
Aging-Related and Gender Specific Albumin Misfolding in Alzheimer’s Disease
Abstract
Aging-related protein misfolding and aggregation may play critical roles in the pathogenesis of numerous diseases. In the brain, extracellular aggregated amyloid-β (Aβ) is closely related to the death of neurons in individuals with Alzheimer’s disease (AD). Albumin-Aβ binding is important in preventing Aβ fibril aggregation. However, because albumin is the most abundant and important antioxidant in the circulation, aging-related oxidative stress could have a significant effect on the molecular conformation and binding capacities of albumin. To investigate the link between misfolded albumin and AD, we developed fluorescent assays to determine the effects of misfolded albumin on membrane integrity in the presence of a lipolytic, inflammatory response-like enzyme, secretory phospholipase A2 (sPLA2). We found that misfolded albumin increased degradation of phospholipids in highly fluid bilayer membranes in the presence of sPLA2 due to hydrophobic effects of misfolded albumin. High amounts of misfolded albumin were present in sera of elderly (average 74 years) versus young (average 24 years) subjects (p < 0.0001). Albumin in cerebrospinal fluid (CSF) of elderly subjects, though present in small concentrations, had a 2- to 3-fold increased capacity to promote sPLA2-catalyzed membrane phospholipid degradation as compared with the same amount of albumin in serum (p < 0.0001). In addition, the fatty acid binding capacity of albumin in CSF from female subjects was considerably lower than values obtained for men, especially for individuals diagnosed with AD (p = 0.0006). This study suggests that inflammation, misfolded albumin and/or other dysfunctional proteins, and changes in membrane fluidity could alter cell membrane integrity and homeostasis and contribute to the pathogenesis of aging-related dementia and AD.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive disorder caused by central nervous system (CNS) neuronal death that leads to gradual loss of memory and relentless decline in other cognitive functions. AD is characterized pathologically by the formation of extracellular amyloid-β (Aβ) plaque and intracellular tau protein tangles in microtubules in the neurons that leads to cell death. The Aβ fibrils may block cell-to-cell signaling, trigger inflammation, and block and disrupt the flow of nutrients to the cells, and the tau tangles abolish microtubule function, which is needed to transport nutrients and other essential substances among neurons [1]. The Aβ peptides are derived from proteolytic cleavage of an amyloid-β protein precursor (AβPP) which is concentrated in the plasma membranes of neuronal synapses. In AD, misfolded Aβ peptides are predisposed to seed polymerization that leads to aggregation of the peptides into soluble oligomers both intracellularly and extracellularly [2]. The Aβ oligomers may further incorporate into plaques, due to their hydrophobic effects on plasma membranes, and create membrane pores that can lead to neuronal cell membrane leakage and death [3–5]. Although formation of Aβ plaques may involve the complex processes of hydrophobic interactions between misfolded Aβ and membranes, the pathogenic cause of Aβ plaque formation remains unknown.
Over the past two decades, dozens of clinical trials targeting Aβ as a treatment for AD have all failed to have a significant impact on AD progression. It is gradually becoming plausible that other toxic proteins and systemic inflammation need to be considered as important factors in the pathogenesis of AD. In additionally, there is a paucity of knowledge that can explain why women are twice as likely to develop AD as compared to men [6], and methods that may help uncover the basis for this gender disparity are not available. It is expected that nearly 14 million Americans over age 65 could be living with AD in 2050, a dramatic increase from current 4.8 million of people with AD. Additionally, it usually takes one to two decades of incubation time before clinical symptoms of AD appear that lead to its diagnosis. Consequently, by the time AD is clinically diagnosed, the disease is already at advanced stages of untreatable and irreversible disease that lead to severe cognitive dysfunction that can last for years or even decades before patients die. Given the additional psychological and financial burdens on caregivers and family members, a lab test for early determination of AD is urgently needed, not only for better patient care and treatment, but also to alleviate emotional, economic, and social impacts.
Advanced age is the most important risk factor for developing AD. In aging, many proteins may undergo conformational changes in their structures or become increasingly susceptible to misfolding [7]. Some misfolded proteins have been shown to be toxic to cells and have been linked to numerous diseases including neurodegenerative diseases (most prominently AD, Parkinson’s disease, and late-onset multiple sclerosis), type 2 diabetes, certain forms of heart disease, cancers, and cataracts [8–10]. In addition, although not related to advance age, misfolded proteins have been found to be associated with preeclampsia [11, 12]. While oxidative stress and inflammation may play important roles in protein structural modifications and misfolding [13, 14], it is difficult to determine the level of oxidative stress and inflammation in body fluids and link these to disease development.
Albumin is the most abundant antioxidant in the body, and it can undergo structural modifications in association with the aging process and in response to oxidative stress [15, 16]. Albumin can also undergo conformational change by ligand binding [17] and glycosylation [18, 19] under certain pathophysiological conditions. Furthermore, it is known that serum albumin molecules can self-aggregate to form amyloid fibrils at physiological pH in the presence of metal ions including Cu(II) and Zn(II) [20, 21] or when heated [22]. Such modifications of albumin may affect its secondary and tertiary structure and transform albumin into a misfolded or unfolded form. Consequently, albumin misfolding could have a major effect on albumin’s functional activity including its fatty acid (FA) binding properties and exert hydrophobic effects on membranes that affect membrane integrity.
We used a previously developed fluorescent assay with modifications to determine whether albumin misfolding in serum and cerebrospinal fluid (CSF) can be detected in patients with AD. The assay contained fluorescently labeled bilayer membrane liposomes and an inflammatory response-like enzyme, the 14 kDa secretory phospholipase A2 (sPLA2), in the presence of albumin. Albumin’s hydrophobic effects and FA binding activity, which were modulated by sPLA2, could be sensitively determined from changes in fluorescence intensity in the assay [23]. As previously reported [23], oxidation of albumin and ligand binding to albumin greatly affected albumin’s hydrophobic interactions with bilayer membranes and its FA binding activity. We speculate that this assay might be useful to investigate the correlation between albumin misfolding and AD pathogenesis.
MATERIAL AND METHODS
Materials
Porcine pancreatic phospholipase A2 (sPLA2) in ammonium sulfate suspension, dioleoyl phosphatidylcholine (DOPC), phosphatidylglycerol (PG), and fatty acid-free (FAF) albumin were purchased from Sigma-Aldrich, St. Louis, MO, USA. Fluorescent bis-BODIPY® C11-PC (1,2-bis-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-undecanoyl)-sn-glycero-3-phosphocholine) (BODIPY-PC) (flu-PC) and fluorescent C1-BODIPY® C12 (4,4-difluoro-5-methyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoic acid) (BODIPY-FA) (flu-FA) were purchased from Molecular Probes, Eugene, OR, USA. All chemicals used in this study were reagent grade.
Serum and cerebrospinal fluid samples
Sera and CSF were collected from age-matched elderly patients who were healthy and without evidence of cognitive impairment, patients with mild cognitive impairment (MCI), and patients with AD (age 74.1±7.4, n = 72). All these serum and CSF samples were obtained from the Wisconsin Alzheimer’s Disease Research Center. Serum samples were also collected from young, healthy volunteers (age 24.2±4.7, n = 25). All research protocols were approved by the Institutional Review Board of the University of Wisconsin School of Medicine and Public Health (Protocol HSC# 2002-617). All samples were stored at –80C. The concentration of total protein in each sample was determined using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, USA). The amounts of albumin in serum and CSF were determined by the Bromocresol Green (BCG) Assay Kit according to the procedure of the manufacturer (Sigma-Aldrich, St. Louis, MO, USA).
Low membrane fluidity (LMF) flu-PC liposome assay
We used a previously developed fluorescent liposomes-sPLA2 assay [24] to determine serum albumin fatty acid binding activity [23]. Briefly, substrates of unilamellar liposomes (UL) composed of 1 mg of DOPC, 1 mg of PG, and 0.014 mg of BODIPY-PC (50% PC-50% PG) were prepared in 1 ml of liposome buffer (flu-PC UL). The sPLA2 working solution was freshly prepared by diluting 1μl of Sigma-Aldrich sPLA2 product (1.25μg) in 0.25 ml 0.01 M Tris-HCl, pH 7.4 for each day’s assay. Routinely, 4 glass tubes (1.3×10 cm) were placed on ice, and an aliquot of 671μl of assay buffer (0.01 M Tris-HCl buffer, pH 7.4, containing 10 mM CaCl2) was added into tube number 1, and 669μl of buffer to each tubes 2–4. The tube was followed by addition of 9μl of substrate flu-PC UL (18.1μg of liposome phospholipids) and 2.4μl of serum (except tube 1) with agitation after each component was added. An aliquot of 280μl of the mixture in each tube was transferred into wells of a white polystyrene 96-well microplate (Porvair PS White, PerkinElmer Inc., Waltham, MA, USA) set at room temperature (24C) in duplicates. An aliquot of 30μl of sPLA2 working solution was added to a pool of 570-μl assay buffer in a reservoir at room temperature and mixed well. An aliquot of 20μl of diluted sPLA2 solution was withdrawn from the reservoir using an 8-well transfer pipette, and the diluted sPLA2 solution was transferred to each well with agitation (23 ng sPLA2 in each well). The microplate was immediately placed in a temperature-controlled (25C) fluorescent microplate reader (Gen5 Synergy HT, BioTek Instruments, Inc., Winooski, VM, USA). The fluorescence intensity (FI) in each well was recorded every 10 s for 120 cycles at 485 nm excitation and 528 nm emission. An initial reading was recorded as zero time, and the activity was expressed as FI vs. time (min) after the initial reading was subtracted from each subsequent reading (ΔFI). The initial rate of the reaction (FI/min) was determined from the reaction curve fitted with a second-order polynomial equation and the first-degree coefficient.
Determination of misfolded albumin in serum
The experiments were designed to test the hypothesis that presence of misfolded albumin in serum can be determined by the albumin-specific flu-PC liposome assay. The assay was similar to that described above with minor modifications. In these experiments, FAF-albumin or albumin in serum was partially denatured under heating conditions or treated with β-mercaptoethanol (BME). In the albumin heat-treatment experiment, an aliquot of 20μl of FAF-albumin solution (0.325 mg in Tris-HCl buffer, 0.01 M, pH 7.4) in a vial was heated to 40C, 60C, or 80C as specified in a thermal controller for 10 min followed by cooling on ice. The 20μl of heat-treated albumin was then added to a glass tube containing 669μl 0.01 M Tris-HCl (pH 7.4, 10 mM CaCl2) and 9μl of flu-PC UL. Then, an aliquot of 2μl sPLA2 working solution was added to the mixture with agitation. An aliquot of 0.3 ml of the final 0.7 ml assay mixture was transferred to a well of a 96-well microplate in duplicates for assay as described above. Similarly, an aliquot of 10μl serum was mixed with 30μl water in a vial and heated to a desired temperature for 10 min followed by cooling on ice. An aliquot of 9μl of heat-treated serum was applied to a glass tube containing the assay buffer and flu-PC UL prior to the addition of 2μl sPLA2 as described above. An aliquot of 0.3 ml of the final 0.7 ml assay mixture was used for assay in duplicates. In the BME-treatment experiment, an aliquot of 20μl FAF-albumin solution (0.65 mg in Tris-HCl buffer) or 20μl serum was mixed with 1μl of BME (Fisher Scientific) on ice for 1 h. An aliquot of 5 or 10μl of BME-treated albumin or 2.3μl BME-treated serum was applied to the flu-PC liposome assay mixture followed by addition of sPLA2 solution to a final volume of 0.7 ml for duplicate assays, similar to that described above for heat treatment experiments. Untreated albumin or serum was used as control and assayed in parallel.
High membrane fluidity (HMF) flu-PC liposome assay
Assay components were identical to those described in LMF-flu-PC assay above. A 276-μl aliquot of assay buffer pre-equilibrated at room temperature was added to each of two microplate wells, and 266μl of buffer was added to each of the remaining 6 wells of the microplate. An aliquot of 4μl of flu-PC UL was subsequently added to each well with agitation, followed by addition of 10μl of 1/10th dilution of serum (2μl serum diluted to 200μl assay buffer) to each of wells number 3–8. Duplicates were performed for all samples. The microplate was then placed in the 25C-controlled plate reader while the sPLA2 assay solution (30μl working solution in 570-μl assay buffer) was prepared in a reservoir at room temperature, and then an aliquot of 20μl of sPLA2 assay solution was added to each well using a transferpette-8 and the plate was read immediately as described above. The HMF-flu-PC assay was also used for CSF, for which each sample well contained 272-μl assay buffer, 4-μl flu-PC UL, and 4μl CSF. The assays were conducted at 25C in the microplate reader as described above.
Low membrane fluidity (LMF) flu-FA liposome assay
LMF-Flu-FA liposomes were prepared as described above with compositions of 2 mg DOPC, 0.1 mg PG, and 0.016 mg BODIPY-FA (95% PC-5% PG). The assay procedure using flu-FA UL was similar to that described for HMF-flu-PC assay for CSF.
RESULTS
Effect of albumin denaturation on albumin fatty acid binding activity (Alb-FA-BA)
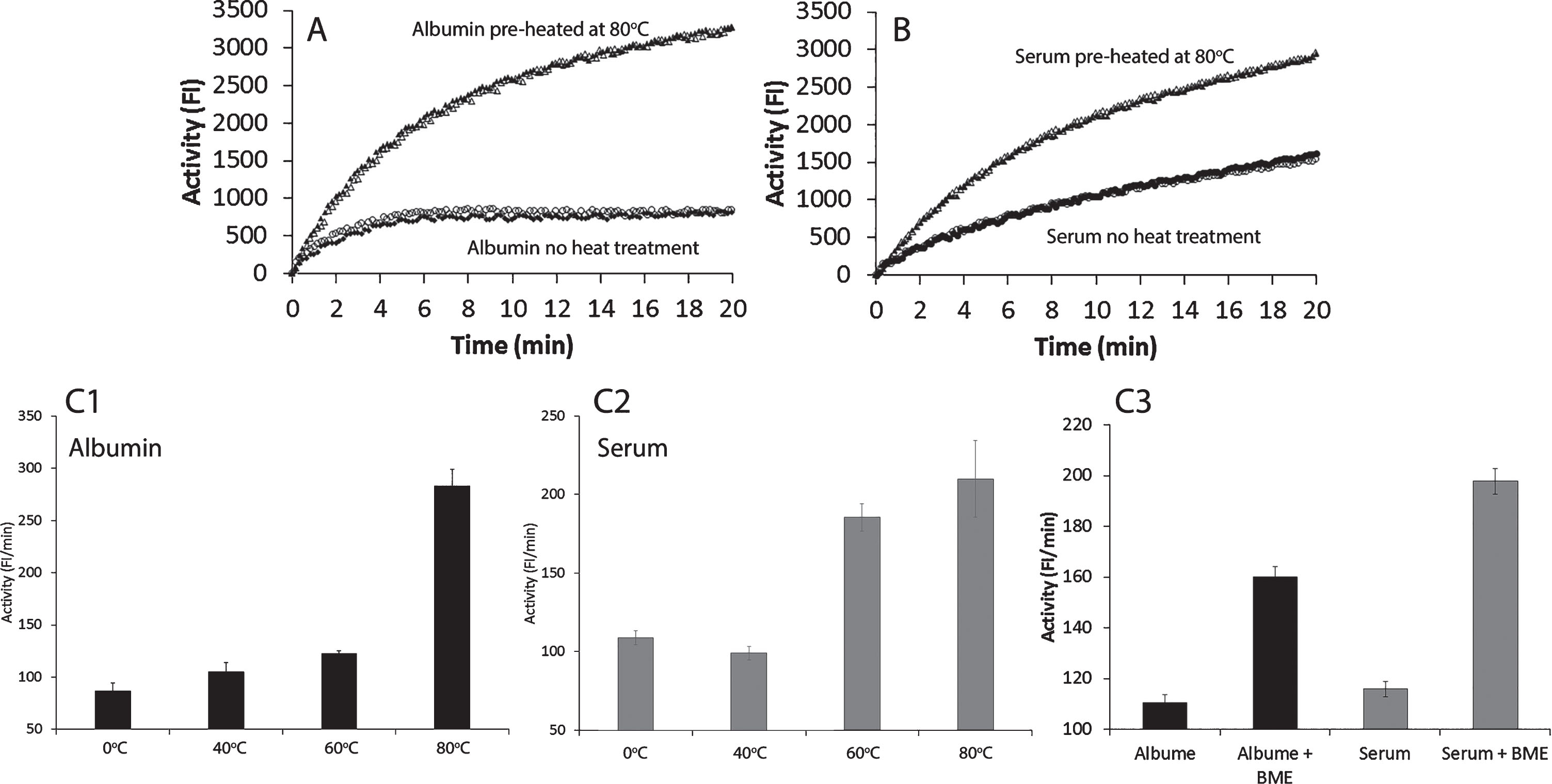
The microplate fluorescent assay contained synthetic bilayer liposomes as substrates and a 14 kDa sPLA2 as the catalytic enzyme [23]. In the presence of FAF-albumin or serum, the assay measured the combination of sPLA2 catalytic reaction, interactions between albumin and liposomes, and albumin-FA binding. However, albumin interaction with membrane and binding with FA were mediated by sPLA2 [23]. We observed that heat- or β-mercaptoethanol (BME)-treated albumin markedly increased the FI in the assay (Fig. 1), which indicated that heat- or BME-treated albumin was converted to denatured, unfolded, and/or misfolded forms. These structural changes expose albumin’s hydrophobic amino acid moieties, which consequently display increased hydrophobic interactions with liposome membranes. The resultant increased membrane fluidity that consequently enhanced sPLA2 catalytic activity and caused an increase in albumin binding to FA and lysoPL.
Fig.1
Effects of heat-treated and β-mercaptoethanol (BME)-treated human fatty acid free (FAF)-albumin and serum on albumin-FA binding activity (Alb-FA-BA). Figures 1A and 1B show the real time activity of FAF-albumin and serum from healthy individuals without heat treatment and after heat treatment at 80°C for 10 min in duplicates. Figures 1C1 and 1C2 show heat effects on FAF-albumin and serum after treatment at 40°C, 60°C, and 80°C for 10 min. Figure 1C3 shows the effects of BME treatment of FAF-albumin and serum on ice for 30 min on the albumin-FA binding activity. The results are presented in Fig. 1C as mean±SEM of triplicates.
Albumin index
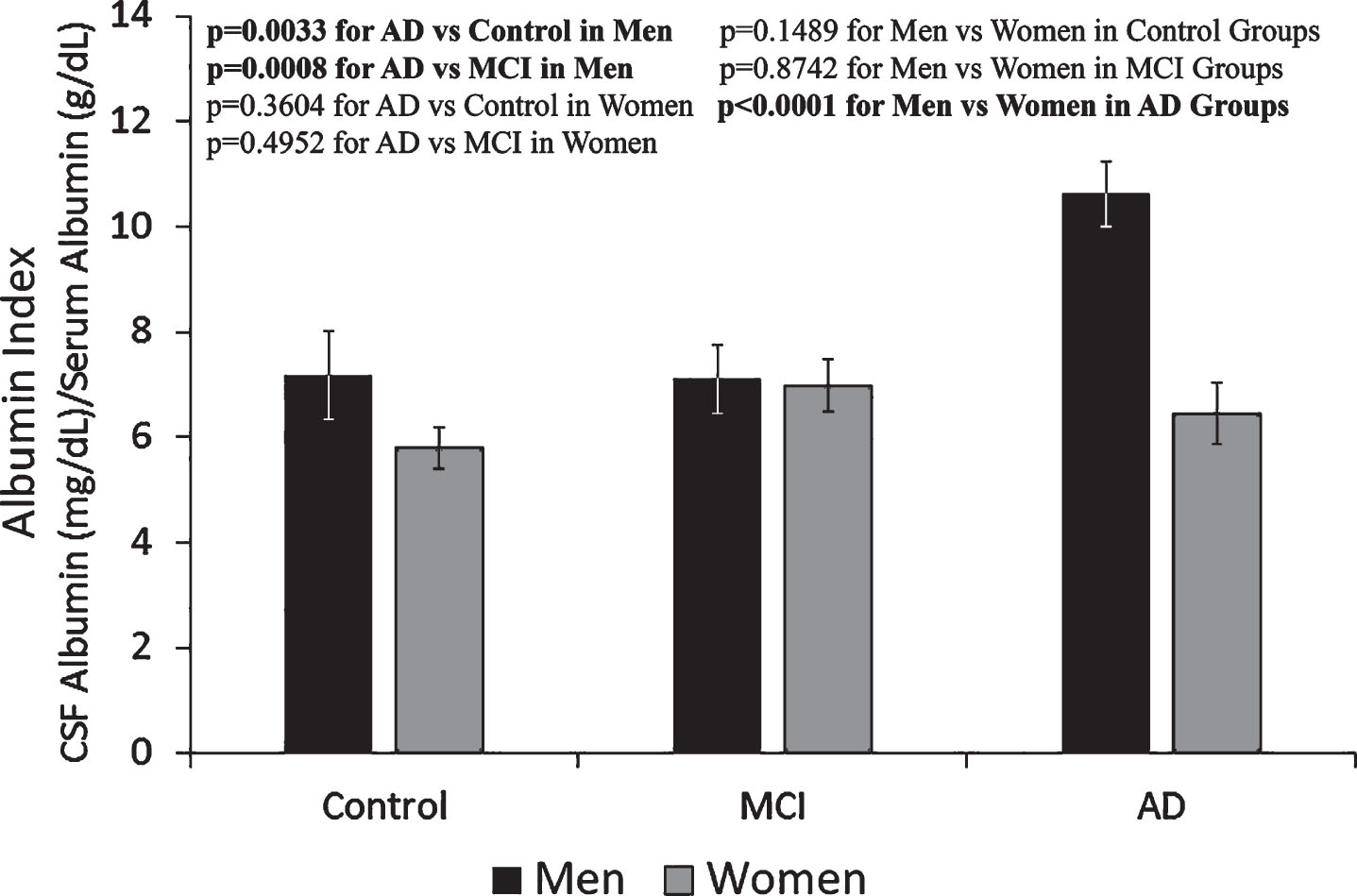
The amounts of total protein and albumin in the serum and CSF of groups of control, MCI, and AD of men and women are listed in Table 1. The amounts of total protein and albumin in CSF were less than 1% of that measured in the serum in all groups. In both serum and CSF in all groups, albumin was approximately 60–65% of the amount of total protein. Among total serum proteins, the groups of young men and women had the highest albumin percentage as compared to the three older groups, whereas the AD group had the lowest. In addition, the albumin level in the CSF of men was generally higher than that in the women, especially in the AD group (Table 1). The albumin index was calculated from the amount of CSF albumin (mg/dl) in each sample divided by the amount of serum albumin (g/dl) in the corresponding sample. The average values for albumin indices of each group show that the albumin index of the male AD group was approximately 60% higher than that of the female AD group as well as the male and female control and MCI groups (Fig. 2).
Table 1
Content of total protein and albumin in serum and CSF
| Control | MCI | AD | Young | ||||||||||||
| Serum Protein (g/dl) | CSF Protein (g/dl) | Serum Albumin (g/dl) | CSF Albumin (mg/dl) | Serum Protein (g/dl) | CSF Protein (g/dl) | Serum Albumin (g/dl) | CSF Albumin (mg/dl) | Serum Protein (g/dl) | CSF Protein (g/dl) | Serum Albumin (g/dl) | CSF Albumin (mg/dl) | Serum Protein (g/dl) | Serum Albumin (g/dl) | ||
| Men | Mean | 8.187 | 0.053 | 5.331 | 38.622 | 9.003 | 0.062 | 5.570 | 39.333 | 8.780 | 0.069 | 5.116 | 53.800 | 8.250 | 5.710 |
| SEM | ±0.370 | ±0.006 | ±0.218 | ±5.290 | ±0.562 | ±0.006 | ±0.153 | ±3.642 | ±0.459 | ±0.008 | ±0.153 | ±2.754 | ±0.515 | ±0.105 | |
| Women | Mean | 8.679 | 0.055 | 5.240 | 30.563 | 8.388 | 0.053 | 5.294 | 36.872 | 8.433 | 0.057 | 5.049 | 32.458 | 8.557 | 5.659 |
| SEM | ±0.724 | ±0.003 | ±0.137 | ±2.429 | ±0.508 | ±0.005 | ±0.126 | ±2.611 | ±0.829 | ±0.004 | ±0.096 | ±2.954 | ±0.493 | ±0.103 | |
The results are presented as mean±SEM of 12 samples for each group of Older Age Control, MCI, and AD; 11 samples for Young Control Men, and 14 samples for Young Control Women. Each sample was assayed in duplicates.
Fig.2
Albumin index determined from serum albumin and CSF albumin levels. Albumin index in the men AD group was approx. 60% higher than the rest of the groups, indicating more blood-brain-barrier leakage in the group of men with AD. The results are presented as mean±SEM of 12 samples for each group. Each sample was assayed in duplicates. The measurements between two groups were compared by t-test. Significance between two values is highlighted in bold.
Determination of Alb-FA-BA in serum by LMF-flu-PC assay and HMF-flu-PC assay
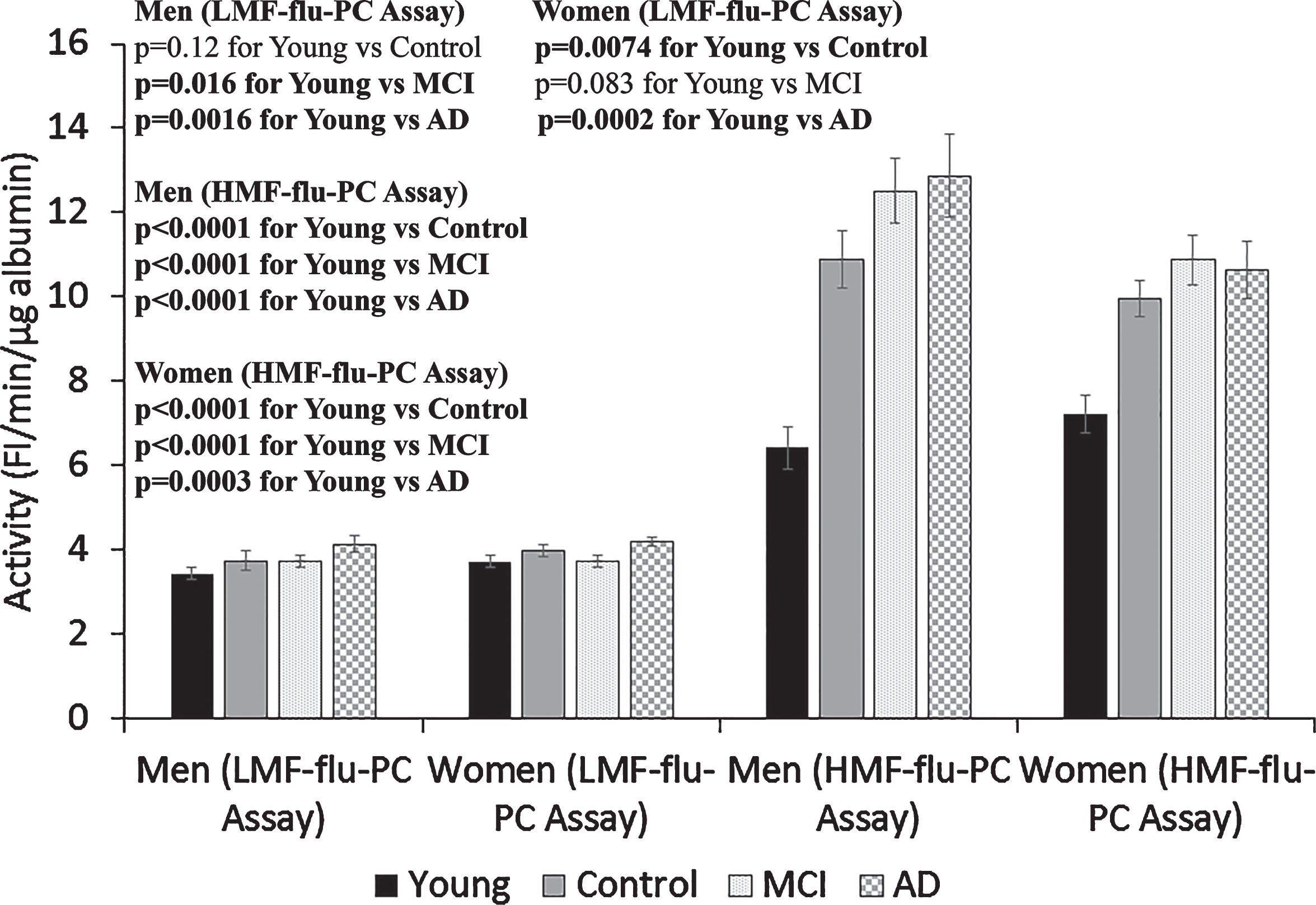
Alb-FA-BA in the serum samples from the Young, Control, MCI, and AD groups were determined by the LMF-flu-PC assay and HMF-flu-PC assay (Fig. 3). In the LMF-flu-PC assay, sera from both the young male and young female groups had significantly lower Alb-FA-BA than the men and women with AD (p 0.0016 and 0.0002, respectively), although the differences were moderate. No significant differences were observed among other groups. However, activities of Alb-FA-BA in all serum samples of both young and older groups determined by HMF-flu-PC assay were 2-3 time higher than that determined by the LMF-flu-PC assay. In addition, the activity in the sera of older Control, MCI, and AD groups was 50% higher than that in the Young group (Fig. 3). This suggests that higher HMF-flu-PC assay activity as compared to LMF-flu-PC assay activity in the Young group samples was due to high temperature effects on liposome membranes causing increased membrane fluidity. However, in addition to the temperature effect on liposome membrane fluidity, the much higher activity in the older group samples versus the young group (Fig. 3) was likely due to the presence of misfolded albumin in the sera.
Fig.3
Effects of liposome membrane fluidity and albumin misfolding effects on serum albumin activity. Albumin-FA binding activity (Alb-FA-BA) was determined in serum obtained from Young Control, Old Control, MCI, and AD groups of males or females by using the low membrane fluidity (LMF)-flu-PC assay and high membrane fluidity (HMF)-flu-PC assay. Serum albumin activities determined by the HMF-flu-PC assay were 2-3-times higher than that determined by the LMF-flu-PC assay, presumably due to membrane fluidity effects. In the HMF-flu-PC assay, the Control, MCI, and AD groups had much higher albumin activity than the Young Control group, which suggests the presence of misfolded albumin in the sera from the older groups. The results are presented as mean±SEM of 12 samples for each Control, MCI, and AD group, 11 samples for Young Men, and 14 samples for Young Women. Each sample was assayed in duplicates. The measurements between two groups were compared by t-test. Significance between two values is highlighted in bold.
Determination of Alb-FA-BA in CSF by HMF-flu-PC assay
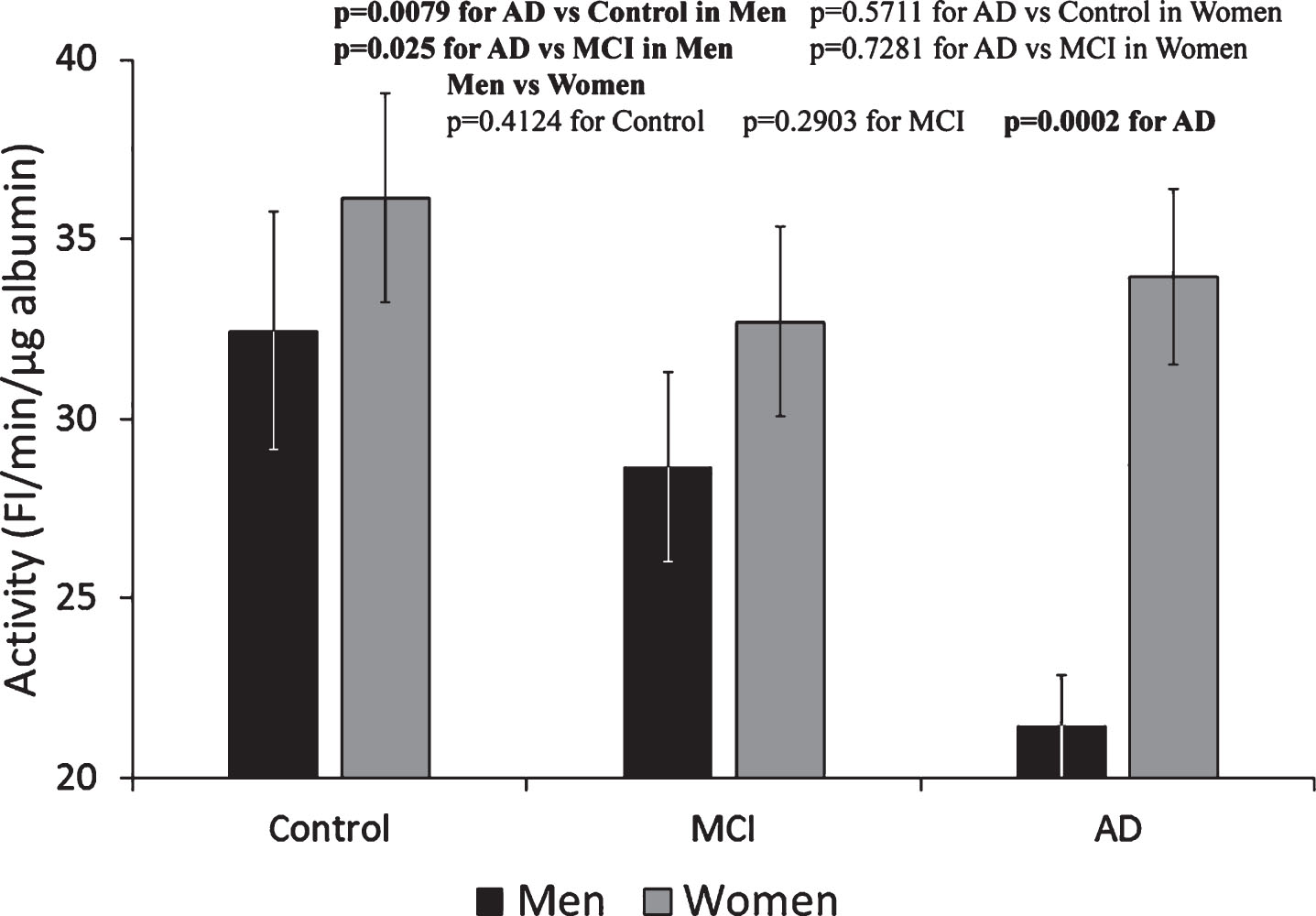
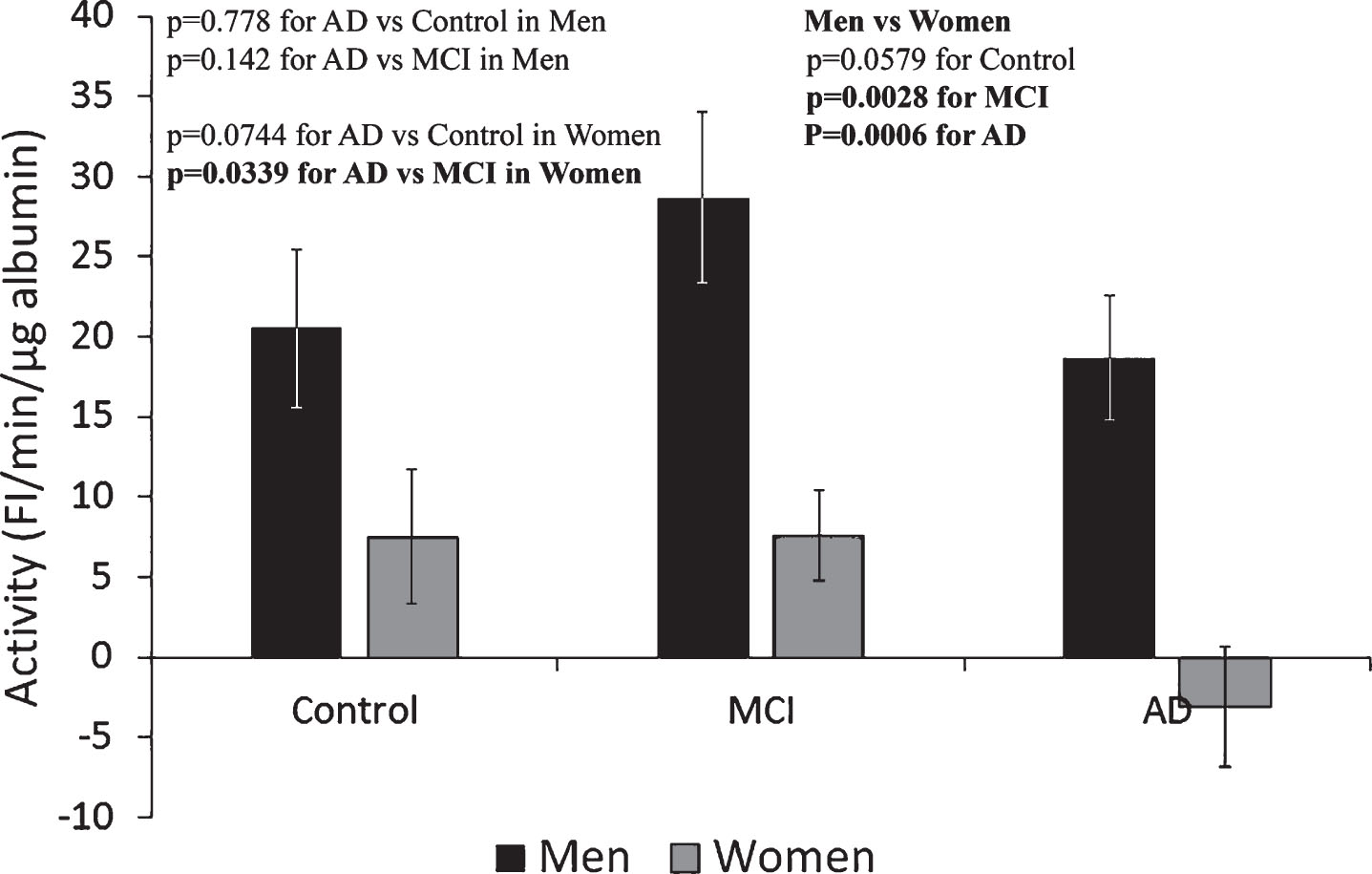
The activity of Alb-FA-BA in CSF could not be determined by the previously established LMF-flu-PC-assay for serum [23]; FI generated from the assay in the presence of CSF gave negative values. Therefore, we developed the HMF-flu-PC assay in which FI increased with time in a fashion similar to that shown in Fig. 1. Using the HMF-flu-PC assay, Alb-FA-BA in CSF was determined and converted to specific activity per μg albumin. Based on the same concentration of μg albumin, Alb-FA-BA in CSF was 2-3-times higher than the specific activity of serum albumin in all three groups of Control, MCI, and AD (both men and women) (Fig. 4). These results suggest that percentage of misfolded albumin was much higher in the CSF than in the sera for both male and female groups of Control, MCI, and AD subjects. Interestingly, Alb-FA-BA decreased significantly from the Control to MCI to AD groups in men. However, Alb-FA-BA remained relatively unchanged in the CSF of women in the Control, MCI, and AD groups. We speculate that Alb-FA-BA in CSF of men with AD might have pre-bound FA that occupied albumin FA binding sites and caused the decrease in Alb-FA-BA. In this case, Alb-FA-BA in CSF samples from women with AD might have FA binding deficiency. To test this hypothesis, we developed the LMF-flu-FA assay.
Fig.4
Specific activity of albumin in CSF determined by HMF-flu-PC Assay. The specific activity of albumin in the CSF of men progressively decreased from Control to MCI to AD subjects. In contrast, there were no significant changes in the CSF among the Control, MCI, and AD female groups. These results indicate a gender-specific pattern of Alb-FA-BA response of albumin conformational changes in CSF. The results are presented as mean±SEM of 12 samples for each group. Each sample was assayed in duplicates. The measurements between two groups were compared by t-test. Significance between two values is highlighted in bold.
Determination of Alb-FA-BA in CSF by LMF-flu-FA assay
Alb-FA-BA in CSF of men and women was determined by the LMF-flu-FA assay using liposomes with low membrane fluidity (95% DOPC-5% PG) and labeled with fluorescent FA. In this assay, FI increased in a time-dependent manner when flu-FA was removed from liposomal membranes by albumin during the reaction. The substrate of low membrane fluidity liposome could also restrict the interactions between albumin and membranes and minimize the sPLA2 catalytic reaction. Additionally, the liposome substrate labeled with fluorescent FA also masked the sPLA2 effect on FI change because of FA being a sPLA2 enzymatic reaction product [23]. Albumin in the CSF of male Control and MCI groups removed flu-FA from liposome membranes 3-4-times more than the albumin in the corresponding female groups (Fig. 5). In the female AD group albumin almost completely failed to remove flu-FA from liposome membranes. The results show that FA binding capacity of albumin in the CSF of women was impaired in all three groups of Control, MCI, and AD, and FA binding capacity appeared to be totally absent in the AD group.
Fig.5
Determination of albumin activity in CSF of men and women using liposomes with low membrane fluidity (95% DOPC-5% PG) labeled with fluorescent fatty acid (LMF-flu-FA Assay). The ability of albumin to remove flu-FA from liposome membranes in CSF from male groups was significantly increased as compared to the female groups. These results show that CSF albumin-FA binding capacity was impaired in women. The ability of albumin to remove flu-FA from liposome membranes was totally absent in the female AD group. The results are presented as mean±SEM of 12 samples for each group. Each sample was assayed in duplicates. The measurements between two groups were compared by t-test. Significance between two values is highlighted in bold.
DISCUSSION
Albumin is the most abundant antioxidant in circulation and can be subjected to structural modifications and conformational changes that impair its important functional capacity as an antioxidant. In this study, we used a fluorescent liposome-sPLA2 assay model to determine the hydrophobic effects of misfolded albumin on bilayer membranes. In the assay, we demonstrated that heat- or BME-denatured albumin markedly increased FI released from the fluorescent membranes of unilamellar liposomes when catalyzed by sPLA2. The increase in FI was likely in part due to increased hydrophobic interactions between denatured (unfolded/misfolded or aggregated) albumin and liposome membranes that provided a physical environment suitable for enhancing the catalytic activity of sPLA2. These findings suggest that the liposome-sPLA2 assay could be used to determine whether misfolded albumin was present in serum and/or CSF from patients. In this study, we found that with increasing liposome membrane fluidity (HMF-flu-PC assay), the strikingly higher Alb-FA-BA in the sera of older age groups versus younger groups strongly suggests the presence of significantly elevated levels of misfolded albumin in the sera of older subjects.
Dysfunction of the blood-brain-barrier (BBB) is associated with the pathogenesis of AD and other neurodegenerative diseases [25]. Among the male groups of Control, MCI, and AD subjects, the significantly increased albumin index in the AD group suggests that the BBB may be dysfunctional in men with AD and allows more albumin to enter the CSF. However, the dysfunctional BBB permeability did not appear to be present in the female cohort with AD versus the female Control and MCI groups. The cause of the albumin level differences in CSF between male and female AD groups is unclear.
Although the amount of albumin in the CSF was only 0.5–1% of the level of serum albumin in all male and female study groups, albumin activity in the CSF could be sensitively determined for all groups by the HMF-flu-PC assay, although not by using the LMF-flu-PC assay (data not shown). This suggests that high liposomal membrane fluidity was critical for measuring the albumin activity in the CSF, which was due to increased hydrophobic interactions between misfolded albumin and fluid liposomal membranes. The significant decline of Alb-FA-BA in CSF from the male AD group versus the male Control and MCI groups suggests that albumin in the AD group might have pre-bound FA that prevented FA binding in the assay reaction [23]. Albumin with pre-bound FA can undergo allosteric reorientation that may influence albumin’s binding capacities [26]. This decline was not observed in serum (Fig. 3), which was probably due to the large albumin pool in circulating blood. It is possible that the albumin pre-bound FA in CSF may be derived from degraded membrane phospholipids from neurons, and an inflammatory response sPLA2 has been found to be present in the CSF of patients with AD [27, 28]. In addition, inflammation may also contribute to the high level of arachidonic acid released from neuronal membrane phospholipids by group IVA-PLA2 in the hippocampus of a mouse model of AD and in individuals with AD [29]. Our assay model shows that sPLA2 and misfolded albumin in CSF may provide an environment that enhances the ability of sPLA2 to break down neuronal cell membranes under conditions of systemic inflammation. Thus, the high content of misfolded albumin in the CSF of men due to BBB leakage may be a useful marker for AD. Unlike males, albumin activity in the CSF of the female AD group remained relatively unchanged as compared to the female Control and MCI groups. We speculate that due to albumin-FA binding deficiency, albumin in the CSF of women with AD may not have pre-bound FA as found for CSF from males with AD.
The hypothesis that a FA binding deficiency of albumin may exist in the CSF of women with AD was tested by the LMF-flu-FA assay. This assay used liposomes that contained 95% DOPC and 5% PG plus traces of fluorescent FA, and this mixture of DOPC and PG provided greater rigidity to membrane phospholipid alignment and lower membrane fluidity than the liposomes made of 50% DOPC-50% PG used in the assays shown in Fig. 4. In the LMF-flu-FA assay, sPLA2 catalytic activity was not detectable, and the FI increase was caused solely via removal of FA from liposomal membranes by albumin [23]. Consequently, the markedly lower albumin activities in the CSF from women in the Control, MCI, and AD groups as compared to the values for the corresponding male groups support the presence of FA binding impairment for albumin in CSF obtained from females. The albumin-FA binding impairment was even greater in the female AD group as compared to the Control and MCI female groups (Fig. 5). High albumin activity in female groups as shown in Fig. 4 was likely due to hydrophobic interactions between misfolded albumin and liposome membranes rather than albumin-FA binding.
Although albumin is an essential FA transporter that supports cellular metabolism, it also serves as a scavenger that binds and removes FAs derived from degraded cell membranes, which maintains membrane homeostasis [23]. If FAs that are produced via membrane phospholipid degradation under inflammatory conditions are not removed or properly regulated, hydrophobic FAs left in cell membranes may become cytotoxic. A combination of membrane phospholipid degradation and FA accumulation could disrupt membrane structure and fluidity and alter cell-cell signaling [30, 31]. Insertion of free FA into membranes could also create membrane pores [32], a phenomenon that can facilitate pathogen destruction in concert with antimicrobial peptides and amyloid proteins [3]. Hydrophobic Aβ may act as an antimicrobial peptide in the brain to combat microbes that gain access to CSF due to increased permeability caused by BBB disruption [33]. However, by acting as a double-edged sword, Aβ fragments could also create toroidal pores in neuronal membranes that injure neurons [5, 34–37]. We speculate that degradation of plasma membrane phospholipids and/or retention of FFA in the membrane could increase hydrophobic interactions between Aβ fragments and membranes that may lead to membrane pore formation and seed Aβ aggregation, thereby contributing significantly to Aβ plaque formation leading to cellular toxicity [38]. Furthermore, the presence of misfolded albumin in CSF causing deficiency of albumin-FA binding in women might add an additional layer of risk to the development of AD and may explain why women are more likely to develop AD and then decline more rapidly than men after AD is diagnosed.
The results depicted in Figs. 4 and 5 show that the CSF of both aged men and women had much higher levels of misfolded albumin forms than were detected in serum, presumably due to more oxidation and other types of albumin modification, and albumin oxidation in the plasma and CSF of patients with AD has been reported [39]. Our study suggests that albumin in CSF of older women (average 74.1 years) might have different conformational changes than albumin found in the CSF from males (average 74.2 years). It has been shown that the ability of estrogenic antioxidants to provide protective mechanisms in the neuronal mitochondria of women declined with aging [40]. However, gender differences in albumin misfolding and binding capacity were not seen in serum (Fig. 3), probably because differences were masked by the large pool of albumin in sera. Nevertheless, this study showed that albumin misfolding and its effects on albumin function appeared to be gender-specific.
It is known that under normal conditions over 95% of the Aβ peptides in plasma and CSF are bound to albumin, which prevents Aβ peptide aggregation [41–45]. It is possible that albumin, after being subjected to conformational changes that cause FA binding impairment, could also fail to bind Aβ, which could lead to Aβ aggregation and the formation of toxic oligomers. In this regard, we speculate that age-dependent albumin misfolding that impairs its binding properties may be initiated and progress differently in healthy subjects versus individuals with AD, and detection and quantitation of misfolded albumin could provide a useful marker that can detect the onset of AD. We also suggest the toxic effects of misfolded albumin associated with membrane poration and Aβ aggregation could be reduced by administration of native and fully functional albumin. Interestingly, studies of plasma exchange and albumin replacement given to patients with AD to restore albumin antioxidant capacity have shown promising benefits that have led to a number of clinical trials of such a therapeutic approach [39, 46, 47].
Although the levels of serum albumin misfolding in the groups of older adult control, MCI, and AD were insignificantly different, the onset of albumin misfolding in the subjects of these three groups could be different, with possibly taking place much earlier in patients with AD. If this is true, then, the fluorescent liposome assays described in this study may be useful for determination of the early stages of AD development. This study also show that combination of misfolded albumin, abnormal membrane fluidity, and inflammation could disrupt membrane and lipid homeostasis in the central nervous system. For that, it may shed new light on AD pathogenesis, and similar changes could be involved in the pathogenesis of other neurodegenerative diseases including multiple sclerosis and Parkinson’s disease.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank Rachel A. Krause for preparing serum and CSF samples provided by the Wisconsin Alzheimer’s Disease Research Center (ADRC) for the study. This study was supported in part by the George and Julie Mosher Research Fund. Recruitment and study of the young participants were supported by the HL118154 grant from the National Institutes of Health to Jill N. Barnes.
REFERENCES
[1] | Querfurth HW , LaFerla FM ((2010) ) Alzheimer’s disease. N Engl J Med 362: , 329–344. |
[2] | Sakono M , Zako T ((2010) ) Amyloid oligomers: Formation and toxicity of Aβ oligomers. FEBS J 277: , 1348–1358. |
[3] | Last NB , Miranker AD ((2013) ) Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc Natl Acad Sci U S A 110: , 6382–6387. |
[4] | Andreasen M , Lorenzen N , Otzen D ((2015) ) Interactions between misfolded protein oligomers and membranes: A central topic in neurodegenerative diseases? Biochim Biophys Acta 1848: , 1897–1907. |
[5] | Gilbert RJC , Serra MD , Froelich CJ , Wallace MI , Anderluh G ((2014) ) Membrane pore formation at protein–lipid interfaces. Trends Biochem Sci 39: , 510–516. |
[6] | Ferretti MT , Iulita MF , Cavedo E , Chiesa PA , Dimech AS , Chadha AS , Baracchi F , Girouard H , Misoch S , Giacobini E , Depypere H , Hampel H ((2018) ) Sex differences in Alzheimer disease — the gateway to precision medicine. Nat Rev Neurol 14: , 457–469. |
[7] | David DC ((2012) ) Aging and the aggregating proteome. Front Genet 3: , 1–6. |
[8] | Hartl FU ((2017) ) Protein misfolding diseases. Annu Rev Biochem 86: , 21–26. |
[9] | Sweeney P , Park H , Baumann M , Dunlop J , Frydman J , Kopito R , McCampbell A , Leblanc G , Venkateswaran A , Nurmi A , Hodgson R ((2017) ) Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl Neurodegener 6: , 1–13. |
[10] | Moreau KL , King JA ((2012) ) Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med 18: , 273–82. |
[11] | Cheng SB , Nakashima A , Sharma S ((2016) ) Understanding pre-eclampsia using Alzheimer’s etiology: An intriguing viewpoint. Am J Reprod Immunol 75: , 372–381. |
[12] | Catera JH , Kumitac JR , Abdallaha RZ , Zhaod G , Bernardo-Gancedoc A , Henrye A , Winatae W , Chia M , Grenyera BSF , Townsenda ML , Ransona M , Buhimschii CS , Charnock-Jonesj DS , Dobsonc CM , Wilsona MR , Buhimschid IA , Wyatta AR ((2019) ) Human pregnancy zone protein stabilizes misfolded proteins including preeclampsia- and Alzheimer’s associated amyloid beta peptide. Proc Natl Acad Sci U S A 116: , 6101–6110. |
[13] | Scheinost JC , Witter DP , Boldt GE , Wentworth P Jr ((2010) ) Role of oxidative stress in protein misfolding and/or amyloid formation. In Protein Misfolding Diseases: Current and Emerging Principles and Therapies, Ramirez-AlvaradoM, KellyJW, DobsonCM, eds. John Wiley & Sons, Inc., pp. 615–630. |
[14] | Lipton SA , Gu Z , Nakamura T ((2007) ) Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders. Int Rev Neurobiol 82: , 1–27. |
[15] | Oettl1 K , Stauber RE ((2007) ) Physiological and pathological changes in the redox state of human serum albumin critically influence its binding properties. Br J Pharmacol 151: , 580–590. |
[16] | Luna C , Alique M , Navalmoral E , Noci M , Bohorquez L , Carracedo J , Ramirez R ((2016) ) Aging-associated oxidized albumin promotes cellular senescence and endothelial damage. Clin Interv Aging 11: , 225–236. |
[17] | Ishii T , Ito S , Shigenori Kumazawa S , Sakurai T , Yamaguchi S , Mori T , Nakayama T , Uchida K ((2008) ) Site-specific modification of positively-charged surfaces on human serum albumin by malondialdehyde. Biochem Biophys Res Commun 371: , 28–32. |
[18] | Dolhofer R , Wieland OH ((1980) ) Increased glycosylation of serum albumin in diabetes mellitus. Diabetes 29: , 417–422. |
[19] | Shaklai N , Garlick RL , Bunn HF ((1984) ) Nonenzymatic glycosylation of human serum albumin alters its conformation and function. J Biol Chem 259: , 3812–3817. |
[20] | Stirpe A , Pantusa M , Rizzuti B , Sportelli L , Bartucci R , Guzzi R ((2011) ) Early stage aggregation of human serum albumin in the presence of metal ions. Intern J Biol Macromol 49: , 337–342. |
[21] | Pandey NK , Ghosh S , Dasgupta S ((2010) ) Fibrillation in human serum albumin is enhanced in the presence of copper(II). J Phys Chem B 114: , 10228–10233. |
[22] | Sancataldo G , Vetri V , Fodera V , Di Cara G , Militello V , Leone M ((2014) ) Oxidation enhances human serum albumin thermal stability and changes the routes of amyloid fibril formation. PLOS One 9: , 1–11. |
[23] | Tsao FHC , Xiang Z , Meyer KC ((2015) ) Fluorescent determination of secretory phospholipase A2 (sPLA2)-mediated human serum albumin binding activity with membrane phospholipids and fatty acids. Transl Med 5: , 1–9. |
[24] | Tsao FH , Shanmuganayagam D , Zachman DK , Khosravi M , Folts JD , Meyer KC ((2007) ) A continuous fluorescence assay for the determination of calcium-dependent secretory phospholipase A2 activity in serum. Clin Chim Acta 379: , 119–126. |
[25] | Cai Z , Qiao PF , Wan CQ , Cai M , Zhou NK , Li Q ((2018) ) Role of blood-brain barrier in Alzheimer’s disease. J Alzheimers Dis 63: , 1223–1234. |
[26] | Jafari N , Ahmed R , Gloyd M , Bloomfield J , Britz-McKibbin P , Melacini G ((2016) ) Allosteric sensing of fatty acid binding by NMR: Application to human serum albumin. J Med Chem 59: , 7457–7465. |
[27] | Chalbot S , Zetterberg H , Blennow K , Fladby T , Grundke-Iqbal I , Iqbal K ((2009) ) Cerebrospinal fluid secretory Ca2+-dependent phospholipase A2 activity is increased in Alzheimer disease. Clin Chem 55: , 2171–2179. |
[28] | Chalbot S , Zetterberg H , Blennow K , Fladby T , Andreasen N , Grundke-Iqbal I , Iqbal K ((2011) ) Blood-cerebrospinal fluid barrier permeability in Alzheimer’s disease. J Alzheimers Dis 25: , 505–515. |
[29] | Sanchez-Mejia RO , Newman JW , Toh S , Yu GQ , Zhou Y , Halabisky B , Cisse M , Scearce-Levie K , Cheng IH , Gan L , Palop JJ , Bonventre JV , Mucke L ((2008) ) Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat Neurosci 11: , 1311–1318. |
[30] | Nishizuka Y ((1983) ) Phospholipid degradation and signal translation for protein phosphorylation. Trans Biochem Sci 8: , 13–16. |
[31] | Ibarguren M , López DJ , Escribá PV ((2014) ) The effect of natural and synthetic fatty acids on membrane structure,microdomain organization, cellular functions and human health. Biochim Biophys Acta 1838: , 1518–1528. |
[32] | Belosludtsev KN , Belosludtseva NV , Agafonov AV , Penkov NV , Samartsev VN , Lemasters JJ , Mironov GD ((2015) ) Effect of surface-potential modulators on the opening of lipid pores in liposomal and mitochondrial inner membranes induced by palmitate and calcium ions. Biochim Biophys Acta 1848: , 2200–2205. |
[33] | Wellinga MM , Nabuursa RJA , van der Weerd L ((2015) ) Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimers Dement 11: , 51–57. |
[34] | Peters C , Bascuñán D , Opazo C , Aguayo LG ((2016) ) Differential membrane toxicity of amyloid-β fragments by pore forming mechanisms. J Alzheimers Dis 51: , 689–699. |
[35] | Serra-Batiste M , Ninot-Pedrosa M , Bayoumi M , Gairí M , Maglia G , Carulla N ((2016) ) Aβ42 assembles into specific β-barrel pore-forming oligomers in membrane-mimicking environments. Proc Natl Acad Sci U S A 113: , 10866–10871. |
[36] | Kandel N , Zheng T , Huo Q , Tatulian SA ((2017) ) Membrane binding and pore formation by a cytotoxic fragment of amyloid β peptide. J Phys Chem B 121: , 10293–10305. |
[37] | Di Scala C , Yahi N , Boutemeur S , Flores A , Rodriguez L , Chahinian H , Fantini J ((2016) ) Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci Rep 6: , 28781. |
[38] | Ahmed R , Akcan M , Khondker A , Rheinstadter MC , Bozelli JC Jr , Epand RM , Huynh V , Wylie RG , Boulton S , Huang J , Verschoord CP , Melacini G ((2019) ) Atomic resolution map of the soluble amyloid beta assembly toxic surfaces. Chem Sci 10: , 6072–6082. |
[39] | Costa M , Horrillo R , Ortiz AM , Perez A , Mestre A , Ruiz A , Boada M , Grancha S ((2018) ) Increased albumin oxidation in cerebrospinal fluid and plasma from Alzheimer’s disease patients. J Alzheimers Dis 63: , 1395–1404. |
[40] | Vina J , Lloret A ((2010) ) Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-beta peptide. J Alzheimers Dis 20: , S527–S533. |
[41] | Finn TE , Nunez AC , Sunde M , Easterbrook-Smith SB ((2012) ) Serum albumin prevents protein aggregation and amyloid formation and retains chaperone-like activity in the presence of physiological ligands. J Biol Chem 287: , 21530–21540. |
[42] | Stanyon HF , Viles JH ((2012) ) Human serum albumin can regulate amyloid-peptide fiber growth in the brain interstitium. J Biol Chem 287: , 28163–28168. |
[43] | Milojevic J , Costa M , Ortiz AM , Jorquera JI , Melacini G ((2014) ) In vitro amyloid-β binding and inhibition of amyloid-β self-association by therapeutic albumin. J Alzheimers Dis 38: , 753–765. |
[44] | Algamal M , Ahmed R , Jafari N , Ahsan B , Ortega J , Melacini G ((2017) ) Atomic-resolution map of the interactions between an amyloid inhibitor protein and amyloid β (Aβ) peptides in the monomer and protofibril states. J Biol Chem 292: , 17158–17168. |
[45] | Ezra A , Rabinovich-Nikitin I , Rabinovich-Toidman P , Solomon B ((2016) ) Multifunctional effect of human serum albumin reduces Alzheimer’s disease related pathologies in the 3xTg mouse model. J Alzheimers Dis 50: , 175–188. |
[46] | Boada M , Anaya F , Ortiz P , Olazaran J , Shua-Haim JR , Obisesan TO , Hern'andez I , Munoz J , Buendia M , Alegret M , Lafuente A , Tarraga L , Nunezh L , Torres M , Grifols JR , Ferrer I , Lopez OL , Paez A ((2017) ) Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-β concentrations and cognition outcomes in Alzheimer’s disease patients: A multicenter, randomized, controlled clinical trial. J Alzheimers Dis 56: , 129–143. |
[47] | Boada M , Lopez O , Nunez L , Szczepiorkowski ZM , Torres M , Grifols C , Paez A ((2019) ) Plasma exchange for Alzheimer’s disease management by albumin replacement (AMBAR) trial: Study design and progress. Alzheimers Dement (N Y) 5: , 61–69. |


