
Alignment of Alzheimer’s Disease Amyloid-β Peptide and Herpes Simplex Virus-1 pUL15 C-Terminal Nuclease Domain
Abstract
Background:
The cause of Alzheimer’s disease (AD) is poorly understood. Neurotropic microbes, particularly herpesviruses, might set off chronic neuroinflammation. Amyloid-β (Aβ) has antimicrobial properties and could represent a brain defense against infection.
Objective:
We searched for protein sequence alignment between herpes simplex virus type I (HSV-1) HSV-2, and Aβ.
Methods:
Protein data bank (pdb) structures for Aβ, HSV-1, and HSV-2 were searched on the RCSB Protein Data Bank. The protein structures were superimposed and aligned on PYMOL v 2.3.4.
Results:
For HSV-1 and Aβ, amino acid residues ser549 – his569 of HSV-1 aligned closely with residues asp7 - asn27 of Aβ. For HSV-2 and Aβ, amino acid residues of HSV-2 aligned less closely than those of HSV-1 with residues of Aβ.
Conclusion:
Conjugating and binding to the same alpha helix in the HSV-1 protease, Aβ could be marking HSV-1 for attack by the immune system, providing a rapid inherited immune response to a destructive neurotropic virus that would otherwise require the more time-consuming involvement of T-cells, B-cells, and the adaptive immune system. But older people do not respond to viral infections as well as younger individuals. When HSV-1 infection advances in an old person, more and more amyloid is produced, forming an adhesive web. As the brain tries to hold the pathologic process in check, neuroinflammation increases and spreads. Progressive neurodegeneration and cognitive decline are the outcome.
INTRODUCTION
The cause of Alzheimer’s disease (AD) is poorly understood. The disease process is associated with amyloid-β (Aβ) plaques, tau neurofibrillary tangles, and neuroinflammation. In 1991, the amyloid hypothesis postulated that Aβ accumulation is a key element [1]. Aβ was supposed to stimulate both the development of tau neurofibrillary tangles and neuroinflammation. Aβ, tau, and inflammation each led to the malfunction and destruction of neurons and synapses. It follows that clearing the brain of Aβ would be beneficial, which has not been the case. Therefore, Aβ is likely a result, not a cause, of AD [2, 3].
A newer disease model suggests that infection stimulates neuroinflammation, resulting in AD. Neurotropic microbes, particularly herpesviruses, might set off chronic neuroinflammation.
Multiple reports link herpes simplex virus type 1 (HSV-1) to AD [4, 5]. DNA of HSV-1 is identified more often in the brains of individuals with AD than in healthy controls; and the viral DNA is especially prominent within Aβ plaques [6]. In addition, a herpes simplex virus glycoprotein B fragment with homology to Aβ induces fibril formation and is neurotoxic, suggesting a possible role for this infectious agent in the pathophysiology of sporadic cases of AD [7].
Aβ has antimicrobial properties [8] and could represent a brain defense against infection [9]. To evaluate this possibility, we searched for protein sequence alignment between HSV-1 and Aβ.
METHODS
Protein data bank (pdb) entries were searched on the RCSB Protein Data Bank. We identified three that allowed us to examine the relationship of Aβ to HSV-1, and HSV-2.
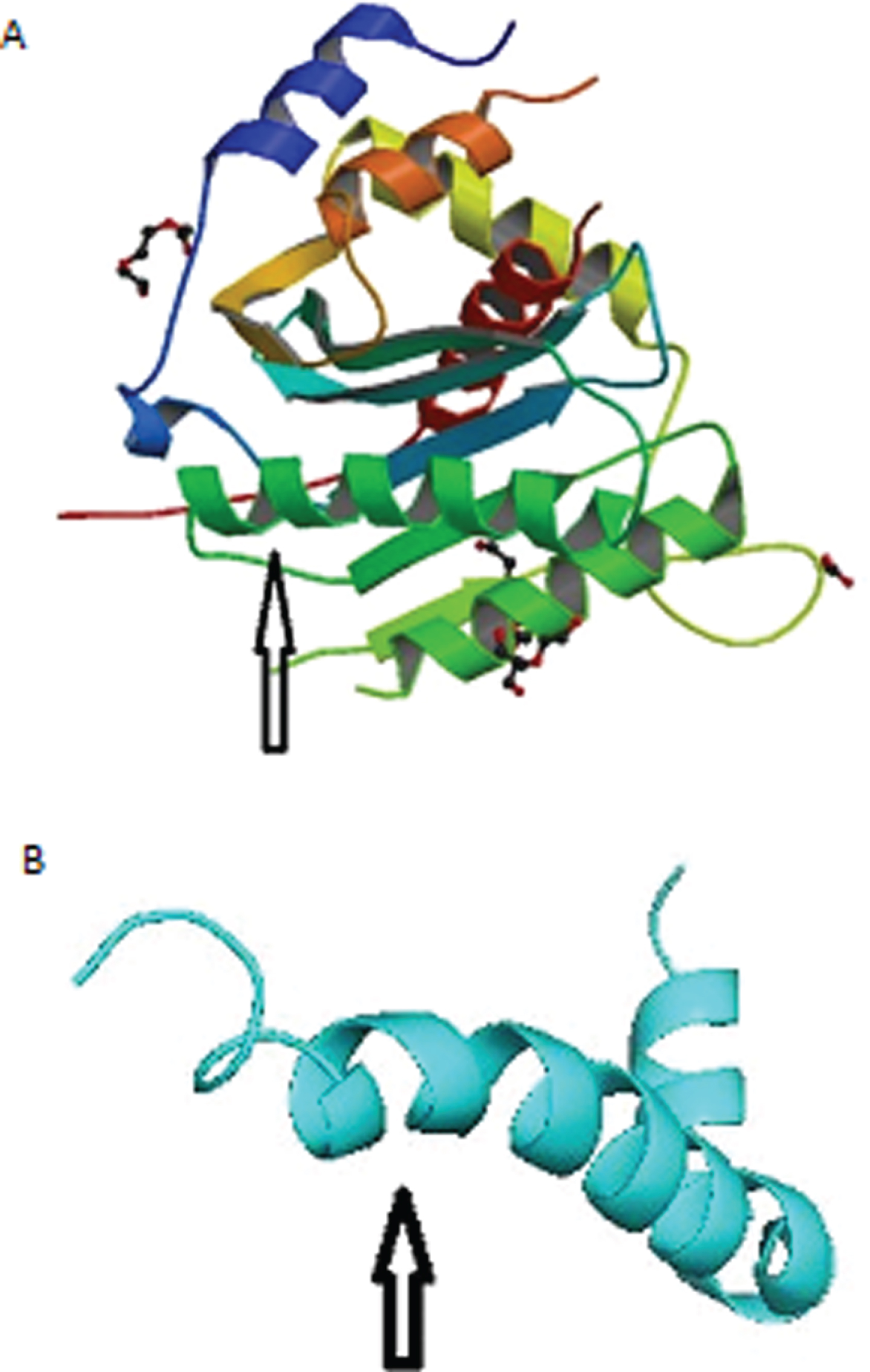
• 1IYT (Fig. 1A). Solution structure of the AD Aβ peptide, Method: solution NMR, Deposited: 2002-09-06 Released: 2003-02-11 [10].
• 4IOX (Fig. 1B). The structure of the herpes simplex virus (HSV-1) DNA-packaging motor pUL15 C-terminal nuclease domain, Method: X-Ray diffraction, Resolution: 2.46 Å, deposited: 2013-01-08 released: 2013-05-01 [11]. pUL15C, the C-terminal nuclease domain, is required for herpesvirus genome processing and packaging. pUL15 and its homologues are highly conserved and have attracted considerable interest as drug targets [12].
• 1AT3 (Fig. 3A, B). Herpes simplex virus (HSV-2) protease. Method: X-Ray diffraction, Resolution: 2.50 Å, deposited: 1997-08-16, released: 1998-10-14 [13].
Fig.1
A) Structure of the herpes simplex virus (HSV-1) DNA-packaging motor pUL15 C-terminal nuclease domain. Arrow points to alpha helix that aligns with Aβ-peptide. B) Solution structure of the Alzheimer’s disease Aβ-peptide. Arrow points to alpha helix that aligns with HSV-1.
The protein structures were superimposed and aligned on Pymol v 2.3.4. Pymol is an open source molecular visualization system. Pymol can produce high-quality 3D images of small molecules and biological macromolecules, such as proteins.
We utilized the Pymol super command, which super aligns two protein selections. Super does a sequence-independent structure-based dynamic programming alignment (unlike the align command) followed by a series of refinement cycles intended to improve the fit by eliminating pairing with high relative variability. The super command is more reliable than the align command for proteins with low sequence similarity.
To evaluate conservation and alignment of the Aβ genome across species, we used BLAT, the Blast-Like Alignment Tool of the UCSC Genome Browser [14]. BLAT can align a user sequence of 25 bases or more to the genome. Because some level of mismatch is tolerated, cross-species alignments may be performed provided the species have not diverged too far from each other; this capability allowed comparison of the Mouse Mammary Tumor Virus genome to the human genome [15]. BLAT calculates a percent identity score to indicate differences between sequences without a perfect match (i.e., without 100% identity). The differences include mismatches and gaps [16].
RESULTS
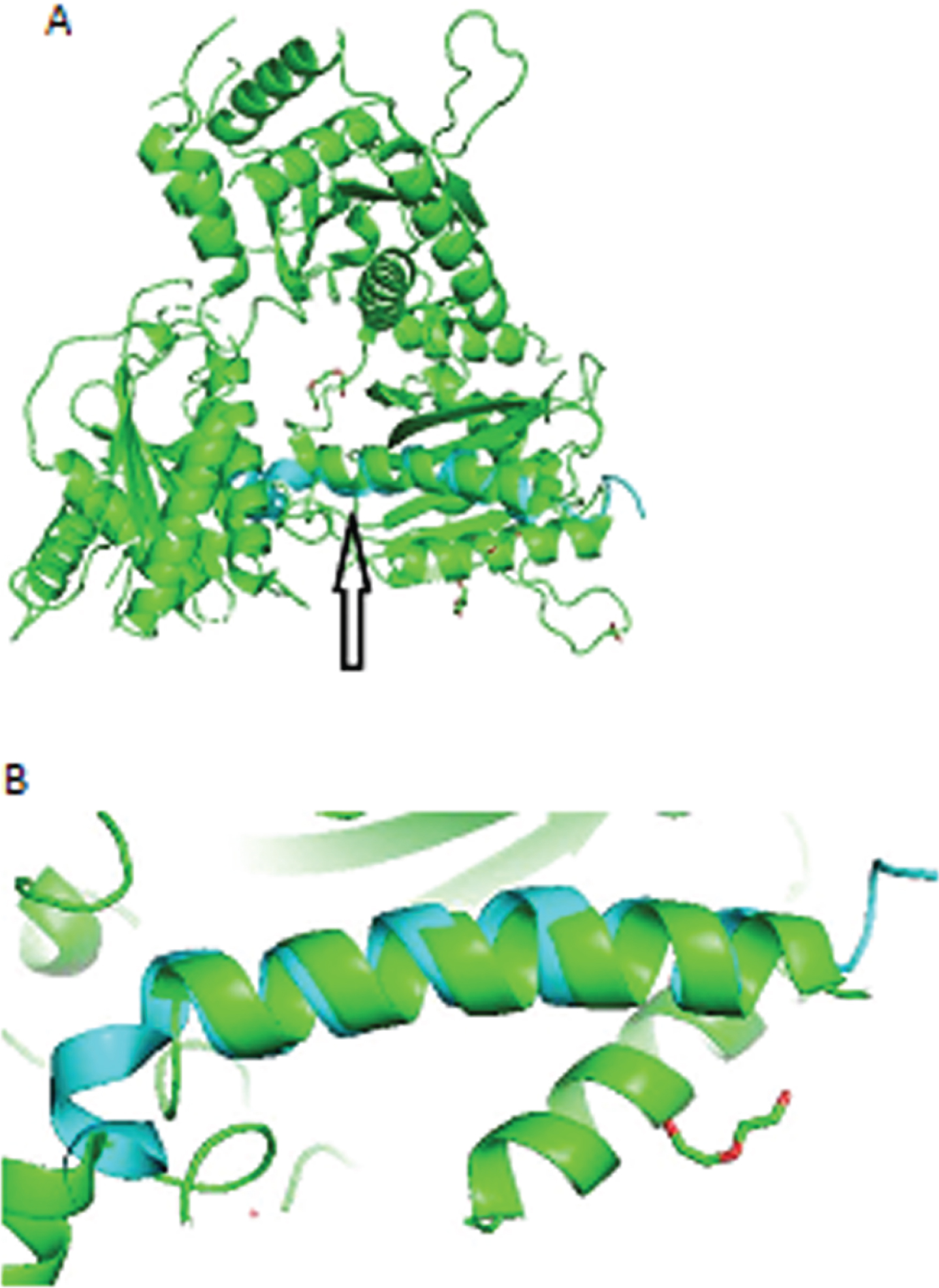
For HSV-1 and Aβ, Pymol performed 6 cycles of calculations on 124 aligned atoms, final root mean square deviation of atomic positions (RMSD) = 0.937Å for 94 atoms (Table 1). Amino acid residues ser549 – his569 of the HSV-1 protease aligned closely with residues asp7 - asn27 of Aβ (Fig. 2A, B).
Table 1
Pymol alignment cycles and RMSD (root mean square deviation of atomic positions) score for HSV-1 and Aβ. The root-mean-square deviation of atomic positions is the measure of the average distance (Å) between the atoms of superimposed proteins. The RMSD (0.937Å) indicates good alignment of the Aβ and HSV-1 residues
| MatchAlign: aligning residues (864 versus 42) |
| MatchAlign: score 77.391 |
| ExecutiveAlign: 124 atoms aligned. |
| ExecutiveRMS: 9 atoms rejected during cycle 1 (RMSD = 2.08). |
| ExecutiveRMS: 8 atoms rejected during cycle 2 (RMSD = 1.63). |
| ExecutiveRMS: 6 atoms rejected during cycle 3 (RMSD = 1.32). |
| ExecutiveRMS: 5 atoms rejected during cycle 4 (RMSD = 1.12). |
| ExecutiveRMS: 2 atoms rejected during cycle 5 (RMSD = 0.97). |
| Executive: RMSD = 0.937Å (94 to 94 atoms) |
Fig.2
HSV-1 aligned with Aβ. A) Arrow points to closely aligned (RMSD = 0.937Å) alpha helices of HSV-1 (green) and Aβ (blue). B) Closeup of aligned alpha helices of HSV-1 and Aβ.
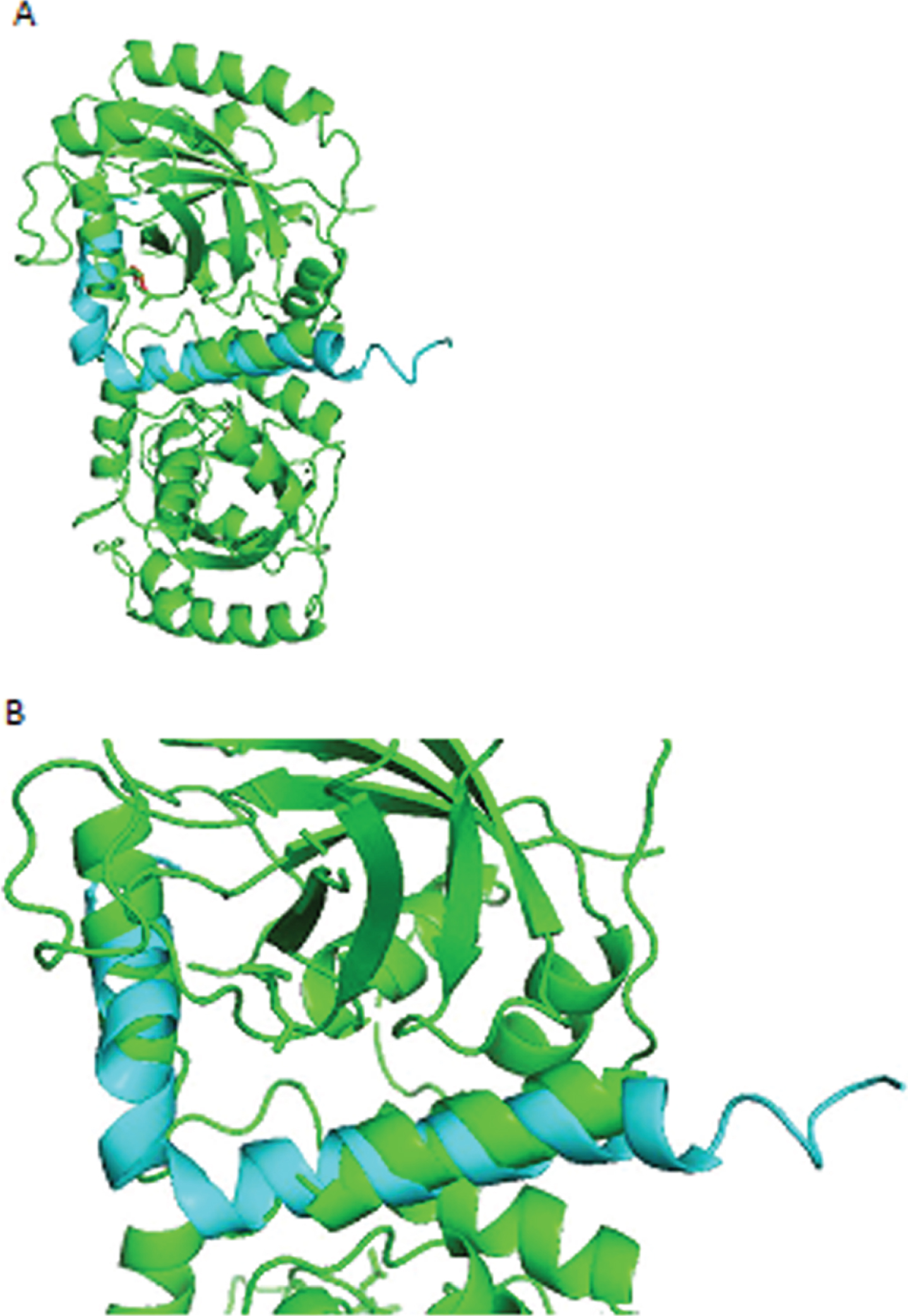
For HSV-2 and Aβ, Pymol performed 6 cycles of calculations on 124 aligned atoms, final RMSD = 2.67Å for 94 atoms. Amino acid residues of HSV-2 aligned less closely than those of HSV-1 with residues of Aβ (Fig. 3A, B).
Fig.3
A) HSV-2 aligned with Aβ. Amino acid residues of HSV-2 aligned less closely (RMSD = 2.67Å) than those of HSV-1 with residues of Aβ. B) Closeup of aligned alpha helices of HSV-2 and Aβ.
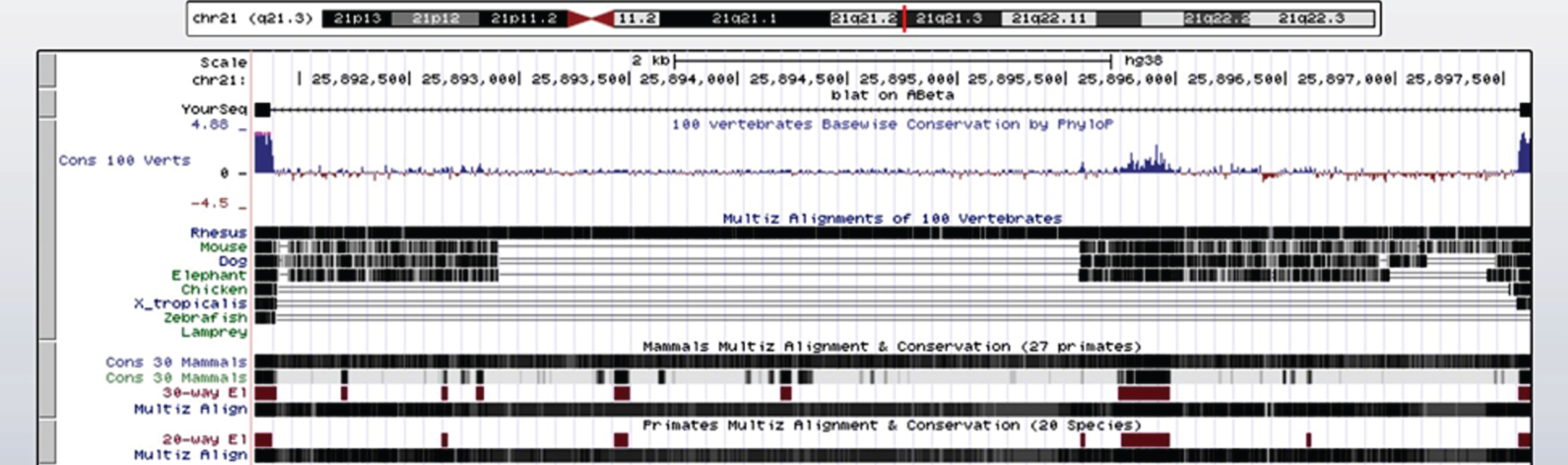
Results of the cross-species comparison of Aβ are shown in Fig. 4. There is a high degree of alignment and conservation of human Aβ (chr 21q21.3) in Aβ of the rhesus monkey and 27 other primates, but much less alignment and conservation in the mouse, dog, and elephant, even less in the chicken, western clawed frog (Xenopus tropicalis), zebrafish, and lamprey. The rhesus macaque diverged from ancestors of Homo sapiens about 25 million years ago [17].
Fig.4
Alignment of 42 amino acid residue human Aβ across species in the UCSC genome browser. There is a high degree of alignment and conservation of human Aβ (chr 21q21.3) in Aβ of the rhesus monkey (solid black horizontal line) and 27 other primates, but much less alignment and conservation in the mouse, dog, and elephant, even less in the chicken, western clawed frog (Xenopus tropicalis), zebrafish, and lamprey. The high degree of conservation of Aβ in primates suggests that Aβ is important for survival.
DISCUSSION
Aβ is an ancient neuropeptide expressed in vertebrates. Many vertebrate species share the human Aβ sequence (Fig. 4), which has been highly conserved over millions of years and therefore must be important for survival [18]. The antimicrobial properties of Aβ could be one of the reasons for Aβ conservation. In vivo, Aβ may behave like ubiquitin, and some studies have suggested that a relationship between Aβ and ubiquitin that may be involved in AD [19].
Ubiquitin is a small (8.6 kDa) regulatory protein present in most tissues of eukaryotic organisms, that is, it occurs ubiquitously [20]. The conjugation and binding of ubiquitin to a substrate protein is called ubiquitination. Ubiquitination affects proteins in many ways; one is especially important: Ubiquitin can mark a protein for degradation by the proteasome, a protein complex which destroys unneeded or damaged proteins by proteolysis, breaking peptide bonds [21].
Similarly, conjugating and binding to an alpha helix in the HSV-1 protease, Aβ could be marking HSV-1 for attack by the immune system. Aβ could evoke a rapid immune response to a destructive neurotropic virus that would otherwise require the more time-consuming involvement of T-cells, B-cells, and the adaptive immune system.
A weakness in our hypothesis is that HSV-1 is an enveloped virus. If Aβ behaves as an immunoglobulin/opsonin (ubiquitin) for phagocytic cells, the virus envelope should be destroyed first. But Aβ could have a dual action: 1) interacting with the membrane and destroying it, probably by pore formation, as many studies suggest [22–25], and then 2) acting as an immunoglobulin/opsonin (ubiquitin) for phagocytic cells.
Aβ did not align as well with HSV-2 as with HSV-1. This finding may be related to other studies indicating a closer HSV-1 than HSV-2 association with AD. Few studies suggest an HSV-2 AD link.
Older people do not handle viral infections as well as younger individuals. A common denominator of age-associated frailty is increased baseline inflammation, called inflammaging, that is present in older individuals. Recent studies have shown that the presence of excessive inflammation can inhibit immunity in both animals and humans, increasing the morbidity and mortality of viral infections [26].
As HSV-1 infection advances in an old person, more and more amyloid is produced, forming an adhesive web, as the brain tries to hold the pathologic process in check. Meanwhile, neuroinflammation increases and spreads. Progressive neurodegeneration and cognitive decline are the outcome.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[2] | Castellani RJ , Lee HG , Zhu X , Perry G , Smith MA ((2008) ) Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol 67: , 523–531. |
[3] | Castellani RJ , Perry G ((2014) ) The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem Pharmacol 88: , 671–676. |
[4] | Itzhaki RF ((2018) ) Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front Aging Neurosci 10: , 324. |
[5] | Mancuso R , Sicurella M , Agostini S , Marconi P , Clerici M ((2019) ) Herpes simplex virus type 1 and Alzheimer’s disease: Link and potential impact on treatment. Expert Rev Anti Infect Ther 17: , 715–731. |
[6] | Komaroff AL ((2020) ) Can infections cause Alzheimer disease? JAMA, doi: 10.1001/jama.2020.4085 |
[7] | Cribbs DH , Azizeh BY , Cotman CW , LaFerla FM ((2000) ) Fibril formation and neurotoxicity by a herpes simplex virus glycoprotein B fragment with homology to the Alzheimer’s A beta peptide. Biochemistry 39: , 5988–5994. |
[8] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[9] | Moir RD , Lathe R , Tanzi RE ((2018) ) The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14: , 1602–1614. |
[10] | Crescenzi O , Tomaselli S , Guerrini R , Salvadori S , D’Ursi AM , Temussi PA , Picone D ((2002) ) Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem 269: , 5642–5648. |
[11] | Selvarajan Sigamani S , Zhao H , Kamau YN , Baines JD , Tang L ((2013) ) The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol 87: , 7140–7148. |
[12] | Masaoka T , Zhao H , Hirsch DR , D’Erasmo MP , Meck C , Varnado B , Gupta A , Meyers MJ , Baines J , Beutler JA , Murelli RP , Tang L , Le Grice SF ((2016) ) Characterization of the C-terminal nuclease domain of herpes simplex virus pUL15 as a target of nucleotidyltransferase inhibitors. Biochemistry 55: , 809–819. |
[13] | Hoog SS , Smith WW , Qiu X , Janson CA , Hellmig B , McQueney MS , O’Donnell K , O’Shannessy D , DiLella AG , Debouck C , Abdel-Meguid SS ((1997) ) Active site cavity of herpesvirus proteases revealed by the crystal structure of herpes simplex virus protease/inhibitor complex. Biochemistry 36: , 14023–14029. |
[14] | Kuhn RM , Haussler D , Kent WJ ((2013) ) The UCSC genome browser and associated tools. Brief Bioinform 14: , 144–161. |
[15] | Lehrer S , Rheinstein PH ((2019) ) Mouse mammary tumor viral env sequences are not present in the human genome but are present in breast tumors and normal breast tissues. Virus Res 266: , 43–47. |
[16] | Bhagwat M , Young L , Robison RR ((2012) ) Using BLAT to find sequence similarity in closely related genomes. Curr Protoc Bioinformatics Chapter 10: , Unit 10. |
[17] | Rhesus MGS , Gibbs R , Rogers J , Katze M , Bumgarner R , Weinstock G , Mardis E , Remington K , Strausberg R , Venter J ((2007) ) Evolutionary and biomedical insights from the rhesus macaque genome. Science 316: , 222–234. |
[18] | Moir RD , Tanzi RE ((2019) ) Low evolutionary selection pressure in senescence does not explain the persistence of Abeta in the vertebrate genome. Front Aging Neurosci 11: , 70. |
[19] | Hong L , Huang HC , Jiang ZF ((2014) ) Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol Res 36: , 276–282. |
[20] | Pickart CM , Eddins MJ ((2004) ) Ubiquitin: Structures, functions, mechanisms. Biochim Biophys Acta 1695: , 55–72 . |
[21] | Gong B , Radulovic M , Figueiredo-Pereira ME , Cardozo C ((2016) ) The ubiquitin-proteasome system: Potential therapeutic targets for Alzheimer’s disease and spinal cord injury. Front Mol Neurosci 9: , 4. |
[22] | Kandel N , Zheng T , Huo Q , Tatulian SA ((2017) ) Membrane binding and pore formation by a cytotoxic fragment of amyloid beta peptide. J Phys Chem B 121: , 10293–10305. |
[23] | Di Scala C , Yahi N , Boutemeur S , Flores A , Rodriguez L , Chahinian H , Fantini J ((2016) ) Common molecular mechanism of amyloid pore formation by Alzheimer’s beta-amyloid peptide and alpha-synuclein. Sci Rep 6: , 28781. |
[24] | Bourgade K , Dupuis G , Frost EH , Fulop T ((2016) ) Anti-viral properties of amyloid-beta peptides. J Alzheimers Dis 54: , 859–878. |
[25] | Fernandez-Perez EJ , Peters C , Aguayo LG ((2016) ) Membrane damage induced by amyloid beta and a potential link with neuroinflammation. Curr Pharm Des 22: , 1295–1304. |
[26] | Akbar AN , Gilroy DW ((2020) ) Aging immunity may exacerbate COVID-19. Science 369: , 256–257. |


