i.e., to establish an early EOfAD brain transcriptomic signature). We exploit zebrafish as a model organism for this work as the EOfAD genes are conserved in zebrafish, and large families containing both EOfAD-like mutant fish and their wild type siblings can be generated from single mating events between single pairs of fish (reviewed in [15]). Analyzing the transcriptomes of sibling fish raised in the same environment (aquaria within a single recirculated water system) reduces genetic and environmental sources of variation (noise) and allows subtle effects of mutations to be identified. After introducing EOfAD-like mutations into the endogenous, zebrafish orthologues of EOfAD genes (i.e., knock-in models), we analyze their effects on brain transcriptomes in heterozygous fish to mimic the genetic state of such mutations in the human disease. We term this experimental strategy Between Sibling Transcriptome (BeST) analysis. We have previously performed BeST analyses for EOfAD-like mutations in psen1 [16–19] and sorl1 [20, 21] and for a complex (but possibly EOfAD-like) mutation in psen2: psen2S4Ter [22].
In the work described in this report, we performed a BeST analysis using a family of sibling fish generated by mating two fish with genotypes psen2T141 _ L142delinsMISLISV/+ and psen2N140fs/+. The family included fish heterozygous for the mutations psen2T141 _ L142delinsMISLISV (for simplicity, hereafter referred to as “EOfAD-like”) and psen2N140fs (for simplicity, hereafter referred to as “FS”) and their wild type siblings, all raised together in the same tank until 6 months of age. This strategy allowed direct comparisons of the brain transcriptomes from the two psen2 mutant genotypes to their wild type siblings while reducing confounding effects from genetic and environmental variation. Despite this, considerable gene expression variation (noise) is still observed between individuals and the effects of the mutations are subtle so that few genes are identified initially as differentially expressed. However, when analyzed at the pathway level, heterozygosity for each mutation shows distinct transcriptomic effects. The sets of genes involved in oxidative phosphorylation, long-term potentiation, and the cell cycle are only altered significantly in the EOfAD-like brains, while the FS mutant brains shows apparent changes in Notch and MAPK signaling, focal adhesion, and ECM receptor interaction. Both mutation states affected genes encoding ribosomal subunits, but the effect of each mutation was in opposite directions. Our results are consistent with the concept that only dominant, in-frame mutations of the PRESENILIN genes cause EOfAD and support that effects on oxidative phosphorylation may be a common signature of such mutations.
METHODS
Zebrafish lines and ethics statement
Generation of the psen2T141 _ L142delinsMISLISV and psen2N140fs lines of zebrafish used in this study is described in [13]. All experiments involving zebrafish were conducted under the auspices of the Animal Ethics Committee of the University of Adelaide, permit numbers S-2017–089 and S-2017–073.
Breeding strategy
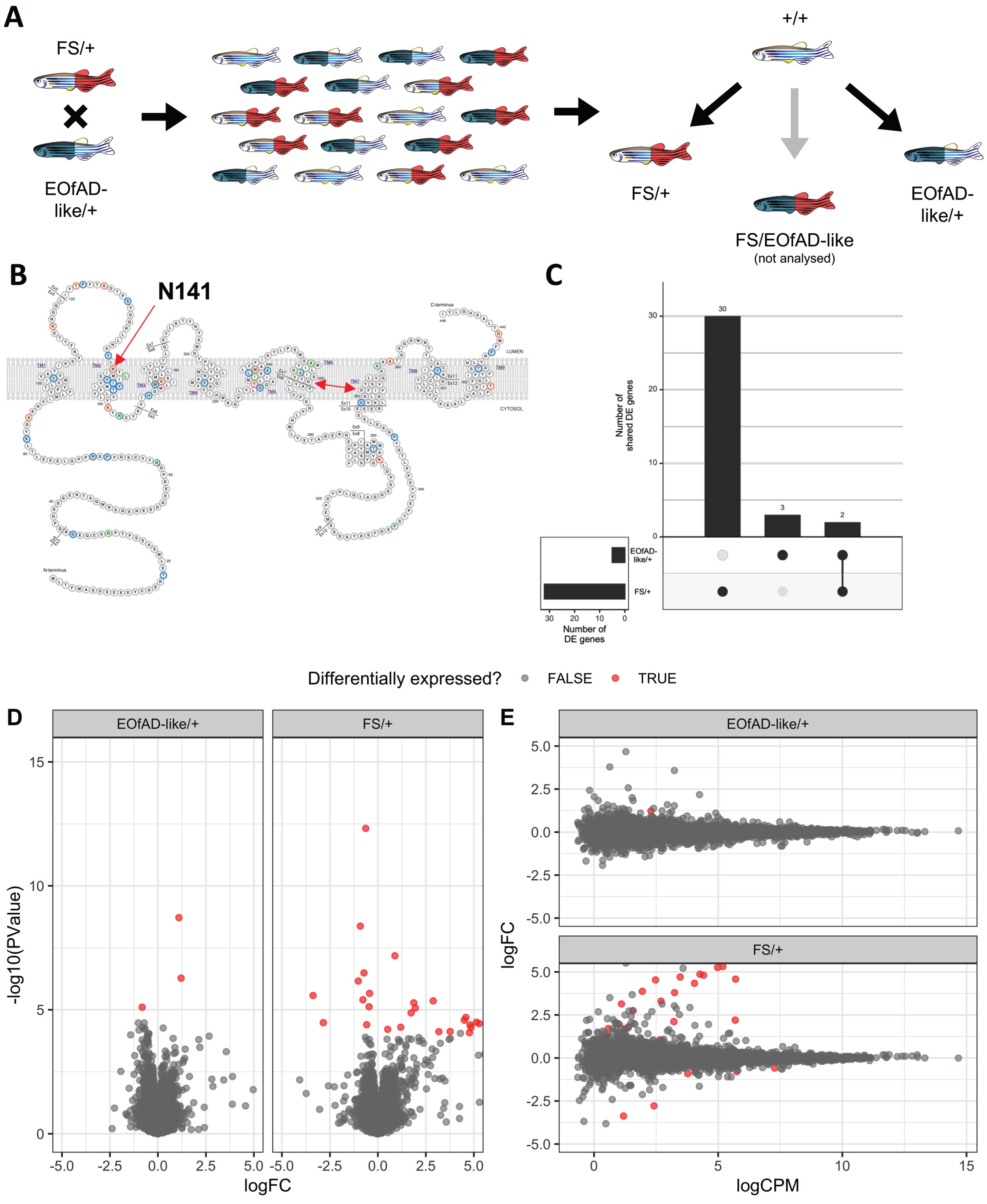
Two fish heterozygous for these mutations were mated to generate a family of sibling fish with psen2 genotypes EOfAD-like/+, FS/+, EOfAD-like/FS (transheterozygous) or wild type (Fig. 1A). This family was raised together in the same tank until 6 months of age, at which time 24 fish were randomly selected and sacrificed in a loose ice slurry (to allow for n = 5 of each desired genotype in the RNA-seq analysis). The heads of these fish were removed and stored in 600μL of RNAlater Stabilization Solution (Invitrogen, Carlsbad, CA, USA), while their tails were removed for genomic DNA extraction and genotyping by polymerase chain reactions (PCRs).
Fig. 1
A) Mating strategy and experimental design. B) Schematic of the human PSEN2 protein (adapted from https://www.alzforum.org/mutations/psen-2 with permission from FBRI LLC (Copyright © 1996–2020 FBRI LLC. All Rights Reserved. Version 2.7 –2020)). Amino acid residues are color-coded to indicate whether substitutions have been observed to be pathogenic (orange), non-pathogenic (green) or of uncertain pathogenicity (blue). The site of the human EOfAD mutation N141I, in the second transmembrane domain (TMD) is indicated by the red single-headed arrow. The aspartate residues critical for γ-secretase catalysis are indicated by a red double-headed arrow. C) Upset plot indicating the number of differentially expressed (DE) genes in each comparison of the psen2 mutant genotypes to wild type noting that only 2 genes appear to be DE in common between both comparisons. D) Mean-difference (MD) plot and E) volcano plot of DE genes in each comparison. Note that the logFC axis limits in D) and E) are constrained to -5 and 5 for visualization purposes.
PCR genotyping
Allele-specific genotyping PCRs were performed on genomic DNA extracted from fin biopsies as described in [20]. The allele-specific primer sequences can be found in Table 1. Primers were synthesized by Sigma Aldrich (St. Louis, MO, USA).
Table 1
Genotyping primer sequences
| Primer Name | Sequence (5’ to 3’) |
| Wild type specific forward primer | TGAATTCGGTGCTCAACACTC |
| T141_L142delinsMISLISV specific forward primer | TGAATTCGGTGCTCAACATG |
| N140fs specific forward primer | TGCTGAATTCGGTGCTCTG |
| Common reverse primer | TCACCAAGGACCACTGATTCAGC |
RNA-seq data generation and analysis
Preparation of RNA for RNA-seq and the subsequent analysis of the data was performed mostly as in [20]. Briefly, brains of n = 5 fish of wild type, EOfAD-like/+ and FS/+ genotypes (each group contained 3 females and 2 males) were carefully removed from the heads preserved RNAlater Stabilization Solution (Invitrogen, Carlsbad, CA, USA), followed by total RNA extraction using the mirVana™ miRNA Isolation Kit (Ambion, Austin, TX, USA) and DNase treatment using the DNA-free™ Kit (Ambion). Total RNA was then delivered to the Genomics service at the South Australian Health and Medical Research Institute (SAHMRI, Adelaide, AUS) for polyA+, stranded library preparation and RNA sequencing using the Illumina Nextseq platform.
Processing of the demultiplexed fastq files provided by SAHMRI (75 bp single-end reads) was performed as in [20]. However, the pseudo-alignment using kallisto (v0.43.1) [23] was performed using a modified version of the zebrafish transcriptome (Ensembl release 94) which additionally included the two novel mutant psen2 transcript sequences.
Transcript counts as estimated by kallisto were imported into R [24] using tximport [25], summing the transcript-level counts to gene-level counts. We also imported the transcript-level counts using the catchKallisto function from the package edgeR [26] to assess simply the allele-specific expression of the different psen2 transcripts.
We removed genes considered as undetectable in all three genotypes of interest (less than one count per million in at least one third of the samples) from downstream analysis, leaving library sizes ranging between 17 million and 27 million reads. We normalized for these differences in library sizes using the trimmed mean of M-values (TMM) method [27], followed by removal of one factor of unwanted variation using the RUVg method RUVseq [28] as described in [20].
Differential gene expression analysis was performed using a generalized linear model (GLM) and likelihood ratio test using edgeR [26], specifying a design matrix with an intercept as the wild type genotype, and the coefficients as the FS/+ and EOfAD-like/+ genotypes, and the W_1 coefficient from RUVg. Genes were considered differentially expressed (DE) in each specific comparison if the FDR-adjusted p-value was below 0.05.
Enrichment analysis was performed on the KEGG [29] and GO [30, 31] gene sets from the Molecular Signatures Database (MSigDB) [32], with GO terms excluded if the shortest path back to the root node was < 3 steps. We also tested for possible iron dyshomeostasis in the RNA-seq data by performing an enrichment analysis on the iron-responsive element (IRE) gene sets described in [18]. To test for over-representation of these gene sets within the DE gene lists, we used goseq [33], using the average transcript length per gene to estimate sampling bias for DE genes. For enrichment testing on the entire list of detectable genes, we calculated the harmonic mean p-value [34] of raw p-values from fry [35], camera [36] and fgsea [37, 38]. Calculation of the harmonic mean p-value is a method robust to dependent tests [34] and has been validated previously on simulated datasets [20]. Gene sets were considered significantly altered if the FDR-adjusted harmonic mean p-value was below 0.05. Visualization of RNA-seq data analysis was performed using ggplot2 [39], pheatmap [40], and UpSetR [41].
Reproducibility and data availability
The raw fastq files and the gene-level counts have been deposited in the GEO database with accession number GSE158233. All code to reproduce this analysis can be found at https://github.com/UofABioinformaticsHub/20181113_MorganLardelli_mRNASeq.
RESULTS
We first inspected the transcript-level counts to ensure that expression of the psen2 alleles was as expected (Supplementary File 1). Consistent with observations in [13], expression of the EOfAD-like transcript was at a similar level to that of the wild type psen2 transcript in the EOfAD-like/+ brains, while the decreased expression of the FS allele of psen2 relative to the wild type allele was suggestive of nonsense-mediated decay (NMD). We observed that one of the samples had been incorrectly genotyped during sample preparation and was actually transheterozygous for the EOfAD-like and FS alleles of psen2. Therefore, we omitted this sample from subsequent analyses. We also examined the relationship between samples by principal component analysis (PCA) on the gene-level counts. Samples did not cluster by genotype in the PCA plot, supporting that heterozygosity for the psen2 mutations in this study does not give widespread effects on the brain transcriptome. Additionally, samples did not cluster by sex, consistent with our previous observations that sex does not show extensive effects in zebrafish brain transcriptomes [20, 21, 42]. In the PCA plot, we noticed that one of the wild type brain samples (12_WT_4) separated greatly from the others, appearing to be an outlier (Supplementary File 1). Sample weights, as calculated using the voomWithQualityWeights algorithm from the limma package [43] on the gene-level counts, revealed that this wild type sample was highly down-weighted. Therefore, we also omitted this sample from subsequent analyses.
Heterozygosity for an EOfAD-like or an FS mutation in psen2 results in limited differential expression of genes
To identify which genes were dysregulated due to heterozygosity for the EOfAD-like or FS alleles of psen2, we performed differential gene expression analysis using a GLM and likelihood ratio test with edgeR. This revealed 32 differentially expressed (DE) genes due to the FS mutation, and 5 DE genes due to the EOfAD-like mutation relative to wild type (Fig. 1, Supplementary File 2). Only two genes were seen to be significantly upregulated in both comparisons relative to wild type: AL929206.1 and BX004838.1. However, these genes are currently not annotated and have no known function. We tested for over-representation within the DE genes using goseq (using the average transcript length per gene as the predictor variable) of gene ontology (GO) [30, 31] terms which, as the name suggests, use ontologies to annotate gene function; KEGG [29] gene sets which can give insight into changes in various biological pathways and reactions; and our recently defined IRE gene sets [18] which can give insight to possible iron dyshomeostasis. However, due to the low numbers of DE genes in both comparisons, no significantly over-represented GO terms or gene sets were identified. (For the top 10 most significantly over-represented GO terms and gene sets in the DE genes, see Supplementary File 3).
Heterozygosity for an EOfAD-like or an FS mutation in psen2 has distinct effects on biological processes in young adult zebrafish brains
We next performed enrichment analysis on the entire list of detectable genes in the RNA-seq experiment to obtain a more complete view on the changes to gene expression due to heterozygosity for the EOfAD-like or FS mutations in psen2. Due to the highly overlapping nature of GO terms (many GO terms include many of the same genes), we tested only the KEGG and IRE gene sets. We found statistical evidence for 8 KEGG gene sets in the EOfAD/+ brains and 6 KEGG gene sets in the FS/+ brains to be significantly altered as a group. Notably, no IRE gene sets were found to be significantly altered (Fig. 2A). Genes in the KEGG_RIBOSOME gene set were significantly altered in both comparisons. However, the genes were mostly downregulated in EOfAD-like/+ brains and upregulated in FS/+ brains (Fig. 2B). For additional visualizations of our enrichment analysis, see Supplementary File 4.
Fig. 2
A) Heatmap showing the significant KEGG [29] gene sets in psen2 heterozygous mutant brains, clustered based on their Euclidean distance. The columns represent the gene sets which are significant in at least one comparison, and the rows are the two comparisons. The color of each cell represents the significance value with more significant gene sets appearing lighter. The numbers within each cell are the FDR-adjusted harmonic mean p-values (HMP). ns, not significant. B) Heatmap indicating the logFC of genes in the KEGG_RIBOSOME gene set in EOfAD/+ and FS/+ brains. C) Upset plot showing the overlap of leading edge genes from the fgsea algorithm in gene sets found to be altered in EOfAD-like/+ brains, and D) FS/+ brains. See the online version for color.
![A) Heatmap showing the significant KEGG [29] gene sets in psen2 heterozygous mutant brains, clustered based on their Euclidean distance. The columns represent the gene sets which are significant in at least one comparison, and the rows are the two comparisons. The color of each cell represents the significance value with more significant gene sets appearing lighter. The numbers within each cell are the FDR-adjusted harmonic mean p-values (HMP). ns, not significant. B) Heatmap indicating the logFC of genes in the KEGG_RIBOSOME gene set in EOfAD/+ and FS/+ brains. C) Upset plot showing the overlap of leading edge genes from the fgsea algorithm in gene sets found to be altered in EOfAD-like/+ brains, and D) FS/+ brains. See the online version for color.](https://content.iospress.com:443/media/adr/2021/5-1/adr-5-1-adr200279/adr-5-adr200279-g002.jpg)
To determine whether the statistical significance of the enrichments of the gene sets was being driven by the expression of the same genes, we inspected the genes from the “leading edge” from fgsea. (These can be thought of as the core genes driving the enrichment of each gene set). The leading edge genes in each comparison were mostly independent of one another, except for the KEGG gene sets for oxidative phosphorylation, Parkinson’s disease and Alzheimer’s disease, which share 39 genes (Fig. 2C, D).
DISCUSSION
Here, we aimed to identify similarities and differences in the effects on brain transcriptomes of heterozygosity for an in-frame (presumably EOfAD-like) mutation in psen2 compared to a frameshift (presumably not EOfAD-like) mutation in this gene. We previously generated zebrafish models of these two types of mutations: the EOfAD-like mutation psen2T141 _ L142delinsMISLISV, an in-frame mutation similar to that of human PSEN2N141I-, and psen2N140fs, a frameshift mutation in the same region in the zebrafish genome [13]. Although zebrafish psen2T141 _ L142delinsMISLISV is not a direct equivalent of the human PSEN1N141I mutation it models, we have assumed that the EOfAD-relevant effects of the two mutations on brain transcriptomes will be similar, (under the “reading-frame preservation rule” of EOfAD mutations in PSEN genes). However, it must be recognized that the population prevalence of EOfAD mutations in PSEN2 is less than one tenth that of such mutations in PSEN1 despite that the coding sequences of both genes represent mutational targets of similar size [44, 45]. Therefore, an additional factor or factors (such as subcellular locations of the protein products of the two genes [46] or other differences in function [47, 48]) must differentiate the pathogenicity of EOfAD mutations in PSEN1 and PSEN2. It is possible that the specific protein structure caused by the PSEN2N141I mutation (compared to the psen2T141 _ L142delinsMISLISV mutation at the equivalent position) is required for its pathogenicity.
Our BeST analysis strategy involves analysis of sibling fish raised together in an identical environment. This reduces confounding effects in direct comparisons of two heterozygous psen2 mutations relative to wild type, allowing detection of subtle transcriptome state differences. We observed subtle, but highly statistically significant, changes to the transcriptomes of the psen2 mutant fish relative to wild type. At the single gene level, relatively few genes were observed to be statistically significantly DE in each mutant genotype, and differentially enriched biological pathways were not detected in these DE gene lists (Supplementary File 3). As a comparison, heterozygosity for an EOfAD-like mutation in zebrafish psen1, (orthologous to the human gene most commonly mutated in EOfAD [49]), gives 251 DE genes in young adult zebrafish brains [19]. However, we were able to identify statistically significant changes in the psen2 mutant brains by performing enrichment analysis on the entire list of detectable genes in the RNA-seq experiment using an ensemble approach. This involves combining raw p-values by calculation of a harmonic p-value—a strategy thought to be less restrictive than other methods of combining p-values. An additional benefit of this method is that it has been shown specifically to be robust to dependent p-values [34]. The subtlety of the psen2 mutation effects is consistent with a generally later age of onset, and variable penetrance of EOfAD mutations in PSEN2 relative to EOfAD mutations in PSEN1 and AβPP.
Interestingly, the pathways affected by the two psen2 mutations showed very little overlap. Gene sets significantly altered in EOfAD-like/+ brains were not significantly altered in the FS/+ brains and vice versa. Only one biological process appears to be significantly affected by both mutations: ribosome function (the KEGG_RIBOSOME gene set). However, in general, the direction of differential expression of genes in this gene set was observed to be opposite between the two mutants, implying different mechanisms of action of each mutation (Fig. 2B). The low concordance between the brain transcriptome effects of these two mutations supports that presenilin EOfAD mutations do not act through a simple loss of function but, instead, through a gain of function mechanism connected with encoding an abnormal, but “full-length” protein [14]. Notably, the EOfAD-like mutation of psen2 is predicted to affect the process of long-term potentiation thought to be critical for memory formation. We also observed the dysregulation of genes involved in controlling the cell cycle. Genes driving the enrichment of this gene set (i.e., the leading edge genes) include the minichromosome maintenance (mcm) genes (see Supplementary File 4), which we observed to be significantly dysregulated due to heterozygosity for an EOfAD-like mutation in psen1 in 7 day old zebrafish larvae [17]. Together, these results provide support for the “two-hit hypothesis” of Mark Smith and colleagues [53], which postulates that “cell cycle events” (CCEs) involving inappropriate attempts at cell division by neurons are critical for development of AD. The second “hit” of this hypothesis is oxidative stress, which is an anticipated outcome of disturbance of oxidative phosphorylation. Notably, the EOfAD-like mutation of psen2 is predicted to affect oxidative phosphorylation, which is in common with all the other EOfAD-like mutations of genes we have examined in zebrafish [16, 18–22].
One of the most characterized functions of presenilin proteins is their role as the catalytic core of gamma-secretase (γ-secretase) complexes. γ-secretase activity is responsible for cleavage of a wide range of transmembrane proteins including AβPP and NOTCH. We observed that genes involved in the Notch signaling pathway were only significantly altered in FS/+ genotype brains, indicating that γ-secretase activity may be altered by the FS mutation. It is difficult to discern from these data whether γ-secretase activity is increased or decreased, as both up- and downregulation of Notch target genes is observed (Supplementary File 4). The FS mutation results in a truncated protein lacking critical aspartate residues needed for γ-secretase activity (Fig. 1B). Therefore, a simplistic view would be that γ-secretase activity would be reduced. However, a minor, (although statistically non-significant) increase in expression of psen1 is observed in FS/+ brains (also in EOfAD-like/+ brains) (Supplementary File 4) raising the possibility that transcriptional adaptation (formerly known as “genetic compensation”) might explain an apparent increase in γ-secretase activity [54]. Since presenilin holoproteins are known to form multimeric complexes, we have suggested previously that this may be involved in the regulation of the conversion of the holoprotein into its γ-secretase-active form by endoproteolysis [14]. N-terminal fragments of presenilin proteins are also known to multimerize [55], and the possibility exists that these may disrupt holoprotein multimer formation/stability. In our previous publication identifying the EOfAD-like and FS mutations of psen2 analyzed here, we saw that both mutations reduce adult melanotic surface pigmentation when homozygous [13]. This implies that both mutant alleles encode Psen2 proteins with reduced intrinsic γ-secretase activity, at least in melanosomes. Further research is required to discern whether γ-secretase activity is increased or decreased in psen2 mutant brains.
In conclusion, our results support that frameshift and in-frame mutations in the presenilins have distinct effects on the brain transcriptome, consistent with the reading frame preservation rule of presenilin EOfAD-causative mutations. The data presented here, along with our growing collection of BeST analyses (summarized in [56]), indicates that changes to mitochondrial and ribosomal functions are effects-in-common of heterozygosity for EOfAD-like mutations in different genes. These may be early cellular stresses which eventually lead to AD pathology, and warrant investigation for discovery of therapeutic targets.
ACKNOWLEDGMENTS
The authors would like thank FBRI LLC for use of the presenilin protein schematic in Fig. 1 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) for permission to display their Pathway Maps. This work was supported with supercomputing resources provided by the Phoenix HPC service at the University of Adelaide, and by grants GNT1061006 and GNT1126422 from the National Health and Medical Research Council of Australia (NHMRC). KB was supported by an Australian Government Research Training Program Scholarship. HJ was supported by an Adelaide Scholarship International scholarship from the University of Adelaide.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-200279.
REFERENCES
[1] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL ((2013) ) Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12: , 357–367. |
[2] | Masters CL , Bateman R , Blennow K , Rowe CC , Sperling RA , Cummings JL ((2015) ) Alzheimer’s disease. Nat Rev Dis Primers 1: , 15056. |
[3] | Cummings J , Lee G , Ritter A , Sabbagh M , Zhong K ((2020) ) Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y) 6: , e12050. |
[4] | Elmaleh DR , Farlow MR , Conti PS , Tompkins RG , Kundakovic L , Tanzi RE ((2019) ) Developing effective Alzheimer’s disease therapies: Clinical experience and future directions. J Alzheimers Dis 71: , 715–732. |
[5] | Modrego P , Lobo A ((2019) ) A good marker does not mean a good target for clinical trials in Alzheimer’s disease: The amyloid hypothesis questioned. Neurodegener Dis Manag 9: , 119–121. |
[6] | Kunkle BW , Grenier-Boley B , Sims R , Bis JC , Damotte V , Naj AC , Boland A , Vronskaya M , van der Lee SJ , Amlie-Wolf A , et al. ((2019) ) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51: , 414–430. |
[7] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , et al. ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[8] | Dourlen P , Kilinc D , Malmanche N , Chapuis J , Lambert JC ((2019) ) The new genetic landscape of Alzheimer’s disease: From amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol 138: , 221–236. |
[9] | Neuner SM , Tcw J , Goate AM ((2020) ) Genetic architecture of Alzheimer’s disease. Neurobiol Dis 143: , 104976. |
[10] | Barthelson K , Newman M , Lardelli M ((2020) ) Sorting out the role of the sortilin-related receptor 1 in Alzheimer’s disease. J Alzheimers Dis Rep 4: , 123–140. |
[11] | Mutations, https://www.alzforum.org/mutations, Accessed 1 November. |
[12] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K , et al. ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[13] | Jiang H , Newman M , Lardelli M ((2018) ) The zebrafish orthologue of familial Alzheimer’s disease gene PRESENILIN 2 is required for normal adult melanotic skin pigmentation. PLoS One 13: , e0206155. |
[14] | Jayne T , Newman M , Verdile G , Sutherland G , Münch G , Musgrave I , Moussavi Nik SH , Lardelli M ((2016) ) Evidence for and against a pathogenic role of reduced γ-secretase activity in familial Alzheimer’s disease. J Alzheimers Dis 52: , 781–799. |
[15] | Newman M , Ebrahimie E , Lardelli M ((2014) ) Using the zebrafish model for Alzheimer’s disease research. Front Genet 5: , 189. |
[16] | Barthelson K , Dong Y , Newman M , Lardelli M ((2021) ) PRESENILIN 1 mutations causing early-onset familial Alzheimer’s disease or familial acne inversa differ in their effects on genes facilitating energy metabolism and signal transduction. bioRxiv, 2021.2001.2026.428321. |
[17] | Dong Y , Newman M , Pederson SM , Barthelson K , Hin N , Lardelli M ((2021) ) Transcriptome analyses of 7-day-old zebrafish larvae possessing a familial Alzheimer’s disease-like mutation in psen1 indicate effects on oxidative phosphorylation, ECM and MCM functions, and iron homeostasis. BMC Genomics 22: , 211. |
[18] | Hin N , Newman M , Pederson SM , Lardelli MM ((2020) ) Iron Responsive Element (IRE)-mediated responses to iron dyshomeostasis in Alzheimer’s disease. bioRxiv, 2020.2005.2001.071498. |
[19] | Newman M , Hin N , Pederson S , Lardelli M ((2019) ) Brain transcriptome analysis of a familial Alzheimer’s disease-like mutation in the zebrafish presenilin 1 gene implies effects on energy production. Mol Brain 12: , 43. |
[20] | Barthelson K , Pederson SM , Newman M , Lardelli M ((2020) ) Brain transcriptome analysis reveals subtle effects on mitochondrial function and iron homeostasis of mutations in the SORL1 gene implicated in early onset familial Alzheimer’s disease. Mol Brain 13: , 142. |
[21] | Barthelson K , Pederson SM , Newman M , Lardelli M ((2021) ) Brain transcriptome analysis of a protein-truncating mutation in sortilin-related receptor 1 associated with early-onset familial Alzheimer’s disease indicates early effects on mitochondrial and ribosome function. J Alzheimers Dis 79: , 1105–1119. |
[22] | Jiang H , Pederson SM , Newman M , Dong Y , Barthelson K , Lardelli M ((2020) ) Transcriptome analysis indicates dominant effects on ribosome and mitochondrial function of a premature termination codon mutation in the zebrafish gene psen2. PLoS One 15: , e0232559. |
[23] | Bray NL , Pimentel H , Melsted P , Pachter L ((2016) ) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: , 525–527. |
[24] | R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ |
[25] | Soneson C , Love MI , Robinson MD ((2015) ) Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 4: , 1521. |
[26] | Robinson MD , McCarthy DJ , Smyth GK ((2010) ) edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: , 139–140. |
[27] | Robinson MD , Oshlack A ((2010) ) A scaling normalization method for differential expression analysis of RNA-seq data.R. Genome Biol 11: , 25. |
[28] | Risso D , Ngai J , Speed TP , Dudoit S ((2014) ) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32: , 896–902. |
[29] | Kanehisa M , Goto S ((2000) ) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: , 27–30. |
[30] | Gene Ontology Consortium ((2021) ) The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res 49: , D325–d334. |
[31] | Ashburner M , Ball CA , Blake JA , Botstein D , Butler H , Cherry JM , Davis AP , Dolinski K , Dwight SS , Eppig JT , Harris MA , Hill DP , Issel-Tarver L , Kasarskis A , Lewis S , Matese JC , Richardson JE , Ringwald M , Rubin GM , Sherlock G ((2000) ) Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: , 25–29. |
[32] | Liberzon A ((2014) ) A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol Biol 1150: , 153–160. |
[33] | Young MD , Wakefield MJ , Smyth GK , Oshlack A ((2010) ) Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol 11: , R14. |
[34] | Wilson DJ ((2019) ) The harmonic mean p-value for combining dependent tests. Proc Natl Acad Sci U S A 116: , 1195–1200. |
[35] | Wu D , Lim E , Vaillant F , Asselin-Labat ML , Visvader JE , Smyth GK ((2010) ) ROAST: Rotation gene set tests for complex microarray experiments. Bioinformatics 26: , 2176–2182. |
[36] | Wu D , Smyth GK ((2012) ) Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res 40: , e133. |
[37] | Sergushichev AA ((2016) ) An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv, 060012. |
[38] | Subramanian A , Tamayo P , Mootha VK , Mukherjee S , Ebert BL , Gillette MA , Paulovich A , Pomeroy SL , Golub TR , Lander ES , Mesirov JP ((2005) ) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: , 15545–15550. |
[39] | Wickham H ((2016) ) ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag, New York. |
[40] | Kolde R ((2019) ) pheatmap: Pretty Heatmaps. R package version 1.0.12. https://CRAN.R-project.org/package=pheatmap. |
[41] | Conway JR , Lex A , Gehlenborg N ((2017) ) UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 33: , 2938–2940. |
[42] | Newman M , Nik HM , Sutherland GT , Hin N , Kim WS , Halliday GM , Jayadev S , Smith C , Laird AS , Lucas CW , Kittipassorn T , Peet DJ , Lardelli M ((2020) ) Accelerated loss of hypoxia response in zebrafish with familial Alzheimer’s disease-like mutation of presenilin 1. Hum Mol Genet 29: , 2379–2394. |
[43] | Ritchie ME , Phipson B , Wu D , Hu Y , Law CW , Shi W , Smyth GK ((2015) ) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: , e47. |
[44] | Ryman DC , Acosta-Baena N , Aisen PS , Bird T , Danek A , Fox NC , Goate A , Frommelt P , Ghetti B , Langbaum JB , Lopera F , Martins R , Masters CL , Mayeux RP , McDade E , Moreno S , Reiman EM , Ringman JM , Salloway S , Schofield PR , Sperling R , Tariot PN , Xiong C , Morris JC , Bateman RJ ((2014) ) Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 83: , 253–260. |
[45] | Pottier C , Hannequin D , Coutant S , Rovelet-Lecrux A , Wallon D , Rousseau S , Legallic S , Paquet C , Bombois S , Pariente J , Thomas-Anterion C , Michon A , Croisile B , Etcharry-Bouyx F , Berr C , Dartigues JF , Amouyel P , Dauchel H , Boutoleau-Bretonnière C , Thauvin C , Frebourg T , Lambert JC , Campion D ((2012) ) High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry 17: , 875–879. |
[46] | Sannerud R , Esselens C , Ejsmont P , Mattera R , Rochin L , Tharkeshwar AK , De Baets G , De Wever V , Habets R , Baert V , Vermeire W , Michiels C , Groot AJ , Wouters R , Dillen K , Vints K , Baatsen P , Munck S , Derua R , Waelkens E , Basi GS , Mercken M , Vooijs M , Bollen M , Schymkowitz J , Rousseau F , Bonifacino JS , Van Niel G , De Strooper B , Annaert W ((2016) ) Restricted location of PSEN2/γ-secretase determines substrate specificity and generates an intracellular Aβ pool. Cell 166: , 193–208. |
[47] | Escamilla-Ayala A , Wouters R , Sannerud R , Annaert W ((2020) ) Contribution of the Presenilins in the cell biology, structure and function of γ-secretase. Semin Cell Dev Biol 105: , 12–26. |
[48] | Mastrangelo P , Mathews PM , Chishti MA , Schmidt SD , Gu Y , Yang J , Mazzella MJ , Coomaraswamy J , Horne P , Strome B , Pelly H , Levesque G , Ebeling C , Jiang Y , Nixon RA , Rozmahel R , Fraser PE , St George-Hyslop P , Carlson GA , Westaway D ((2005) ) Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma-secretases. Proc Natl Acad Sci U S A 102: , 8972–8977. |
[49] | Cruts M , Theuns J , Van Broeckhoven C ((2012) ) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33: , 1340–1344. |
[50] | Thordardottir S , Rodriguez-Vieitez E , Almkvist O , Ferreira D , Saint-Aubert L , Kinhult-Ståhlbom A , Thonberg H , Schöll M , Westman E , Wall A , Eriksdotter M , Zetterberg H , Blennow K , Nordberg A , Graff C ((2018) ) Reduced penetrance of the PSEN1 H163Y autosomal dominant Alzheimer mutation: A 22-year follow-up study. Alzheimers Res Ther 10: , 45. |
[51] | Rossor MN , Fox NC , Beck J , Campbell TC , Collinge J ((1996) ) Incomplete penetrance of familial Alzheimer’s disease in a pedigree with a novel presenilin-1 gene mutation. Lancet 347: , 1560. |
[52] | Sherrington R , Froelich S , Sorbi S , Campion D , Chi H , Rogaeva EA , Levesque G , Rogaev EI , Lin C , Liang Y , Ikeda M , Mar L , Brice A , Agid Y , Percy ME , Clerget-Darpoux F , Piacentini S , Marcon G , Nacmias B , Amaducci L , Frebourg T , Lannfelt L , Rommens JM , St George-Hyslop PH ((1996) ) Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet 5: , 985–988. |
[53] | Zhu X , Lee HG , Perry G , Smith MA ((2007) ) Alzheimer disease, the two-hit hypothesis: An update. Biochim Biophys Acta 1772: , 494–502. |
[54] | El-Brolosy MA , Kontarakis Z , Rossi A , Kuenne C , Günther S , Fukuda N , Kikhi K , Boezio GLM , Takacs CM , Lai S-L , Fukuda R , Gerri C , Giraldez AJ , Stainier DYR ((2019) ) Genetic compensation triggered by mutant mRNA degradation. Nature 568: , 193–197. |
[55] | Cervantes S , Gonzàlez-Duarte R , Marfany G ((2001) ) Homodimerization of presenilin N-terminal fragments is affected by mutations linked to Alzheimer’s disease. FEBS Lett 505: , 81–86. |
[56] | Barthelson K , Newman M , Lardelli M ((2021) ) Comparative analysis of Alzheimer’s disease knock-in model brain transcriptomes implies changes to energy metabolism as a causative pathogenic stress. bioRxiv, 2021.2002.2016. 431539. |


