
Vaccination Prevented Short-Term Memory Loss, but Deteriorated Long-Term Spatial Memory in Alzheimer’s Disease Mice, Independent of Amyloid-β Pathology
Abstract
Background:
Soluble oligomeric amyloid-β (Aβ), rather than Aβ plaques, seems to be the culprit in Alzheimer’s disease (AD). Accordingly, a new concept vaccine of small cyclic peptide conjugates, selectively targeting oligomeric Aβ, has been developed.
Objective:
Study the therapeutic potential of this new vaccine in a mouse model for AD.
Methods:
J20 mice, overexpressing human amyloid precursor protein, were validated for an AD-like phenotype. Then, J20 mice were vaccinated at 2, 3, and 4 months of age and AD phenotype was evaluated at 6, 9, and 12 months of age; or at 9, 10, and 11 months with evaluation at 12 months. Effects on Aβ pathology were studied by plaque load (immunohistochemistry; 6E10) and antibody titers against Aβ (ELISA). AD behavioral phenotype was evaluated by performance in a battery of cognitive tests.
Results:
J20 mice displayed age-related Aβ plaque development and an AD-like behavioral phenotype. A consistent antibody response to the cyclic peptides was, however, not extended to Aβ, leaving plaque load unaffected. Nevertheless, immunization at young ages prevented working- and short-term spatial memory loss, but deteriorated long-term spatial learning and memory, at 12 months of age. Immunization at later ages did not affect any measured parameter.
Conclusion:
J20 mice provide a relevant model for AD to study potential anti-Aβ treatment. Early vaccination prevented short-term memory loss at later ages, but deteriorated long-term spatial memory, however without affecting Aβ pathology. Later vaccination had no effects, but optimal timing may require further investigation.
INTRODUCTION
Alzheimer’s disease (AD) has been described for the first time in 1906 by Dr. Alois Alzheimer as a peculiar disease. Since then, AD has become the most prevalent neurodegenerative disease of adult-onset, causing up to 70% of all dementia worldwide [1]. Patients suffering from AD show progressive loss of cognitive functions resulting from severe neurodegeneration of the brain. Age is the major risk factor for AD, and as yet there is no effective treatment.
AD is characterized by an accumulation of amyloid-β (Aβ) and tau proteins, leading to extracellular Aβ plaques and intracellular neurofibrillary tau-tangles [2]. The plaques consist of Aβ, generated from the amyloid-β protein precursor (AβPP) [3]. These proteins can undergo conformational change, subsequently aggregating into fibrils and ultimately deposited into plaques. It was until recently that the neuronal dysfunction in AD was attributed to toxic effects of these Aβ plaques [4]. However, the correlation between plaques and cognitive function appears to be poor, and evidence is accumulating that the oligomeric soluble misfolded Aβ, rather than the fibrillary plaques, are the culprit in AD [5]. Plaque formation then may even be regarded as a protective mechanism of the body to stabilize these toxic peptides [6]. Accordingly, attention has shifted to the oligomeric Aβ as potential therapeutic target. Attempts have been undertaken to decrease neurodegeneration caused by Aβ peptides through immunization against Aβ [7, 8]. In this regard, active immunization would be preferred above passive immunization, because a long-term effect could be established with few injections, and the patient’s own immune system is programmed to remove the misfolded Aβ peptides. Despite the logic of this approach, there are no major breakthroughs yet. In 2002, the first active AD vaccine (AN1792), containing the full-length Aβ1–42 peptide, was developed. This vaccine went through a phase IIa clinical trial and showed beneficial effects, including less cognitive decline, but had to be discontinued because 6% of the subjects developed meningoencephalitis [9–11]. The vaccine could have evoked an immune response against the pathological misfolded Aβ, but also against the physiological proper linear peptide. Although the physiological role of Aβ has not yet been clearly revealed, recent papers suggest to specifically target the proven toxic forms of Aβ [7, 8]. Accordingly, Hoogerhout and coworkers [12] developed a concept vaccine that was selectively directed to the misfolded oligomeric Aβ, by mimicking the bend in the molecular structure with small cyclic peptides. They showed that immunization with these small cyclic peptides conjugated to tetanus toxoid (TTd) induces antibodies to Aβ in Balb/c mice. The antibodies were highly specific for misfolded Aβ, as they did not recognize the homologous unmodified linear peptide nor AβPP [12]. Moreover, the antibodies cross-reacted in vitro with amyloid plaques and oligomeric and fibrillar Aβ1–42 in mouse and human brain tissue. In a Nucleus Basalis of Meynert lesion model [13], C57BL/6 mice that were immunized with these small cyclic peptides showed significantly less cholinergic fiber loss upon stimulation with oligomeric Aβ than mock-immunized mice. Although the effect was rather small, the results were considered highly promising for further research. The next step would be to study the effects of this vaccination procedure in an appropriate AD mouse model. Transgenic J20 mice overexpress the human APP [14], resulting in spontaneous development of Aβ plaques and cognitive deficits, starting from the age of 6 months [15]. The aim of the present study is to investigate whether active immunization at young age prevents or delays the development of cognitive decline and plaque load in J20 mice at later ages. Secondly, effects of active immunization at later ages, when plaque load and cognitive impairment are established, are evaluated.
METHODS
Animals and housing
Experiments were performed using 220 male J20 mice that expresses the mutant form of the human amyloid protein precursor (APP) bearing both the Swedish (K670N/M671L) and the Indiana (V717F) mutations, under control of the human platelet derived growth factor B polypeptide promoter. This strain, originally obtained from Jackson (USA), is bred in our own facility (Groningen, The Netherlands). In addition, 25 male wild type littermate mice from the J20 breeding colony were included. At the age of 7 weeks, all mice were housed individually in macrolon type I L cages (length 30,0 cm, width 12 cm, height 13 cm; Bayer, Germany) with sawdust as bedding, a cardboard tunnel as cage enrichment and shredded cardboard as nesting material. Housing occurred in climate rooms (temperature 22±1°C, humidity 50±10% and a reversed light/dark schedule of 12 h light (±50 lux) and 12 h dark, normal tap water and food (standard rodent chow: RMHB/ 2180, Arie Block BV, Woerden, NL) ad libitum. All mice were weighted every 2 weeks and checked daily for health/activity/food/water and abnormal behavior. Cages were cleaned once every two weeks. All procedures were in accordance with the regulation of the ethical committee for the use of experimental animals of the University of Groningen, The Netherlands. Mice were genotyped at inclusion and at sacrifice; only mice that display the right phenotype at both occasions were included in the analyses.
Experimental outline
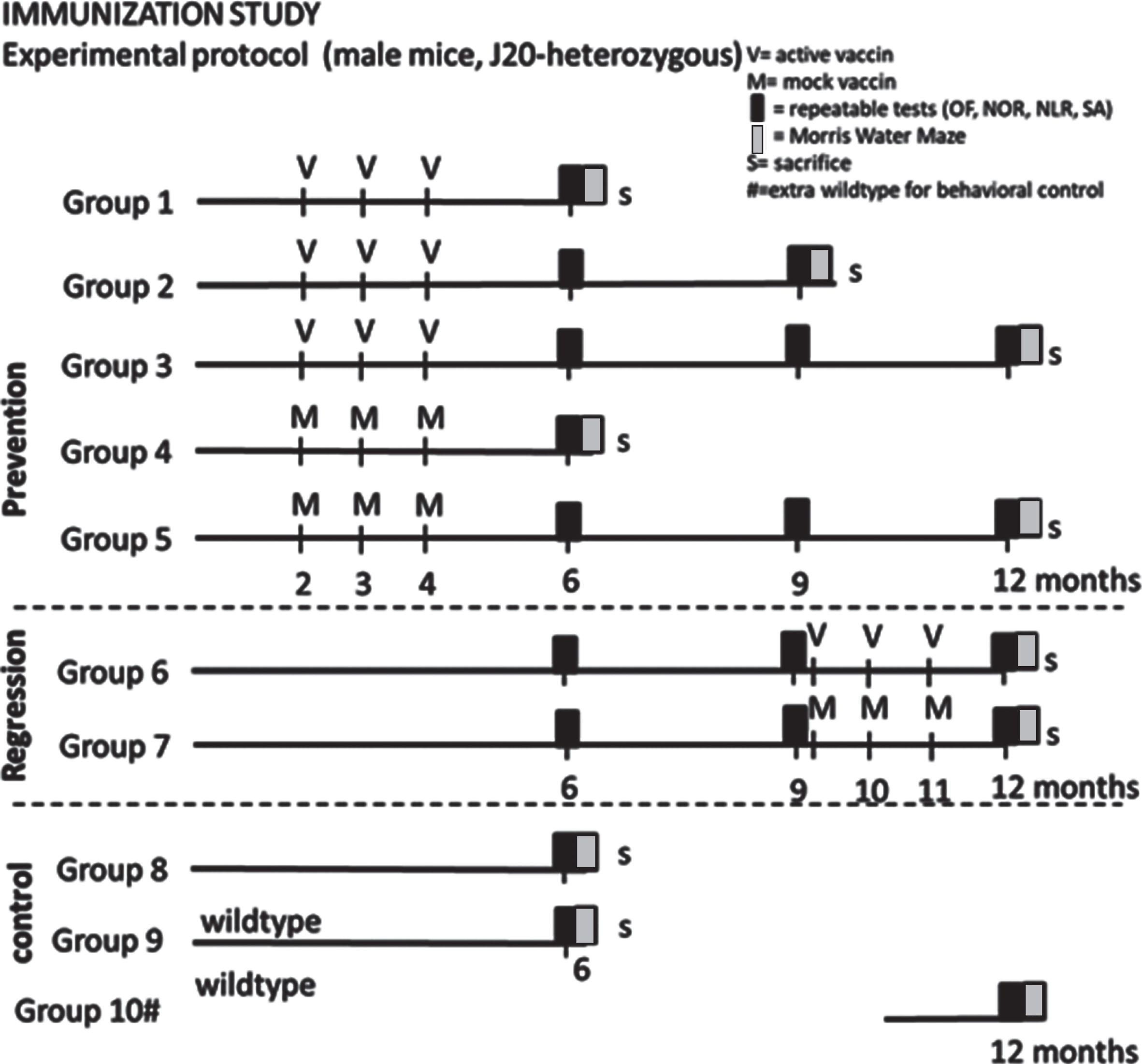
Mice at the age of ∼7 weeks were randomized into groups 1–9 (dependent on genotype) presented in Fig. 1 (35 mice per vaccination group; 25 mice per mock treated group; 15 non-treated J20; 15 wild type littermates). The mice were habituated to the climate room and housing conditions for at least seven days before receiving the first round of immunization. Mice were immunized either before plaque development, or after established plaque formation [14], while control mice received mock vaccine. According to the schedule in Fig. 1, mice that were vaccinated/mock treated early, received 3 vaccine/mock injections at 2, 3, and 4 months of age (prevention groups 1–5) and were tested at 6, 9, and 12 months of age, while delayed vaccination/mock treated mice received injections at 9, 10, and 11 months of age (regression groups 6–7) and were tested at 12 months of age. Non-treated 6-month-old J20 and littermate wild type mice were included to verify a strain-related behavioral phenotype. At a later phase, a 12-month-old non-treated wild type group (littermates of the included J20 mice) was added to confirm effectiveness of our behavioral tests for mice this age. Since the latter group was added at the age of around 10 months and hence did not follow the same procedures as the other mice, this group was not used for statistical comparison.
Fig.1
Experimental outline of the study. OF, Open Field; NOR, Novel Object Recognition; NLR, Novel Location Recognition; SA, Spontaneous Alternation; V, active vaccine; M, mock vaccine; S, sacrifice #: group 10 has been added later to verify suitability of behavioral tests for 12-month-old mice.
Two weeks after the last injection of vaccine or mock, serum samples were collected by tail cut to obtain titers for antibody responses against oligomeric Aβ.
Before the repeatable behavioral tests, mice were habituated to the procedure 5, 4, and 3 days ahead. The repeatable behavioral tests were performed; at 6 months (group 1–9), at 9 months (group 2, 3, 5, 6, and 7) and at 12 months of age (groups 3, 5, 6, 7, and 10). These repeatable behavioral tests consisted of: Open Field (OF), Novel Object Recognition (NOR), Novel Location Recognition (NLR), and Spontaneous Alternation (SA) test. One week before sacrifice, a Morris Water Maze (MWM) test was performed; at the age of 6 months (group 1, 4, 8, and 9), at the age of 9 months (group 2) or at the age of 12 (group 3, 5, 6, 7, and 10).
At sacrifice, mice were deeply anesthetized with pentobarbital. Blood samples were collected by cardiac puncture, serum was derived and stored at –20°C before sending to Intravacc to verify antibody responses against oligomeric Aβ and cyclic peptides. The remaining blood sample were pipetted into a 1.5 ml Eppendorf tube with 20 μl ethylenediaminetetraacetic acid (EDTA) and kept on ice. Thereafter, plasma was derived by centrifuging for 15 min at 4°C at 2000 g, and stored at –80°C for later use. Brains were dissected and processed for further immunohistochemical analysis.
Vaccination and verifying immunization
The vaccine consisted of peptides cyclo[Aβ22–28-YNGK’], cyclo[Aβ23–29-YNGK’], and cyclo[Aβ22–29-YNGK’] conjugated to tetanus toxoid (TTd). As adjuvant an aqueous suspension of aluminum hydroxide (Alhydrogel 2%, 10.3 mg Al3 +/ml) from Brenntag (Frederikssund, Denmark) and monophosphoryl lipid A from Salmonella enterica serotype Minnesota Re 595 (Re mutant) was added. The mock vaccine was prepared by capping bromoacetylated-TTd with 2-aminoethanethiol and mixing purified capped TTd with adjuvant. The vaccine and mock vaccine were prepared, purified, and provided by the Institute for Translational Vaccinology (Intravacc) (Bilthoven, The Netherlands). For each batch, cyclic peptides from the same original preparation were freshly conjugated to TTd and adjuvant was added.
Mice were immunized by three subcutaneous injections in the groin with either 0.3 ml trivalent peptide-TTd conjugate vaccine with adjuvant (referred to as ‘immunized’ mice or vaccine) or 0.3 ml mock-TTd with adjuvant (referred to as ‘mock-immunized’ mice, or mock). In our previous study, using this procedure, almost 80% of the immunized mice displayed a specific immune response against misfolded Aβ (13). Fourteen days after the mice had received their third immunization round, blood samples were collected by a tail cut (10–20 μl per mouse). Group 8–10 underwent no tail cut blood sampling. At sacrifice blood samples were collected after heart puncture (300 μl). The blood samples were pipetted into a SARSTEDT microvette CB 300 Z (SARSTEDT AG & CO, Nümbrecht, Germany). Thereafter, serum was derived from the blood samples by centrifuging for 15 min at 4°C at 2000 g. Subsequently the serum was stored at –20°C and sent to Intravacc (Bilthoven, The Netherlands) for an enzyme-linked immunosorbent assay (ELISA) to determine the anti-oligo-Aβ1–42 titers in tail samples and anti-cyclic peptide titers as well as anti-oligo-Aβ1–42 titers in heart samples at sacrifice.
In a subset of mice, plasma levels of Aβ1–42 were measured by ELISA (code 292-64501, WAKO, USA), according to manufacturer’s procedures; The kit is constructed as a sandwich ELISA format with two kinds of antibodies. The monoclonal antibody BNT77, which epitope is Aβll–28, is coated on 96 well surfaces of a separable microplate and acts as a capture antibody for Aβx–42. Captured Aβx–42 is recognized by another antibody, BC05 (Fab’ fragment), which is specifically detects the C-terminal portion of Aβx–42, labeled with HRP. TMB is used as a read-out and the reaction is terminated by the addition of stop solution. The absorbance is then measured at 450 nm. For this, plasma from mice with highest or with lowest Aβ antibody titers within the immunized groups was selected to evaluate plasma levels of Aβ of high responders (= high titers) versus low responders (= low titers). Samples of wild type mice served as negative control.
Behavioral tests
Behavioral tests were performed in the active phase of the mice (dark period) excluding the first and last 2 h of the active phase. All behavioral tests were performed in dim light conditions. The mice were habituated starting 5 days before the repeated behavioral tests, also at 9 and 12 months, by transporting the cage to the room and by handling the mouse for 30 s. This was repeated for 3 sequential days, once a day. The tests were always performed in the same sequence; OF, SA, NOR/NLR, MWM. Mice were tested in random order. All tests were recorded by EthoVision Version: 11.5 (Noldus Information Technology, Wageningen, The Netherlands). The OF and MWM test were also live-tracked using EthoVision Version: 11.5.
Day 1: Open field (OF)
The OF test was included for general exploratory behavior (anxiety and locomotor activity). The open field box (50×50×35 cm) was divided in 3 zones; border (30×10 cm), corners (10×10 cm), and center (30×30 cm). The mouse was placed in the middle of the box and exploring behavior was recorded for 5 min after the mouse had left the center area. After each trial the box was cleaned with 70% ethanol and dried before the next trial. The distance the mouse moved was used as a measure for exploratory behavior. The percentage of time the mice spent in the safe zone; the corners was taken as the degree of anxiety.
Day 2: Spontaneous alternation (SA)
The SA test was included to give a reflection of the spatial working memory. The SA test was performed in a Y-maze with transparent arms with a length of 32 cm and a diameter of 6.5 cm each. The center of the Y-maze consists of a circular area with a diameter of 11 cm. Each mouse was placed in the center of the Y-maze and exploring behavior was recorded for 10 minutes, starting when the mouse entered the first arm. An entry was considered complete when the mouse has entered the arm with 4 legs and exits with 4 legs. Entries were recorded manually and the correct alternations were calculated. A successful alternation was defined as three consecutive entries into a new arm before returning to the two previously visited arms. After each trial the Y-maze was cleaned with 70% ethanol and dried before starting the next trial. Percentage of successful alternations was calculated by dividing the correct alternations by the possible alternations (defined as the number of the total arm entries minus two) and multiplied by 100. Mice that had less than 20 entries in the 10 min test are excluded from analysis.
Day 3: Novel object/Novel location recognition (NOR/NLR)
The NOR/NLR test is representative for short-term object recognition and spatial recognition. The same box as for the OF test is used. Repeated NOR/NLR tests in the same mouse required different sets of objects, resulting in the use of three pairs during the whole experiment. The NOR/NLR tests consist of four trials of 5 min each: Habituation, Baseline, Novel Object Recognition, and Novel location Recognition. The mouse stayed in the box for the whole test. In between the trials a pause of 45 s is introduced, to clean the objects. The trial starts with habituation; the mouse is introduced in the middle of the box and allowed to explore for 5 min. Subsequently, two similar objects were introduced for the mouse to explore. Then in random order, either one of the objects is replaced by a novel one; NOR, or one of the objects was relocated to another position; NLR. The time spent exploring the two objects was recorded for 5 min. The box was cleaned and dried before the first trials and the objects were cleaned with 70% ethanol and dried before introduction in the box. The time exploring the objects was analyzed in E-line. Preference was calculated by dividing the time spent exploring the novel object or relocated object by the time spent exploring both objects and multiplied by 100. Data of mice that had a total exploration time less than 3% at baseline, or climbed on one of the objects were excluded from further analyses.
Days 4–5: Morris water maze (MWM)
The MWM was performed to test long-term spatial learning and memory as well as cognitive flexibility. The MWM test was only performed in the week before sacrifice, as this test cannot be repeated without bias. For the MWM, a pool with a diameter of 140 cm was used, which was virtually divided into 4 quadrants. Temperature of the water was maintained at 23±1°C throughout the test. A white dye was added to the water to visually distinguish the black mouse from the water. A hidden platform, submerged 1–2 cm below the water level, with a diameter of 15.5 cm was placed in one of the quadrants; the target quadrant for the training phases, and in the opposite quadrant for the reversal test. The MWM consists of 8 training blocks, 1 probe trial, 1 relearning block, and 3 reversal blocks. The first day the mice had 2 training blocks with 4 h in-between. The following two days, three training blocks with 3 h in-between were given. Each training block consists of 3 trials, starting at three different quadrants, excluding the target quadrant containing the platform. The starting positions were randomly selected but differed between the trials. Mice entered the trials with their nose facing the wall of the pool. For each trial a maximum of 2 min exploration was allowed. If the mice reached the platform, it remained there for 10 s before taking it out of the pool. When the mice did not find the platform within the 2 min, the mice were gently guided to the platform and maintained there for 10 s. Mice were towel-dried between trials. After finishing one block the mice were put under a heating lamp for ±15 min. A learning curve is constructed from the average latencies to reach the platform per block (training session). The Area Under the learning Curve (AUC) is used to statistically compare the different groups. A non-successful learner is a mouse that could not find the platform in at least 5 out of the 8 training sessions, from which at least 2 in the last 3 sessions. The probe trial test was performed 24 h after the last training block. In the probe trial the platform was removed and the mice were put in the pool for 100 s. All the mice entered the pool at the quadrant opposing the target quadrant. The time spent by the mouse in the target quadrant and the number of platform crossings was measured. Immediately after the probe trial, a relearning block was performed to train the mice to search for the platform again. In the relearning, the platform was placed back in the target quadrant and 1 training block with 3 trials was executed. The last day a reversal test is performed. Reversal learning is considered to examine cognitive flexibility. In the reversal learning, the platform was moved to the opposite quadrant. Subsequent, three blocks with each 3 trials, excluding the new target quadrant as start position, were executed. Memory consolidation was calculated as the difference between the latency of the relearning block and the first reversal training block.
Sacrifice
At sacrifice, mice were transcardially perfused to collect brain tissue free of blood. For that, mice were deeply anesthetized with pentobarbital; the heart was punctured to collect blood samples. Subsequently, mice are perfused with saline until the liver discolored, followed by perfusion with 4% paraformaldehyde in 0.1 M PBS. Brains were dissected and post-fixated for 24 h in 4% paraformaldehyde in 0.1 M PBS and rinsed for 3 days (4 times) in 0.01 M phosphate buffered saline (PBS, pH 7.4). To prevent freezing damage, brains were dehydrated in 30% sucrose in PBS overnight at room temperature before freezing with liquid nitrogen. Brains were stored at –80°C for further processing.
Immunohistochemistry
Plaque load
To examine the effect of active immunization on plaque formation in the brain, a 6E10 staining was performed. 6E10 binds to amino acid residues 1–16 of Aβ, and hence does not distinguish between the different forms of Aβ (AβPP, linear, oligomeric fibrillary). The epitope lies within residues 3–8 (EFRHDS). A monoclonal mouse anti-amyloid beta 1–16 antibody was purchased from Biolegend (San Diego, CA, USA). The Biolegend’s beta amyloid antibody clone 6E10 binds to the abnormally processed isoforms, as well as precursor forms. Secondary goat anti-mouse was purchased from Jackson ImmunoResearch Inc (West Grove, PA, USA). After the staining, stitched photographs (50×) were processed for manual analyses of plaque load; plaques were counted regarding number, size and location (Image-Pro Plus version 6, Media Cybernetics, Rockville MD, USA). Plaque load was calculated as number times size per section.
Neuroinflammation
Neuroinflammation was investigated by immunohistochemistry in subsets of mice. Mouse brains were cut in 20 μm thick sections. Prefrontal cortex (PFC) (Bregma 2.34 – 1.54) and dorsal hippocampus (DHC) (Bregma –1.34 –2.18) sections were stained to visualize microglia. To examine the morphological features of microglia in the mouse brain, DHC and PFC sections were stained using antibodies against ionized calcium binding adaptor molecule 1 (Iba 1), a 17 kDa protein which in the central nervous system is specific for microglia. Sections were washed 3 times in 0.01 M PBS, endogenous peroxidases were blocked using a 0.3% H2O2 (Merck Darmstadt, Germany) solution in 0.01 M PBS for 30 min. Sections were washed in 0.01 M PBS and subsequently incubated for 72 h at 4°C in Rabbit anti-Iba-1 (Wako Pure Chemical Industries, Osaka, Japan) at 1:2500 dilution in 1% BSA (Sigma-Aldrich, St. Louis MO, USA) in PBS containing 0.1% Triton X-100 (Merck Darmstadt, Germany). After washing, Biotin-SP-Goat anti-Rabbit antibody (Jackson ImmunoResearch, Suffolk, UK) in a 1:500 dilution in 0.01 M PBS was added for 2 h at room temperature. After washing, Vectastain Elite ABC HRP Kit (Vector Laboratories, Burlingame, Ca, USA) in 1:500 dilution in PBS which was added to the sections and after washing again, DAB (Sigma-Aldrich, St. Louis, MO, USA) was used for counterstaining. Images were taken using an Olympus BH2 microscope with a Leica DFC280 camera, software used was Leica Qwin V3. Iba1 photos were taken at 200 magnification of the CA1, CA3, dentate gyrus inner blade, hilus, and in the prefrontal cortex. Iba-1 pictures were analyzed using Image-Pro Plus version 6 image software (Media Cybernetics, Rockville MD, USA) according to the protocol as described previously [16]. Microglia activity, calculated as cell body area/total cell area, was obtained as a measure for neuroinflammation.
Neurogenesis
Neurogenesis was obtained from doublecortin (DCX) staining of dorsal hippocampal sections [17]. Sections, 20 μm, were washed twice in 0.01 M PBS before blocking endogenous peroxidases by 20 min incubation in 3% H2O2 in PBS after which the sections were washed again 4 times in 0.01 M PBS. Sections were blocked by 1 h incubation in 5% normal rabbit serum (NRS) in 0.01 M PBS, which is removed, followed by adding the primary antibody, goat anti doublecortin (DCX, Santa Cruz, Santa Cruz, CA, USA) at 1:4000 dilution in 0.1% Triton X-100, 1% BSA, 1% NRS in 0.01 M PBS. Primary antibody is incubated at 37°C for 3 h, followed by overnight incubation at room temperature, followed by 3 nights at 4°C. After washing 5 times the secondary antibody, rabbit anti goat (Jackson ImmunoResearch, Suffolk, UK) was added at a 1:500 dilution in 0.1% Triton X-100, 1% BSA in 0.01 M PBS and incubated overnight at 4°C. Washed 5 times and added Vectastain Elite ABC HRP at 1:500 dilution in 0.01 M PBS for 2 h at room temperature, washed 3 times, left for 2 h and incubated overnight at 4°C. DAB enhanced with ammonium nickel sulfate hexahydrate was used for counterstaining. DCX photos were taken at 50 magnification using the Leica microscope system, imaging the entire dentate gyrus at this magnification. The area of DCX positive cells per length of the dentate gyrus was used as measure for neurogenesis.
Brain derived neurotrophic factor
Brain function was estimated from brain-derived neurotrophic factor (BDNF) expression in non-treated 6 months mice. BDNF was stained in free-floating dorsal hippocampus slices, which were first incubated for 20 min at room temperature with 3% H2O2 and subsequently washed with 0.01 PBS. Slices were then incubated for 1 h at room temperature with 0.01 PBS containing 2% normal goat serum (Bio-Connect Life Sciences, Huissen, The Netherlands) and 1% BSA to reduce aspecific staining. Slices were subsequently incubated for 3 h at 37°C with rabbit anti-BDNF antibody (Alomone, Jerusalem, Israel) in a 1:1000 dilution in PBS containing 1% BSA and 1% normal goat serum. Afterwards, slices were kept overnight at room temperature after which they were transferred to a cold room of 4°C. After three nights, slices were washed and incubated overnight at 4°C with secondary goat anti-rabbit antibody in a 1:500 dilution in PBS containing 1% BSA. The following day, slices were washed and incubated at room temperature for 2 h with avidin-biotin complex in PBS. Slices were then washed and kept overnight at 4°C. The next day, DAB was added and activated with 0.1% H2O2. Slices were incubated until specific staining could be observed and were mounted on slides the next day. Pictures were taken at a 100× magnification. Within the dorsal hippocampus, pictures were taken of the CA1 and CA3 regions. Optical density of the BDNF staining in the stratum pyramidale of the CA1 and CA3 region was measured using Leica LAS Macro Editor software. The optical density of the stratum radiatum of the CA1 and CA3 region was used as a measure of background staining and was subtracted from the optical density of the stratum pyramidale.
Data analyses
Statistical analyses were performed using IBM SPSS Statistics Version 22. To verify J20 mice as model for AD, comparison of results of 6-month-old non-treated J20 with wild type were analyzed using an Independent-Samples T-test. Overall comparison of early treated groups was performed with a 2-way ANOVA, with age and treatment as factors. In case of significant effects in repeatable tests, effects were subsequently confirmed by comparing appropriate groups in an ANOVA for repeated measurements. A One-Sample T-test was used for calculating the difference of each group from chance level in the NOR/NLOR tests. Data of the later immunized and mock-immunized mice (regression study) were compared using an Independent-sample T-test. Behavioral data from the prevention study (early vaccination) at 12 months, and the treatment study (delayed vaccination) at 12 months were compared with a 2-way ANOVA, with prevention/regression and treatment as factors. All error bars shown in the figures represent the Standard Error of the Mean (SEM) and p < 0.05 was considered statistically significant.
As mice from some different experimental groups had exactly the same experience at certain time points, for these time points data were pooled. For this reason, 6 months repeated tests outcomes in treatment groups 1, 2, and 3 are combined and will be referred to as the 6 months vaccine groups, while treatment groups 4 and 5 will represent the mock group. Similarly, data of group 6, 7, and 8 will be referred to as 6 months J20 non-treated mice. Group 9 is representative for wild type 6 months in the repeated behavioral test. At 9 months, data for repeated tests of group 2 and 3 (vaccine) were combined and compared to data of group 5 mock.
As group 10 (wild type) has been added later to check whether 12-month-old mice could perform in our behavioral tests, although litter mate mice did not undergo the exact same procedures; for that this group is not statistically compared to other groups.
RESULTS
General
From the 257 mice collected from the breeding facility, 2 mice did not look healthy and were omitted before entering the study. As anticipated, 255 mice were included in the study. Seventeen mice died and one mouse had to be sacrificed prematurely, during the course of the experiments. Table 1 presents mortality among the different groups. Deaths occurred at different stages of the protocol; before entering experiment (n = 6), before vaccination rounds (n = 8), after vaccination rounds (n = 2), and in-between behavioral testing (n = 2). No striking differences were observed between groups. Six mice were excluded because of inconclusive genotype. Two mice in the non-treated wild type group displayed a human APP band in genotyping after sacrifice, and again by retry, and were hence translocated to the non-treated J20 group.
Table 1
Premature mortality among the experimental groups
| Group | Treatment | Premature mortality |
| 1 | 6 months J20 vaccine prevention | 1/35 (2.9%) |
| 2 | 9 months J20 vaccine prevention | 5/35 (14.3%) |
| 3 | 12 months J20 vaccine prevention | 2/35 (5.7%) |
| 4 | 6 months J20 mock prevention | 1/25 (4.0%) |
| 5 | 12 months J20 mock prevention | 2/25 (8.0%) |
| 6 | 12 months J20 vaccine regression | 2/35 (5.7%) |
| 7 | 12 months J20 mock regression | 5/25 (20.0%) |
| 8 | 6 months J20 non-treated | 0/15 (0%) |
| 9 | 6 months wild type non-treated | 0/15 (0%) |
J20 mice as model for AD
In literature, J20 mice could display signs of cognitive problems from the age of 6 months. To verify that our J20 mice could indeed be used for evaluating the effects of immunization, firstly non-treated J20 mice (group 6, 7, and 8 in case of repeatable tests and only group 8 for the MWM) at 6 months of age were compared to their non-treated wild type littermates (group 9), regarding their AD phenotype. Results from behavioral tests are summarized in Fig. 2, and show hyperactivity without increased anxiety in the OF test; hyperactivity, but impaired working memory in the SA test; no impaired NOR, but impaired NLR; and impaired spatial learning (longer latency) and memory in the MWM test. As spatial learning and memory were severely impaired in J20 mice, results from the reversal test in the MWM would not provide a relevant measure for cognitive flexibility, and therefore was not presented here. Hence, results confirm AD-like phenotype in J20 mice at 6 months of age. Moreover, these 6-month-old J20 mice showed plaque formation, and displayed significantly lower neurogenesis and BDNF levels compared to wild type, but at similar microglia activity (Table 2).
Fig.2
Evaluation of behavioral comparison between non-treated J20 mice and their wild type littermates at 6 months of age. A, B) Exploratory behavior and preference for the safe area of the open field, respectively. C, D) Total number of arms entered and % correct alternations, resp., in the spontaneous alternation test. E) Preference for the novel object in the Novel Object Recognition test (NOR) and F) preference for the relocated object in the Novel Location Recognition test (NLR). G) Area under the spatial learning curve as latency to find the platform (AUC). H) Spatial memory as the time spent in the target quadrant of the Morris Water maze test. *significant difference between J20 and wild type.
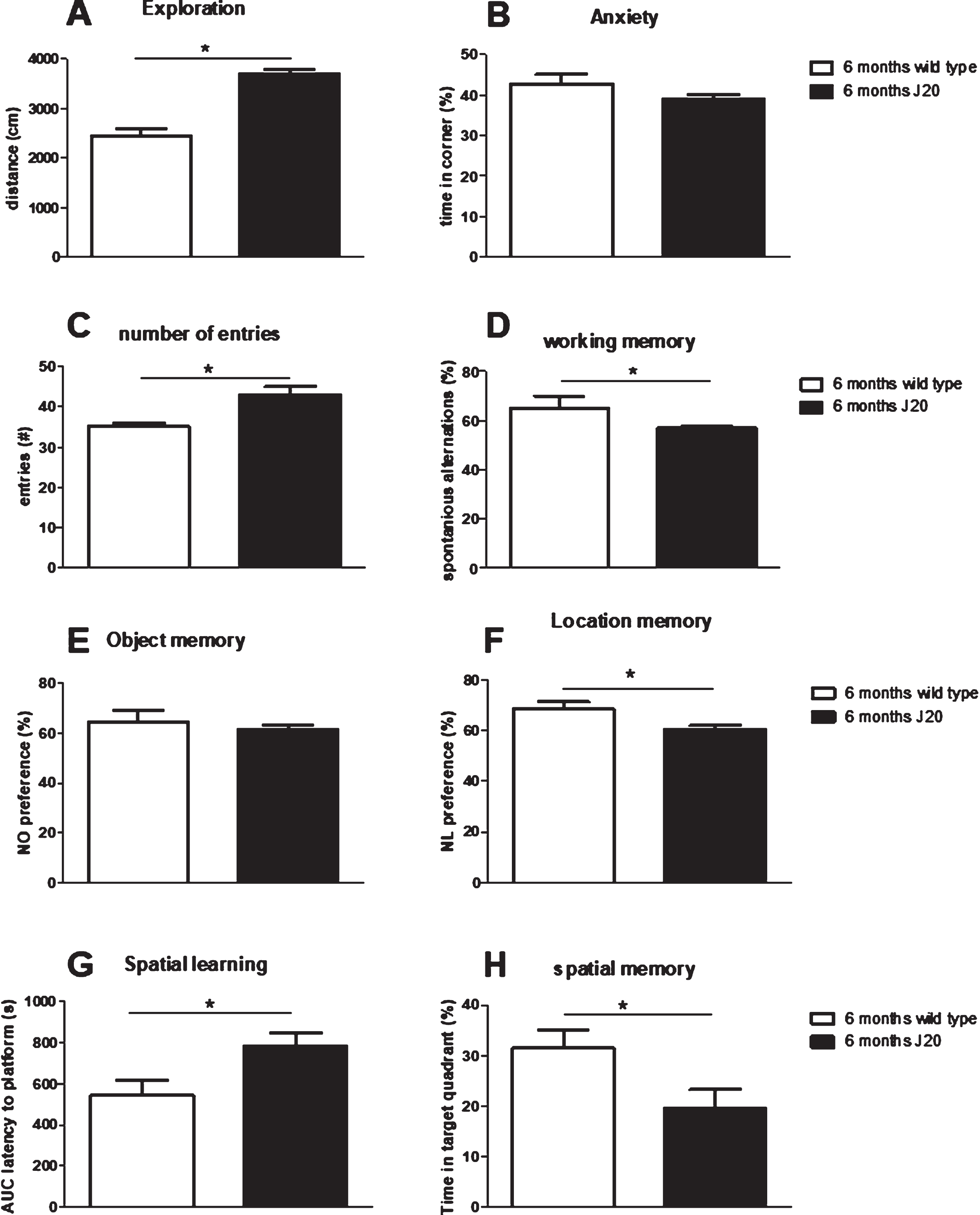
Table 2
Immunohistochemical parameters obtained in the hippocampus of 6-month-old non-treated J20 and wild type littermates
| Wild type | J20 | |
| N | 11 | 12 |
| Microglia activity hippocampus (cellbody/cellsize %) | 10.0±0.3 | 10.9±0.4 |
| Double Cortin (area/length) | 2.5±0.2 | 1.8±0.2* |
| BDNF (OD) | 43.3±4.9 | 32.3±4.6* |
OD, optical density; BDNF, brain-derived neurotrophic factor. *significantly different from wild type.
Effects of early vaccination on behavior
Exploration and anxiety in the open field (OF)
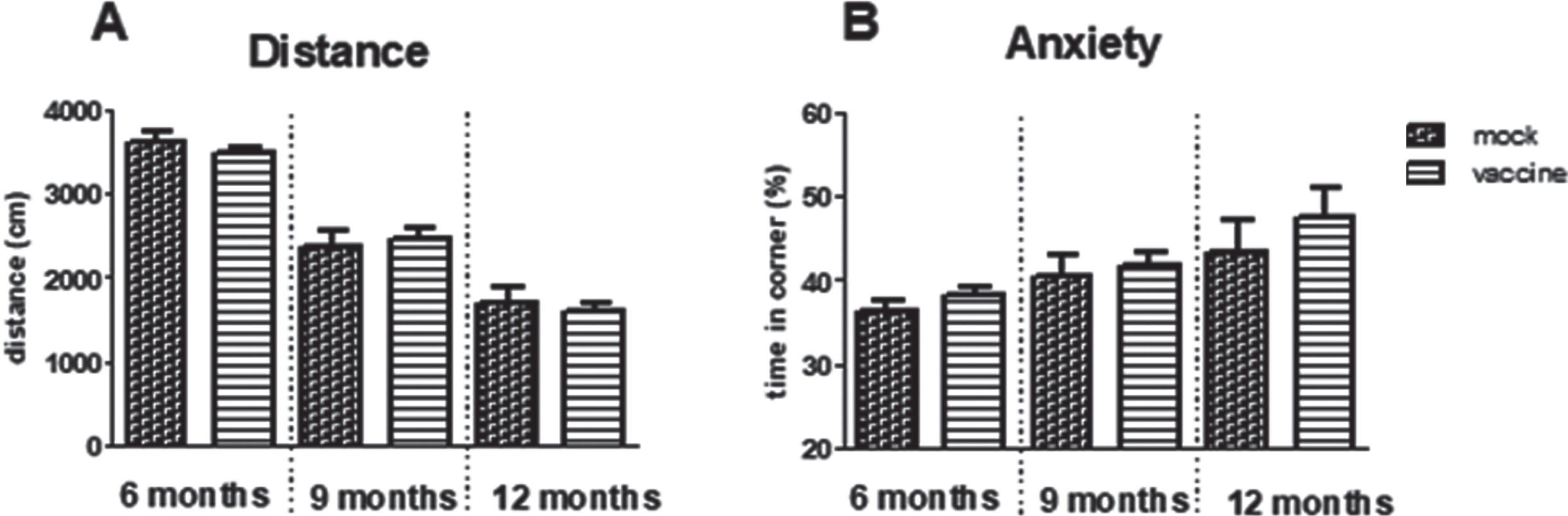
The way the mice act in the open field expresses general exploratory and anxiety behavior. Time spent in the safe zone, the corner, was significantly increased with aging (p = 0.009) but no effect of treatment could be observed. Similarly, exploration measured as distance moved, was significantly decreased with aging (p < 0.001) without effect of treatment (Fig. 3).
Fig.3
Results of the open field test in immunized and mock-immunized mice at different ages. A significant effect of aging was observed for the distance moved (A) as well as for preference for the safe zone; the corner (B), however, without effects of treatment.
Working memory: Spontaneous alternation (SA)
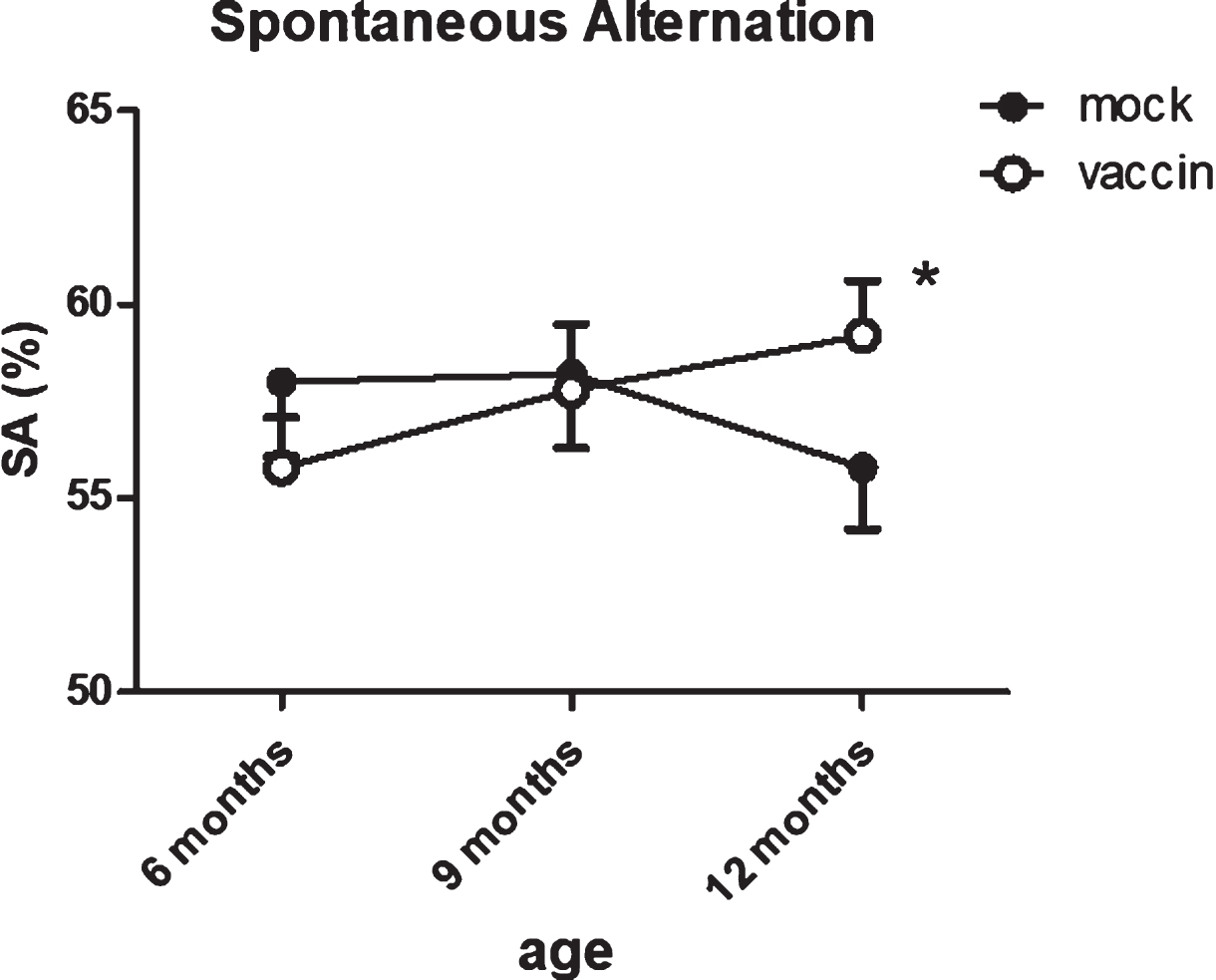
The number of entries showed a slight, but significant (p = 0.050) age-related reduction, with no effect of treatment. Correct alternations as percentage of number of entries was used as a measure for working memory. The percentage of correct alternations appeared significantly improved by immunization (p = 0.029). The effect of treatment was confirmed with ANOVA for repeated measurements within the same mice (group 3 versus group 5; p = 0.04) (Fig. 4), with the vaccinated mice performing almost at the level of 12-month-old wild type mice (SA = 61.7±2.2%).
Fig.4
Serial measurements of spontaneous alternations as measure for working memory in the same mice shows a significant improvement after vaccination at 12 months. The number of arms entered in the spontaneous alternation test showed significant age-related decline, without effect of treatment. *significant difference between vaccinated and mock treated mice.
Short-term memory: NOR/NLR
Short-term memory function was obtained from a NOR and NLR test. In neither test, significant differences regarding age or treatment were observed. However, whereas J20 could recognize the Novel Object, irrespective of age or treatment (6 months mock: 58±3%; 6 months vaccine: 60±3%; 9 months vaccine: 61±3%; 12 months mock: 60±3%; 12 months vaccine: 57±3%, all significantly different from 50%), Novel Location Recognition was better at all ages (repeated measurements) in 12 months vaccinated compared to mock treated mice (64±3; 60±3; 61±3% in 6-, 9-, and 12-month-old vaccinated mice, resp. n = 31, versus 56±4; 57±3; 58±3% in 6-, 9-, and 12-month-old mock treated mice, resp., n = 22).
Spatial learning in the MWM
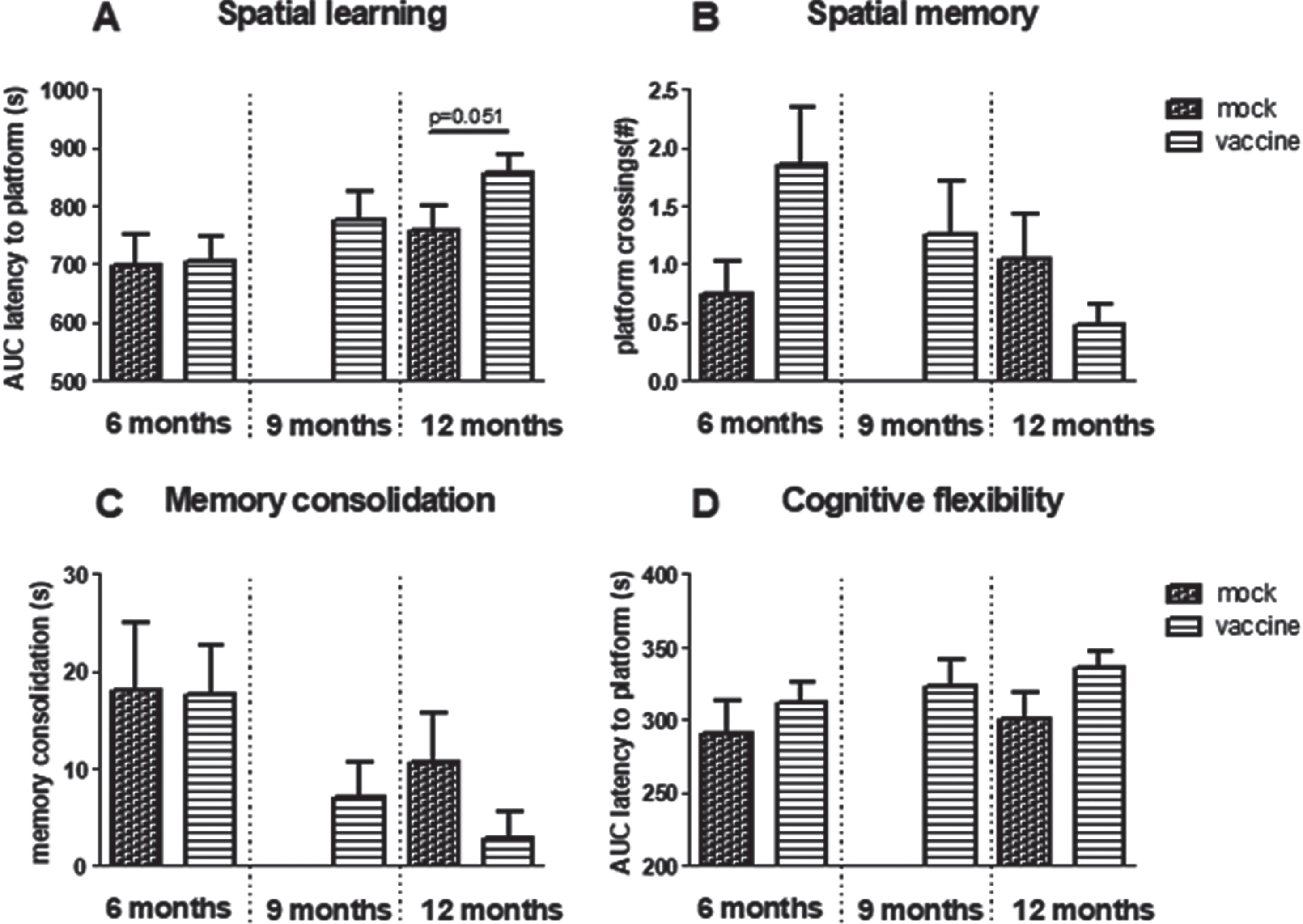
When spatial learning curves of mice that received vaccination or mock (group 1–5) were compared, results showed a significant effect of age (p = 0.009), but no effect of treatment, nor a significant interaction between the two factors. Although borderline statistically significant (p = 0.051), 12-month-old vaccinated J20 mice perform worse than 12-month-old mock mice (Fig. 5A). After learning, vaccinated mice found the platform in 104±6 s, while mock mice found it in 86±9 s. Furthermore, the percentage of mice that successfully could learn in the MWM declined with age; whereas at 6 months, 62% of vaccinated and 65% of mock treated mice could learn the position of the platform, at 12 months, 48% of mock and only 25% of vaccinated mice were successful. For comparison, AUC of the learning curve of 12-month-old wild type mice was 700±63 s, with 78% successful learners.
Fig.5
Results obtained in the Morris Water Maze. A) Spatial learning as area under the curve (AUC) of latency to find the platform and B) spatial memory as the number of times mice swam over the previous position of the platform. C) Memory consolidation. D) AUC of latency to find the relocated platform in reversal learning. Note that although not statistically significant, all parameters indicate worse performance in 12-month-old vaccinated compared to mock treated mice.
Spatial memory (MWM)
Overall comparison of spatial memory in the groups treated with early vaccination or mock did not reveal any statistical differences regarding effects of aging, treatment, or interaction, neither in time in target quadrant, nor in number of crossing over the previous position of the platform. However, although not statistically significant, the results regarding platform crossings may indicate improvement at 6 months, but deterioration at 12 months of age (Fig. 5B) in vaccinated mice. Time in target quadrant did not exceed chance level (25%) for any of the treated groups, whereas wild type mice could remember the platform position at 6 months (32±4% in target quadrant and 3.3±1.1 platform crossings; N = 13) as well as at 12 months (39±7% in target quadrant and 2.2±1.0 platform crossings). Nevertheless, if mice do not learn the position of the platform successfully (Fig. 5A), it is not surprising that they also would not remember its position in the probe trial. However, if only mice that actually learned the position of the platform are considered, overall performance improved, but vaccinated mice still performed worse than mock.
Memory consolidation (MWM)
If mice had learned the position of the platform well, they may be more confused when it is relocated to another position; memory consolidation. Memory consolidation showed a significant age-related decline (p = 0.017), without a significant effect of vaccination versus mock (Fig. 5C).
Cognitive flexibility (MWM)
Cognitive flexibility is obtained from the AUC of the learning curve in the reversal learning trial, in which the platform is relocated to the opposite quadrant. Comparing results of all early treated groups, no effect of age was observed, but treatment gave a trend (p = 0.079) with vaccinated mice performing worse compared to mock (Fig. 5D). No interaction effect was seen.
The relevance of this test, however, is questionable as groups showed already differences in learning and memory of the position of the platform.
Immunohistochemistry
Aβ plaques
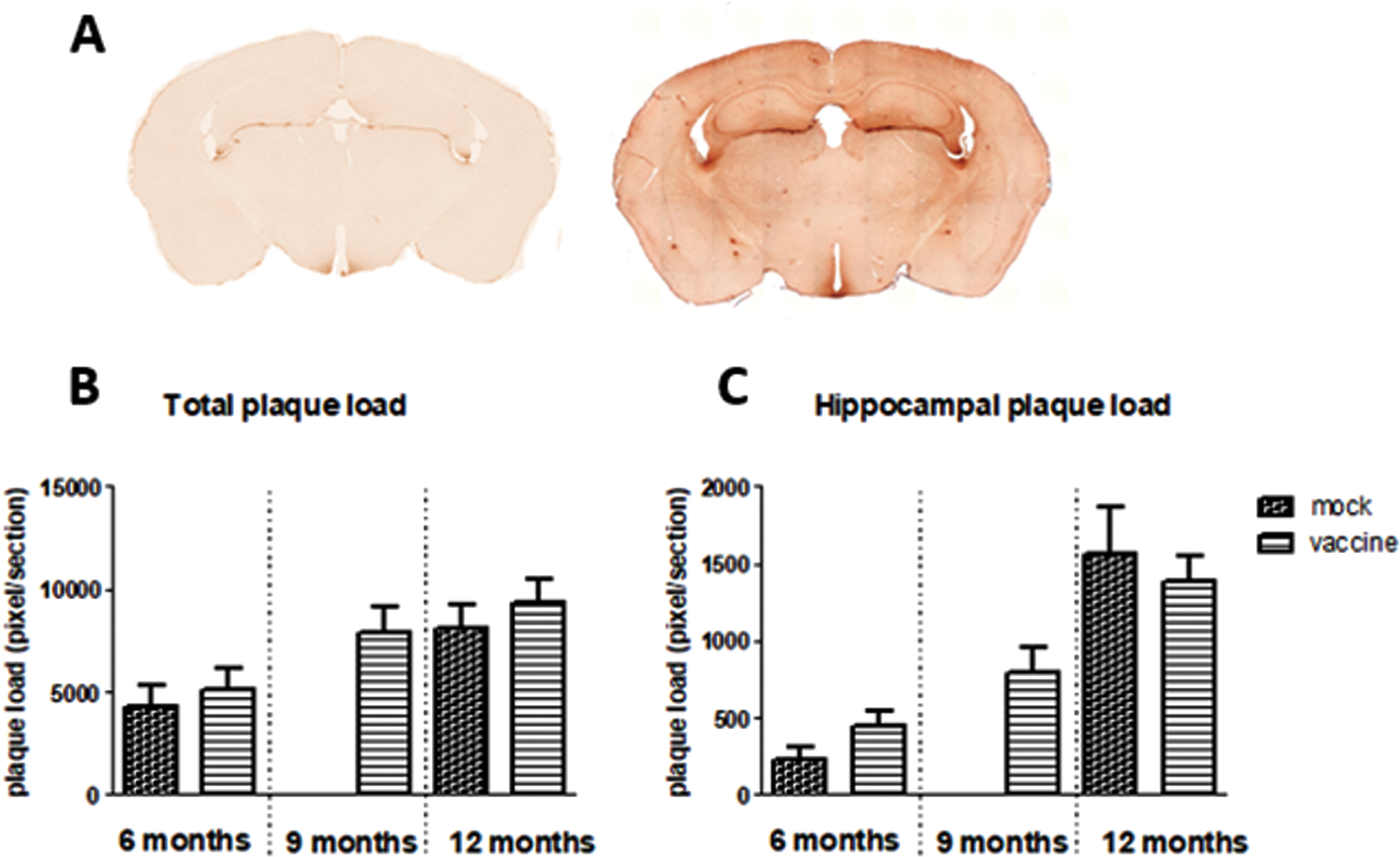
Aβ presence was stained with 6-E10. This stains all forms of Aβ, from the precursor to the actual Aβ plaques. An example is shown in Fig. 6A. The figure shows background staining; probably AβPP, as well as dark spots; Aβ plaques, in different brain areas in J20 mice. Plaques mainly occurred in the hippocampus and cortex. Figure 6B and C shows plaque load in the whole brain section, as well as specifically in the hippocampal area. There is a significant age-related increase in plaque load (p = 0.002), but no significant effects of treatment, nor interaction. Changes in plaque load could be attributed to both increases in size as well as in number of plaques.
Fig.6
A) Example of 6-E10 staining of Aβ in brain sections of wild type mice (left panel) and J20 mice (right panel), showing more background staining and appearance of Aβ plaques in J20 mice. Total plaque load (B) and specific plaque load in the hippocampus (C) in the different experimental groups is presented in the lower panel.
Neither total plaque load nor hippocampal plaque load was significantly correlated to any of the cognitive parameters. However, hippocampal plaque load was significantly correlated to parameters obtained in the open field test (see Table 3).
Table 3
Regressions between open field (OF) test parameters, and total plaque load and hippocampal plaque load. Bold results indicate significant correlations
| Hippocampal plaque load | Total plaque load | |||
| R | p | r | p | |
| OF distance | –0.38 | 0.000 | –0.14 | 0.090 |
| OF corner | 0.17 | 0.045 | 0.11 | 0.107 |
| OF center | –0.19 | 0.028 | –0.19 | 0.025 |
Neurogenesis
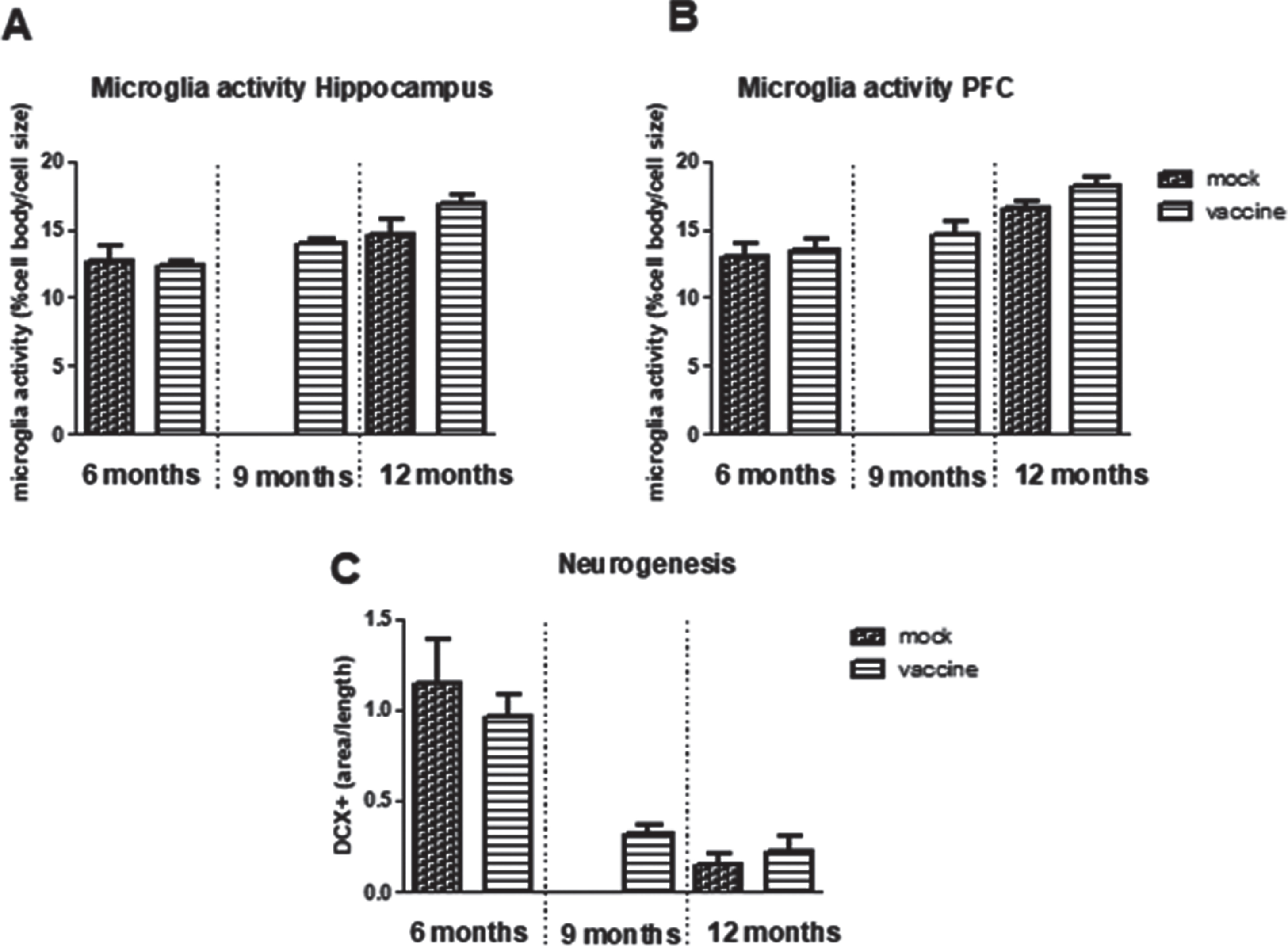
Neurogenesis is determined by the double cortin (DCX) positive area in the hippocampus (Fig. 7C). Neurogenesis was significantly decreased with aging (p < 0.001) without effect of treatment, nor interaction. Neurogenesis was significantly correlated negatively to total plaque load (r = –0.34, p = 0,033), but not with hippocampal plaque load. Moreover, neurogenesis was not correlated to any of the parameters of the cognitive tests, but appeared significantly correlated to distance in the open field (r = 0.43, p = 0.006).
Fig.7
Neuroinflammation measured as microglia activity in hippocampus (A) and prefrontal cortex (PFC) (B) in the different experimental groups (n = 5–10 per group). C) Measurement of Double Cortin (DCX) positive area in hippocampus, as measure for neurogenesis (n = 8 per group).
Neuroinflammation
Neuroinflammation is measured as microglia activity. This parameter was calculated from morphological changes as cell body area/total cell size. In the hippocampus as well as in the prefrontal cortex, a significant age-related increase in microglia activity was observed, without significant effects of treatment nor interaction (Fig. 7A, B). Microglia activity in the hippocampus was negatively correlated to neurogenesis (r = –0.46, p = 0.019) and positive to plaque load (r = 0.55, p = 0.001). Whereas for all mice no associations were observed for microglia activity and behavioral performance, when focusing on only the 12-month-old vaccinated mice and their mock controls, a highly significant correlation was seen between hippocampal microglia activity and AUC of the MWM learning curve (r = 0.78, p = 0.008), which could completely be attributed to microglia activity in the Hilus (r = 0.93, p < 0.000).
Aβ metabolism
Antibody titers for the cyclic peptides as well as for Aβ1–42 were measured at Intravacc. Results are presented in Table 4, and show a good and persistent response to the peptides at sacrifice in all age groups, though significantly declining with aging. However, antibody responses toward Aβ in immunized mice at sacrifice did not exceed the background values seen in mock treated mice. Aβ antibody titers measured in tail samples, collected 2 weeks after the last immunization in all mice treated at young age, revealed a slight positive response in favor of immunized mice (mock:1.17±0.22 versus vaccinated 1.63±0.16, p = 0.047). Immunization at later ages, regression study, showed the same results (Supplementary Table 2). When taking the results of tail samples from all immunized mice together, including early as well as delayed vaccination, the significantly increased Aβ antibody titers in immunized mice versus mock (1,69±0.14, N = 126 versus 1.22±0.18, n = 64; p = 0.019) suggest a positive Aβ response of only short duration (2 weeks).
Table 4
Antibody titers of responses against cyclic peptides or Aβ in serum samples collected at the time of sacrifice
| 6 months | 9 months | 12 months | ||||
| mock | vaccine | mock | vaccine | mock | vaccine | |
| C(22–28)BSA | 2.1±0.3 | 4.7±0.1 | NA | 4.3±0.1 | 1.0±0.3 | 3.8±0.1 |
| C(23–29)BSA | 2.1±0.4 | 4.9±0.1 | NA | 4.5±0.1 | 1.4±0.3 | 4.1±0.1 |
| C(22–29)BSA | 1.5±0.4 | 4.6±0.1 | NA | 4.2±0.1 | 1.3±0.3 | 3.9±0.1 |
| Aβ | 2.2±0.3 | 1.9±0.3 | NA | 3.0±0.2 | 2.5±0.3 | 2.4±0.3 |
To further elucidate on the effects of Aβ, in subgroups of mice; 6-month-old wild type (random), 6-month-old non-treated J20 (random), 6-month-old mock treated J20 (random), 6-month-old vaccine treated J20 (6 mice with the highest and 6 mice with lowest anti- Aβ antibody titers), 12-month-old mock treated mice (random), and 12-month-old vaccinated mice (6 mice with the highest and 6 mice with lowest anti- Aβ antibody titers) circulating levels of Aβ1–42 were measured. Results showed a strain-related increase in circulating Aβ, independent of age, treatment or antibody titers (Table 5).
Table 5
Plasma levels of Aβ 1–42 in the different experimental groups. Vaccinated mice were separated into mice with high and mice with low Aβ titers
| 6 months | |
| Wild type (n = 3) | 6.4±1.5 pM |
| Non-treated J20 (n = 12) | 25.8±1.3 pM |
| J20 mock (n = 12) | 25.1±1.3 pM |
| J20 vaccine low titers (n = 7) | 23.9±2.3 pM |
| J20 vaccine high titers (n = 6) | 24.2±1.9 pM |
| 12 months | |
| J20 mock (n = 11) | 22.3±1.1 pM |
| J20 vaccine low titers (n = 6) | 25.4±1.6 pM |
| J20 vaccine high titers (n = 7) | 25.9±1.6 pM |
Effects of late vaccination
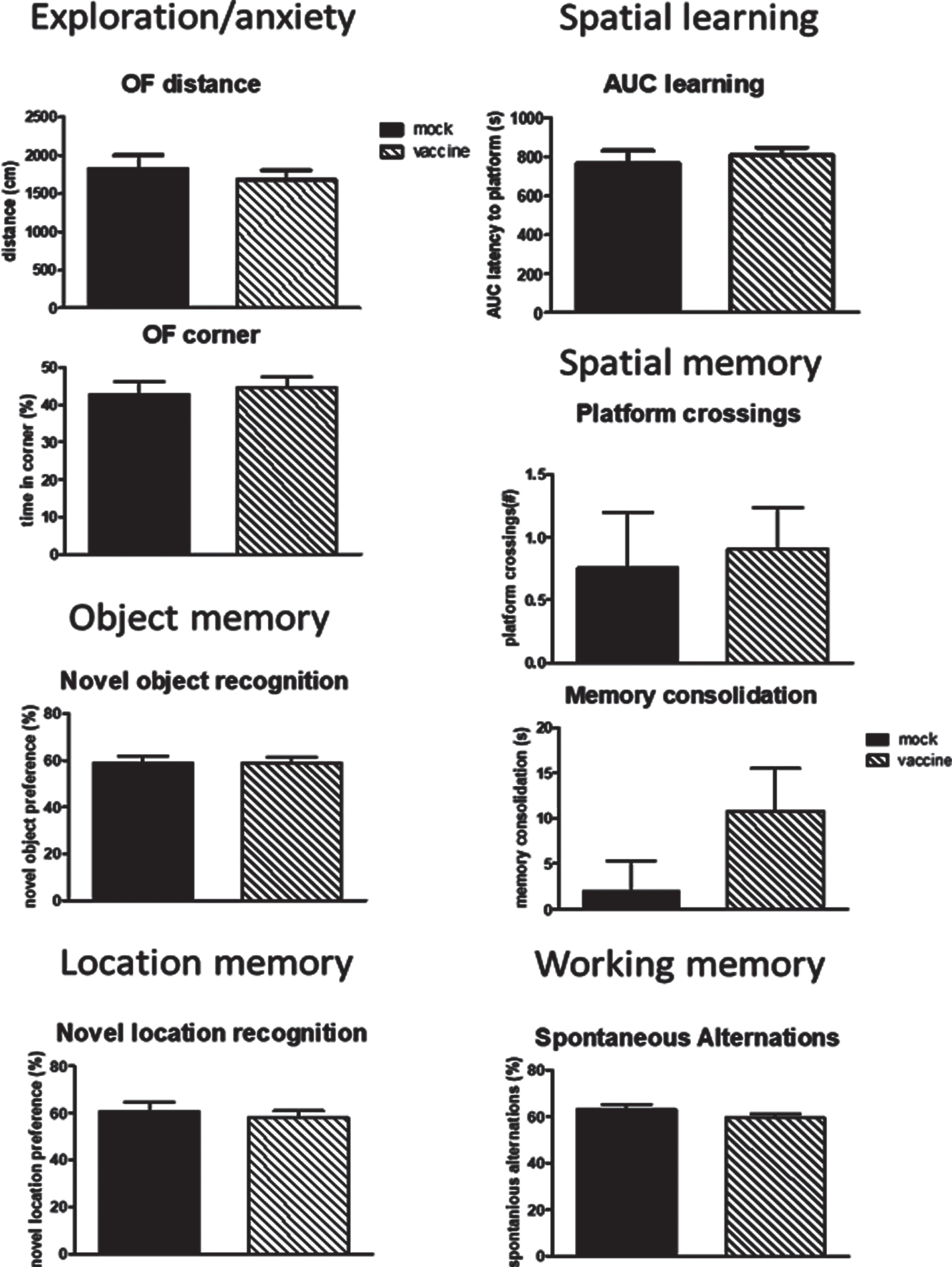
In order to investigate whether vaccination still may have an effect once cognitive decline and plaque formation has been established, J20 mice were vaccinated later in life and compared to mock vaccination. No difference between mock and vaccine mice were seen for any of the behavioral parameters (Fig. 8). Moreover, no significant differences were observed when comparing these delayed treated mice to the early treated mice at 12 months of age. Similarly, no differences were observed in immunohistochemical parameters (Supplementary Table 1). Similar to early vaccination, a good antibody response to the cyclic peptides without antibodies against Aβ were observed (Supplementary Table 2).
Fig.8
Behavioral data of the mice vaccinated or mock treated at 9, 10 and 11 months of age in order to study effects on regression. No effects of late vaccination were observed. AUC, area under the curve; NO, Novel Object; NL, Novel Location.
DISCUSSION
Active immunization against oligomeric Aβ could be a promising step in the development of therapy against AD. A novel concept vaccine of small cyclic peptides mimicking the specific molecular bend in the structure of oligomeric Aβ, conjugated to TTd, induced antibodies specifically against the misfolded oligomeric Aβ in mice [12]. A subsequent in vivo study in mice [13], confirmed a specific immune response against misfolded Aβ, and significantly less oligomeric Aβ–induced cholinergic fiber loss, after vaccination with these peptides. In the present study, the therapeutic potential of these cyclic Aβ peptide conjugates was investigated in J20 mice as model for AD. J20 mice showed an age-dependent AD-like phenotype; cognitive decline and Aβ plaques, supporting the relevance of this mouse model for our study on the effects of active immunization against oligomeric Aβ.
Vaccination at young ages seemed to prevent short-term working memory loss, but deteriorated long-term spatial learning and memory, at later ages. The absence of a significant antibody response to Aβ despite a persisting response to the cyclic peptides, may exclude a mediating role of Aβ in these cognitive effects. Active immunization at later ages, when the AD-like phenotype was established, did not affect cognition nor Aβ pathology. Hence, we conclude that despite the clear and straightforward rational behind the development of the concept vaccine used in the present study, it does not appear to have the anticipated anti-Aβ effects. Still, cognitive performance was affected by early, but not late, vaccination. For late vaccination, the period between vaccination and behavioral testing may have been too short to exert effects on cognitive performance.
J20 mice as model for AD
The behavioral phenotype of our J20 mice was compared to their wild type littermates to verify an AD-like phenotype that could be demonstrated with our test battery. Indeed, in agreement with literature [14], 6-month-old J20 mice displayed overall cognitive decline. Significant plaque development support presence of enhanced Aβ expression. No age-related mortality was observed up to 12 months. Results from wild type littermates indicated that mice ranging from 6 to 12 months of age could perform well in the behavioral tests, supporting that the tests are suitable for this age range. Moreover, for most parameters an age-related deterioration was observed in J20 mice, indicating age-related development of an AD phenotype. The observed features of the model are in general agreement with previous studies [18–21]. From the above results we concluded that the J20 mice provide a relevant model to study the effects of active immunization against oligomeric Aβ.
The concept vaccine
A promising way to prevent AD is to promote the removal of aggregated Aβ proteins through vaccination against oligomeric Aβ [22]. When Aβ proteins aggregate, they undergo a conformational change from a relatively linear protein to a protein with a bended conformation. This misfolded oligomeric Aβ has been shown to directly promote apoptosis of neurons in the brain, resulting in the neurodegeneration in AD [23]. Furthermore, accumulation of oligomeric Aβ leads to oxidative stress, neuroinflammation, loss of synapses and synaptic plasticity in the brain [5, 23]. These soluble Aβ peptides can form fibrils, that arrange themselves into β-pleated sheets to form insoluble fibers that are deposited as extracellular and perivascular amyloid plaques. Active immunization against the oligomeric Aβ could potentially delay or reverse the aggregation processes and hence diminish the cognitive dysfunction [7, 8]. Hoogerhout and co-workers [12] developed a concept vaccine of cyclic Aβ peptides that mimic the bend in misfolded oligomeric Aβ. As this bend is not present in AβPP or normal linear Aβ, indeed the vaccine was shown to be specific for the aggregated and pathological forms of Aβ [12]. This specificity prevents (over)activation of the immune system when no aggregated Aβ is present, and limits interference with the normal physiological function of Aβ. The linear form of Aβ may even have neuroprotective properties, as it can modulate long-term potentiation and plays a role in the innate immune system [24–26]. (Over)activation of the immune system has been an issue with previous vaccines. A phase II trial using the Aβ vaccine AN1792 was discontinued as 6% of patients developed meningoencephalitis [9, 27]. This was linked to T-cell mediated inflammation, as Aβ1–42 vaccine-activated T-cells infiltrated into the brains of patients [28]. In our previous study, immunization with the new concept vaccine [12] diminished neurodegeneration induced by oligomeric Aβ in mice [13]. In the present study, we investigated whether the protective effects of this concept vaccine could be extended in a mouse model for AD.
Active immunization to prevent AD-like phenotype
Cognition
To examine whether vaccination at young ages can delay or prevent an AD-like behavioral phenotype at later ages, J20 mice were immunized by 3 injections, at 2, 3, and 4 months of age. Cognitive behavior was evaluated at 6, 9, and 12 months of age. Since our J20 mice showed an age-dependent cognitive decline from 6 to 12 months of age, improved cognitive performance was anticipated in vaccinated over mock-vaccinated mice. Whereas, at 6 and 9 months of age no differences were observed between vaccinated and mock-treated mice yet, at 12 months differences became more apparent. At 12 months of age, working memory, measured as percentage correct alternations in the Y-maze, was significantly improved by vaccination. An improved working memory may be supported by the findings in the novel location test for short term spatial memory; though not significantly different from mock, vaccinated mice performed significantly above chance level, whereas their 12-month-old mock control did not. In contrast, spatial learning, as latency to find the platform in the MWM, seemed to deteriorate faster with aging in vaccinated than in mock-treated mice, resulting even in an impaired learning curve at 12 months in vaccinated compared to mock mice. Moreover, the number of mice capable of learning this task anyway was about twice as low in the vaccinated compared to the mock group. As the mice did not learn the position of the platform, subsequent spatial memory (platform crossings) was also impaired, and hence results from a reversal trial have no relevance for cognitive flexibility. Interestingly, the mixed cognitive effects observed in the present study showed striking resemblance with the competitive short-term and long-term spatial memory processes described by Sanderson and Bannerman [28]. An improved gluA1 AMPA receptor dependent working memory, and a deteriorated gluA1-independent MWM performance [29], suggests a role for gluA1 AMPA receptors in our cognitive findings. Intracellular levels of oligomeric Aβ have been reported to regulate gluA1 subunit of AMPA receptors [30]. Although we did not observe effects of vaccination on extracellular Aβ (plaques or circulating levels), we cannot exclude effects through changes in intracellular Aβ levels though. Although in literature, most of the studies present positive effect of vaccination [31–35], accurate comparison is hampered by the use of different models, different time schedules, different vaccines and/or adjuvants, and last but not least different behavioral tests. Moreover, a publication bias, negative outcomes are less often published, may have contributed to the overall indicated positive effects. Still, potential deleterious effects of vaccination through interference with the immune system are also recognized [32]. Nevertheless, in the present study a mixed positive/negative effect of vaccination at 2, 3, and 4 months of age on cognition became apparent only at 12 months of age.
Aβ metabolism
Vaccination started at 2 months of age, anticipating no substantial plaques load yet [14]. Whereas at 6, 9 and 12 months, a significant antibody response to the cyclic peptides confirmed accuracy of the vaccination protocol, antibody responses to Aβ did not exceed background levels. Only 2 weeks after the last vaccine/mock injection a significant but mild Aβ response in favor or immunized mice was observed, suggesting at the most a short-lasting Aβ response. This is in agreement with our previous study [13], showing a slight, but significant increase in antibody titers against Aβ, 12 days after the last immunization round. While plaque load showed an age-dependent increase from 6–12 months of age, no effect of vaccination was found. Hence, our hypothesis that vaccination would reduce oligomeric Aβ levels, and hence decrease plaque load, can be rejected. In contrast, Schenk and co-workers [33, 34] did show effects on amyloid pathology after vaccination, using synthetic human Aβ42.
Although overexpression of AβPP is supposed to be brain-specific in the J20 mice [14], plasma levels of Aβ1–42 were increased as well, and Aβ1–42 may have been transported out of the brain. If we consider an equilibrium between circulating and brain Aβ, we would expect age differences, as plaque load increased with age. If circulating levels represent the amount of Aβ that is extruded from the brain, absence of effects of age or treatment on plasma levels may then indicate that this Aβ transport in J20 mice may have reached its limits already at 6 months of age.
Finally, no correlation was found between Aβ pathology and cognition, suggesting no causal relationship between the two AD hallmarks. A similar lack of association has been described in other transgenic mouse models [36] as well as in patients.
Neuroinflammation/neurogenesis
Proliferation and activation of microglia in the brain is a prominent feature of AD [37]. Microglia activation associated with plaque formation could be harmful to neurons, by means of synaptic pruning and/or (over)production of pro-inflammatory cytokines, but microglia activity may also protect against AD, since decreased microglia activity is associated with increased AD risk [37]. Neuroinflammation in the present study is obtained from morphological measurement of microglia activity as cell body to cell size ratio [16]. In 6-month-old J20 mice, significant plaque load with concomitantly decreased cognitive function, did not coincide with microglia activation yet. In contrast, Wright and co-workers [21] suggested that neuroinflammation precedes plaques deposition. Yet similar to the present study, a very recent study indicated no microglia activity at 6 months, but also not at 12 months of age [38]. As expected, microglia activity increased with age, however irrespective of treatment. If anything, at 12 months, microglia activity is higher (p = 0.079) in vaccinated versus mock mice. Although activated microglia can cluster around Aβ plaques, their role is not completely clear. In an elegant study in mice, Baik and coworkers [39] showed that apart from eating the plaque away, microglia could also contribute to plaque growth by consolidation of newly generated plaque clusters in the vicinity of existing plaques. Hippocampal microglia activity was negatively correlated to spatial learning in the MWM suggesting a detrimental rather than a supporting role for microglia activity in the present study. Furthermore, no overall correlations were observed with immunohistochemical parameters and cognitive behavior. Hippocampal microglia activity was negatively correlated to neurogenesis, and neurogenesis was significantly correlated to plaque load, suggesting neurodegenerative processes coupled within the brain. Nevertheless, since neither circulating levels of Aβ, nor plaque load were affected by vaccination, behavioral changes seem to be attributed to mechanisms other than altered Aβ pathology.
Active immunization to reverse already developed AD-like phenotype; regression
For this part of the study, J20 mice were vaccinated or mock treated at 9, 10, and 11 months of age and evaluated at 12 months old. For none of the measured parameters significant differences between immunized and mock immunized were observed. This result may indicate that by active immunization, an already developed AD-like phenotype in J20 mice cannot be rescued. These results are in contrast to effects of Enoxaparin treatment [40] or immunization with Aβ1–42 [33] in mice with sustained plaque formation. On the other hand, time between vaccination and evaluation (1 month) may have been too short to exert an effect. Based on the studies of Wright et al. [21], we anticipated cognitive improvement and plaque reduction after early vaccination, to be seen at 6 months of age, and either persisting or waning at later ages. However, it took 8 months for cognitive improvement to become visible after early vaccination. Accordingly, a longer time between delayed vaccination and analyses could have provided a more complete answer on effectiveness of delayed vaccination.
Clinical perspective
Although we do realize that translation of data from animal studies into the human situation is at the least challenging [41] as AD-like phenotypes in mice are usually artificially induced and mimic only one or a few aspects of AD in humans, we are still able to learn from these results.
Active immunization makes use of the subjects own immune system to remove the unwanted factors. An important advantage of this prophylactic therapy is that the senescent immune system present in elderly is circumvented. In a proper immune response, the new antigen induces specific activation of effector-T-cells, with a short life span, but a subset of these T-cells has the potential for long-term survival; the memory-T-cells [42]. Essential features for these cells are: prior activation by a novel antigen, persistence in absence of cognate antigens and enhanced functional activity upon re-exposure. In case of the present study, we speculate that the memory T-cells may not function optimally, as none of the above features may be optimal; no clear first-time presentation of the antigen during vaccination, as the oligomeric Aβ (epitope) may already be present in low levels; no persistence in the absence of antigen as the oligomeric Aβ is continuously present and may even accumulate; and no clear re-exposure as the exposure persisted all the time. Hence, a proper antibody response to oligomeric Aβ may be hampered by the continuous presence of the antigen, and might have induced a form of immune tolerance, as oligomeric Aβ may have been regarded as “self”. This reasoning would explain the good response to the cyclic peptides; prior activation upon a novel antigen in the mouse, subsequent absence of this antigen, displaying a proper response in titer measurements when re-exposed, but not to Aβ. Moreover, it also may explain the significant effects of vaccination in our previous study [13], in which wild type mice were vaccinated, with presumably no oligomeric Aβ present, followed by a period of absence of antigens, and re-exposed to Aβ in their brains. Absolute antibody titers indeed appear higher than in the present study (3.5 versus 1.6). That the epitope of the cyclic peptides is shared with oligomeric Aβ was confirmed by the responses in mice and human AD tissue [12], in both cases a first-time presentation. Finally, if presence of even low levels of oligomeric Aβ in AD patients is hampering proper antibody responses after vaccination, this may explain the poor outcome of some of the clinical studies. In line with this, recent papers indicate that an effective vaccine should be administered as early as possible and be as specific as possible, will it stand a change in the treatment of AD [7, 8, 27, 43]. That is at least before first appearance of oligomeric Aβ.
Conclusion
J20 mice provide a relevant model to test interference with Aβ pathology. The cyclic peptide-based vaccine, mimicking the oligomeric Aβ structure, did not evoke a long-lasting antibody response to Aβ, and accordingly did not reduce plaque load. Nevertheless, vaccination at young ages seemed to prevent working- and short-term spatial memory loss at later ages, but deteriorated long-term spatial learning and memory. Hence cognitive effects cannot be attributed to altered Aβ pathology. Later vaccination had no effect within the studied timeframe. Translation of the message regarding mechanism and timing of vaccination into the clinical setting of AD patients will need further investigation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank Wanda Douwenga, Jan Keijser, Kunja Slopsema, and Wichard Tilstra for their technical assistance. Moreover, masterstudents Vlad Constantinescu, Siebren Faber, Ingeborg Frentz, Luud Roorda, and Sander Nijhof are well appreciated for their help with behavioral testing and immunohistochemistry.
The study is partly funded by Intravacc (Bilthoven, the Netherlands). As Intravacc is an institution only developing vaccines, without direct commercial targets, the authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-200213.
REFERENCES
[1] | Fiest KM , Roberts JI , Maxwell CJ , Hogan DB , Smith EE , Frolkis A , Cohen A , Kirk A , Pearson D , Pringsheim T , Venegas-Torres A , Jette N ((2016) ) The prevalence and incidence of dementia due to Alzheimer’s disease: A systematic review and meta-analysis. Can J Neurol Sci 43: (Suppl 1), S51–82. |
[2] | Tiraboschi P , Hansen LA , Thal LJ , Corey-Bloom J ((2004) ) The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62: , 1984–1989. |
[3] | Xu X ((2009) ) Gamma-secretase catalyzes sequential cleavages of the AbetaPP transmembrane domain. J Alzheimers Dis 16: , 211–224. |
[4] | Hardy J ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[5] | Selkoe DJ ((2008) ) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behaviour. Behav Brain Res 192: , 106–113. |
[6] | Smith MA , Casadesus G , Joseph JA , Perry G ((2002) ) Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med 33: , 1194–1199. |
[7] | Bachmann MF , Jennings GT , Vogel M ((2019) ) A vaccine against Alzheimer’s disease: Anything left but faith? Expert Opin Biol Ther 19: , 73–78. |
[8] | Herline K , Drummond E , Wisniewski T ((2018) ) Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev Vaccines 17: , 707–721. |
[9] | Orgogozo J- , Gilman S , Dartigues J , Laurent B , Puel M , Kirby LC , Jouanny P , Dubois B , Eisner L , Flitman S , Michel BF , Boada M , Frank A , Hock C ((2003) ) Subacute meningoencephalitis in a subset of patients with AD after Aβ 42 immunization. Neurology 61: , 46–54. |
[10] | Bayer AJ , Bullock R , Jones RW , Wilkinson D , Paterson KR , Jenkins L , Millais SB , Donoghue S ((2005) ) Evaluation of the safety and immunogenicity of synthetic Aβ 42 (AN1792) in patients with AD. Neurology 64: , 94–101. |
[11] | Gilman S , Koller M , Black RS , Jenkins L , Griffith SG , Fox NC , Eisner L , Kirby L , Rovira MB , Forette F , Orgogozo J ((2005) ) Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64: , 1553–1562. |
[12] | Hoogerhout P , Kamphuis W , Brugghe HF , Sluijs JA , Timmermans HAM , Westdijk J , Zomer G , Boog CJP , Hol EM , van den Dobbelsteen GP ((2011) ) A cyclic undecamer peptide mimics a turn in folded Alzheimer amyloid β and elicits antibodies against oligomeric and fibrillar amyloid and plaques. PLoS One 6: , e19110. |
[13] | Mulder CK , Dong Y , Brugghe HF , Timmermans HA , Tilstra W , Westdijk J , van Riet E , van Steeg H , Hoogerhout P , Eisel UL ((2016) ) Immunization with small amyloid-beta-derived cyclopeptide conjugates diminishes amyloid-beta-induced neurodegeneration in mice. J Alzheimers Dis 52: , 1111–1123. |
[14] | Mucke L , Masliah E , Yu GQ , Mallory M , Rockenstein EM , Tatsuno G , Hu K , Kholodenko D , Johnson-Wood K , McConlogue L ((2000) ) High-level neuronal expression of Abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci 20: , 4050–4058. |
[15] | Webster SJ , Bachstetter AD , Nelson PT , Schmitt FA , Van Eldik LJ ((2014) ) Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioural changes in 10 mouse models. Front Genet 5: , 88. |
[16] | Hovens IB , Nyakas C , Schoemaker RG ((2014) ) A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: Cell body to cell size ratio. Neuroimmunol Neuroinflamm 1: , 82–82. |
[17] | Hovens IB , Schoemaker RG , van der Zee EA , Absalom AR , Heineman E , van Leeuwen BL ((2014) ) Postoperative cognitive dysfunction: Involvement of neuroinflammation and neuronal functioning. Brain Behav Immun 38: , 202–210. |
[18] | Beauquis J , Vinuesa A , Pomilio C , Pavia P , Saravia F ((2014) ) Hippocampal and cognitive alterations precede amyloid deposition in a mouse model for Alzheimer’s disease. Medicina (B Aires) 74: , 282–286. |
[19] | Beauquis J , Vinuesa A , Pomilio C , Pavia P , Galvan V , Saravia F ((2014) ) Neuronal and glial alterations, increased anxiety, and cognitive impairment before hippocampal amyloid deposition in PDAPP mice, model of Alzheimer’s disease. Hippocampus 24: , 257–269. |
[20] | Karl T , Bhatia S , Cheng D , Kim WS , Garner B ((2012) ) Cognitive phenotyping of amyloid precursor protein transgenic J20 mice. Behav Brain Res 228: , 392–397. |
[21] | Wright AL , Zinn R , Hohensinn B , Konen LM , Beynon SB , Tan RP , Clark IA , Abdipranoto A , Vissel B ((2013) ) Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One 8: , e59586. |
[22] | Wang CY , Wang PN , Chiu MJ , Finstad CL , Lin F , Lynn S , Tai YH , De Fang X , Zhao K , Hung CH , Tseng Y , Peng WJ , Wang J , Yu CC , Kuo BS , Frohna PA ((2017) ) UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (N Y) 3: , 262–272. |
[23] | Canter RG , Penney J , Tsai LH ((2016) ) The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539: , 187–196. |
[24] | Puzzo D , Privitera L , Leznik E , Fa M , Staniszewski A , Palmeri A , Arancio O ((2008) ) Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28: , 14537–14545. |
[25] | Giuffrida ML , Caraci F , Pignataro B , Cataldo S , De Bona P , Bruno V , Molinaro G , Pappalardo G , Messina A , Palmigiano A , Garozzo D , Nicoletti F , Rizzarelli E , Copani A ((2009) ) Beta-amyloid monomers are neuroprotective. J Neurosci 29: , 10582–10587. |
[26] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[27] | Fettelschoss A , Zabel F , Bachmann MF ((2014) ) Vaccination against Alzheimer disease. Hum Vaccin Immunother 10: , 847–851. |
[28] | Sanderson DJ , Bannerman DM ((2011) ) Competitive short-term and long-term memory processes in spatial habituation. J Exp Psychol Anim Behav Process 37: , 189–199. |
[29] | Sanderson DJ , McHugh SB , Good MA , Sprengel R , Seeburg PH , Rawlins JN , Bannerman DM ((2010) ) Spatial working memory deficits in GluA1 AMPA receptor subunit knockout mice reflect impaired short-term habituation: Evidence for Wagner’s dual-process memory model. Neuropsychologia 48: , 2303–2315. |
[30] | Whitcomb DJ , Hogg EL , Regan P , Piers T , Narayan P , Whitehead G , Winters BL , Kim DH , Kim E , St George-Hyslop P , Klenerman D , Collingridge GL , Jo J , Cho K ((2015) ) Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep 5: , 10934. |
[31] | Janus C , Pearson J , McLaurin J , Mathews PM , Jiang Y , Schmidt SD , Chishti MA , Horne P , Heslin D , French J , Mount HT , Nixon RA , Mercken M , Bergeron C , Fraser PE , St George-Hyslop P , Westaway D ((2028) ) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408: , 979–982. |
[32] | Morgan D , Diamond DM , Gottschall PE , Ugen KE , Dickey C , Hardy J , Duff K , Jantzen P , DiCarlo G , Wilcock D , Connor K , Hatcher J , Hope C , Gordon M , Arendash GW ((2028) ) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408: , 982–985. |
[33] | Schenk D , Barbour R , Dunn W , Gordon G , Grajeda H , Guido T , Hu K , Huang J , Johnson-Wood K , Khan K , Kholodenko D , Lee M , Liao Z , Lieberburg I , Motter R , Mutter L , Soriano F , Shopp G , Vasquez N , Vandevert C , Walker S , Wogulis M , Yednock T , Games D , Seubert P ((1999) ) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: , 173–177. |
[34] | Bard F , Cannon C , Barbour R , Burke RL , Games D , Grajeda H , Guido T , Hu K , Huang J , Johnson-Wood K , Khan K , Kholodenko D , Lee M , Lieberburg I , Motter R , Nguyen M , Soriano F , Vasquez N , Weiss K , Welch B , Seubert P , Schenk D , Yednock T ((2000) ) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: , 916–919. |
[35] | DeMattos RB , Bales KR , Cummins DJ , Dodart JC , Paul SM , Holtzman DM ((2001) ) Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 98: , 8850–8855. |
[36] | Harris JA , Devidze N , Halabisky B , Lo I , Thwin MT , Yu G-Q , Bredesen DE , Masliah E , Mucke L ((2010) ) Many neuronal and behavioural impairments in transgenic mouse models of Alzheimer’s disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci 30: , 372–381. |
[37] | Hansen DV , Hanson JE , Sheng M ((2018) ) Microglia in Alzheimer’s disease. J Cell Biol 217: , 459–472. |
[38] | Ameen-Ali KE , Simpson JE , Wharton SB , Heath PR , Sharp PS , Brezzo G , Berwick J ((2019) ) The time course of recognition memory impairment and glial pathology in the hAPP-J20 mouse model of Alzheimer’s disease. J Alzheimers Dis 68: , 609–624. |
[39] | Baik SH , Kang S , Son SM , Mook-Jung I ((2016) ) Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia 64: , 2274–2290. |
[40] | Timmer NM , van Dijk L , van der Zee CE , Kiliaan A , de Waal RM , Verbeek MM ((2010) ) Enoxaparin treatment administered at both early and late stages of amyloid β deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Aβ levels. Neurobiol Dis 40: , 340–347. |
[41] | Reardon S ((2018) ) Frustrated Alzheimer’s researchers seek better lab mice. Nature 563: , 611–612. |
[42] | Rosenblum MD , Way SS , Abbas AK ((2016) ) Regulatory T cell memory. Nat Rev Immunol 16: , 90–101. |
[43] | Agadjanyan MG , Petrovsky N , Ghochikyan A ((2015) ) A fresh perspective from immunologists and vaccine researchers: Active vaccination strategies to prevent and reverse Alzheimer’s disease. Alzheimers Dement 11: , 1246–1259. |

