
Neuroprotective Approach of Anti-Cancer Microtubule Stabilizers Against Tauopathy Associated Dementia: Current Status of Clinical and Preclinical Findings
Abstract
Neuronal microtubule (MT) tau protein provides cytoskeleton to neuronal cells and plays a vital role including maintenance of cell shape, intracellular transport, and cell division. Tau hyperphosphorylation mediates MT destabilization resulting in axonopathy and neurotransmitter deficit, and ultimately causing Alzheimer’s disease (AD), a dementing disorder affecting vast geriatric populations worldwide, characterized by the existence of extracellular amyloid plaques and intracellular neurofibrillary tangles in a hyperphosphorylated state. Pre-clinically, streptozotocin stereotaxically mimics the behavioral and biochemical alterations similar to AD associated with tau pathology resulting in MT assembly defects, which proceed neuropathological cascades. Accessible interventions like cholinesterase inhibitors and NMDA antagonist clinically provides only symptomatic relief. Involvement of microtubule stabilizers (MTS) prevents tauopathy particularly by targeting MT oriented cytoskeleton and promotes polymerization of tubulin protein. Multiple in vitro and in vivo research studies have shown that MTS can hold substantial potential for the treatment of AD-related tauopathy dementias through restoration of tau function and axonal transport. Moreover, anti-cancer taxane derivatives and epothiolones may have potential to ameliorate MT destabilization and prevent the neuronal structural and functional alterations associated with tauopathies. Therefore, this current review strictly focuses on exploration of various clinical and pre-clinical features available for AD to understand the neuropathological mechanisms as well as introduce pharmacological interventions associated with MT stabilization. MTS from diverse natural sources continue to be of value in the treatment of cancer, suggesting that these agents have potential to be of interest in the treatment of AD-related tauopathy dementia in the future.
INTRODUCTION
Alzheimer’s disease (AD) is acknowledged as a progressive neurodegenerative disorder causing significant disruption of normal brain structure and function including degeneration beginning in the medial temporal lobe. Specifically, the disease starts in the entorhinal cortex and hippocampus and the major fiber tracts that connect it to the cerebral cortex (fornix and cingulum), amygdala, cingulate gyrus, and thalamus, manifested by cognitive and memory deterioration and characterized by accumulation of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs), destruction of cholinergic neurons in cerebral cortex and hippocampus, microgliosis and astrocytosis, oxidative stress, neuroinflammation, and excitotoxicity [1–3]. AD has been classified into two groups, depending on its onset: the first classification is familial AD, related to genetic alterations of amyloid precursor protein (APP), presenilin1 (PSEN1), and presenilin 2 (PSEN2). This group represents approximately about 3% of the diseased patients. The other classification group is late-onset AD (LOAD), also known as sporadic, which is due to polymorphisms of apolipoprotein E (APOE) ɛ4, accounts for the remaining 97% of the cases [4, 5]. The sign and symptoms are categorized into mild, moderate, and severe, including memory loss, language problems, mood swings, behavioral changes, unable to learn new info, agitation, aggression, ataxia (loss of muscle control and balance), aphasia (impairment of language), amnesia (forgetfulness), loss of visuospatial function (ability to recognize faces and objects), praxis (ability to perform purposeful movements), confusion, and motor disturbances [6, 7]. Prevalence of AD in India is >4 million people and worldwide around 46.8 million people [8]. Currently, the cause of AD remains poorly understood and no medications are available to stop or reverse its progression [9]. Currently available treatments like acetylcholinesterase inhibitors (rivastigmine, galantamine, donepezil) and N-methyl D-aspartate (NMDA) receptor antagonist (memantine) show least impact on the disease and contribute symptomatic relief only but unsuccessful to provide definite cure [10, 11]. To render this disease, new therapeutic targets are available to tackle AD directly. MT destabilization is accompanied by tau hyperphosphorylation. Tau proteins that are abundant in nerve cells perform the function of stabilizing MT [12]. Compounds targeting MT have been massively successful clinically as chemotherapeutic agents [13]. MT targeting agents have been shown to have the potential to treat neurodegenerative disease.
ETIOPATHOLOGICAL FACTORS
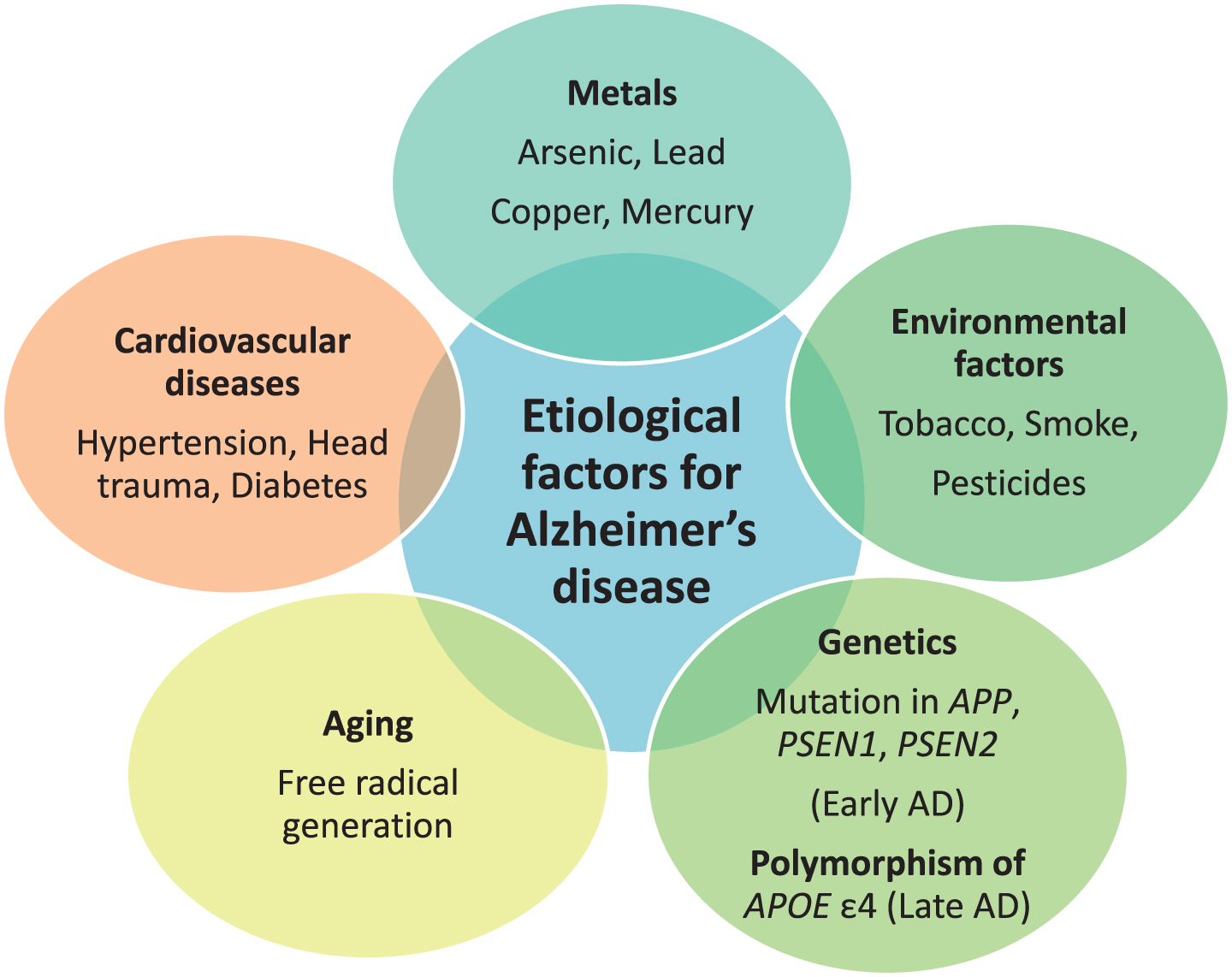
The etiology behind AD is multifactorial. Exposure to neurotoxic metals like arsenic, lead, copper, mercury, and aluminum have been involved in AD due to their tendency to increase phosphorylation of tau protein and increase Aβ peptide. Other putative etiological factors involve environmental pollutants, genetic factors, disease state, and aging. Environmental pollutants including tobacco, smoke, and pesticides, like deltamethrin and carbofuran, are responsible for disrupting tau function [14]. Head trauma, hypertension, and diabetes mellitus are disease conditions that can lead to AD [15, 16]. Genetics play a major role in causing AD. Mutations in APP located on chromosome 21 q, PSEN1 on chromosome 14 q, and PSEN2on chromosome 1qare associated with early-onset AD, or familial AD, and development of the disease before the age of 65 year. Polymorphism of APOE with the ɛ4 allele is responsible for causing late-onset AD [17]. APOE is located on chromosome 19 q. ApoE is associated with cholesterol transport in the brain. Common alleles of APOE are ɛ2, ɛ3, and ɛ4. APOE ɛ4 is responsible for LOAD risk, whereas APOE ɛ2 shows a protective role. The predominant risk factor for sporadic AD is aging. Aging also impacts AD through two mechanisms as free radicals generated during cellular respiration in aging lead to AD and another mechanism is mutation in messenger RNA of amyloid-β protein precursor (AβPP) and ubiquitin B [18]. The majority of AD cases typically have an onset after 65 years of age [19]. The various etiological factors are mentioned in Fig. 1.
Fig.1
Etiological factors responsible for Alzheimer’s disease (AD).
NEUROPATHOLOGY OF ALZHEIMER’S DISEASE ASSOCIATED WITH TAU DYSFUNCTION
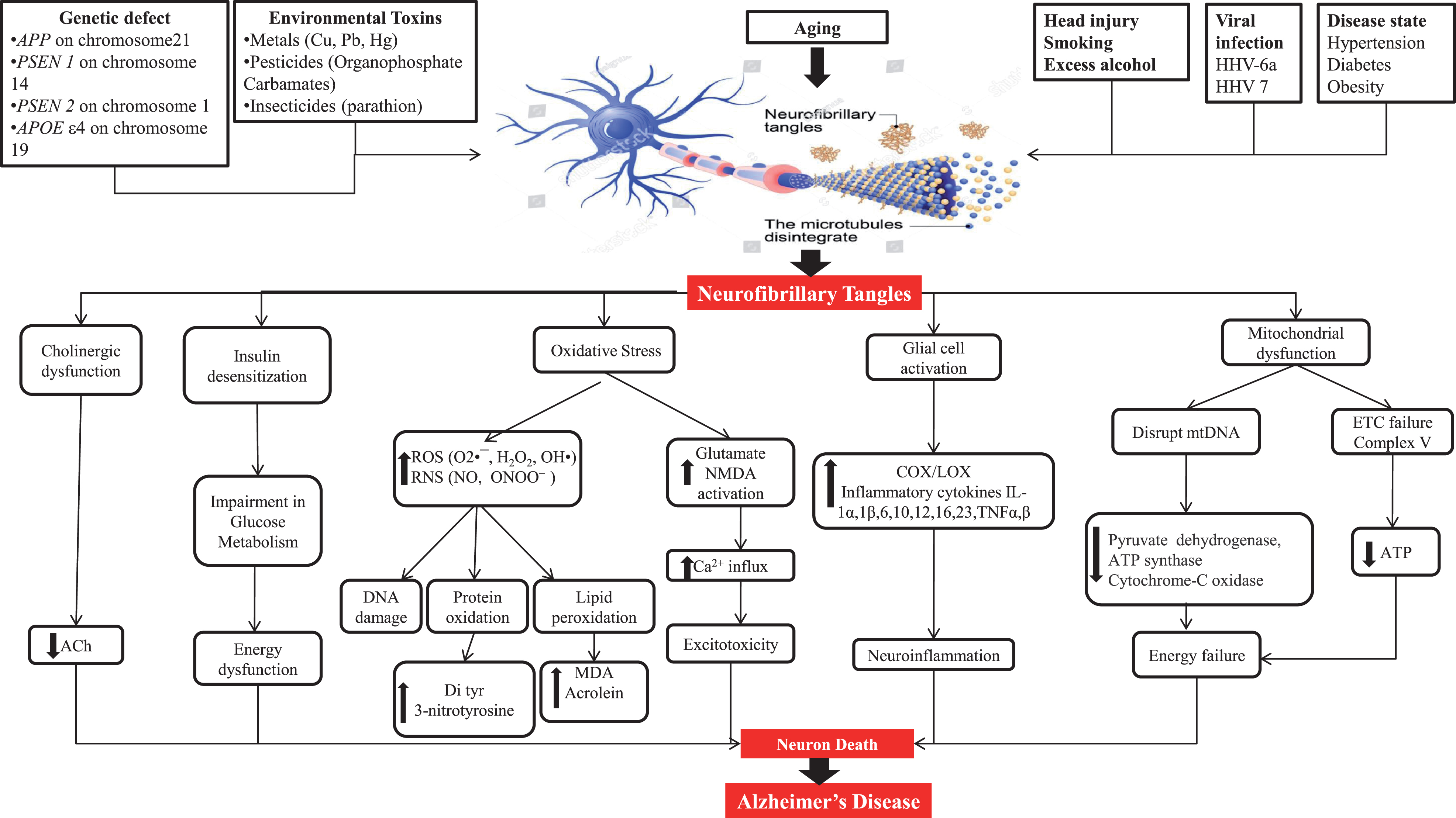
Pathogenesis gives information about the cause of AD as well as therapeutic targets. Some plausible etiological factors like genetic defects of APP, PSEN1, PSEN2, and APOEɛ4; environmental toxins like metals, pesticides, and insecticides; head injury; smoking; excess alcohol; viral infection through viruses like HHV-6a and HHV 7; and diseases like hypertension, diabetes, and obesity are somewhat responsible for AD hallmarks including amyloid plaques and NFT [5, 14–16]. NFT are formed due to hyperphosphorylation of tau protein and its further processing. Figure 2 shows a representation of the mechanism of tau pathology and the way it additionally causes pathological alterations inside cells like cholinergic dysfunction, oxidative stress, mitochondrial dysfunction, and glial cell activation.
Fig.2
Neuropathological factors engaged to cause Alzheimer’s disease.
Tau pathogenesis
Tau protein is most profusely expressed in axons of central nervous system (CNS) neurons but can also be found in the somatodendritic part of neurons, oligodendrocytes, and non-neural tissues [20, 21]. The most important role of tau protein is to promote assembly and stability of MT [20, 22]. In AD, hyperphosphorylation of certain amino acids in tau proteins causes the proteins to detach from the MT, disturbing the cytoskeleton of neurons and transport system, resulting in starvation of neurons and, ultimately, cell death. Hyperphosphorylated tau thus plays a crucial role in intracellular neurofibrillary alterations and the pathogenesis of AD and related tauopathies [23, 24]. Before the tangle formation, tau undergoes a series of post-translational modifications, including hyperphosphorylation, acetylation, N-glycosylation and truncation, which is different from the normal tau that is seen in healthy brains [25, 26]. Post-translational modifications of tau interfere with tau–MT binding and promote tau misfolding [26].
Steps lagging behind post-translational modifications of tau
Hyperphosphorylation of tau protein
With the identification of tau as the primary component of AD-associated NFTs, the aggregated tau was believed to be hyperphosphorylated. Abnormal phosphorylation of the tau protein affects its ability to bind tubulin and promote MT assembly [26, 27]. In AD, the pattern of phosphorylation changes as the disease progresses. Phosphorylation at sites such as Ser199, Ser202/205, Thr231, and Ser262 seems to be linked with pre-tangles in the neuronal processes [28]. Tau level in somatodendritic compartment increases. The reason behind the hyperphosphorylation of tau protein is an imbalance of phosphorylation and dephosphorylation of tau. This imbalance is associated by a reduced tau protein dephosphorylation or by an overactivity of the phosphorylating protein kinases. The degree of phosphorylation reflects abnormal activity of both protein kinases and phosphatases. Levels of active cyclin-dependent-like kinase 5 (CDK5), glycogen synthase kinase-3β (GSK-3β), and its regulator c-Jun N-terminal kinase (JNK) are associated with neurofibrillary pathology and are upregulated in AD [29]. Protein phosphatase 2A (PP2A) is one of only a few tau phosphatases and is responsible for the total tau phosphatase activity. The total phosphatase activity and the activities of PP2A and PP5 toward tau were significantly decreased, whereas that of PP2B was increased in AD brain [30]. Expression of PP2A and its activators is significantly reduced in the brains of individual suffering from AD compared with age-matched controls, whereas PP2A inhibitors are upregulated. Interestingly, PP2A also regulates GSK-3β, CDK5, and JNK, providing a surplus route to influence tau phosphorylation [31].
Acetylation of tau protein
In addition to being hyperphosphorylated, tau from patients with AD and other tauopathies is more acetylated than that within the brains of cognitively healthy individuals. Lysine acetylation contends phosphorylation in regulating diverse cellular functions, including energy metabolism, signaling from the plasma membrane, and cytoskeleton dynamics. Enzymes that transfer an acetyl group to the protein are called histone acetyltransferase or lysine acetyltransferase. Enzymes that eliminate an acetyl group from the protein are called histone deacetylases or lysine deacetylases [32]. Like phosphorylation, tau acetylation can arise through multiple mechanisms, including histone acetyltransferase p300, cAMP-responsive element-binding protein, or auto-acetylation, with sirtuin 1 and histone deacetylases 6 acting to deacetylate tau [33]. Improper functioning of this process produces dysfunction in multiple systems, thereby leading to neurodegeneration. Tau pathology is due to contribution of acetylation which leads to tau cleavage, preventing ubiquitin binding and inhibiting tau turnover.
Carboxy-terminal truncation of tau protein
Caspase activation is detected in the AD brain, and active caspases are found within tangle-bearing neurons. Furthermore, tau is cleaved by caspase-3. Besides caspase-3, other caspases are capable of cleaving tau at Asp421 [34]. During the characterization of paired helical filaments isolated from the brains of individuals with AD, it was discovered that tau had undergone carboxy-terminal truncation by caspase 3 [26]. Using a rabbit polyclonal antibody directed against the caspase-cleaved carboxyl-terminus of tau after cleavage at Asp421, analysis of temporal cortex brain lysates revealed that caspase-cleaved tau was found in mild cognitive impairment and AD but not in controls. In the case of AD, Aβ promotes caspase activation. In addition to tau cleavage at Asp421, other caspase cleavage products of tau have been identified in the AD brain. Caspase-6 cleaved tau has been identified in intracellular tangles, extracellular tangles, pretangles, neuropil threads, and neuritic plaques. In additional to C-terminal caspase cleavage of tau, tau can be cleaved at the N-terminus by caspase-6 [34].
O-GlcNAcylation and N-glycosylation of tau protein
O-GlcNAcylation, a type of O-glycosylation, seems to be protective against tauopathies. O-GlcNAcylation is a process regulated by glucose metabolism markedly decrease in AD. Impaired brain glucose metabolism leads to decrease in O-GlcNAcylation which further leads to abnormal phosphorylation of tau and neurofibrillary degeneration via downregulation of tau O-GlcNAcylation in AD. Decreased O-GlcNAcylation induces hyperphosphorylation of tau [35]. N-glycosylation of tau, which is thought to increase phosphorylation and pathological conformational changes, is increased in AD [26].
Neuropathological cascade associated with tau dysfunction
Oxidative stress and tau hyperphosphorylation
Oxidative stress takes place when reactive oxygen species (ROS) and reactive nitrogen species (RNS) production are not adequately counterbalanced by an endogenous antioxidant defense system [36]. Accumulation of hyperphosphorylated tau causes oxidative stress, but ROS have also been shown to stimulate tau hyperphosphorylation [37]. Accumulation of a truncated tau fragment has been described in sporadic AD cases, and cultured cortical neurons from a transgenic rat model expressing this truncated protein showed high levels of oxidative stress markers and an increased susceptibility to ROS [38]. Oxidative stress due to free radicals generate which attack phospholipid polyunsaturated fatty acids in cellular membrane can lead to lipid peroxidation, thereby results in formation of major end products 4-hydroxyl-2-nonenal (HNE), acrolein, and malondialdehyde, causing neurotoxicity to neurons [39]. HNE reacts with proteins forming stable covalent adducts to lysine, cysteine, and histidine residues, thereby producing carbonyl functionalities to the proteins, leading to oxidative damage called protein oxidation, and these protein carbonyls are mostly abundant in frontal, temporal, occipital, hippocampus, and inferior parietal lobe [40, 41]. RNS-like peroxynitrite causes tyrosine nitration of protein (3-nitrotyrosine and di tyrosine). Expanded dimensions of these proteins can be found in hippocampus and cortical areas. Increased levels of protein 3-nitrotyrosine and protein carbonyl result in alteration of antioxidant enzymes like glutathione peroxidase, glutathione reductase, and catalase [42].
Glutamate excitotoxicity and tau hyperphosphorylation
Glutamate is an excitatory neurotransmitter abundantly present in cortical and hippocampal regions involved in CNS functions like learning and memory and has low micromolar concentrations during normal conditions, but during synaptic transmission, its concentration increases from μM to mM because of its tendency having synaptic plasticity i.e., long-term potentiation (LTP) [43]. Glutamate receptors are ionotropic and metabotropic. Ionotropic receptors are transmembrane molecules that open or close a channel that allow small particles to travel in and out of cell. The ions that can travel through these receptors are K+, Na+, Cl–, and Ca2 +. Metabotopic receptors do not have channel that open or close [44]. Rather, they are connected to small chemical called G-protein. Once activated G-proteins go on and activate another molecule called secondary messenger. Ionotropic receptors are divided into α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), NMDA, and kainite [28]. NMDA receptors (NMDARs) are mainly essential for LTP induction. The resting potential of healthy neuron is normally around –70 mV, and at this potential, the Ca2 + channel of NMDAR is blocked by Mg2 + ions. Induction of LTP causes strong and prolonged release of glutamate in the synaptic cleft, and glutamate uptake is severely impaired due to pathological changes induced by ROS and lipid peroxidation end product HNE in excitatory amino acid transporter 2 (EAAT2), which is concentrated in perisynaptic astrocytes cell membrane and plays a role in the removal of excess glutamate from extracellular fluid, thereby limiting NMDAR activation. This glutamate excess will lead to overactivation of NMDARs [43, 45]. During LTP induction, glutamate binds to AMPA receptors leading to Na+ influx through AMPA receptors causing depolarization of the postsynaptic compartment and leading to activation of NMDARs to permit Ca2 + influx. In AD, there is overactivation of NMDARs which further leads to more influx of Ca2 + ions, and there is Ca2 + overload, which ultimately causes excitotoxicity [46]. Increased levels of mGluR2 are found in the hippocampus of individuals suffering from AD, and this correlates with elevated phosphorylated tau levels [47, 48]. Increased mGluR2 expression may result in increased intracellular Ca2 +, which may activate tau kinases [49]. Fyn kinase can regulate glutamatergic NMDAR activation by triggering phosphorylation and subsequent interaction of the N-methyl D-aspartate receptor subtype 2B (NR2B) subunit of the receptor with postsynaptic density protein 95 (PSD-95). The NR2B/PSD-95/Fyn complex that is formed when NMDARs are activated increase Ca2 + influx, which can activate two key tau kinases, GSK-3β and CDK5 [50, 51]. It is therefore possible that NMDAR activation stabilizes the NR2B/PSD-95/Fyn complex, resulting in a constant activation of the NMDAR channel and increasing tau phosphorylation either by direct phosphorylation of tau via Fyn kinase or by activating other tau kinases, such as GSK-3β or CDK5 [52].
Insulin desensitization and tau hyperphosphorylation
Insulin plays multiple roles in the periphery, most especially in the regulation of tissue metabolism by controlling cellular glucose uptake. In contrast, the brain was previously considered insulin insensitive. However, it is now known that insulin does play several roles within the CNS at the cellular levels, including the regulation of neuronal survival and cognition [53]. Insulin is primarily derived from the periphery, and it can be shown that changes in peripheral insulin levels lead to alterations in CNS insulin signaling and could contribute to cognitive decline and pathogenesis. Impairment of CNS insulin signaling is due to decrease in brain insulin levels, e.g., due to decreased peripheral production or altered capacity for insulin to cross the blood-brain barrier (BBB), or a change in CNS insulin receptor (IR) sensitivity (e.g., IR desensitization due to elevated insulin levels). IRs is differentially expressed in various parts of brain like hippocampus, hypothalamus, cerebral cortex, and amygdala [54]. Under normal physiological conditions when insulin binds to the IR, a cascade regulates key downstream serine/threonine kinases such as, protein kinases B (AKT/PKB), mechanistic target of rapamycin (mTOR), and extracellular signal-regulated kinases (ERK) that eventually phosphorylate serine/threonine residues of insulin receptor substrate (IRS), and inhibiting insulin signaling in a negative feedback regulation. In neurons, the phosphoinositide 3-kinase (PI3K), AKT, GSK-3β, BCL-2 agonist of cell death (BAD), mTOR, and the mitogen activated protein kinase (MAPK) pathways are essential for cell survival signaling and are undertaken by the activity of the IR [55]. Therefore, alteration of the physiological activity of these pathways might be the source of alteration in normal neuronal performance, showing that brain insulin resistance could promote LOAD, precisely by inhibition of these pathways. The decrease in gene expression and protein levels of insulin, insulin-like growth factor-1 (IGF-1) receptors, and other downstream molecules leads to impaired acetylcholine production and cognitive performance in LOAD brain [56]. IR activation, through phosphorylated IRS proteins, results in activation of signaling pathways including PI3K and ERK [57]. Activation of PI3K⟶Akt cascade increase neuronal growth and survival [58]. Akt inactivates GSK-3β, which further inhibits tau phosphorylation [59]. There are number of studies that show insulin-regulated tau phosphorylation and increased rate of NFT formation [60, 61]. Insulin andIGF-1 regulate tau phosphorylation through the inhibition of GSK-3βvia the PI3K-protein kinase B (PI3K-PKB) signaling pathway [62]. Insulin and IGF-1 signaling normally prevents tau hyperphosphorylation in the brain [63]. In type 2 diabetes, increased GSK-3β activity might lead to an elevation of and increased tau phosphorylation [64]. IRS-1deficiency leads to insulin resistance in diabetes. Significantly, there is reduction in levels of IRS-1 and IRS-2occurring in AD brain, accompanied by elevated cytosolic phospho-IRS-1 (Ser 312 and 316) [65, 66]. Phosphorylation of IRS-1 (Ser 312 and 316) inhibits the regulation of insulin on GSK-3β activity, leading to further increase in hyperphosphorylation of tau [67]. Insulin resistance in the hippocampus might induce a neuroplasticity deficit, including deficits in spatial learning and memory.
Glial cell overactivation and tau hyperphosphorylation
Tau pathology is likely to induce microglial/astrocytic activation that is present near neurons and prone to directly favor the progression of neuroinflammation [68]. Microglial cells are the intrinsic macrophages of the CNS and thereby are responsible for monitoring and responding to injury and insult in the surrounding brain and serve as the brain’s natural defense mechanism [69]. Activated microglia express different types of cell surface molecules, including Fc receptors, scavenger receptors, cytokine and chemokine receptors, and major histocompatibility complex (MHC) molecules and have a wide variety of pattern recognition receptors from the Toll-like receptor group (TLR) that detect microbial intruders [70]. After stimulation, TLRs initiate a signal cascade that involves myeloid differentiation primary response 88 and the stimulation of transcription factors including nuclear factor κB (NF-κB) and Activator protein-1. Following activation, microglia is able to trigger a pro-inflammatory cascade resulting in the release of cytotoxic molecules such as cytokines, complement factors [71]. Microglia release cytokines [(interleukin (IL)-1β, IL-1, IL-6, IL-10, IL-12, IL-16, IL-23, tumor necrosis factor-α (TNF-α)]; chemokines [CC (CCL2/MCP-1, CCL3/MIP-1 β, CL4/MIP-1, CCL5/RANTES); CXC (CXCL8/IL8, CXCL9/MIG, CXCL10/IP-10, CXCL12/SDF-1α); CX3 C (CX3CL1/fractalkine)]; matrix metalloproteinases (MMP-2, MMP-3, MMP-9); and eicosanoid (Prostaglandin D2, leukotriene C4, cathepsins B and L, and complement factors C1, C3, and C4), which also induce astrocyte chemotaxis around NFT. Microglial activation brings out the proliferation of astrocytes. Astrocytes require activation, which involves Interferon-γ (IFN-γ) that induces expression of class I or II MHC molecules; the microglia present antigens to CD8+ cells while the astrocytes present them to CD4+ cells. Microglia is more effective as antigen-presenting cells (APCs) when they are previously stimulated with IFN-γ. However, astrocytes are considered non-professional APCs [72]. Overactivation of microglia cells result in neuroinflammation.
Mitochondrial dysfunction and tau hyperphosphorylation
MT dysfunction can be due to hyperphosphorylated tau, which plays a pathological role, in addition to impairing axonal transport of organelles including mitochondria and results in synaptic dysfunction [73]. Tau, a major MT-associated protein, plays a consequential role in neuronal processes. Between the MT-binding domain and projection domain lays a basic proline-rich region, which contains ample phosphorylation sites. The interaction of the proline-rich region of tau with the MT-surface leads to MT stabilization [74]. Phosphorylation as the most chief post-translational modification of tau plays a crucial role in the dynamic equilibrium of tau with the MT. It has been found that the serine/threonine-directed phosphorylation of tau directly regulates the binding affinity of tau for MT [75]. The MT plays an essential role in axonal transport. Dysfunction of MT leads to abnormal axonal transport and synaptic dysfunction. By binding the MT, tau has profound effects on axonal transport, which allows signaling molecules, trophic factors, and essential organelles including mitochondria and so on, to travel along the axons [76]. To meet high energy demands and regulate calcium buffering of neuronal cells, efficient delivery of mitochondria in neurons is essential. The delivery of mitochondria is the task of MT with the help of proteins like kinenins and dynenins. Mitochondria are commodities that are delivered by MT-associated proteins, including tau, across axons into synapses. Balanced mitochondrial fission/fusion dynamics are important to meet high energy demands and enhance neuroprotective effects [77]. A group of guanosine triphosphatases (GTPases) has been found to govern mitochondrial fission and fusion processes. Dynamin- like protein 1 (DLP-1 or Drp1) and a small molecule fission protein-1(Fis1) participate in the regulation of the fission process. The fusion process is regulated by mitofusin 1, mitofusin 2, and optic atrophy protein 1. Abnormal interaction between hyperphosphorylated tau and Drp1 causes an excessive mitochondrial fission process and further leads to the degeneration of mitochondria and synapses in brain [78]. Mitochondrial dynamics within the neuronal environment are regulated by the above-listed proteins. Over expression and hyperphosphorylation of tau impair distribution of mitochondria which further cause defects in axonal function and loss in synapses. It was filamentous, rather than soluble, forms of hyperphosphorylated tau that inhibited anterograde fast axonal transport by activating GSK-3 and axonal protein phosphatase. Defects in mitochondrial function are manifested by a variety of indicators, including decreased ATP synthesis, increased ROS production, impaired oxidative phosphorylation system complexes and antioxidant enzymes [73]. An inhibition of oxidative phosphorylation depolarizes mitochondria, which further impairs the ability of cells to buffer calcium loads. Elevated calcium interferes with mitochondrial action, which reduces ATP production. Neurons require a constant supply of energy for its normal functioning. Neurons have a restricted glycolytic capacity and they therefore depend on mitochondrial aerobic oxidative phosphorylation for energy needs [79]. Interestingly, oxidative phosphorylation is an important source of endogenous toxic free radicals, inclusive of hydrogen peroxide (H2O2), hydroxyl (OH–), and superoxide (O2–) radicals that are byproducts of normal cellular respiration. These ROS generated are constantly neutralized by several efficient enzymatic processes like superoxide dismutase (SOD), glutathione peroxidase, superoxide reductase, and catalase (CAT). If there is an excess of ROS generation, which overcomes the antioxidant capacity to neutralize them, it can lead to oxidative stress followed by mitochondrial dysfunction. ROS are produced by mitochondria target mitochondrial components such as lipids, proteins, and DNA causing lipid and protein peroxidation of cell membrane [80, 81].
Cholinergic dysfunction and tau hyperphosphorylation
Acetylcholine is a major neurotransmitter in the brain, having activity throughout the cortex, basal ganglia, and basal forebrain [82]. Acetylcholine is released from neurons projecting to a wide range of cortical and subcortical sites. These projections can be divided into two groups: the basal forebrain cholinergic system and the brainstem cholinergic system. The basal forebrain cholinergic system consists of cells located in the medial septal nucleus, the vertical and horizontal limbs of the diagonal band of Broca, and the nucleus basalis magnocellularis or nucleus basalis of Meynert (NBM) [83]. These structures send both cholinergic and non-cholinergic projections to a wide range of sites in the neocortex as well as limbic cortices such as cingulate cortex, entorhinal cortex, and hippocampus and other structures including the basolateral amygdala and the olfactory bulb. The brainstem cholinergic system includes neurons situated in the pedunculopontine tegmental nucleus and laterodorsal pontine tegmentum principally innervating the thalamus and basal ganglia but also innervating the basal forebrain and serving as a minor component of the cholinergic innervation of cortical structures [84]. Cholinergic neuron density in the thalamus, striatum, limbic system, and neocortex is high, which suggests that cholinergic transmission is likely to be important for memory, learning, attention, and other higher brain functions. Neurofibrillary degeneration in the basal forebrain is believed to be the primary cause for the dysfunction and death of forebrain cholinergic neurons, giving rise to a widespread pre-synaptic cholinergic dysfunction [85]. NBM is in the basal forebrain which is the source of cortical cholinergic innervations and undergoes severe neurodegeneration in AD. Nicotinic ionotropic receptors and muscarinic metabotropic receptors of the cerebral cortex also undergo changes [86]. Most studies show a loss of nicotinic receptors in the cerebral cortex. There is a decrease of postsynaptic nicotinic receptors on cortical neurons. With respect to muscarinic receptors of the cerebral cortex, it is interesting that the muscarinic M1 receptors (mostly postsynaptic) are not decreased, whereas the M2 receptors (mostly presynaptic) are decreased. However, there is evidence that the remaining postsynaptic M1 receptors of the cerebral cortex may be dysfunctional. Loss of cortical cholinergic innervations is probably provoked by NFTs in the NBM. Cholinergic depletion then contributes to the cognitive impairment which ultimately leads to neuron death [82].
EXPERIMENTAL ANIMAL MODELS (TOXIN-INDUCED) FOR DECIPHERING THE PATHOGENESIS OF ALZHEIMER-TYPE DEMENTIAS
Animal models are used to study the development and progression of diseases and to test novel treatments before they reach clinically. By the beginning of the twentieth century, the use of animal modeling had escalated, especially in rodents, and become the mandatory method of demonstrating biological significance. Rats and mice both play a vital role in understanding the etiology, pathophysiology, and pharmacology of neuropsychiatric diseases, and a careful examination of both organisms is necessary before a choice of model for a translational study can be made. Strains derived from Mus musculus and Rattus norvegicus are used in the inevitable majority of animal research for biomedical purposes [87]. Another frequently used technique is intracerebral cannula implantation, in which a small cannula is implanted into the brain. This can be used to locally administer a drug directly into a specific brain region, allowing the role of this brain region in a behavioral phenotype to be examined. More techniques for brain imaging in animals are being developed, based on functional magnetic resonance imaging and positron emission tomography (PET). Rats have more benefits as compared to mice in cognitive tests, since rats, like humans, have six isoforms of the tau protein. Hyperphosphorylation of tau is involved in the formation of tangles, an essential pathological hallmark of AD, and the similarity in isoforms between humans and rats could indicate a higher degree of similarity in tangle formation as well [88]. There are two types of AD, including familial (5% of all AD) and sporadic, but the transgenic model does not show the complete model of AD, especially the sporadic form of AD, which accounts for 95% of AD cases. Agents such as colchicine, scopolamine, okadaic acid (OKA), streptozotocin, and trimethyltin are used to induce AD in animal model [89].
The Colchicine model
Colchicine is an alkaloid isolated from Colchicum autumnale having properties of anti-gout and anti-inflammatory actions. Decades later, it was used for preventing amyloidosis [89]. Colchicine blocks the axonal transport via depolymerization of MT and without inhibiting protein synthesis [90–92]. Colchicine is a cytotoxic agent that binds irreversibly to tubulin molecules and in result stops the aggregation of tubulin dimers to the fast-growing end, causing interruption of MT polymerization. By blocking axoplasmic transport, colchicine critically damages hippocampal granule cells, ultimately leading to neuronal loss, which manifests with cognitive impairment and spontaneous motor activity. Intracerebroventricular (ICV) injection (15 μg in 5 μL/7.5 μg in 10 μL) of colchicine in rats could induce AD-like pathology with consequent cognitive and behavioral alterations similar to AD [89, 93–95]. The drug selectively blocks acetylcholine transferase in the basal forebrain and hippocampus, which are regions responsible for memory [90]. When colchicine penetrates to the subarachnoid space, symptoms begin to show, including jumpy and irritable behavior, aggression, and loss of body weight. Colchicine administration induced lipid peroxidation, decreased glutathione (GSH) and acetylcholine levels in the brains of rats, and led to consequential oxidative damage resulting in cognitive impairment. Impairment of memory and neurodegeneration was characterized as a sporadic in the AD model after colchicine administration in rodents [89]. Decrease in appetite, and transient diarrhea, adipsia, and aphasia after 7–10 days of its administration are some of the limitations of colchicine-induced memory impairment [94].
The Scopolamine model
Scopolamine is a muscarinic cholinergic receptor antagonist that is practiced for cognitive dysfunction in experimental animals. Injection of scopolamine (1 mg/kg, 0.5 mg/kg intraperitoneal) raised cholinergic dysfunction and impaired cognition in rats [96–98]. Scopolamine is an anti-cholinergic drug that causes amnesia in humans and also impairs learning in animals. Hence, it is widely utilized as a model imitating human dementia in general and AD in particular [99]. Scopolamine caused reduced activity of choline acetyltransferase (the enzyme responsible for synthesis of acetylcholine in the cortex of AD patients. Scopolamine induced cerebral blood flow and glucose metabolism changes, which have been studied with PET and single photon emission-computed tomography. Scopolamine increased blood flow in the left orbitofrontal and the lateral occipital cortex regions bilaterally and decreased regional cerebral blood flow in the region of the right thalamus, the precuneus and the right and left lateral premotor areas [89]. ICV scopolamine-induced amnesia is connected with increased oxidative stress in structures associated with learning and memory. Oxidative stress, in turn, is a critical impairment factor leading to neuroinflammation and loss of cognitive function in AD [100]. This model has limitation that they fail to replicate the pathological aspects and the progressive degenerative nature of AD [101].
The Streptozotocin model
Streptozotocin (STZ) is synthesized by Streptomycetes achromogenes, soil bacteria. It was first used as an antibiotic and later used as an anticancer agent and drug therapy for neuroendocrine tumors [89, 102]. STZ, a glucosamine derivative of nitrosourea and preferentially toxic to pancreatic β-cells, being taken up via the glucose transporter Solute Carrier Family 2 Member 2, has been commonly used to induce diabetes in experimental animals [93]. In the periphery, STZ causes selective pancreatic β-cell toxicity due to the drug’s chemical structure which allows it to enter the cell via the GLUT2 glucose transporter. After peripheral administration, STZ causes alkylation of β-cell DNA which triggers activation of poly ADP ribosylation, leading to depletion of cellular NADH and ATP. When given intraperitoneally in high doses (45–75 mg/kg), STZ is toxic for insulin producing/secreting cells, which induces experimental diabetes mellitus type 1. Low doses (20–60 mg/kg) of STZ given intraperitoneally in neonatal rats damages IR and alters IR signaling and causes diabetes mellitus type 2 [7]. The mechanism of central STZ action and its target cells/molecules have not yet been clarified but a similar mechanism of action in the periphery has been recently suggested. GLUT2 may also be responsible for the STZ-induced effects in the brain, as GLUT2 also is reported to have regional specific distribution in the mammalian brain. ICV-STZ injection in rats has provided a relevant animal model for sporadic AD, as both the animal model and the human disease are characterized by progressive deterioration of cognition, oxidative stress, metabolic disorders, and insulin resistance [103]. Various studies have shown that injection of STZ (3 mg/kg) in rat brain results in cognitive decline, decreased brain weight, increased Aβ and tau levels in the hippocampus [89]. The central administration of STZ causes dysregulated brain insulin signaling and abnormalities in cerebral glucose utilization/metabolism accompanied by an energy deficit [104]. An experimental rat model was developed using STZ administered ICV in doses of up to 100 times lower (per kg body weight) than those used peripherally to induce an insulin resistant brain state [7, 105]. This model is specific for tau hyperphosphorylation.
The okadaic acid model
The OKA animal model is comparable to the STZ animal model. OKA, a polyether C38 fatty acid extracted from a black sponge, Hallichondria okadaii, has been extensively used, since it is a potent and selective inhibitor of the serine/threonine phosphatases 1 (PP1) and 2A (PP2A) and also, although at higher concentrations, of the Ca2 +/calmodulin-dependent PP2B (calcineurin) [93, 106, 107]. The reduced activity of PP2A has been linked with the pathology of AD and was supposed to be involved in hyperphosphorylation of tau [54]. ICVOKA injection develops memory impairment in rats, making it suitable for identification as a potential AD model. OKA caused lack of memory and elevation of Ca2 + that has a relationship with neurotoxicity. Increased intracellular Ca2 + resulted in the accumulation of Aβ, hyperphosphorylation of tau, and neuronal death [89]. ICV infusion of OKA (70 ng/day; for up to 4 months) could lead to some AD-associated pathologies including hyperphosphorylation of tau (at Ser-202/Thr-205) and apoptotic cell death within 2 weeks, as well as cortical deposition of non-fibrillar Aβ within 6 weeks of infusion. Formation of paired helical filaments of tau following intrahippocampal injection of OKA (1 mM, 0.5 ml); (1 μl bilateral infusion of 100 ng OKA) has been confirmed. It is noteworthy, that in this model, hyperphosphorylated tau aggregates do not develop into NFTs [93, 108]. Administration of OKA caused significant increase in PP2A, tau, Ca2 + calmodulin-dependent protein kinase II, and Calpain mRNA expression in cerebral cortex and hippocampus in rats. Increased phosphorylation resulted in reduction of the normal tau stabilization of MT thereby leading to neuronal dysfunction [89].
Out of the above experimental models, STZ-induced AD is the best suitable model for inducing tau hyperphosphorylation which is one of the main hallmarks in AD. Taking care of the target site MTs and NFTs due to tau hyperphosphorylation, STZ-induced tau hyperphosphorylation is the appropriate animal model for tauopathy dementias (Table 1).
Table 1
Pre-clinical experimental animal model using neurotoxin streptozotocin ethidium-mediated tau hyperphosphorylation
| S. No. | Dose and Route | Stereotaxic Co-ordinates | Key Points | Behavioral Parameters | Biochemical Parameters | Conclusion | Reference |
| 1 | ICV-STZ3 mg/kg bilaterally | AP: 0.8 mm ML: 1.5 mm DV: 4.0 mm | •SIRT1 •Tau phosphorylation ERK1/2 •Streptozotocin | Morris water maze test | •Western blotting •NAD/NADH ratio assay •Co-immunoprecipitation •Measurement activity of SIRT1 deacetylase | •Inactivation of SIRT1, tau hyperphosphorylation,and memory impairment occurred in ICV-STZ-treated rats •Activation of SIRT1 by Resveratrol attenuated tau hyperphosphorylation and memory impairment via inhibiting ERK1/2 activity | [238] |
| 2 | ICV-STZ 3 mg/kg bilaterally Volume = 10 μl | AP: 0.8 mm ML: 1.5 mm DV: 3.6 mm | •Xanthoceraside •Learning and memory •Hyperphosphorylated tau •PI3K, Akt, protein kinases •Phosphatases | •Y-Maze test •Novel object recognition test | •Western blot •Phosphorylation level of PI3K,GSK-3β,PP2A | •Xanthoceraside has protective effect against learning and memory impairments •Inhibits tau hyperphosphorylationin the hippocampus through the inhibition of the PI3K/Akt-dependent GSK-3β signaling pathway and increases phosphatases activity. | [239] |
| 3. | ICV-STZ 3 mg/kg bilaterally Volume = 0.5 μl/min | AP: –0.5 mm ML: 1.1 mm DV: –2.8 mm | •AD •Streptozotocin •Nox2 •Cytokines | Object recognition test | •Analysis of cytokines like IL-4,IL-1β,IL-2,10,IFN-γ,TNF-α,IL-12/23 •Nox2 mRNA expression evaluated by RT-PCR in hippocampus •Level of Ox-42 protein expression, a microglial cell marker. •Analysis of GFAP, an astrocyte marker •Level of 4-HNE during lipid peroxidation •Level of 3-NT,marker of oxidative damage induced by tyrosine nitration •Expression ofapoptosis-inducing factor | •Nox2-dependent oxidative stress increase. •Nox2 deletion prevented tau phosphorylation, Aβ expression and decreased neuroinflammation in hippocampus | [240] |
| 4. | ICV-STZ 3 mg/kg bilaterally Volume = 10 μl | AP: 0.8 mm ML: 1.5 mm DV: 4.0 mm | •Adiponectin •Glycogen synthase kinase-3β •AD | •Morris water maze test | •Golgi stain •Western blotting of Tau205, Tau396, Tau404 and Tau5, GSK-3β, GSK-3β (Ser9) and GSK-3β (Tyr216), Akt, p-Akt, PI3K and p-PI3K | •Adiponectin supplements attenuate ICV-STZ-induced spatial cognitive deficits and tau hyperphosphorylation •Inactivate GSK-3β by increasingPI3K/Akt activity | [241] |
| 5. | ICV-STZ 3 mg/kg bilaterally in both lateral ventricles | AP: 0.8 mm ML: 1.5 mm DV: 3.6 mm | •ICV-STZ •Oxidative stress •Tau hyperphosphorylation •Tenuigenin | Morris water maze test | •Nissl staining •Activities of SOD, glutathione peroxidase, and MDA in the hippocampus were measured | •Western blotting: Analysis of 4-HNE protein and phosphorylated tau protein. •SOD activity, glutathione peroxidase, and malondialdehyde contents in the hippocampus were estimated. | [242] |
| 6. | ICV-STZ 3 mg/kg bilaterally in both lateral ventricles Volume = 1 μl/ventricle | AP: 0.8 mm ML: 1.6 mm DV: 4.0 mm | •Streptozotocin •Edaravone •Oxidative stress •Tau hyperphosphorylation | •Balance beam test •Morris water maze test | •Total protein assay •Nissl staining •Western blot was •used to assay the levels of 4-HNE protein adducts, tau, ser396- •phosphorylated tau and thr181-phosphorylated tau | •The analysis of T-SOD, MDA,glutathione, H2O2 and OH, andprotein carbonyl levels were performed | [243] |
| 7. | ICV-STZ3 mg/kg bilaterally Volume = 10 μl on each site | AP: 0.8 mm ML: 1.5 mm DV: 4.0 mm | •PI3K/Akt pathway •GSK-3β •Magnesium sulphate •Tau hyperphosphorylation | •Morris water maze test | •Long term potentiation •Atomic absorption spectroscopy •Golgi staining •Immunohistochemistry •Real-Time Quantitative PCR •Western blotting •Protein expression of IR •Expression of postsynaptic PSD95, PSD93, GLUR1,GLUR2 •Level of GSK-3β,Akt •Expression of synapsin | •GSK-3β and PP2A regulate tau phosphorylation. •ICV-STZ induces inactivation of PI3K/Akt signaling pathway and activation of GSK-3β and cause insulin desensitization. •Magnesium could promote the protein expression of IR,the mRNA levels of insulin and IRimproved synaptic efficacy, and prevented memory and learning impairments | [244] |
| 8. | ICV-STZ 3 mg/kg bilaterally | AP: 0.8 mm ML: 1.5 mm DV: 4.0 mm | •AD •AMPK •Diabetes mellitus Mitochondria tau | •Morris water maze test | •Mitochondrial membrane potential, complex I •activity, and ATP levels assays •Western blotting- p-AMPK,Tau 5 and α-tubulin •Measurement activity of SIRT1 deacetylase •ROS measurement •SOD assay •Estimation of SOD, ROS, Mitochondrial membrane potential, complex I activity, and ATP levels assays | •AMPK isa serine/threonine protein kinase maintainscellular energy balance in mammalian cells. •Reduction in AMPK inducestau hyperphosphorylation in ICV-STZ rats. •Restoring AMPK with its specific activator AICAR can attenuate mitochondria dysfunction, redox dysregulation, cleaved caspase-3, tau hyperphosphorylation, and cognitive impairment. | [245] |
| 9. | ICV-STZ 3 mg/kg injected bilaterally Volume = 1.5 μl was injected in each hemisphere | AP: –0.5 mm ML: 1.1 mm DV: –2.8 mm | •AD •Aβ •Neurofilaments •Streptozotocin •Synapsin •Tau protein | Novel Object Recognition test | •Immunoblotting •Level of ChAT protein •Level of synapsin •Level of NF-L in hippocampus •Estimation of GSK-3β, tyrosineregulated kinase 1A, P25/cdk5, and MAPK, PP1, PP2A, and PP5 | •Synapsin is a phosphoprotein related to synaptic vesicles. •Increase activity protein kinases such as glycogen synthase kinase 3β, dual-specific tyrosine regulated kinase 1A, P25/cdk5, MAPK have been observed in AD brains •Decrease in activity of phosphatases such as PP1, PP2A, and PP5, Aβ and increasephos phorylation of Tau, and decreased synapsin expression levels at 14 days after icv injection of STZ | [246] |
| 10. | ICV-STZ 3 mg/kg bilaterally in both lateral ventricles Volume = 5 μl on each site | AP: 0.8 mm ML: 1.5 mm DV: 3.8 mm | •AD •Naringenin •Aβ •Tau •PPARγ | •Morris water maze test •Place navigation test •Body weight •Brain weight | •Glucose measurements •Western-blot analysis- assess the levels of Tau, phospho-Tau, total GSK-3β, phospho-GSK-3β (ser9), IDE and β-actin •Immunohistochemical staining studies •Real-time quantitative RT-PCR=PPARγ and IDE were measured •Expression of AChE •Activity of GSK-3β | •The balance between kinase and phosphatase activities determines the level of p-Tau. •GSK-3 is the S/T kinase that has two isozymes, GSK-3β and GSK-3α. •The GSK-3α regulates amyloidogenic processing of AβPP and GSK-3βkinase in PI3 K/AKT/GSK-3 signaling is regulated by insulin. GSK-3β regulates tau phosphorylation •Increased GSK-3β activity could promote the hyperphosphorylation of tau via inhibiting insulin signaling. | [247] |
| 11. | ICV-STZ 3 mg/kg in one lateral ventricle, i.e., unilaterally Volume = 10 μl | AP: 0.8 mm ML: 1.5 mm DV: 3.6 mm | •AD •Geniposide •Glucagon-like •peptide-1 receptor •Glycogen synthase kinase-3β •Tau hyperphosphorylation •Type 2 diabetes | •Morris water maze test | •Immunohistochemistry •Western blot Assay •Observe PHF using TEM •Estimation of GSK-3β | •GSK-3β is key regulator in glycogen synthesis. •Phosphorylation at tyrosine-216 in GSK-3β or tyrosine-279 in GSK-3α enhances the enzymatic activity, while phosphorylation of serine-9 in GSK-3β or serine-21 in GSK-3α decreases the activity. •Geniposide may serve as a GSK-3 inhibitor to exert its neuroprotective effect in the AD animal model. | [248] |
| 12. | ICV-STZ 3 mg/kg unilaterally in left ventricle only Volume = 3 μl | AP: –1.0 mm ML: 0.3 mm DV: –2.5 mm | •Streptozotocin 3xTg-ADmice •Cognitive deficits •Tau phosphorylation •Aβ •Synaptic proteins •Neuroinflammation •Insulin signaling | •Elevated Plus Maze •Open field •One-Trial Object Recognition Task •Accelerating Rotarod Test •Morris water maze | •Western blot analysis •Immunohistochemical staining •Levels of IR, insulin-like growth factor-1 receptor, insulin receptor substrate-1,PI3K, 3 phosphoinositide dependent protein Kinase-1, AKT, and GSK-3. | •The 3xTg-AD mice develop numerous NFTs after the age of 12 months, but hyperphosphorylation of tau occurs at earlier age. •Tau phosphorylate at multiple phosphorylation sites in the 3xTg-AD mice at theage of 7–8 months. •STZ treatment causes increase in tau hyperphosphorylation and neuroinflammation, a insulin signaling dysfunction, decrease in synaptic plasticity. •Intranasal insulin treatment will improve cerebral glucose metabolism and cognition. | [249] |
| 13. | ICV-STZ 3 mg/kg unilaterally in right lateral ventricle Volume = 10 μl | AP: –0.8 mm ML: 1.5 mm DV: –3.6 mm | •Insulin signaling •Intranasal insulin •Tau hyperphosphorylation •Microglial activation | •Open field test •Morris water maze | •Western blotting •Immunohistochemical staining •Doublecortin evaluation •Evaluation of tau kinases, GSK-3, cdk5 and its activator p35, MAPK/ERK, JNK, and calcium/calmodulin- dependent protein kinase II | •Intranasal delivery of insulin is a non-invasive technique that bypasses the blood-brain barrier and delivers insulin from the nasal cavity to the CNS via intraneuronal pathway. •Intranasal insulin restore the dysregulation of tau kinases in the hippocampus of ICV-STZ rats | [250] |
| 14. | ICV-STZ 3 mg/kg bilaterally Volume = 10 μl in both ventricles | AP: 0.8 mm ML: 1.5 mm DV: 3.6 mm | •AD •LX2343 •Cognitive deficits •Oxidative stress •Tauopathy •Mitochondria •GSK-3β inhibitor •Neuroprotection | •Morris water maze test | •Mitochondrial membrane potential assay- mitochondrial membrane potential was determined •Mitochondrial function assay- concentration of ATP was measured •Transmission electron microscopy-based assay •GSK-3β enzymatic activity assay •Western blot-Cytochrome c estimationTUNEL assay- Cell death in animal brain tissue was detected •Immunohistochemistry- detect theexpression of P396-Tau protein | •Protein levels of PSD95, synaptophysin, and vesicle-associated membrane protein 2, which are crucial for neurotransmission and synaptic plasticity, were detected by western blot. •Protective effect of LX2343 involves JNK/p38 pathway inhibition. | [251] |
| 15. | ICV-STZ 3 mg/kg unilaterally in right lateral ventricle Volume = 5 μl | AP: 1.0 mm ML: 0.5 mm DV: 2.5 mm | •AD •Liraglutide •Tau neurofilaments •Streptozotocin | •Morris water maze test | •Western blot analysis-Protein concentration were measured •Microtubule binding assay •Immunohistochemistry staining-Detect NF and tau phosphorylation •Immunofluorescence staining- phosphorylation of NF-M/H detected by antibody SMI31 | •Protein concentrations of the samples were measured by BCA Protein Assay Kit. •Microtubule binding assay tells about amount of tau and tubulin | [252] |
| 16. | ICV-STZ 3 mg/kg bilaterally in both lateral ventricles | AP: –0.3 mm ML: –1.0 mm DV: –2.5 mm | •AD •Astrocyte activation •FPR2 •Tau phosphorylation | •Accelerating Rotarod test •Open field •Morris water maze | •Western blot- Protein concentrations were determined •Immunofluorescence staining | •FPR2 is known to be involved in host defense and inflammation. •As a receptor for Aβ, FPR2 could uptake and clear Aβ, suggesting a protective effect in brain. After being activated by Aβ, FPR could also induce glial cells to release proinflammatory factors, indicating its harmful effect on brain. •FPR2 deletion could improve cognitive function, hyperphosphorylation of tau, and the activation of astrocytes | [253] |
| 17. | ICV-STZ 3 mg/kg bilaterally Volume = 2 μl/ventricle | AP: 0.8 mm ML: 1.5 mm DV: 3.6 mm | •Memory deficit •Tau hyperphosphorylation •GSK-3β •PP2A •Streptozotocin •Hippocampus | •Autoshaping Learning Task •Food Magazine and Autoshaping Training | •Western blot-levels of p-tau, levels of p-GSK-3β estimated | •Insulin and IR are selectively distributed in the brain, including olfactory bulb, hypothalamus, cerebral cortex, amygdala and hippocampus. •IR expression is involved in memory formation. •Binding of insulin to IR induces activating signal transduction cascade of the PI3K pathway in turn activates Akt/PKB, which phosphorylate the GSK-3 results in its inactivation causing tau hyperphosphorylation. •Disruption of IR-PI3K-Akt/PKB signaling cascade leads to the dephosphorylation in Ser9 of GSK-3β confirming GSK-3β, a major kinase that phosphorylate in vivo tau in several sites; hyperphosphorylated in PHF. | [254] |
| 18. | ICV-STZ 3 mg/kg bilaterally Volume = 1 μl/ min | AP: 0.8 mm ML: 1.4 mm DV: 3.6 mm | •Nicotinamide •AD •SLN •Tau protein •PS | •Spatial learning and memory test | •ELISA tests- calculate the total tau protein amount (T-tau) and phosphorylated tau 231 •Histopathology of animals brains-cresyl violetstaining used | •Nicotinamide is ahistone deacetylase inhibitor. •The i.p. administration of nicotinamide loaded PS-SLN showed better memory improvement, preserved more neuronal cells and reduced the tau hyperphosphorylation in experimented animals comparing to its non-formulated conventional administration in the early stage of AD | [255] |
| 19. | ICV-STZ 3 mg/kg bilaterally Volume = 5 μl/ventricle | AP: 0.8 mm ML: 1.5 mm DV: 3.6 mm | •TMP •Memory impairment •GSK-3β •Tau hyperphosphorylation •Cholinergic neuron | •Inhibitory avoidance task assay •Morris water maze assay | •Western blot-Total protein concentration was determined •Analysis of cholinergic function- ChAT and AChE activity was measured spectrophotometrically | •ICV-STZ, a good sporadic AD model causes spatial memory and fear memory impairments, impaired insulin signaling and overactivation of GSK-3β. •Activation of GSK-3β accounts for memory impairment, tau hyperphosphorylation, increased Aβ production, and neuroinflammation, all of which are hallmark characteristics of AD. •TMP was shown to decrease GSK-3β expression at both protein and RNA levels, and its analogue CXC195 could protect against cerebralischemia/reperfusion induced apoptosis through the PI3K/Akt/GSK-3β pathway. | [256] |
4-HNE, 4-hydroxyl-2-nonenal; AChE, acetylcholinesterase; AD, Alzheimer’s disease; AKT, protein kinase B; AMPK, AMP-activated protein kinase; APP, amyloid-β proteinprecursor; Aβ, amyloid-β; cdk5, cyclin-dependent kinase 5; ChAT, choline acetyltransferase; CNS, central nervous system; ERK, extracellular signal regulated kinase; FPR2, formyl peptide receptor 2; GSK, glycogen synthase kinase; ICV-STZ, intracerebroventricular streptozotocin; IDE, insulin degrading enzyme; IR, insulin receptor; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated proteinkinase; MDA, malondialdehyde; NF, neurofilaments; NFTs, neurofibrillary tangles; PHF, paired helical filaments; PI3K, phosphatidylinositide 3-kinase; PP2A, protein phosphatase 2A; PPARγ, peroxisome proliferator-activated receptor gamma; PS, phosphatidylserine; PSD, postsynaptic density protein; ROS, reactive oxygen species; SLN, solid lipid nanoparticle; SOD, superoxide dismutase; TEM, transmission electron microscopy; TMP, tetramethylpyrazine.
MICROTUBULE STABILIZATION, A PLAUSIBLE THERAPEUTIC TARGET SITE CENTERED BEHIND RESOLUTION OF AD
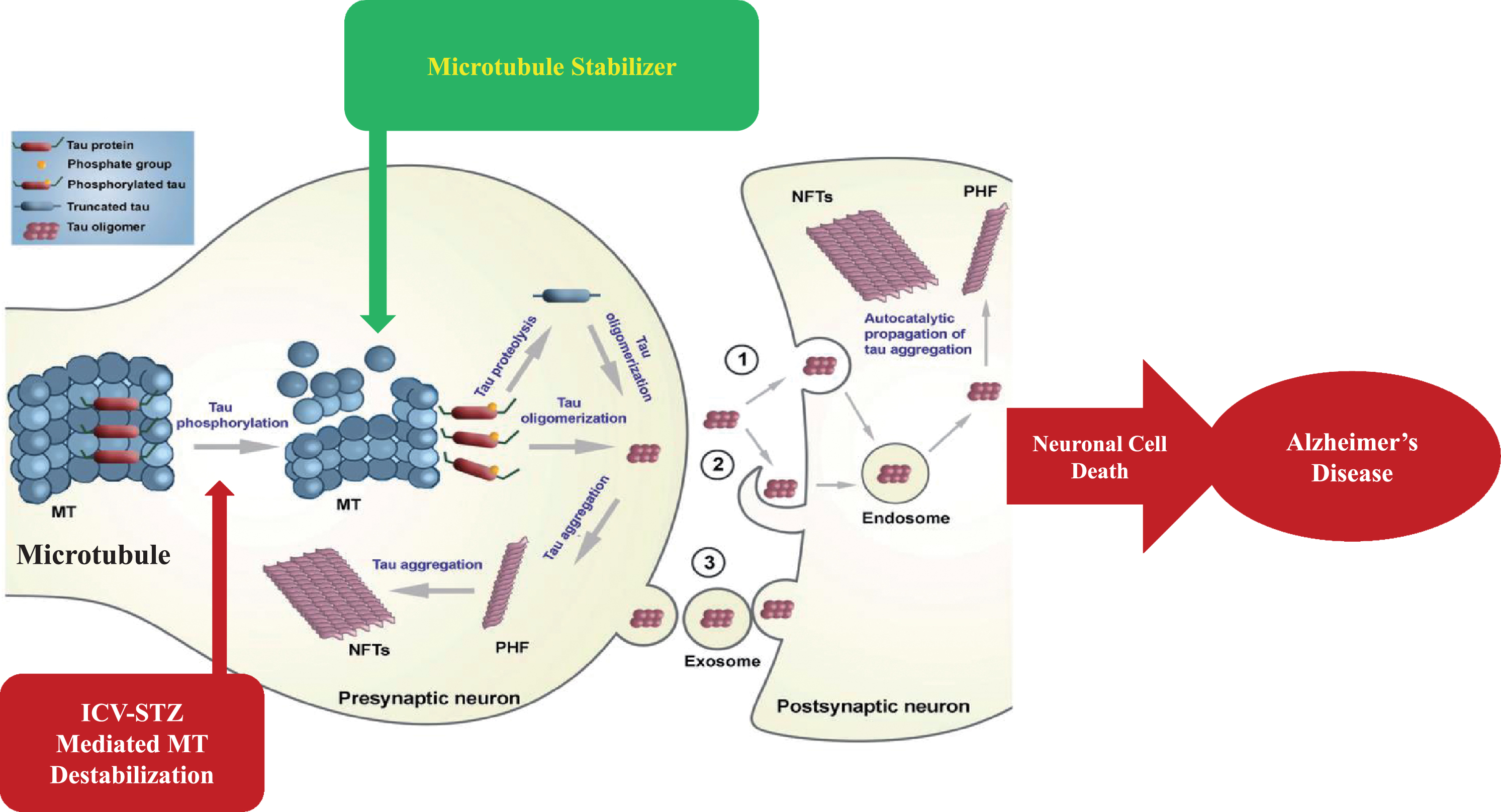
MTs are dynamic components of the intracellular neuronal cytoskeleton that alternate between polymerization and depolymerization phases forming polarized linear hollow tubing with diameter 24 nm extending their (+) ends facing toward the synapse and their (–) ends toward the cell body in the axons of neurons and composed of α-tubulin and β-tubulin heterodimers, which are micrometer long and play a role in maintenance of cell shape and transport of vesicles and organelles like mitochondria and cell division [109]. Axonal transport is carried out by two motor proteins kinesin and dynenins that transport cargo throughout neuron-like kinesin motors transport cargo toward axon terminals (anterograde) and dynein motors carry cargo away from axon tips (retrograde). For proper transport of neuronal contents and organelles, functional axonal MTs are required, and in many neurodegenerative diseases axonal transport is impaired in one way or another [110]. Tau is a class of proteins that are ample in nerve cells and perform the function of stabilizing the MT. In certain neuropathological situations, tau proteins become defective and fail to adequately stabilize the MT, which can result in MT destabilization further leading to detachment of tau from MT and being responsible for generation of abnormal masses known as NFTs that are toxic to neurons. NFTs are remarked in entorhinal cortex, limbic, and neocortex over the course of clinical progression in AD brains. Alterations in the stability of the MTs due to abnormal phosphorylation of tau proteins often precede damage to intracellular axonal transport that leads to neurotransmitter deficit [111]. The main challenge for this dementing disorder is to identify a preventive drug therapy that typically blocks the progression of AD. Moreover, in this review, we are focusing on one of the major pathological hallmarks of AD that is MT destabilization associated with NFT formed due to tau hyperphosphorylation, which may prove to be a preventive target in AD. There is availability of various treatment drugs for behavioral complications observed in AD. However, no particular drug therapy is showing remarkable improvement directly linked with tauopathy. Microtubule stabilizers (MTS) are potential neuroprotective agents to treat AD by restoring axonal function [109]. Therefore, involvement of MTS in prevention of tau abnormal phosphorylation may give hope for dementia patients. In Fig. 3, we present a schematic illustration of post-translational modifications of tau after inducing neurotoxin and use of an MTS as a plausible intervention to prevent MT destabilization.
Fig.3
Schematic illustration of post-translational modifications of tau after inducing neurotoxin and microtubule stabilizer as plausible intervention to prevent microtubule destabilization.
CURRENT PHARMACOLOGICAL INTERVENTION FOR ALZHEIMER-TYPE DEMENTIAS
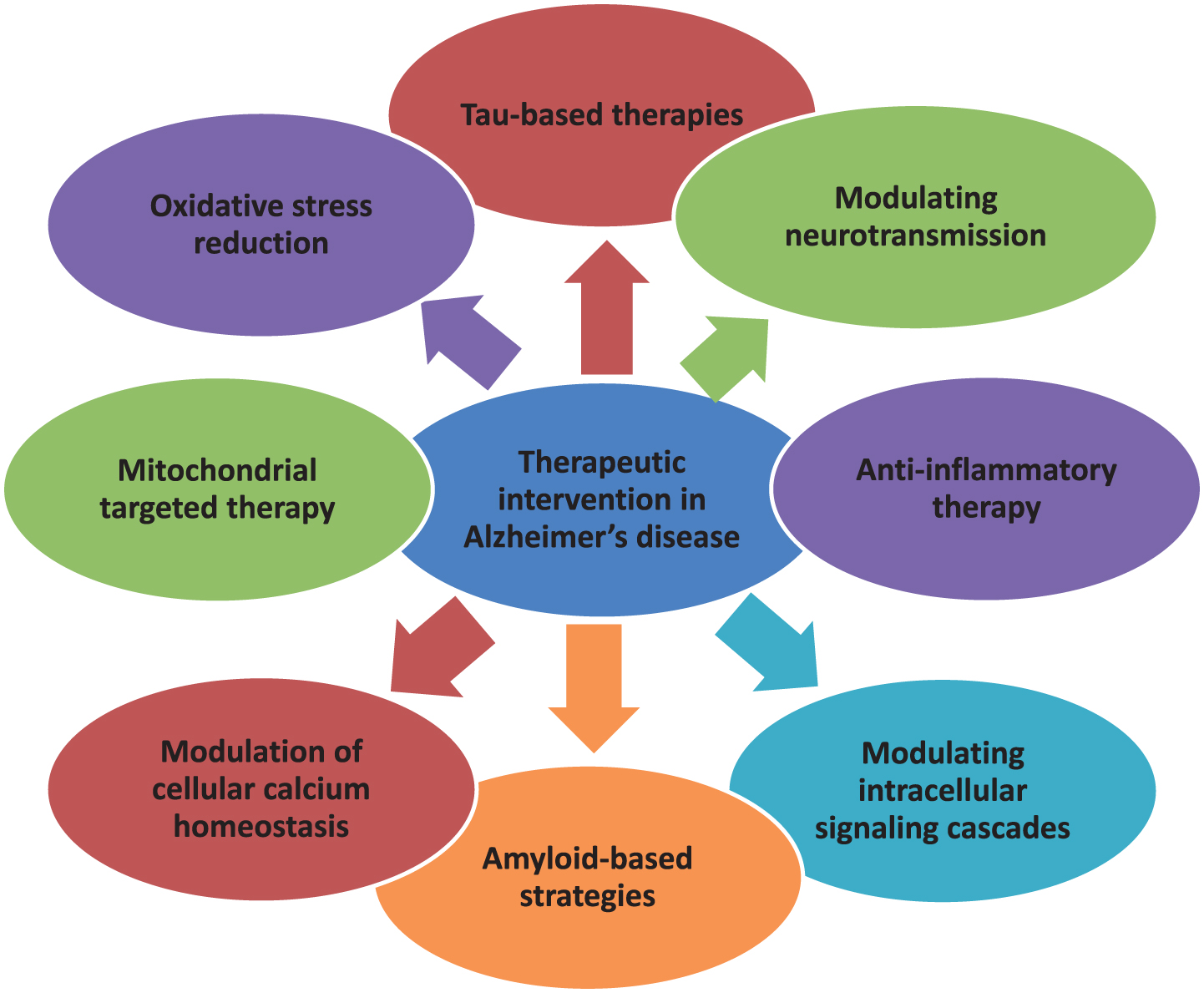
Drugs available clinically are for symptomatic relief only. No intervention is currently available as a preventive therapy. In the decades since Aβ and tau were identified, development of therapies for AD has primarily focused on Aβ, but tau has received more attention in recent years, in part because of the failure of various Aβ-targeting treatments in clinical trials [26, 112]. There are various therapeutic targets focusing which interventions are formed. The currently available treatment drugs include acetylcholinesterase inhibitors and NMDA receptor antagonists which are United States Food and Drug Administration (US FDA) approved. In order to ameliorate the disease, novel strategies have been developed [10]. In this regard, major focus is targeted to Aβ- and tau-based therapeutics, which is a major key to unlocking this disease in the near future. The various mechanisms involved in the pathogenesis of AD create enough difficulty in producing an effective treatment. On the basis of different therapeutic targets as characterized in Fig. 4, interventions are prepared and their safety and efficacy are confirmed in different preclinical and clinical trials.
Fig.4
Multifarious Target-based Intervention.
Tau-based therapies in clinical trials
Table 2 summarizes the pre-clinical and clinical data of recent tau-based therapies, phases of clinical trial, and related dose and route used in these trials. This review paper primarily focuses on deteriorating tau hyperphosphorylation, one of the main hallmarks of AD, so accumulating data of tau-related therapy is key. Instead of focusing on one hallmark, it also throws some light on ameliorating various pathogenesis consequences of AD by revealing new drug candidates focusing on other pathogenesis outcomes like neurotransmitter deficit, oxidative stress, and neuroinflammation.
Table 2
Post translational modified tau based interventions with preclinical and clinical status
| Tau based therapies | |||||
| S.no. | Intervention | Dose and route | Category | Mechanism of Action | Reference |
| 1. | Memantine | •2 mg/kg orally in Wistar rats •1 μM–5 μM in cell cultures | Phosphatase Modifier | •N-methyl-d-aspartate receptor antagonist •Memantine enhances PP2A activity by blocking inhibitor 2. | [257–260] |
| 2. | Sodium selenate | •1.5 μg/μL subcutaneous injection in mice •100 μM in cell culture •Phase IIa randomized trial, 30 mg/day orally | Phosphatase Modifier | •Increases PP2A activity via activation of the regulatory B subunit and reduce sphosphorylation of tau | [261–263] |
| 3. | Acyclovir, Penciclovir, Foscarnet | 50 μM–100 μM in cell culture | Phosphorylated tau protein reducers | Target viral DNA replication. Inhibition of HSV1 DNA replication | [264–266] |
| 4. | Flavopiridol, Roscovitine | 5 μM and 50 μM | Kinase inhibitor, i.e., CDK5 | Compete with ATP for binding to CDK5, resulting in reduced activation of this kinase | [267–270] |
| 5. | Tideglusib | Phase IIa and b, 400–1000 mg/day orally | GSK-3β inhibitor | •Does not compete with ATP binding •Reduces tau phosphorylation, Aβ plaque burden, memory deficits, cell death, and astrocytosis | [271–274] |
| 6. | Lithium chloride | 300 μg orally in AD patients Phase II | GSK-3β inhibitor | Lithium treatment significantly reduced phospho-tau levels in CSF and improved cognitive performance | [275–277] |
| 7. | Salsalate | •225 mg/kg in mice •Phase Ib 3,000 mg total daily by mouth | Tau acetylation inhibitor | •Reduced p300 HAT activity •Inhibits acetylation of tau at Lys174 by p300 HAT | [278, 279] |
| 8. | MK-8719 | •10–100 mg/kg in transgenic mouse model •Phase I 1200 mg orally | Tau deglycosylation inhibitor O-GlcNAcase inhibitor | Inhibitor of the O-GlcNAcaseenzyme | [297, 280] |
| 9. | Methylthioninium chloride or Methylene Blue | 138 mg/day orally effective Higher dose 228 mg ineffective as causes decreases in red cell count and hemoglobinand increases in methemoglobin Discontinued for AD | Tau aggregation inhibitor | Blocks the polymerization of tau in vitro by trapping the tau monomers in an aggregation incompetent conformation | [281] |
| 10. | TRx0237 | •0.16 μM in cell culture •5–75 mg/kgorally in vivo •3 Phase III study 200 mg/day orally 150, 250 mg/day 200 mg/day Active Placebo –8 mg/day | Tau aggregation inhibitor | •Reduced form of methylthioninium. •Blocks the polymerization of tau by trapping the tau monomers in an aggregation incompetent conformation | [215, 282, 283] |
| 11. | TRx-0014 | NA | Tau aggregation inhibitor | Inhibitory properties for monoamine oxidase, nitric oxide production and as blocker of Tau aggregation | [26, 150] |
| 12. | Thiamet G | 500 mg/kg orally in mice | O-GlcNAcase inhibitor | Inhibition of O-GlcNAcase, the enzyme responsible for the removal of O-GlcNAc modification, shown to reduce tau pathology | [284] |
| 13. | Curcumin | 5 μM in N2a/APP695swe cells | P-Tau (ser404) /Tau expression suppressor | •Curcumin could inhibit the abnormal excessive phosphorylation of Tau byinactivating GSK-3 •Protect the nerve in AD through adjusting the cytoskeleton balance and maintain microtubule function | [285] |
| 14. | Rolipram | Human trials fail after Phase II narrow therapeutic window and gastrointestinal adverse effects like emetic effect | PDE4 inhibitor | •Increases cAMP levels, enhances proteasome function and reduces the accumulation of tau •Reduce amounts of total and insoluble tau including phosphorylated tau | [286–288] |
| 15. | BPN14770 | •Phase I 10 and 20 mg orally given twice day •Phase II is on the way to initiate | PDE4 inhibitor | •Increases cAMP levels,enhances proteasome function and reduces the accumulation of tau •Reduce amounts of total and insoluble tau including phosphorylated tau | [289, 290] |
| 16. | Bapineuzumab | •Phase II 0.15, 0.5, 1 mg/kg by intravenous infusion •Phase III had no effect on tau pathology | Tau immunotherapy | Humanized monoclonal antibody that specifically targets these N-terminal residues ofAβ peptides and reduce tauhyperphosphorylation | [291, 292] |
| 17. | AADvac1 vaccine | •Transgenic rat given subcutaneous injection of (100 μg of peptide-KLH conjugate/dose) with PBS in a final dose volume of 300 μl •Phase II 40 μg Axon peptide 108 (coupled to KLH) using aluminum hydroxide (containing 0.5 mg Al3+) as adjuvant, in a phosphate buffer | Anti-tau vaccine Active immunotherapy | NA | [215, 259, 293–295] |
| 18. | ACI-35 vaccine | •200 μL subcutaneous injection in mice •Phase I completed | Anti-tau vaccine Active immunotherapy | •Contains antibodies that selectively bound pSer396/404 over the non-phosphorylated version of the epitope and could detect tau pathology by immunostaining. •Display a significant reduction in soluble tau phosphorylated at Ser396 but not at other amino acid residues | [296, 297] |
| 19. | RG7345 | •30 mg/kg antibodyintraperitoneally •PhaseI trial and data is not released. Drug has been discontinued because of an unfavorable pharmacokinetic profile, because no safety or efficacy concerns seem to have been raised during the trial. | Passive immunotherapy | Recognizes and bind to tau phosphorylated at Ser422, enter neurons and may potentially interfere with immunodetection of tau/pS422 by antibody and reduce tau pathology | [298, 299] |
| 20. | BMS-986168 | •60 mg/kg in mice •0.5–20 mg/kg in monkeys •Phase I IV infusion of 700, 2100, and 4200 mg | Passive immunotherapy | •Humanized IgG4P monoclonal antibody that recognizes full length tau and N- terminal tau fragments which are secreted by neurons and found in extracellular interstitial fluid and CSF. •BMS-986168 mediates removal of e tau and reduces progression of disease. Total tau and free tau (not bound to BMS-986168) were measured | [159, 300, 301] |
| 21. | ABBV-8E12 | •10 or 50 mg/kg intraperitoneal in mice •Phase I 2.5, 7.5, 15, 25, and 50 mg/kg intravenously •Phase II started | Passive immunotherapy | Recognizes amino acids 25–30 of the tau protein and work extracellular reduced the levels of aggregated and hyper phosphorylated tauand improved cognition | [302, 303] |
| 22. | RO 7105705 | •3, 10, or 30 mg/kg in mice •Phase I IV dose of 225 mg–16.8 g; subcutaneous administration of 1,200 mg •Phase II is recruiting | Passive immunotherapy | •Act primarily on pathological tau because it targets extracellular forms of the protein •Recognizes tau’s N-terminus and reacts with all six isoforms of human and primate tau, but not mouse tau | [304, 305] |
| 23. | LY3303560 | •Phase I completed •Phase II is recruiting | Passive immunotherapy | Bind and neutralize soluble tau aggregate | [306, 307] |
| 24. | JNJ-63733657 | Phase I recruiting | Passive immunotherapy | •Recognizes the mid-region of tau potentially interfere with cell-to-cell propagation of pathogenic, aggregated tau •Antibodies targeting the tau N-terminus and eliminate pathogenic tau “seeds” in a cell-based assay and inhibit spread of tau pathology | [308, 309] |
| 25. | UCB0107 | 300 nM in cell culture | Passive immunotherapy | •Antibody binds to amino acids 235–246 in the proline-rich region of tau •Antibody was effective in preventing seeding and spreading of pathological tau | [309, 310] |
AD, Alzheimer’s disease; Aβ, amyloid-β; CSF, cerebrospinal fluid; GSK, glycogen synthase kinase; HAT, histone acetyltransferase; KLH, keyhole limpet hemocyanin; PDE4, phosphodiesterase 4; PP2A, protein phosphatase 2A.
Anti-amyloid drugs in clinical trials
Mismetabolism of AβPP and the impaired clearance of Aβ generate a cascade of events including hyperphosphorylated tau mediated breakdown of microtubular assembly and resultant synaptic failure, leading to AD. Intracellular assembly states of the oligomeric and protofibrillar species may promote tau hyperphosphorylation, disruption of proteasome and mitochondria function, dysregulation of calcium homeostasis, synaptic failure, and cognitive dysfunction [113, 114]. Aβ generation from AβPP forms via a two-step proteolytic process involving β- and γ-secretases. The β-site AβPP cleaving enzyme (BACE1) first cleaves AβPP to produce a membrane bound soluble C-terminal fragment. A consequential cleavage of the C-terminal fragment by the γ-secretase activity further generates Aβ40 and Aβ42. Both types of peptide are found in amyloid plaques, but Aβ42 is evidently more directly neurotoxic and has a greater propensity to aggregate [113, 115]. Solanezumab, a vaccine acting on the soluble monomeric forms of the protein, did not significantly affect cognitive decline in a Phase III trial [116, 117]. Aducanumab, which is a human monoclonal IgG1 antibody anti-Aβ derived from an AD patient, was successful in a Phase Ib trial, and a Phase II trial is recruiting and the Phase III is active but not recruiting [118–120]. Gantenerumab, a fully human monoclonal antibody that binds aggregated Aβ and removes Aβ plaques by Fc receptor-mediated phagocytosis, is in the Phase III recruiting stage [121, 122]. Some other monoclonal antibodies which are recently observed are BAN-2401 [123, 124]; UB-311 [125, 126]; Crenezumab [127, 128]; Ponezumab [129, 130]; Octagam [131, 132]; SAR-228810 [133, 134]; MEDI-1814 [135]; KHK-6640 [136, 137]; Lu-AF-20513 [138–140] and TTP4000 [133, 141]. Some approaches to inhibition of enzymes, i.e., β- and γ-secretases involved in AβPP cleavage, resulted in Aβ peptide formation. BACE1 is the β secretase implicated in AD and inhibitors of this enzyme are verubecestat MK8931 [142, 143]; AZD-3293(LY-3314814) [144, 145]; AtabecestatJNJ-54861911 [146, 147]; E-2609 [148]; BI-1181181 [145, 149]; inhibitor of γ-secretaseareEVP-0962 [150, 151]; and BMS-932481 [152]. Recently developed compounds which prevent aggregation of Aβ are GV-971 [153]; ALZT-OP1 [154]; Phenserine [155]; Posiphen [156, 157]; Scyllo-Inositol [158, 159]; ALZ-801 [160]; SAN-61 [161] and Exebryl-1 [162].
Anti-inflammatory drugs in clinical trials reducing inflammatory biomarkers
AD pathogenesis is not confined to the neuronal compartment but strongly interacts with immunological mechanisms in the brain. Misfolded and aggregated proteins bind to pattern recognition receptors on microglia and astroglia and initiate an innate immune response, characterized by the release of inflammatory mediators, which contribute to disease progression [163]. AAD-2004 is an inhibitor of the formation of cytokines that is in Phase I clinical trials [164]. Sargramostim enhances the microglial phagocytosis of Aβ and suppresses the generation of pro-inflammatory cytokines and is now in Phase II clinical trials [165]. Angiotensin II, a key hormone peptide that binds to angiotensin II type 1 and type 2 receptors (AT1R and AT2R, respectively) expressed in neurons, microglia, and astrocytes, has pleiotropic roles in the brain, including mediation of inflammation [166]. Blockage of AT1R signaling directed the microglia toward a less pro-inflammatory stage [167]. Two antagonists of AT1R candesartan and telmisartan are now in Phase II clinical trials for AD [165, 167]. Purinoceptor 6 (P2Y6) is a purinergic receptor expressed on microglia that facilitates inflammation regulating microglial activation and phagocytosis. The uracil nucleotide uridine 5′-diphosphate is a specific ligand for P2Y6 receptor that can be released from damaged neurons to employ microglia to phagocytose cell debris [168]. GC021109, a small molecule reported to bind to P2Y6 receptor to stimulate microglial phagocytosis and inhibit pro-inflammatory cytokine release from microglia, completed Phase Ia trials yielding positive results [165]. Receptor for advanced glycation end products (RAGE) is a pattern-recognition receptor expressed on microglia and astrocytes that mediate pro-inflammatory or cytotoxic responses; it is also expressed on brain endothelial cells [169, 170]. RAGE is elevated in astrocytes and microglia in the hippocampus [171]. Azeliragon, a small antagonist of RAGE, has been evaluated in clinical trials [172]. Non-steroidal anti-inflammatory drugs were one of the early classes of anti-inflammatory drugs subjected to AD drug development. The main actions of non-steroidal anti-inflammatory drugs are thought to be initiated through the inhibition of cyclooxygenase (COX) activity [173]. In several AD transgenic mouse models, levels of arachidonic acid, COX-2, and prostaglandins are elevated in the hippocampus [174, 175]. Ibuprofen and indomethacin exert their effects by acting as agonists of peroxisome proliferator-activated receptor-γ (PPARγ), while tarenflurbil (MPC-7869) and CHF5074 improve cognition and reduce brain inflammation [176–178]. Combination therapy of ibuprofen together with cromolyn (ALZT-OP1) targets the early stages of AD and ameliorates neuroinflammatory responses [165]. TNF-α mediates inflammation through binding with TNF receptor-1, which initiate the activation of NF-κB, JNK, and p38 MAPK signaling [179]. Etanercept functions as a decoy receptor for binding to TNF-α and inhibit TNF signaling. Phase II clinical trials of etanercept for AD did not significantly improve cognition or behavior [180]. Thalidomide inhibits the release of TNF-α in monocytes. Thalidomide not only inhibits TNF-α production by microglia and astrocytes, but also exhibits a neuroprotective effect in the hippocampus in an inflamed AD mouse model [181]. PPARγ is a nuclear hormone receptor that acts as a transcription factor in the regulation of inflammatory gene expression [182]. Agonists targeting PPARγ suppress the expression of proinflammatory genes. Pioglitazone and rosiglitazone are agonists of PPARγ. Pioglitazone and rosiglitazone failed in Phase III clinical trials owing to a lack of efficacy [183]. Minocycline, an antibiotic that can permeate the BBB, exhibits anti-inflammatory and neuroprotective effects [184]. A recent trend in drug research is discovering the use of epigenetic drugs for AD treatment [185]. Two epigenetic drugs are currently undergoing clinical trials: ORY-2001 and vorinostat. ORY- 2001 is undergoing a Phase II clinical trial and is an epigenetic drug that selectively inhibits the activity of the lysine (K)-specific demethylase 1A and monoamine oxidase B [186]. Vorinostat, a histone deacetylase 2 inhibitor, is undergoing Phase I clinical trials [187]. Herpesviruses such as HSV-1, HHV-6A, and HHV-7 have been detected in the brains of AD patients and are concerned with the promotion of amyloid plaque deposition in AD progression [188, 189]. Valaciclovir, an antiviral drug, is undergoing two Phase II clinical trials for early AD treatment [190].
Antioxidant therapy reducing oxidative stress parameters
Oxidative stress is defined as the production of free radicals in a diseased state. This condition instructs the production of necessary antioxidants. Excessive free radicals occur in the case where there is a shortage of antioxidants [191]. The most prevalent antioxidants in the cell is glutathione, which exists as thiol-reduced (GSH) and disulfide-oxidized states. GSH can react with free radicals either independently or in the reaction catalyzed by glutathione peroxidase to form glutathione disulfide (GSSG), which can be converted back to the reduced state by glutathione reductase. The GSH/GSSG ratio is used as an indicator of cell redox potential and oxidative stress [192]. Other antioxidant enzymes are SOD and catalase, which catalyzes the disproportionation of superoxide to molecular oxygen and peroxide and the conversion of H2O2 to water and oxygen. The activity of these enzymes has been reported to be reduced in AD [193]. Several antioxidants, such as N-acetylcysteine, curcumin, resveratrol, vitamin E, ferulic acid, coenzyme Q (CoQ), selenium, and melatonin, have been tested for their potential to improve cognitive performance in healthy individuals [194–196]. Despite potent effects of these compounds on cellular oxidative status in vitro and in vivo, the convincing evidence of their therapeutic potential in humans is lacking. In vitro and in vivo studies have demonstrated the neuroprotective potential of CoQ10 in AD [197]. Idebenone, an analog of CoQ10, consists of short chains of isoprene units, crosses the BBB easily, is well tolerated in humans, and possesses good antioxidant properties [198]. Another potent antioxidant acting as an effective inhibitor of mitochondrial permeability transition pore is creatine. Creatine supplementation has been shown to protect against neuronal death caused by NMDA, malonate, Aβ, and ibotenic acid [199, 200]. Due to the importance of mitochondrial dysfunctions in the pathogenesis of various disorders, scientists have focused on the engineering of therapeutic molecules that could accumulate in mitochondria. One such therapeutic compound is MitoQ, which is the most widely used mitochondria-targeting antioxidant. MitoQ exhibits neuroprotection by scavenging peroxynitrite and superoxide and protects mitochondria against lipid peroxidation [201]. Another antioxidant, mitotocopherol, protects mitochondria from oxidative stress via inhibition of lipid peroxidation [202, 203]. MitoTEMPOL functions as a SOD mimetic [204].
APPROACHABLE DRUG THERAPY FOR ALZHEIMER’S DISEASE (TABLE 3)
Table 3
Approachable drug therapy with their clinical and preclinical status in Alzheimer’s disease
| Drug | Clinical adverse effect | Mechanism of Action | Dose & Route Clinical | Pre-clinical | References Clinical | Pre-clinical |
| Donepezil (Memory enhancer) | Hepatotoxicity GI adverse events, bradycardia, nausea, diarrhea, insomnia, vomiting, asthenia/fatigue and anorexia, weight loss | AcetylcholinesteraseinhibitorAcetylcholine at cholinergic synapses | 5,10 mg orally;5,10,23 mg orally;10 mg orally;5,10, 23 mg orally;23 mg orally | 0.3 mg/kgp.o.;1 mg/kg i.p.;3 mg/kg i.p.;0.1, 0.3 mg/kg p.o.;1, 3 mg/kg i.p. | [311–315] | [316–319] |
| Memantine (Anti-excitatory) | Fatigue, pain, hypertension, dizziness, headache, constipation, vomiting, back pain, confusion, somnolence, hallucination, coughing, dyspnea, agitation, fall, inflicted injury, urinary incontinence, diarrhea, bronchitis, insomnia, urinary tract infection, influenza-like symptoms, abnormal gait, depression, upper respiratory tract infection, peripheral edema, anorexia, and arthralgia | NMDA receptor antagonistInhibit influx of Ca2 + ions | 20 mg orally;5, 10, 20 mg orally;10 &20 mg orally;5 &23 mg orally;10 &20 mg orally; | 20 mg/kg i.p.;5, 10 mg/kg i.p;10, 20 mg/kg i.p;0.1, 1 mg/kg i.p;30 mg/kg i.p. | [320–325] | [325–330] |
| Epothilone D (Microtubule stabilizer) | NA | Inhibit microtubule destabilizationIncrease polymerization of microtubules | 0.003,0.01, 0.03 mg/kg infusion; 0.003,0.01, 0.03 mg/kg infusion | 1, 3 mg/kg i.p.;1 mg/kg i.p;1,0.3,3 mg/kg i.p | [214, 215, 331, 332] | [210, 333–337] |
| Minocycline (Antibiotic,anti-inflammatory andTau/betaaggregation inhibitor) | Nausea, vertigo and mild dizziness | Suppress microglial activation and inhibit oxygen radicals | 200 mg orally | 10 mg/kg i.p.;45 mg/kg i.p.;90 mg/kg i.p.;90–180 mg/kg i.p. | [338–343] | [339, 344–348] |
| Lycopene (Antioxidant) | Lycopenemia with high intakes of lycopene | Reactive oxygen species scavengerPrevent cellular lipid and protein oxidation | 6.5, 15, 30 mg orally;15 mg orally;15, 30 mg orally;25 mg orally | 10 mg/kg p.o.;5 mg/kg p.o.;2,4 mg/kg p.o.;2.5,5,10 mg/kg p.o.;5, 10 mg/kg p.o. | [349–353] | [351, 354–357] |
Donepezil (acetylcholinesterase inhibitor)
Donepezil was US FDA approved in 1996 for use in mild, moderate, and severe AD. Donepezil hydrochloride is a piperidine derivative and acts as a reversible inhibitor of acetylcholinesterase. Acetylcholinesterase is an enzyme that degrades acetylcholine when it is released from the pre-synapse. Donepezil binds reversibly to acetylcholinesterase and inhibits the hydrolysis of acetylcholine, thus resulting in increasing the availability of acetylcholine at the synapses, enhancing cholinergic transmission [205, 206]. Other cholinesterase inhibitors in clinic use are rivastigmine and galantamine. They have efficacy against the three major domains of AD symptoms, namely functional ability, behavior, and cognition [207]. Donepezil upregulates the nicotinic receptors present in the cortical neurons. Insomnia, muscle cramps, fatigue, and anorexia are common side effects with higher doses; other side effects include bradycardia because of its vagotonic properties, rhabdomyolysis, and nightmares [206].
Memantine (NMDA antagonist)
Memantine is approved as a therapeutic drug in moderate to severe AD in 2003 by the US FDA. Memantine was discovered in 1968 and patented by Eli Lilly. It is a low to moderate affinity NMDAR antagonist. Clinically it shows small positive effect on cognition, global functioning, activities of daily living, and neuropsychiatric symptoms [208, 209]. The most common adverse events are headache, dizziness, diarrhea, hypertension, anxiety, and influenza [209].
Epothilone D (microtubule stabilizer)
MT stabilizing drugs that are used in the treatment of cancer may have utility in the treatment of tauopathies. Epothilone D is brain-penetrant MT-stabilizing agent and treatment with Epothilone D resulted in significant improvements of MT density, axonal integrity, fast axonal transport, and cognitive performance, without the onset of side effects in vivo. It failed clinically due to adverse effects after Phase I and the study ended in 2013 and evaluation of BMS-241027 for AD was discontinued [212–215].
Minocycline (anti-inflammatory, anti-apoptotic)
Minocycline, a tetracycline derivative, has potent anti-inflammatory, anti-apoptotic, and neuroprotective properties. Minocycline easily crosses the BBB and effectively delays disease progression and reduces neuronal death in mouse models. Minocycline treatment results in inhibition of insoluble tau aggregate levels and tau phosphorylation [216]. Minocycline can productively reduce the generation of caspase-3-cleaved tau fragments. It is reported to inhibit protein kinases involved in tau, including CDK5, Akt/GSK-3, and p38 [217, 218].
Lycopene (antioxidant)
Lycopene is a red plant pigment mostly found in tomatoes, apricots, watermelons, etc. As one of the carotenoids, it is an effective antioxidant with a single-oxygen quenching capacity 47 and 100 times stronger than 50 that of vitamin E and beta-carotene [219]. Lycopene is a strong antioxidant with an ability to reduce oxidative damage to lipids, proteins, and DNA. Lycopene alone could reduce tau phosphorylation at Ser262 site and lycopene/vitamin E combination decreased the phosphorylation of tau protein at Ser262 and Ser396 epitopes [220].
CURRENT STATUS OF MICROTUBULE STABILIZERS
Neurological disorders are basically associated with MT anomalies, and efforts to reposition MT-targeting chemotherapeutic agents for treatment of neurodegenerative and psychiatric illnesses are underway [221]. MT destabilization associated with tau hyperphosphorylation results in reduction of axonal transport and MTS could normalize MT and axonal transport in tauopathies [222–224]. MTS are under exploration for treatment of a variety of brain disorders. MTS that are utilized in oncology may prove to be potential therapeutics for AD and other tauopathies [225]. We figure out the classes of MTS in Table 4 utilized for cancer treatment, their brain penetrance properties and clinical use with appropriate dose and route, and depict efforts to enforce these agents for treatment of brain related dysfunctions. Taxane derivative, paclitaxel isolated from the stem bark in 1960s of the Taxus brevifolia (Western yew), was found to exhibit potent anti-tumor properties [109]. However, MT-stabilizing properties of this compound remained unknown until 1979. Furthermore, paclitaxel was the first MT-stabilizing agent to be investigated in an animal model of neurodegenerative tauopathies. Lack of brain penetration of paclitaxel prevents further investigations of this compound in mouse models of tauopathies [225]. Dictyostatin was first isolated from a Maldives marine sponge Spongia sp and was found to be highly potent against a variety of human cancer cell lines and is a brain-penetrant MT-stabilizing agent having the capability to improve MT density of PS19 mice and decrease axonal abnormalities as well as reduce tau pathology resulting in hippocampal neuron survival [109, 226, 227]. Cabazitaxel, a dimethoxy derivative of docetaxel extracted from European yew needles, approved by FDA for castration-resistant prostate cancer in 2010, has the ability to bypass efflux transporters articulate in the BBB as compared to first generation taxanes like paclitaxel and docetaxel [228, 229]. Pre-clinical data has shown that Cabazitaxel has a potential role in CNS tumors due to better BBB penetration, indicating its ability to be used in neurological disorders [230, 231]. TPI-287 belongs to third generation taxane and is a semi-synthetic derivative of abeo-taxane used in cancer therapy. It was investigated as a drug to treat breast cancer that had metastasized to brain and has the ability to cross the BBB [221]. TPI-287 is in Phase I clinical trial for treatment of primary 4-repeat tauopathies, corticobasal syndrome, and progressive Supranuclear palsy [232]. Davunetide (NAP) is an intranasal neuropeptide obtained from the activity-dependent neuroprotective protein. NAP, when administered intranasally, intraperitoneally, or intravenously shows good brain penetration and neuroprotective action at very low concentrations [233]. Clinically, NAP exhibited efficacy in prodromal AD patients (Tau3R/4R tauopathy). A Phase II study in patients with predicted tauopathies (NCT01056965) was completed in July 2017 [234]. Peloruside was isolated from the marine sponge Mycale hentscheli in New Zealand [109, 235]. Peloruside A was active against murine leukemia cells [236]. Peloruside A had no significant effect on its own on phosphorylation of tau but significantly increased the levels of acetyl tubulin in neuronal cell culture [237].
Table 4
Microtubule stabilizer status in Alzheimer’s disease
| S.no. | Drug | Clinical use | Dose and route | Side effect | Blood-brain barrier penetration | AD Status | Reference |
| 1. | Paclitaxel | Ovarian, breast, and lung cancer, as well as Kaposi’s sarcoma | Preclinical •35 mg/m2 or 175 mg/m2 IV over 3 h every 3 weeks in ovarian cancer; 200 mg/m2IV over 3 h in lung cancer; 100 mg weekly, IV inKaposi’s sarcoma •10 or 25 mg/m2 i.p injections in mice for 12 weeks restored fast axonal transport in spinal axons | Neutropenia and peripheral neuropathy, neurotoxicity, and myelosuppression | Poor blood-brain barrier penetration | In vitro •10 nM in cell culture of mt-human-tau-induced hallmark cellular pathologies of AD •Restore axonal length at a low concentration 5 nm and decrease cell survival after OA treatment Preclinical •5 mg/kg i.p. in mice •10 or 25 mg/m2 i.p. in transgenic mice restored fast axonal transport in axons, wherein MT numbers and stable tubulin were increased | [358–366] |
| 2. | Dictyostatin | Breast cancer, lung cancer | In vitro 10 and 100 nM in human lung adenocarcinoma and human breast cell lines. | Gastrointestinal complications and body weight loss | Good blood-brain barrier penetration | Preclinical •0.1 mg/kg was better tolerated in PS19 tau Tg mouse model •i.p. administration to CD1 mice (5 mg/kg) resulted in MT-stabilization | [367–372] |
| 3. | Cabazitaxel | Refractory metastatic prostate cancer; Pediatric patient with refractory solid CNS tumors | Clinical •30 mg/m2 pediatric patients with CNS tumors •20 and 25 mg/m2 IV in patients with CRPC Preclinical •30 or 90 mg/m2 IV infusion in female CD2F1/CrlBR mice •15 or 60 mg/m2 in female Sprague–Dawley rats •15 mg/m2 in female Beagle dogs •15, 30, 45, or 90 mg m2 IV infusion in female mice •15 and 60 mg m2 IV infusion in rat •9,15, or 25 mg/kg i.p. in pediatric brain tumors | Febrile neutropenia, hypersensitivity reactions, thrombocytopenia, peripheral neuropathy | Good blood-brain barrier penetration. Poor substrate for the P-glycoprotein | NA | [373–376] |
| 4. | TPI 287 | Brain metastatic breast cancer | Preclinical •20 mg/kg IV in rats and mice Clinical Phase I20 mg/m2 IV | Peripheral neuropathy, weight loss | Good blood-brain barrier penetration | Clinical 2 mg/m2 in Patients with Primary Four Repeat Tauopathies | [377–379] |
| 5. | Davunetide | Schizophrenia | Clinical 5 mg and 30 mg intranasal | NA | Good blood-brain barrier penetration Applicable to early stages of AD | Preclinical •2 μgNAP/mouse/day intranasally 2.5 μl for each nostril for 3 or 6 months •s.c. injections included l, 0.5 g/mouse/day •Intranasal 0.5 μg/5 μl/mouse/day 2.5 μl/each nostril Clinical •Phase II study •5 mg q.d and 15 mg b.i.d. intranasal in subjects with amnesic mild cognitive impairment and tauopathy | [380–388] |
| 6. | CNDR-51657 | Advanced Malignant Solid Tumors | Clinical Phase 1 22.5 and 18 mg/m2 in patients with refractory solid tumors | Neutropenia fever | Good blood-brain barrier penetration | Preclinical •1 or 5 mg/kg in female mice •10 mg/kg orally in mice •3 mg/kg or 10 mg/kg i.p. in mice for3 months In vitro Increased levels of acetylated tubulin at 1 μM and 10 μM in rat cortical neurons | [389–392] |
| 7. | Peloruside A | Murine leukemic cells, | In vitro Cytotoxic to P388 murine leukemia cells at 10 ng/mL (18 nM) | NA | NA | In vitro 10 nM in neuronal culture | [393–396] |
| 8. | Mapreg | Anti-depressant | Preclinical •50 mg/kg/day orally in tree shrews | Safe profile except body weight loss | NA | In vitro •1 μM PREG in neuronal culture •Tau (25 μg/ml) was incubated with 100 nM [3H]PREG | [397–399] |
AD, Alzheimer’s disease; CNS, central nervous system; CRPC, castration-resistant prostate cancer; i.p., intraperitoneal; IV, intravenous; MT, microtubule.
CONCLUSION
As discussed above, current data signal that changes in MT dynamics are associated with several aspects of the neurodegenerative triad of AD and may explain and connect some of the pathological events, which occur during the disease. MT destabilization appears to underlie axonopathic changes and might lead to disturbance of axonal transport due to hyperphosphorylation of MAP tau, which contributes to oxidative stress, neuroexcitotoxicity, mitochondrial abnormalities, and neuroinflammation. Moreover, experimental evidence shows various neurotoxins induce changes in various post translational modifications of tau protein which damages cytoskeleton of neurons further leading to tauopathy-associated dementia. As much as some open questions and provocations remain out in front, the data reviewed here are encouraging and demonstrate the potential of therapeutic efforts of tau-based strategies for the future treatment of tauopathies, including AD. From recent evidence, MTS have shown a positive potential for treating neurodegenerative diseases. Therefore, MT stabilization by MTS can be a future therapeutic approach to overcome MT degeneration as well as to retard disease progression in Alzheimer-type dementias. Moreover, if successful, such an approach could have the potential to target the hidden connection in the neurodegenerative triad and support the survival and well-functioning of the affected neurons.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors express their gratitude to Chairman, Mr. Parveen Garg and Director, Dr. G.D. Gupta, ISF College of Pharmacy, Moga (Punjab), India for their great vision and support.
The authors received no financial support for the research, authorship, and/or publication of this article.
REFERENCES
[1] | Rajmohan R , Reddy PH ((2017) ) Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J Alzheimers Dis 57: , 975–999. |
[2] | Nelson PT , Braak H , Markesbery WR ((2009) ) Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J Neuropathol Exp Neurol 68: , 1–4. |
[3] | Chopra K , Misra S , Kuhad A ((2011) ) Neurobiological aspects of Alzheimer’s disease. Expert Opin Ther Targets 15: , 535–555. |
[4] | Dai MH , Zheng H , Zeng LD , Zhang Y ((2018) ) The genes associated with early-onset Alzheimer’s disease. Oncotarget 9: , 15132. |
[5] | Bertram L , Tanzi RE ((2012) ) The genetics of Alzheimer’s disease. Prog Mol Biol Transl Sci 107: , 79–100. |
[6] | Korolev IO ((2014) ) Alzheimer’s disease: A clinical and basic science review. Med Student Res J 4: , 24–33. |
[7] | Mehan S , Arora R , Sehgal V , Sharma D , Sharma G ((2012) ) Dementia–A complete literature review on various mechanisms involves in pathogenesis and an intracerebroventricular streptozotocin induced Alzheimer’s disease. In Inflammatory Diseases - Immunopathology, Clinical and Pharmacological Bases, Khatami M, ed. IntechOpen, https://www.intechopen.com/books/inflammatory-diseases-immunopathology-clinical-and-pharmacological-bases/alzheimer-s-disease-an-updated-review-on-pathogenesis-and-intracerebroventricular-streptozotocin-ind |
[8] | Prince M , Comas-Herrera A , Knapp M , Guerchet M , Karagiannidou M ((2016) ) World Alzheimer Report 2016. Improving Healthcare for People Living with Dementia: Coverage, Quality and Costs Now and in the Future. Alzheimer’s Disease International, London, UK. |
[9] | Pimplikar SW ((2014) ) Neuroinflammation in Alzheimer’s disease: From pathogenesis to a therapeutic target. J Clin Immunol 34: , 64–69. |
[10] | Kumar A , Singh A ((2015) ) A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol Rep 67: , 195–203. |
[11] | Glynn-Servedio BE , Ranola TS ((2017) ) AChE inhibitors and NMDA receptor antagonists in advanced Alzheimer’s disease. Consult Pharm 32: , 511–518. |
[12] | Das BC , Pradhan S , Ojha DP , Das A , Hosmane NS ((2018) ) The role of tau protein in diseases. Ann Adv Chem 2: , 001–016. |
[13] | Field JJ , Kanakkanthara A , Miller JH ((2014) ) Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem 22: , 5050–5059. |
[14] | Chin-Chan M , Navarro-Yepes J , Quintanilla-Vega B ((2015) ) Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci 9: , 124. |
[15] | Javanshiri K , Waldö ML , Friberg N , Sjövall F , Haglund M , Englund E ((2018) ) Atherosclerosis, hypertension, and diabetes in Alzheimer’s disease, vascular dementia, and mixed dementia: Prevalence and presentation. J Alzheimers Dis 65: , 1247–1258. |
[16] | Santos CY ((2018) ) Cardiovascular and retinal vascular changes in preclinical Alzheimer’s disease. Open Access Dissertations, Paper 741. https://digitalcommons.uri.edu/oa_diss/741 |
[17] | Kim J , Castellano JM , Jiang H , Basak JM , Parsadanian M , Pham V , Holtzman DM ((2009) ) Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron 64: , 632–644. |
[18] | Munoz DG , Feldman H ((2000) ) Causes of Alzheimer’s disease. CMAJ 162: , 65–72. |
[19] | Swerdlow RH ((2011) ) Brain aging, Alzheimer’s disease, and mitochondria. Biochim Biophys Acta 1812: , 1630–1639. |
[20] | Šimić G , Babić Leko M , Wray S , Harrington C , Delalle I , Jovanov-Milošević N , Wischik C ((2016) ) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: , 6. |
[21] | Jameson L , Frey T , Zeeberg B , Dalldorf F , Caplow M ((1980) ) Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry 19: , 2472–2479. |
[22] | Wischik CM , Crowther RA , Stewart M , Roth M ((1985) ) Subunit structure of paired helical filaments in Alzheimer’s disease. J Cell Biol 100: , 1905–1912. |
[23] | Kocahan S , Doğan Z ((2017) ) Mechanisms of Alzheimer’s disease pathogenesis and prevention: The brain, neural pathology, N-methyl-D-aspartate receptors, tau protein and other risk factors. Clin Psychopharmacol Neurosci 15: , 1–8. |
[24] | Mietelska-Porowska A , Wasik U , Goras M , Filipek A , Niewiadomska G ((2014) ) Tau protein modifications and interactions: Their role in function and dysfunction. Int J Mol Sci 15: , 4671–4713. |
[25] | Kolarova M , García-Sierra F , Bartos A , Ricny J , Ripova D ((2012) ) Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis 2012: , 731526. |
[26] | Congdon EE , Sigurdsson EM ((2018) ) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14: , 399–415. |
[27] | Lindwall G , Cole RD ((1984) ) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259: , 5301–5305. |
[28] | Luna-Munoz J , Chavez-Macias L , Garcia-Sierra F , Mena R ((2007) ) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimers Dis 12: , 365–375. |
[29] | Hilgeroth AP , Tell V ((2013) ) Recent developments of protein kinase inhibitors as potential AD therapeutics. Front Cell Neurosci 7: , 189. |
[30] | Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2005) ) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22: , 1942–1950. |
[31] | Sontag JM , Sontag E ((2014) ) Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci 7: , 16. |
[32] | Min SW , Cho SH , Zhou Y , Schroeder S , Haroutunian V , Seeley WW , Meyers D ((2010) ) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67: , 953–966. |
[33] | Wang Y , Mandelkow E ((2016) ) Tau in physiology and pathology. Nat Rev Neurosci 17: , 5–21. |
[34] | Cotman CW , Poon WW , Rissman RA , Blurton-Jones M ((2005) ) The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 64: , 104–112. |
[35] | Li X , Lu F , Wang JZ , Gong CX ((2006) ) Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci 23: , 2078–2086. |
[36] | Vergallo A , Giampietri L , Baldacci F , Volpi L , Chico L , Pagni C , Bonuccelli U ((2018) ) Oxidative stress assessment in Alzheimer’s disease: A clinic setting study. Am J Alzheimers Dis Other Demen 33: , 35–41. |
[37] | Alavi Naini SM , Soussi-Yanicostas N ((2015) ) Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev 2015: , 151979. |
[38] | Cente M , Filipcik P , Pevalova M , Novak M ((2006) ) Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur J Neurosci 24: , 1085–1090. |
[39] | Pamplona R ((2008) ) Membrane phospholipids, lipoxidative damage and molecular integrity: A causal role in aging and longevity. Biochim Biophys Acta 1777: , 1249–1262. |
[40] | Gonos ES , Kapetanou M , Sereikaite J , Bartosz G , Naparło K , Grzesik M , Sadowska-Bartosz I ((2018) ) Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging (Albany NY) 10: , 868–901. |
[41] | Magalingam KB , Radhakrishnan A , Ping NS , Haleagrahara N ((2018) ) Current concepts of neurodegenerative mechanisms in Alzheimer’s disease. Biomed Res Int 2018: , 3740461. |
[42] | Butterfield DA , Lauderback CM ((2002) ) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med 32: , 1050–1060. |
[43] | Danysz W , Parsons CG ((2012) ) Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br J Pharmacol 67: , 324–352. |
[44] | Hanlon CD , Andrew DJ ((2015) ) Outside-in signaling–a brief review of GPCR signaling with a focus on the Drosophila GPCR family. J Cell Sci 128: , 3533–3542. |
[45] | Zhou Q , Sheng M ((2013) ) NMDA receptors in nervous system diseases. Neuropharmacology 74: , 69–75. |
[46] | Reiner A , Levitz J ((2018) ) Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 98: , 1080–1098. |
[47] | Dewar D , Chalmers DT , Graham DI , McCulloch J ((1991) ) Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer’s disease: An autoradiographic study of the hippocampus. Brain Res 553: , 58–64. |
[48] | Lee HG , Ogawa O , Zhu X , O’Neill MJ , Petersen RB , Castellani RJ , Smith MA ((2004) ) Aberrant expression of metabotropic glutamate receptor 2 in the vulnerable neurons of Alzheimer’s disease. Acta Neuropathol 107: , 365–371. |
[49] | Revett TJ , Baker GB , Jhamandas J , Kar S ((2013) ) Glutamate system, amyloid β peptides and tau protein: Functional interrelationshis and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 38: , 6–23 . |
[50] | Lesort M , Jope RS , Johnson GV ((1999) ) Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3β and Fyn tyrosine kinase. J Neurochem 72: , 576–584. |
[51] | Hernandez P , Lee G , Sjoberg M , Maccioni RB ((2009) ) Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Aβ 25–35: Involvement of lipid rafts. J Alzheimers Dis 16: , 149–156. |
[52] | Lee G , Thangavel R , Sharma VM , Litersky JM , Bhaskar K , Fang SM , Ksiezak-Reding H ((2004) ) Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J Neurosci 24: , 2304–2312. |
[53] | Tumminia A , Vinciguerra F , Parisi M , Frittitta L ((2018) ) Type 2 diabetes mellitus and Alzheimer’s disease: Role of insulin signalling and therapeutic implications. Int J Mol Sci 19: , E3306. |
[54] | Griffith CM , Eid T , Rose GM , Patrylo PR ((2018) ) Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology 136: , 202–215. |
[55] | Laplante M , Sabatini DM ((2012) ) mTOR signaling in growth control and disease. Cell 149: , 274–293. |
[56] | Folch J , Ettcheto M , Busquets O , Sánchez-López E , Castro-Torres R , Verdaguer E , Beas-Zarate C ((2018) ) The implication of the brain insulin receptor in late onset Alzheimer’s disease dementia. Pharmaceuticals 11: , E11. |
[57] | Saltiel AR ((2001) ) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104: , 517–529. |
[58] | Rodgers EE , Theibert AB ((2002) ) Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci 20: , 187–197. |
[59] | Zhao L , Teter B , Morihara T , Lim GP , Ambegaokar SS , Ubeda OJ , Cole GM ((2004) ) Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J Neurosci 24: , 11120–11126. |
[60] | Lesort M , Johnson GVW ((2000) ) Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience 99: , 305–316. |
[61] | Schubert M , Brazil DP , Burks DJ , Kushner JA , Ye J , Flint CL , Corfas G ((2003) ) Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci 23: , 7084–7092. |
[62] | Hong M , Lee VMY ((1997) ) Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem 272: , 19547–19553. |
[63] | Cheng CM , Tseng V , Wang J , Wang D , Matyakhina L , Bondy CA ((2005) ) Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology 146: , 5086–5091. |
[64] | Sims-Robinson C , Kim B , Rosko A , Feldman EL ((2010) ) How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 6: , 551–559. |
[65] | Talbot K , Wang HY , Kazi H , Han LY , Bakshi KP , Stucky A , Arvanitakis Z ((2012) ) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122: , 1316–1338. |
[66] | Yarchoan M , Toledo JB , Lee EB , Arvanitakis Z , Kazi H , Han LY , Arnold SE ((2014) ) Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol 128: , 679–689. |
[67] | Ma QL , Yang F , Rosario ER , Ubeda OJ , Beech W , Gant DJ , Vinters HV ((2009) ) β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J Neurosci 29: , 9078–9089. |
[68] | Laurent C , Buée L , Blum D ((2018) ) Tau and neuroinflammation: What impact for Alzheimer’s disease and tauopathies? Biomed J 41: , 21–33. |
[69] | Dani M , Wood M , Mizoguchi R , Fan Z , Walker Z , Morgan R , Edison P ((2018) ) Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141: , 2740–2754. |
[70] | Doens D , Fernández PL ((2014) ) Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J Neuroinflammation 11: , 48. |
[71] | De Nardo D ((2015) ) Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 74: , 181–189. |
[72] | Cabezas IL , Batista AH , Rol GP ((2014) ) The role of glial cells in Alzheimer disease: Potential therapeutic implications. Neurología 29: , 305–309. |
[73] | Cheng Y , Bai F ((2018) ) The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front Neurosci 12: , 163. |
[74] | He HJ , Wang XS , Pan R , Wang DL , Liu MN , He RQ ((2009) ) The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol 10: , 81. |
[75] | Medeiros R , Baglietto-Vargas D , LaFerla FM ((2011) ) The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther 7: , 514–524. |
[76] | Chevalier-Larsen E , Holzbaur EL ((2006) ) Axonal transport and neurodegenerative disease. Biochim Biophys Acta 1762: , 1094–1108. |
[77] | Woods LC , Berbusse GW , Naylor K ((2016) ) Microtubules are essential for mitochondrial dynamics–fission, fusion, and motility–in dictyostelium discoideum. Front Cell Dev Biol 4: , 19. |
[78] | Ali S , McStay G ((2018) ) Regulation of mitochondrial dynamics by proteolytic processing and protein turnover. Antioxidants 7: , E15. |
[79] | Swerdlow RH ((2018) ) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 62: , 1403–1416. |
[80] | Ramesh S , Govindarajulu M , Jones E , Suppiramaniam V , Moore T , Dhanasekaran M ((2018) ) Mitochondrial dysfunction and the role of mitophagy in Alzheimer’s disease. Alzheimer’s Disease & Treatment, MedDocs Publishers LLC. |
[81] | Bhatti JS , Bhatti GK , Reddy PH ((2017) ) Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta 1863: , 1066–1077. |
[82] | Hampel H , Mesulam MM , Cuello AC , Farlow MR , Giacobini E , Grossberg GT , Khachaturian ZS ((2018) ) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141: , 1917–1933. |
[83] | Ballinger EC , Ananth M , Talmage DA , Role LW ((2016) ) Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91: , 1199–1218. |
[84] | Newman EL , Gupta K , Climer JR , Monaghan CK , Hasselmo ME ((2012) ) Cholinergic modulation of cognitive processing: Insights drawn from computational models. Front Behav Neurosci 6: , 24. |
[85] | Voss K1 , Koren J 3rd , Dickey CA ((2011) ) The earliest tau dysfunction in Alzheimer’s disease? Tau phosphorylated at s422 as a toxic seed. Am J Pathol 179: , 2148–2151. |
[86] | Tiernan CT , Ginsberg SD , He B , Ward SM , Guillozet-Bongaarts AL , Kanaan NM. , Counts SE ((2018) ) Pretangle pathology within cholinergic nucleus basalis neurons coincides with neurotrophic and neurotransmitter receptor gene dysregulation during the progression of Alzheimer’s disease. Neurobiol Dis 117: , 125–136. |
[87] | Ellenbroek B , Youn J ((2016) ) Rodent models in neuroscience research: Is it a rat race? Dis Model Mech 9: , 1079–1087. |
[88] | Drummond E , Wisniewski T ((2017) ) Alzheimer’s disease: Experimental models and reality. Acta Neuropathol 133: , 155–175. |
[89] | Malekzadeh S , Edalatmanesh MA , Mehrabani D , Shariati M ((2017) ) Drugs induced Alzheimer’s disease in animal model. GMJ 6: , 185–196. |
[90] | Nakayama T , Sawada T ((2002) ) Involvement of microtubule integrity in memory impairment caused by colchicine. Pharmacol Biochem Behav 71: , 119–138. |
[91] | Paulson JC , McClure WO ((1975) ) Inhibition of axoplasmic transport by colchicine, podophyllotoxin, and vinblastine: An effect on microtubules. Ann N Y Acad Sci 253: , 517–527. |
[92] | Mukhtar E , Adhami VM , Mukhtar H ((2014) ) Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther 13: , 275–284. |
[93] | Nazem A , Sankowski R , Bacher M , Al-Abed Y ((2015) ) Rodent models of neuroinflammation for Alzheimer’s disease. J Neuroinflammation 12: , 74. |
[94] | More S , Kumar H , Cho DY , Yun YS , Choi DK ((2016) ) Toxin-induced experimental models of learning and memory impairment. Int J Mol Sci 17: , E1447. |
[95] | Khurana S , Jain S , Mediratta PK , Banerjee BD , Sharma KK ((2012) ) Protective role of curcumin on colchicine-induced cognitive dysfunction and oxidative stress in rats. Hum Exp Toxicol 31: , 686–697. |
[96] | Bajo R , Pusil S , López ME , Canuet L , Pereda E , Osipova D , Pekkonen E ((2015) ) Scopolamine effects on functional brain connectivity: A pharmacological model of Alzheimer’s disease. Sci Rep 5: , 9748. |
[97] | Malikowska-Racia N , Podkowa A , Sałat K ((2018) ) Phencyclidine and scopolamine for modeling amnesia in rodents: Direct comparison with the use of Barnes maze test and contextual fear conditioning test in mice. Neurotox Res 34: , 431–441. |
[98] | Saraf MK , Prabhakar S , Khanduja KL , Anand A ((2011) ) Bacopa monniera attenuates scopolamine-induced impairment of spatial memory in mice. Evid Based Complement Alternat Med 2011: , 236186. |
[99] | Zaki HF , Abd-El-Fattah MA , Attia AS ((2014) ) Naringenin protects against scopolamine-induced dementia in rats. Bulletin of Faculty of Pharmacy, Cairo University 52: , 15–25. |
[100] | Tabari SSS , Babri S , Mirzaie F , Farajdokht F , Mohaddes G ((2016) ) Enduring amnesia induced by ICV scopolamine is reversed by sesame oil in male rats. Acta Cir Bras 31: , 520–526. |
[101] | Buccafusco JJ ((2009) ) The revival of scopolamine reversal for the assessment of cognition-enhancing drugs. In Methods of Behavior Analysis in Neuroscience. 2nd edition. CRC Press/Taylor & Francis. |
[102] | Eleazu CO , Eleazu KC , Chukwuma S , Essien UN ((2013) ) Review of the mechanism of cell death resulting from streptozotocin challenge in experimental animals, its practical use and potential risk to humans. J Diabetes Metab Disord 12: , 60. |
[103] | Kimura N ((2016) ) Diabetes mellitus induces Alzheimer’s disease pathology: Histopathological evidence from animal models. Int J Mol Sci 17: , 503. |
[104] | de la Monte SM ((2012) ) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 9: , 35–66. |
[105] | Chao PC , Li Y , Chang CH , Shieh JP , Cheng JT , Cheng KC ((2018) ) Investigation of insulin resistance in the popularly used four rat models of type-2 diabetes. Biomed Pharmacother 101: , 155–161. |
[106] | Tapia R , Peña F , Arias C ((1999) ) Neurotoxic and synaptic effects of okadaic acid, an inhibitor of protein phosphatases. Neurochem Res 24: , 1423–1430. |
[107] | Tachibana K , Scheuer PJ , Tsukitani Y , Kikuchi H , Van Engen D , Clardy J , Schmitz FJ ((1981) ) Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J Am Chem Soc 103: , 2469–2471. |
[108] | Costa AP , Tramontina AC , Biasibetti R , Batassini C , Lopes MW , Wartchow KM , Gonçalves CA ((2012) ) Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav Brain Res 226: , 420–427. |
[109] | Ballatore C , Brunden KR , Huryn DM , Trojanowski JQ , Lee VMY , Smith III AB ((2012) ) Microtubule stabilizing agents as potential treatment for Alzheimer’s disease and related neurodegenerative tauopathies. J Med Chem 55: , 8979–8996. |
[110] | Ballatore C , Smith AB , Lee VMY , Trojanowski JQ , Brunden KR ((2016) ) Microtubule-stabilizing agents for Alzheimer’s and other tauopathies. In Alzheimer’s Disease II. Topics in Medicinal Chemistry, vol 24: , Wolfe M, ed. Springer, Cham, pp. 159–179. |
[111] | Trojanowski JQ , Lee VM ((2002) ) The role of tau in Alzheimer’s disease. Med Clin North Am 86: , 615–627. |
[112] | Salomone S , Caraci F , Leggio GM , Fedotova J , Drago F ((2012) ) New pharmacological strategies for treatment of Alzheimer’s disease: Focus on disease modifying drugs. Br J Clin Pharmacol 73: , 504–517. |
[113] | Mohandas E , Rajmohan V , Raghunath B ((2009) ) Neurobiology of Alzheimer’s disease. Indian J Psychiatry 51: , 55. |
[114] | Zhang Y , McLaughlin R , Goodyer C , LeBlanc A ((2002) ) Selective cytotoxicity of intracellular amyloid β peptide1–42 through p53 and Bax in cultured primary human neurons. J Cell Biol 156: , 519–529. |
[115] | Hemming ML , Selkoe DJ ((2005) ) Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 80: , 37644–37650. |
[116] | Honig LS , Vellas B , Woodward M , Boada M , Bullock R , Borrie M , Case M ((2018) ) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378: , 321–330. |
[117] | Hardy J , De Strooper B ((2017) ) Alzheimer’s disease: Where next for anti-amyloid therapies? Brain 140: , 853–855. |
[118] | Haeberlein SB , Castrillo-Viguera C , Gheuens S , Chen T , O’Gorman J , Chiao P , Nitsch RM ((2018) ) 24-month analysis of change from baseline in clinical dementia rating scale cognitive and functional domains in prime: A randomized phase 1b study of the anti–amyloid beta monoclonal antibody aducanumab. Alzheimers Dement 14: , 242. |
[119] | Sevigny J , Chiao P , Williams L , Chen T , Ling Y , O’Gorman J , Sandrock A ((2015) ) Aducanumab (BIIB037), an anti-amyloid beta monoclonal antibody, in patients with prodromal or mild Alzheimer’s disease: Interim results of a randomized, double-blind, placebo-controlled, phase 1b study. Alzheimers Dement 11: , 277. |
[120] | Sullivan MG ((2017) ) Alzheimer’s candidate drug Aducanumab moves to Phase III. Caring Ages 18: , 18. |
[121] | Ostrowitzki S , Lasser RA , Dorflinger E , Scheltens P , Barkhof F , Nikolcheva T , Klein G ((2017) ) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9: , 95. |
[122] | Jacobsen H , Ozmen L , Caruso A , Narquizian R , Hilpert H , Jacobsen B , Bohrmann B ((2014) ) Combined treatment with a BACE inhibitor and anti-Aβ antibody gantenerumab enhances amyloid reduction in APPLondon mice. J Neurosci 34: , 11621–11630. |
[123] | Tucker S , Möller C , Tegerstedt K , Lord A , Laudon H , Sjödahl J , Satlin A ((2015) ) The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis 43: , 575–588. |
[124] | Söllvander S , Nikitidou E , Gallasch L , Zyśk M , Söderberg L , Sehlin D , Erlandsson A ((2018) ) The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J Neuroinflammation 15: , 98. |
[125] | Wang CY , Wang PN , Chiu MJ , Finstad CL , Lin F , Lynn S , Tseng Y ((2017) ) UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (N Y) 3: , 262–272. |
[126] | Wang CY , Finstad CL , Walfield AM , Sia C , Sokoll KK , Chang TY , Windisch M ((2007) ) Site-specific UBITh® amyloid-β vaccine for immunotherapy of Alzheimer’s disease. Vaccine 25: , 3041–3052. |
[127] | van Dyck CH ((2018) ) Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise. Biol Psychiatry 83: , 311–319. |
[128] | Cummings JL , Cohen S , van Dyck CH , Brody M , Curtis C , Cho W , Quartino A ((2018) ) ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90: , 1889–1897. |
[129] | Landen JW , Zhao Q , Cohen S , Borrie M , Woodward M , Billing CB Jr , Kupiec JW ((2013) ) Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: A phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol 36: , 14–23. |
[130] | Landen JW , Andreasen N , Cronenberger CL , Schwartz PF , Börjesson-Hanson A , Östlund H , Bednar MM ((2017) ) Ponezumab in mild-to-moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimers Dement (N Y) 3: , 393–401. |
[131] | Dodel R , Rominger A , Blennow K , Barkhof F , Wietek S , Haag S , Jessen F ((2011) ) A randomized, double-blind, placebo-controlled dose-finding trial of intravenous immunoglobulin (IVIG; Octagam® 10%, Octapharma AG) in patients with mild to moderate Alzheimer’s disease (GAM10-04). Alzheimers Dement 7: , 55–56. |
[132] | Dodel R , Rominger A , Bartenstein P , Barkhof F , Blennow K , Förster S , Wiltfang J ((2013) ) Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: A phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol 12: , 233–243. |
[133] | Folch López J , Petrov D , Ettcheto M , Abad S , Sánchez-López E , García López ML , Camins Espuny A ((2016) ) Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plasticity 2016: , 1–15. |
[134] | Pradier L , Cohen C , Blanchard V , Debeir T , Barneoud P , Canton T , Cameron B ((2013) ) SAR228810: An antiprotofibrillar beta-amyloid antibody designed to reduce risk of amyloid-related imaging abnormalities (ARIA). Alzheimers Dement 9: , 808–809. |
[135] | Billinton A , Newton P , Lloyd C , Groves M , Welsh F , Bogstedt A , Tan K ((2017) ) Preclinical discovery and development of MEDI1814, a monoclonal antibody selectively targeting beta-amyloid 42 (Aβ42). Alzheimers Dement 13: , P266. |
[136] | Cummings J , Lee G , Ritter A , Zhong K ((2018) ) Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 4: , 195–214. |
[137] | US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02127476. ClinicalTrials.gov Identifier: NCT02127476, Posted on April 30, 2014, Last updated June 1, 2017. |
[138] | Wisniewski T , Goñi F ((2015) ) Immunotherapeutic approaches for Alzheimer’s disease. Neuron 85: , 1162–1176. |
[139] | Lobello K , Ryan JM , Liu E , Rippon G , Black R ((2012) ) Targeting beta amyloid: A clinical review of immunotherapeutic approaches in Alzheimer’s disease. Int J Alzheimers Dis 2012: , 628070. |
[140] | Davtyan H , Ghochikyan A , Petrushina I , Hovakimyan A , Davtyan A , Poghosyan A , Larsen AK ((2013) ) Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: Prelude to a clinical trial. J Neurosci 33: , 4923–4934. |
[141] | Folch J , Ettcheto M , Petrov D , Abad S , Pedrós I , Marin M , Camins A ((2018) ) Review of the advances in treatment for Alzheimer disease: Strategies for combating β-amyloid protein. Neurologia 33: , 47–58. |
[142] | Yan R , Vassar R ((2014) ) Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 13: , 319–329. |
[143] | Kennedy ME , Stamford AW , Chen X , Cox K , Cumming JN , Dockendorf MF , Jhee S ((2016) ) The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 8: , 363ra150. |
[144] | Alexander R , Budd S , Russell M , Kugler A , Cebers G , Ye N , Elsby K ((2014) ) AZD3293 A novel BACE1 inhibitor: Safety, tolerability, and effects on plasma and CSF aβ peptides following single-and multiple-dose administration. Neurobiol Aging 35: , S2. |
[145] | Yan R ((2016) ) Stepping closer to treating Alzheimer’s disease patients with BACE1 inhibitor drugs. Transl Neurodegener 5: , 13. |
[146] | Timmers M , Streffer JR , Russu A , Tominaga Y , Shimizu H , Shiraishi A , Matias-Guiu J ((2018) ) Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimers Res Ther 10: , 85. |
[147] | Streffer J , Börjesson-Hanson A , Van Broeck B , Smekens P , Timmers M , Tesseur I , Aguilar M ((2016) ) Pharmacodynamics of the oral BACE inhibitor JNJ-54861911 in early Alzheimer’s disease. Alzheimers Dement 12: , 199–200. |
[148] | Matijevic M , Watanabe H , Sato Y , Bernier F , McGrath S , Burns L , Oda Y ((2015) ) A single dose of the beta-secretase inhibitor, e2609, decreases CSF bace1 enzymatic activity in cynomolgus monkeys. Alzheimers Dement 11: , 841. |
[149] | Nicolas L , Kammerer KP , Schaible J , Link J , Kleiner O , Borta A , Scholpp J ((2015) ) Pharmacokinetics, pharmacodynamics, and safety of the novel bace inhibitor bi1181181 after oral administration of single ascending doses in healthy subjects. Alzheimers Dement 11: , 740–741. |
[150] | Bachurin SO , Bovina EV , Ustyugov AA ((2017) ) Drugs in clinical trials for Alzheimer’s disease: The major trends. Med Res Rev 37: , 1186–1225. |
[151] | US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01661673. ClinicalTrials.gov Identifier: NCT01661673, Posted on August 9, 2012, Last updated January 13, 2014. |
[152] | Soares HD , Gasior M , Toyn JH , Wang JS , Hong Q , Berisha F , Zheng N ((2016) ) The γ-secretase modulator, BMS-932481, modulates Aβ peptides in the plasma and cerebrospinal fluid of healthy volunteers. J Pharmacol Exp Ther 358: , 138–150. |
[153] | US National Library of Medicine ((2018) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03715114. ClinicalTrials.gov Identifier: NCT03715114, Posted on October 22, 2018, Last updated October 22, 2018. |
[154] | US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02547818. ClinicalTrials.gov Identifier: NCT02547818, Posted on September 11, 2015, Last updated December 5, 2018. |
[155] | Lilja AM , Luo Y , Yu QS , Röjdner J , Li Y , Marini AM , Greig NH ((2013) ) Neurotrophic and neuroprotective actions of (–)-and (+)-phenserine, candidate drugs for Alzheimer’s disease. PLoS One 8: , 54887. |
[156] | Teich AF , Sharma E , Barnwell E , Zhang H , Staniszewski A , Utsuki T , Arancio O ((2018) ) Translational inhibition of APP by Posiphen: Efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement (N Y) 4: , 37–45. |
[157] | US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02925650. ClinicalTrials.gov Identifier: NCT02925650, Posted on October 6, 2016, Last updated May 2, 2019. |
[158] | Lee D , Lee WS , Lim S , Kim YK , Jung HY , Das S , Chung SK ((2017) ) A guanidine-appended scyllo-inositol derivative AAD-66 enhances brain delivery and ameliorates Alzheimer’s phenotypes. Sci Rep 7: , 14125. |
[159] | Ma K , Thomason LA , McLaurin J ((2012) ) Scyllo-inositol, preclinical, and clinical data for Alzheimer’s disease. Adv Pharmacol 64: , 177–212. |
[160] | Hey JA , Jeremy YY , Versavel M , Abushakra S , Kocis P , Power A , Tolar M ((2018) ) Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer’s disease. Clin Pharmacokinet 57: , 315–333. |
[161] | Bachurin SO , Bovina EV , Ustyugov AA ((2018) ) Current trends in the development of drugs for the treatment of Alzheimer’s disease and their clinical trials. Biomed Chem Res Methods 1: , 00015. |
[162] | Snow AD , Cummings J , Lake T , Hu Q , Esposito L , Cam J , Runnels S ((2009) ) Exebryl-1: A novel small molecule currently in human clinical trials as a disease-modifying drug for the treatment of Alzheimer’s disease. Alzheimers Dement 5: , 418. |
[163] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Herrup K ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[164] | Baek IS , Kim TK , Seo JS , Lee KW , Lee Y , Cho J , Han PL ((2013) ) AAD-2004 attenuates progressive neuronal loss in the brain of Tg-betaCTF99/B6 mouse model of Alzheimer disease. Exp Neurobiol 22: , 31–37. |
[165] | Fu WY , Wang X , Ip NY ((2019) ) Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: Mechanisms, drug candidates, and new opportunities. ACS Chem Neurosci 10: , 872–879. |
[166] | Jackson L , Eldahshan W , Fagan S , Ergul A ((2018) ) Within the brain: The renin angiotensin system. Int J Mol Sci 19: , E876. |
[167] | Torika N , Asraf K , Apte RN , Fleisher-Berkovich S ((2018) ) Candesartan ameliorates brain inflammation associated with Alzheimer’s disease. CNS Neurosci Ther 24: , 231–242. |
[168] | Inoue K , Koizumi S , Kataoka A , Tozaki-Saitoh H , Tsuda M ((2009) ) P2Y6-evoked microglial phagocytosis. Int Rev Neurobiol 85: , 159–163. |
[169] | Liu W , Li G , Hölscher C , Li L ((2015) ) Neuroprotective effects of geniposide on Alzheimer’s disease pathology. Rev Neurosci 26: , 371–383. |
[170] | Yan SS , Chen D , Yan S , Guo L , Chen JX ((2012) ) RAGE is a key cellular target for Aβ-induced perturbation in Alzheimer’s disease. Front Biosci (Schol Ed) 4: , 240–250. |
[171] | Lue LF , Yan SD , Stern DM , Walker DG ((2005) ) Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr Drug Targets CNS Neurol Disord 4: , 249–266. |
[172] | Burstein AH , Sabbagh M , Andrews R , Valcarce C , Dunn I , Altstiel L ((2018) ) Development of Azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer’s disease. J Prev Alzheimers Dis 5: , 149–154. |
[173] | Sanchez-Mejia RO , Newman JW , Toh S , Yu GQ , Zhou Y , Halabisky B , Palop JJ ((2008) ) Phospholipase A 2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat Neurosci 11: , 1311–1318. |
[174] | Griffin ÉW , Skelly DT , Murray CL , Cunningham C ((2013) ) Cyclooxygenase-1-dependent prostaglandins mediate susceptibility to systemic inflammation-induced acute cognitive dysfunction. J Neurosci 33: , 15248–15258. |
[175] | Hwang DY , Chae KR , Kang TS , Hwang JH , Lim CH , Kang HK , Goo JS , Lee MR , Lim HJ , Min SH , Cho JY , Hong JT , Song CW , Paik SG , Cho JS , Kim YK ((2002) ) Alterations in behavior, amyloid β-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer’s disease. FASEB J 16: , 805–813. |
[176] | Puhl AC , Milton FA , Cvoro A , Sieglaff DH , Campos JC , Bernardes A , Polikarpov I ((2015) ) Mechanisms of peroxisome proliferator activated receptor γ regulation by non-steroidal anti-inflammatory drugs. Nucl Recept Signal 13: , e004. |
[177] | Wischik CM , Harrington CR , Storey JM ((2014) ) Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88: , 529–539. |
[178] | Porrini V , Lanzillotta A , Branca C , Benarese M , Parrella E , Lorenzini L , Pizzi M ((2015) ) CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience 302: , 112–120. |
[179] | Xu Q , Choksi S , Liu Z ((2018) ) Switching from TNF-induced inflammation to death signaling. Mol Cell Oncol 5: , 1392402. |
[180] | Butchart J , Brook L , Hopkins V , Teeling J , Püntener U , Culliford D , Thomas R ((2015) ) Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 84: , 2161–2168. |
[181] | Shamim D , Laskowski M ((2017) ) Inhibition of inflammation mediated through the tumor necrosis factor α biochemical pathway can lead to favorable outcomes in Alzheimer disease. J Cent Nerv Syst Dis 9: , 1179573517722512. |
[182] | Banno A , Reddy AT , Lakshmi SP , Reddy RC ((2018) ) PPARs: Key regulators of airway inflammation and potential therapeutic targets in asthma. Nucl Receptor Res 5: , 101306. |
[183] | Cheng H , Shang Y , Jiang L , Shi TL , Wang L ((2016) ) The peroxisome proliferators activated receptor-gamma agonists as therapeutics for the treatment of Alzheimer’s disease and mild-to-moderate Alzheimer’s disease: A meta-analysis. Int J Neurosci 26: , 299–307. |
[184] | Budni J , L Garcez M , Medeiros JD , Cassaro E , Bellettini-Santos T , Mina F , Quevedo J ((2016) ) The anti-inflammatory role of minocycline in Alzheimer’s disease. Curr Alzheimer Res 13: , 1319–1329. |
[185] | Maes T , Mascaró C , Ortega A , Lunardi S , Ciceri F , Somervaille TC , Buesa C ((2015) ) KDM1 histone lysine demethylases as targets for treatments of oncological and neurodegenerative disease. Epigenomics 7: , 609–626. |
[186] | Maes T , Mascaró C , Rotllant D , Cavalcanti F , Carceller E , Ortega A , Buesa C ((2016) ) ORY-2001: An epigenetic drug for the treatment of cognition defects in alzheimer’s disease and other neurodegenerative disorders. Alzheimers Dement 12: , P1192. |
[187] | Cacabelos R , Torrellas C ((2014) ) Epigenetic drug discovery for Alzheimer’s disease. Expert Opin Drug Discov 9: , 1059–1086. |
[188] | Eimer WA , Kumar DKV , Shanmugam NKN , Rodriguez AS , Mitchell T , Washicosky KJ , Moir RD ((2018) ) Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99: , 56–63. |
[189] | Readhead B , Haure-Mirande JV , Funk CC , Richards MA , Shannon P , Haroutunian V , Reiman EM ((2018) ) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99: , 64–82. |
[190] | Devanand DP ((2018) ) Viral hypothesis and antiviral treatment in Alzheimer’s disease. Curr Neurol Neurosci Rep 8: , 55. |
[191] | Famitafreshi H , Karimian M ((2018) ) Assessment of improvement in oxidative stress indices with resocialization in memory retrieval in Y-maze in male rats. J Exp Neurosci 12: , 1179069518820323. |
[192] | Wojsiat J , Zoltowska KM , Laskowska-Kaszub K , Wojda U ((2018) ) Oxidant/antioxidant imbalance in Alzheimer’s disease: Therapeutic and diagnostic prospects. Oxid Med Cell Longev 2018: , 6435861. |
[193] | Ansari MA , Scheff SW ((2010) ) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69: , 155–167. |
[194] | Di Domenico F , Barone E , Perluigi M , Butterfield DA ((2015) ) Strategy to reduce free radical species in Alzheimer’s disease: An update of selected antioxidants. Expert Rev Neurother 15: , 19–40. |
[195] | Jiang T , Sun Q , Chen S ((2016) ) Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog Neurobiol 147: , 1–19. |
[196] | Adair JC , Knoefel JE , Morgan N ((2001) ) Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 57: , 1515–1517. |
[197] | Choi H , Park HH , Koh SH , Choi NY , Yu HJ , Park J , Lee KY ((2012) ) Coenzyme Q10 protects against amyloid beta-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 33: , 85–90. |
[198] | Weyer G , Babej-Dölle RM , Hadler D , Hofmann S , Herrmann WM ((1997) ) A controlled study of 2 doses of idebenone in the treatment of Alzheimer’s disease. Neuropsychobiology 36: , 73–82. |
[199] | Dolder M , Walzel B , Speer O , Schlattner U , Wallimann T ((2003) ) Inhibition of the mitochondrial permeability transition by creatine kinase substrates Requirement for microcompartmentation. J Biol Chem 278: , 17760–17766. |
[200] | Chaturvedi RK , Beal MF ((2013) ) Mitochondrial diseases of the brain. Free Radic Biol Med 63: , 1–29. |
[201] | Manczak M , Mao P , Calkins MJ , Cornea A , Reddy AP , Murphy MP , Reddy PH ((2010) ) Mitochondria-targeted antioxidants protect against amyloid-β toxicity in Alzheimer’s disease neurons. J Alzheimers Dis 20: , 609–631. |
[202] | Smith RA , Porteous CM , Coulter CV , Murphy MP ((1999) ) Selective targeting of an antioxidant to mitochondria. Eur J Biochem 263: , 709–716. |
[203] | Kumar A , Singh A ((2015) ) A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front Pharmacol 6: , 206. |
[204] | Zhang J , Wang Q , Xu C , Lu Y , Hu H , Qin B , Wang S ((2017) ) MitoTEMPO prevents oxalate induced injury in NRK-52E cells via inhibiting mitochondrial dysfunction and modulating oxidative stress. Oxid Med Cell Longev 2017: , 7528090. |
[205] | Cacabelos R ((2007) ) Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr Dis Treat 3: , 303–333. |
[206] | Bryson HM , Benfield P ((1997) ) Donepezil. Drugs Aging 10: , 234–239. |
[207] | Knowles J ((2006) ) Donepezil in Alzheimer’s disease: An evidence-based review of its impact on clinical and economic outcomes. Core Evid 1: , 195–219. |
[208] | van Marum RJ ((2009) ) Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat 5: , 237–247. |
[209] | Herrmann N , Li A , Lanctôt K ((2011) ) Memantine in dementia: A review of the current evidence. Expert Opin Pharmacother 12: , 787–800. |
[210] | Zhang B , Carroll J , Trojanowski JQ , Yao Y , Iba M , Potuzak JS , Lee VMY ((2012) ) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32: , 3601–3611. |
[211] | Ismail O , Harrison IF , Holmes H , Colgan N , Wells JA , O’Callaghan JM , Powell NM , Ma D , Ourselin S , Walker-Samuel S , Murray TK ((2015) ) Imaging the efficacy of microtubule stabilizing agent epothilone d in the rtg4510 mouse model of tauopathy. Alzheimers Dement 11: , 348. |
[212] | Anand A , Patience AA , Sharma N , Khurana N ((2017) ) The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur J Pharmacol 815: , 364–375. |
[213] | Amirrad F , Bousoik E , Shamloo K , Al-Shiyab H , Nguyen VH , Montazeri Aliabadi H ((2017) ) Alzheimer’s disease: Dawn of a new era? J Pharm Pharm Sci 20: , 184–225. |
[214] | Bakota L , Brandt R ((2016) ) Tau biology and tau-directed therapies for Alzheimer’s disease. Drugs 76: , 301–313. |
[215] | Panza F , Solfrizzi V , Seripa D , Imbimbo BP , Lozupone M , Santamato A , Daniele A ((2016) ) Tau-centric targets and drugs in clinical development for the treatment of Alzheimer’s disease. Biomed Res Int 2016: , 3245935. |
[216] | Cai Z , Yan Y , Wang Y ((2013) ) Minocycline alleviates beta-amyloid protein and tau pathology via restraining neuroinflammation induced by diabetic metabolic disorder. Clin Interv Aging 8: , 1089–1095. |
[217] | Pi R , Li W , Lee NT , Chan HH , Pu Y , Chan LN , Han Y ((2004) ) Minocycline prevents glutamate-induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J Neurochem 91: , 1219–1230. |
[218] | Tikka T , Fiebich BL , Goldsteins G , Keinänen R , Koistinaho J ((2001) ) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21: , 2580–2588. |
[219] | Liu CC , Huang CC , Lin WT , Hsieh CC , Huang SY , Lin SJ , Yang SC ((2005) ) Lycopene supplementation attenuated xanthine oxidase and myeloperoxidase activities in skeletal muscle tissues of rats after exhaustive exercise. Br J Nutr 94: , 595–601. |
[220] | Yu L , Wang W , Pang W , Xiao Z , Jiang Y , Hong Y ((2017) ) Dietary lycopene supplementation improves cognitive performances in tau transgenic mice expressing P301L mutation via inhibiting oxidative stress and tau hyperphosphorylation. J Alzheimers Dis 57: , 475–482. |
[221] | Varidaki A , Hong Y , Coffey ET ((2018) ) Repositioning microtubule stabilizing drugs for brain disorders. Front Cell Neurosci 12: , 226. |
[222] | Jordan MA , Wilson L ((2004) ) Microtubules as a target for anticancer drugs. Nat Rev Cancer 4: , 253–265. |
[223] | Dumontet C , Jordan MA ((2010) ) Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat Rev Drug Discov 9: , 790–803. |
[224] | Field JJ , Díaz JF , Miller JH ((2013) ) The binding sites of microtubule-stabilizing agents. Chem Biol 20: , 301–315. |
[225] | Brunden KR , Yao Y , Potuzak JS , Ferrer NI , Ballatore C , James MJ , Lee VMY ((2011) ) The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol Res 3: , 341–351. |
[226] | Madiraju C , Edler MC , Hamel E , Raccor BS , Balachandran R , Zhu G , Fukui Y ((2005) ) Tubulin assembly, taxoid site binding, and cellular effects of the microtubule-stabilizing agent dictyostatin. Biochemistry 44: , 15053–15063. |
[227] | Lee HE , Lim D , Lee JY , Lim SM , Pae AN ((2019) ) Development of tau-directed small molecule modulators for Alzheimer’s disease: A recent patent review (2014–2018). Pharm Pat Anal 8: , 15–39. |
[228] | Duran GE , Derdau V , Weitz D , Philippe N , Blankenstein J , Atzrodt J , Sikic BI ((2018) ) Cabazitaxel is more active than first-generation taxanes in ABCB1 (+) cell lines due to its reduced affinity for P-glycoprotein. Cancer Chemother Pharmacol 81: , 1095–1103. |
[229] | Vrignaud P , Sémiond D , Lejeune P , Bouchard H , Calvet L , Combeau C , Bissery MC ((2013) ) Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res 19: , 2973–2983. |
[230] | Ghoochani A , Majernik GH , Sehm T , Wach S , Buchfelder M , Taubert H , Savaskan N ((2016) ) Cabazitaxel operates anti-metastatic and cytotoxic via apoptosis induction and stalls brain tumor angiogenesis. Oncotarget 7: , 38306. |
[231] | Girard E , Ditzler S , Lee D , Richards A , Yagle K , Park J , Olson J ((2014) ) Efficacy of cabazitaxel in mouse models of pediatric brain tumors. Neuro Oncol 17: , 107–115. |
[232] | Panza F , Imbimbo BP , Lozupone M , Greco A , Seripa D , Logroscino G , Daniele A , Colosimo C ((2019) ) Disease-modifying therapies for tauopathies: Agents in the pipeline. Expert Rev Neurother 19: , 397–408. |
[233] | Gozes I , Morimoto BH , Tiong J , Fox A , Sutherland K , Dangoor D , Matsuoka Y ((2005) ) NAP: Research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP). CNS Drugs 11: , 353–368. |
[234] | Jouroukhin Y , Ostritsky R , Assaf Y , Pelled G , Giladi E , Gozes I ((2013) ) NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: Protection against impairments in axonal transport. Neurobiol Dis 6: , 79–94. |
[235] | Hong SW , Singh AJ , Patel V , Russell ER , Field JJ , Miller JH , Northcote PT ((2018) ) Peloruside E (22-Norpeloruside A), a Pelorusane macrolide from the New Zealand marine sponge Mycale hentscheli, retains microtubule-stabilizing properties. J Nat Prod 81: , 2125–2128. |
[236] | Haefner B ((2003) ) Drugs from the deep: Marine natural products as drug candidates. Drug Discov Today 8: , 536–544. |
[237] | Řehulka J , Annadurai N , Frydrych I , Džubák P , Miller JH , Hajdúch M , Das V ((2018) ) Peloruside A-induced cell death in hypoxia is p53 dependent in HCT116 colorectal cancer cells. J Nat Prod 81: , 634–640. |
[238] | Du LL , Xie JZ , Cheng XS , Li XH , Kong FL , Jiang X , Zhou XW ((2014) ) Activation of sirtuin 1 attenuates cerebral ventricular streptozotocin-induced tau hyperphosphorylation and cognitive injuries in rat hippocampi. Age 36: , 613–623. |
[239] | Liu P , Zou LB , Wang LH , Jiao Q , Chi TY , Ji XF , Jin G ((2014) ) Xanthoceraside attenuates tau hyperphosphorylation and cognitive deficits in intracerebroventricular-streptozotocin injected rats. Psychopharmacology 231: , 345–356. |
[240] | Ravelli KG , dos Anjos Rosário B , Vasconcelos AR , Scavone C , Camarini R , Hernandes MS , Britto LR ((2017) ) NADPH oxidase contributes to streptozotocin-induced neurodegeneration. Neuroscience 358: , 227–237. |
[241] | Xu ZP , Gan GS , Liu YM , Xiao JS , Liu HX , Mei B , Zhang JJ ((2018) ) Adiponectin attenuates streptozotocin-induced tau hyperphosphorylation and cognitive deficits by rescuing PI3K/Akt/GSK-3β pathway. Neurochem Res 43: , 316–323. |
[242] | Huang XB , Chen YJ , Chen WQ , Wang NQ , Wu XL , Liu Y ((2018) ) Neuroprotective effects of tenuigenin on neurobehavior, oxidative stress, and tau hyperphosphorylation induced by intracerebroventricular streptozotocin in rats. Brain Circ 4: , 24–32. |
[243] | Zhou S , Yu G , Chi L , Zhu J , Zhang W , Zhang Y , Zhang L ((2013) ) Neuroprotective effects of edaravone on cognitive deficit, oxidative stress and tau hyperphosphorylation induced by intracerebroventricular streptozotocin in rats. Neurotoxicology 38: , 136–145. |
[244] | Xu ZP , Li L , Bao J , Wang ZH , Zeng J , Liu EJ , Zhang Y ((2014) ) Magnesium protects cognitive functions and synaptic plasticity in streptozotocin-induced sporadic Alzheimer’s model. PloS One 9: , 108645. |
[245] | Du LL , Chai DM , Zhao LN , Li XH , Zhang FC , Zhang HB , Zhou XW ((2015) ) AMPK activation ameliorates Alzheimer’s disease-like pathology and spatial memory impairment in a streptozotocin-induced Alzheimer’s disease model in rats. J Alzheimers Dis 43: , 775–784. |
[246] | Ravelli KG , dos Anjos Rosário B , Camarini R , Hernandes MS , Britto LR ((2017) ) Intracerebroventricular Streptozotocin as a model of Alzheimer’s disease: Neurochemical and behavioral characterization in mice. Neurotox Res 31: , 327–333. |
[247] | Yang W , Ma J , Liu Z , Lu Y , Hu B , Yu H ((2014) ) Effect of naringenin on brain insulin signaling and cognitive functions in ICV-STZ induced dementia model of rats. Neurol Sci 35: , 741–751. |
[248] | Gao C , Liu Y , Jiang Y , Ding J , Li L ((2014) ) Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK 3β pathway in streptozotocin-induced Alzheimer rat model. Brain Pathol 24: , 261–269. |
[249] | Chen Y , Liang Z , Tian Z , Blanchard J , Dai CL , Chalbot S , Gong CX ((2014) ) Intracerebroventricular streptozotocin exacerbates Alzheimer-like changes of 3×Tg-AD mice. Mol Neurobiol 49: , 547–562. |
[250] | Guo Z , Chen Y , Mao YF , Zheng T , Jiang Y , Yan Y , Zhang B ((2017) ) Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Sci Rep 7: , 45971. |
[251] | Guo XD , Sun GL , Zhou TT , Wang YY , Xu X , Shi XF , Shen X ((2017) ) LX2343 alleviates cognitive impairments in AD model rats by inhibiting oxidative stress-induced neuronal apoptosis and tauopathy. Acta Pharmacol Sin 38: , 1104–1119. |
[252] | Xiong H , Zheng C , Wang J , Song J , Zhao G , Shen H , Deng Y ((2013) ) The neuroprotection of liraglutide on Alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J Alzheimers Dis 37: , 623–635. |
[253] | Zhang H , Wang D , Gong P , Lin A , Zhang Y , Ye RD , Yu Y ((2019) ) Formyl peptide receptor 2 deficiency improves cognition and attenuates tau hyperphosphorylation and astrogliosis in a mouse model of Alzheimer’s disease. J Alzheimers Dis 67: , 169–179. |
[254] | Ponce-Lopez T , Hong E , Abascal-Díaz M , Meneses A ((2017) ) Role of GSK3β and PP2A on regulation of Tau phosphorylation in hippocampus and memory impairment in ICV-STZ animal model of Alzheimer’s disease. Adv Alzheimers Dis 2017: , 13–31. |
[255] | Vakilinezhad MA , Amini A , Javar HA , Zarandi BFBAB , Montaseri H , Dinarvand R ((2018) ) Nicotinamide loaded functionalized solid lipid nanoparticles improves cognition in Alzheimer’s disease animal model by reducing Tau hyperphosphorylation. Daru 26: , 165–177. |
[256] | Lu F , Li X , Li W , Wei K , Yao Y , Zhang Q , Zhang J ((2017) ) Tetramethylpyrazine reverses intracerebroventricular streptozotocin-induced memory deficits by inhibiting GSK-3β. Acta Biochim Biophys Sin (Shanghai) 49: , 722–728. |
[257] | Chohan MO , Khatoon S , Iqbal IG , Iqbal K ((2006) ) Involvement of in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett 580: , 3973–3979. |
[258] | Li L , Sengupta A , Haque N , Grundke-Iqbal I , Iqbal K ((2004) ) Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett 566: , 261–269. |
[259] | Usmari Moraes M , Gaudet TJ ((2018) ) Immunotherapeutic and pharmacological approaches for the treatment of Alzheimer’s disease. Bioscience Horizons Natl Undergrad Res J 11: , doi: 10.1093/biohorizons/hzy001 |
[260] | Wang X , Blanchard J , Grundke-Iqbal I , Iqbal K ((2015) ) Memantine attenuates Alzheimer’s disease-like pathology and cognitive impairment. PloS One 10: , 0145441. |
[261] | Corcoran NM , Martin D , Hutter-Paier B , Windisch M , Nguyen T , Nheu L , Hovens CM ((2010) ) Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci 17: , 1025–1033. |
[262] | Van Eersel J , Ke YD , Liu X , Delerue F , Kril JJ , Götz J , Ittner LM ((2010) ) Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A 107: , 13888–13893. |
[263] | Malpas CB , Vivash L , Genc S , Saling MM , Desmond P , Steward C , Macfarlane S ((2016) ) A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer’s disease. J Alzheimers Dis 54: , 223–232. |
[264] | Wozniak MA , Frost AL , Preston CM , Itzhaki RF ((2011) ) Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One 6: , 25152. |
[265] | Harris SA , Harris EA ((2018) ) Molecular mechanisms for herpes simplex virus type 1 pathogenesis in Alzheimer’s disease. Front Aging Neurosci 10: , 48. |
[266] | Durães F , Pinto M , Sousa E ((2018) ) Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals 11: , 44. |
[267] | Jorda EG , Verdaguer E , Canudas AM , Jiménez A , Bruna A , Caelles C , Pallàs M ((2003) ) Neuroprotective action of flavopiridol, a cyclin-dependent kinase inhibitor, in colchicine-induced apoptosis. Neuropharmacology 45: , 672–683. |
[268] | Carlson BA , Dubay MM , Sausville EA , Brizuela L , Worland PJ ((1996) ) Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res 56: , 2973–2978. |
[269] | Meijer L , Borgne A , Mulner O , Chong JP , Blow JJ , Inagaki N , Moulinoux JP ((1997) ) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243: , 527–536. |
[270] | Mapelli M , Massimiliano L , Crovace C , Seeliger MA , Tsai LH , Meijer L , Musacchio A ((2005) ) Mechanism of CDK5/p25 binding by CDK inhibitors. J Med Chem 48: , 671–679. |
[271] | Domínguez JM , Fuertes A , Orozco L , del Monte-Millán M , Delgado E , Medina M ((2012) ) Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J Biol Chem 287: , 893–904. |
[272] | Lovestone S , Boada M , Dubois B , Hüll M , Rinne JO , Huppertz HJ , del Ser T ((2015) ) A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis 45: , 75–88. |
[273] | del Ser T , Steinwachs KC , Gertz HJ , Andres MV , Gomez-Carrillo B , Medina M , Leon T ((2013) ) Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J Alzheimers Dis 33: , 205–215. |
[274] | Sereno L , Coma M , Rodriguez M , Sanchez-Ferrer P , Sanchez MB , Gich I , Clarimon J ((2009) ) A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis 35: , 359–367. |
[275] | Stambolic V , Ruel L , Woodgett JR ((1996) ) Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 6: , 1664–1669. |
[276] | Forlenza OV , De-Paula VDJR , Diniz BSO ((2014) ) Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem Neurosci 5: , 443–450. |
[277] | Matsunaga S , Kishi T , Annas P , Basun H , Hampel H , Iwata N ((2015) ) Lithium as a treatment for Alzheimer’s disease: A systematic review and meta-analysis. J Alzheimers Dis 48: , 403–410. |
[278] | Min SW , Chen X , Tracy TE , Li Y , Zhou Y , Wang C , Sohn PD ((2015) ) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 12: , 1154. |
[279] | Medina M ((2018) ) An overview on the clinical development of tau-based therapeutics. Int J Mol Sci 19: , E1160. |
[280] | Sandhu P , Lee J , Ballard J , Walker B , Ellis J , Marcus J , Michener M ((2016) ) Pharmacokinetics and pharmacodynamics to support clinical studies of MK-8719: An O-GlcNAcase inhibitor for progressive supranuclear palsy. Alzheimers Dement 12: , P1028. |
[281] | Wischik CM , Staff RT , Wischik DJ , Bentham P , Murray AD , Storey J , Harrington CR ((2015) ) Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis 44: , 705–720. |
[282] | Cummings J , Lee G , Mortsdorf T , Ritter A , Zhong K ((2017) ) Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 3: , 367–384. |
[283] | Gauthier S , Feldman HH , Schneider LS , Wilcock GK , Frisoni GB , Hardlund JH , Schelter BO ((2016) ) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388: , 2873–2884. |
[284] | Hastings NB , Wang X , Song L , Butts BD , Grotz D , Hargreaves R , Laverty M ((2017) ) Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol Neurodegener 12: , 39. |
[285] | Sun J , Zhang X , Wang C , Teng Z , Li Y ((2017) ) Curcumin decreases hyperphosphorylation of tau by down-regulating caveolin-1/GSK-3β in N2a/APP695swe cells and APP/PS1 double transgenic Alzheimer’s disease mice. Am J Chin Med 45: , 1667–1682. |
[286] | Myeku N , Clelland CL , Emrani S , Kukushkin NV , Yu WH , Goldberg AL , Duff KE ((2016) ) Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med 22: , 46–53. |
[287] | Schneider A , Biernat J , Von Bergen M , Mandelkow E , Mandelkow EM ((1999) ) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38: , 3549–3558. |
[288] | Prickaerts J , Heckman PR , Blokland A ((2017) ) Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs 26: , 1033–1048. |
[289] | NIH. New NIH-funded memory drug moves into Phase 1 clinical study. https://www.nia.nih.gov/alzheimers/clinical-trials/bpn14770-healthy-volunteers. Study start date June 2016, Study end date December 2016. |
[290] | US National Library of Medicine ((2019) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03817684. ClinicalTrials.gov Identifier: NCT03817684, Posted on January 25, 2019, Last updated May 6, 2019. |
[291] | Abushouk AI , Elmaraezy A , Aglan A , Salama R , Fouda S , Fouda R , AlSafadi AM ((2017) ) Bapineuzumab for mild to moderate Alzheimer’s disease: A meta-analysis of randomized controlled trials. BMC Neurol 17: , 66. |
[292] | Khorassani F , Hilas O ((2013) ) Bapineuzumab, an investigational agent for Alzheimer’s disease. P T 38: , 89–91. |
[293] | Kontsekova E , Zilka N , Kovacech B , Novak P , Novak M ((2014) ) First-in-man tau vaccine targeting structural determinants essential for pathological tau–tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res Ther 6: , 44. |
[294] | Novak P , Schmidt R , Kontsekova E , Kovacech B , Smolek T , Katina S , Brunner M ((2018) ) FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res Ther 10: , 108. |
[295] | Godyń J , Jończyk J , Panek D , Malawska B ((2016) ) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68: , 127–138. |
[296] | Theunis C , Crespo-Biel N , Gafner V , Pihlgren M , López-Deber MP , Reis P , Borghgraef P ((2013) ) Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau. P301L mice that model tauopathy. PLoS One 8: , 72301. |
[297] | International Clincal Trials Registry Platform ((2013) ) ISRCT.com http://www.isrctn.com/ISRCTN13033912. ISRCTN13033912, https://doi.org/10.1186/ISRCTN13033912, Posted on October 13, 2015, Last updated November 4, 2015. |
[298] | Sigurdsson EM ((2018) ) Tau immunotherapies for Alzheimer’s disease and related tauopathies: Progress and potential pitfalls. J Alzheimers Dis 66: , 855–856. |
[299] | Alzheimer Research Forum, Drugs in Clinical Trials: RG7345, ALZFORUM https://www.alzforum.org/therapeutics/rg7345. Last updated November 20, 2015. |
[300] | Czerkowicz J , Chen W , Cameron A , Sopko R , Weinreb P , Hering H , Graham D ((2018) ) Anti-tau antibody BIIB092 binds secreted tau in preclinical models and Alzheimer’s disease cerebrospinal fluid. Alzheimers Dement 14: , P1441. |
[301] | Qureshi IA , Tirucherai G , Ahlijanian MK , Kolaitis G , Bechtold C , Grundman M ((2018) ) A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement (N Y) 4: , 746–55. |
[302] | West T , Hu Y , Verghese PB , Bateman RJ , Braunstein JB , Fogelman I , Holtzman DM ((2017) ) Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 4: , 236–241. |
[303] | Budur K , West T , Braunstein JB , Fogelman I , Bordelon YM , Litvan I , Florian H ((2017) ) Results of a phase 1, single ascending dose, placebo-controlled study of ABBV-8E12 in patients with progressive supranuclear palsy and phase 2 study design in early Alzheimer’s disease. Alzheimers Dement 13: , 599–600. |
[304] | Alzheimer Research Forum, Drugs in Clinical Trials: RO 7105705, ALZFORUM https://www.alzforum.org/therapeutics/ro7105705. Last Updated February 20, 2019. |
[305] | US National Library of Medicine ((2017) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02820896. ClinicalTrials.gov Identifier: NCT02820896. Posted on July 1, 2016, Last updated July 21, 2017. |
[306] | US National Library of Medicine ((2018) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03518073. ClinicalTrials.gov Identifier: NCT03518073, Posted on May 8, 2018, Last updated May 22, 2019. |
[307] | US National Library of Medicine ((2018) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02754830. ClinicalTrials.gov Identifier: NCT02754830, Posted on April 28, 2016, Last updated July 23, 2018. |
[308] | US National Library of Medicine ((2018) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03375697. ClinicalTrials.gov Identifier: NCT03375697, Posted on December 18, 2017, Last updated May 20, 2019. |
[309] | Alzheimer Research Forum, Drugs in Clinical Trials: JNJ-63733657, ALZFORUM https://www.alzforum.org/therapeutics/jnj-63733657. Last ipdated April 27, 2018. |
[310] | US National Library of Medicine ((2018) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03464227. ClinicalTrials.gov Identifier: NCT03464227, Posted on March 13, 2018, Last updated December 12, 2018. |
[311] | Fullerton T , Binneman B , David W , Delnomdedieu M , Kupiec J , Lockwood P , Mancuso J , Miceli J , Bell J ((2018) ) A Phase 2 clinical trial of Pf-05212377 (Sam-760) in subjects with mild to moderate Alzheimer’s disease with existing neuropsychiatric symptoms on a stable daily dose of donepezil. Alzheimers Dement 10: , 38. |
[312] | Sabbagh M , Han S , Kim S , Na HR , Lee JH , Kandiah N , Narilastri D ((2016) ) Clinical recommendations for the use of donepezil 23mg in moderate-to-severe Alzheimer’s disease in the Asia-Pacific region. Dement Geriatr Cogn Dis Extra 6: , 382–395. |
[313] | Valis M , Masopust J , Vysata O , Hort J , Dolezal R , Tomek J , Karasova JZ ((2017) ) Concentration of donepezil in the cerebrospinal fluid of AD patients: Evaluation of dosage sufficiency in standard treatment strategy. Neurotox Res 31: , 162–168. |
[314] | Chase TN , Farlow MR , Clarence-Smith K ((2017) ) Donepezil plus Solifenacin (CPC-201) treatment for Alzheimer’s disease. Neurotherapeutics 14: , 405–416. |
[315] | Wirsich J , Rey M , Guye M , Bénar C , Lanteaume L , Ridley B , Rouby F ((2018) ) Brain networks are independently modulated by donepezil, sleep, and sleep deprivation. Brain Topogr 31: , 380–391. |
[316] | Yabuki Y , Matsuo K , Hirano K , Shinoda Y , Moriguchi S , Fukunaga K ((2017) ) Combined memantine and donepezil treatment improves behavioral and psychological symptoms of dementia-like behaviors in olfactory bulbectomized mice. Pharmacology 99: , 160–171. |
[317] | Zheng H , Niu S , Zhao H , Li S , Jiao J ((2018) ) Donepezil improves the cognitive impairment in a tree shrew model of Alzheimer’s disease induced by amyloid-β 1–40 via activating the BDNF/TrkB signal pathway. Metab Brain Dis 33: , 1961–1974. |
[318] | Nyitrai G , Spisák T , Spisák Z , Gajári D , Diószegi P , Kincses TZ , Czurkó A ((2018) ) Stepwise occlusion of the carotid arteries of the rat: MRI assessment of the effect of donepezil and hypoperfusion-induced brain atrophy and white matter microstructural changes. PloS One 13: , e0198265. |
[319] | Bretin S , Krazem A , Henkous N , Froger-Colleaux C , Mocaer E , Louis C , Beracochea D ((2018) ) Synergistic enhancing-memory effect of donepezil and S 47445, an AMPA positive allosteric modulator, in middle-aged and aged mice. Psychopharmacology 235: , 771–787. |
[320] | Amidfar M , Khiabany M , Kohi A , Salardini E , Arbabi M , Roohi Azizi M , Akhondzadeh S ((2017) ) Effect of memantine combination therapy on symptoms in patients with moderate-to-severe depressive disorder: Randomized, double-blind, placebo-controlled study. J Clin Pharm Ther 42: , 44–50. |
[321] | Nakamura Y , Kitamura S , Homma A , Shiosakai K , Matsui D ((2014) ) Efficacy and safety of memantine in patients with moderate-to-severe Alzheimer’s disease: Results of a pooled analysis of two randomized, double-blind, placebo-controlled trials in Japan. Expert Opin Pharmacother 15: , 913–925. |
[322] | Kishi T , Matsunaga S , Oya K , Nomura I , Ikuta T , Iwata N ((2017) ) Memantine for Alzheimer’s disease: An updated systematic review and meta-analysis. J Alzheimers Dis 60: , 401–425. |
[323] | Thomas SJ , Grossberg GT ((2009) ) Memantine: A review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clin Interv Aging 4: , 367–377. |
[324] | Kishi T , Matsunaga S , Iwata N ((2017) ) The effects of memantine on behavioral disturbances in patients with Alzheimer’s disease: A meta-analysis. Neuropsychiatr Dis Treat 13: , 1909–1928. |
[325] | Blanco-Silvente L , Capellà D , Garre-Olmo J , Vilalta-Franch J , Castells X ((2018) ) Predictors of discontinuation, efficacy, and safety of memantine treatment for Alzheimer’s disease: Meta-analysis and meta-regression of 18 randomized clinical trials involving 5004 patients. BMC Geriatr 18: , 168. |
[326] | Amidfar M , Kim YK , Wiborg O ((2018) ) Effectiveness of memantine on depression-like behavior, memory deficits and brain mRNA levels of BDNF and TrkB in rats subjected to repeated unpredictable stress. Pharmacol Rep 70: , 600–606. |
[327] | Bahramian A , Rastegar K , Namavar MR , Moosavi M ((2016) ) Insulin potentiates the therapeutic effect of memantine against central STZ-induced spatial learning and memory deficit. Behav Brain Res 311: , 247–254. |
[328] | Ito K , Tatebe T , Suzuki K , Hirayama T , Hayakawa M , Kubo H , Makino M ((2017) ) Memantine reduces the production of amyloid-β peptides through modulation of amyloid precursor protein trafficking. Eur J Pharmacol 798: , 16–25. |
[329] | Rahman A , Lamberty Y , Schenker E , Cella M , Languille S , Bordet R , Aujard F ((2017) ) Effects of acute administration of donepezil or memantine on sleep-deprivation-induced spatial memory deficit in young and aged non-human primate grey mouse lemurs (Microcebus murinus). PloS One 12: , e0184822. |
[330] | Ettcheto M , Sánchez-López E , Gómez-Mínguez Y , Cabrera H , Busquets O , Beas-Zarate C , Carrera MV ((2018) ) Peripheral and central effects of memantine in a mixed preclinical mice model of obesity and familial Alzheimer’s disease. Mol Neurobiol 55: , 7327–7339. |
[331] | US National Library of Medicine ((2014) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01492374. ClinicalTrials.gov Identifier: NCT01492374, Posted on December 15, 2011, Last updated July 24, 2014. |
[332] | Monk JP , Villalona-Calero M , Larkin J , Otterson G , Spriggs DS , Hannah AL , Hensley ML ((2012) ) A phase 1 study of KOS-862 (Epothilone D) co-administered with carboplatin (Paraplatin®) in patients with advanced solid tumors. Invest New Drugs 30: , 1676–1683. |
[333] | Brunden KR , Ballatore C , Lee VMY , Smith AB , Trojanowski JQ ((2012) ) Brain-penetrant microtubule-stabilizing compounds as potential therapeutic agents for tauopathies. Biochem Soc Trans 40: , 661–666. |
[334] | Barten DM , Fanara P , Andorfer C , Hoque N , Wong PA , Husted KH , Fessler C ((2012) ) Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J Neurosci 32: , 7137–7145. |
[335] | Cartelli D , Casagrande F , Busceti CL , Bucci D , Molinaro G , Traficante A , Cappelletti G ((2013) ) Microtubule alterations occur early in experimental parkinsonism and the microtubule stabilizer epothilone D is neuroprotective. Sci Rep 3: , 1837. |
[336] | Ballatore C , R Brunden K , Q Trojanowski J , M-Y Lee V , B Smith A , M Huryn D ((2011) ) Modulation of protein-protein interactions as a therapeutic strategy for the treatment of neurodegenerative tauopathies. Curr Top Med Chem 11: , 317–330. |
[337] | Brunden KR , Zhang B , Carroll J , Yao Y , Potuzak JS , Hogan AML , Smith AB ((2010) ) Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci 30: , 13861–13866. |
[338] | Plane JM , Shen Y , Pleasure DE , Deng W ((2010) ) Prospects for minocycline neuroprotection. Arch Neurol 67: , 1442–1448. |
[339] | Garrido-Mesa N , Zarzuelo A , Gálvez J ((2013) ) Minocycline: Far beyond an antibiotic. Br J Pharmacol 169: , 337–352. |
[340] | Elewa HF , Hilali H , Hess DC , Machado LS , Fagan SC ((2006) ) Minocycline for short-term neuroprotection. Pharmacotherapy 26: , 515–521. |
[341] | Familian A , Boshuizen RS , Eikelenboom P , Veerhuis R ((2006) ) Inhibitory effect of minocycline on amyloid β fibril formation and human microglial activation. Glia 53: , 233–240. |
[342] | Dodel R , Spottke A , Gerhard A , Reuss A , Reinecker S , Schimke N , Gasser T ((2010) ) Minocycline 1-year therapy in multiple-system-atrophy: Effect on clinical symptoms and [11C](R)-PK11195 PET (MEMSA-trial). Mov Disord 25: , 97–107. |
[343] | Noble W , Garwood C , Stephenson J , Kinsey AM , Hanger DP , Anderton BH ((2009) ) Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J 23: , 739–750. |
[344] | Li C , Yuan K , Schluesener H ((2013) ) Impact of minocycline on neurodegenerative diseases in rodents: A meta-analysis. Rev Neurosci 24: , 553–562. |
[345] | Martín A , Boisgard R , Kassiou M , Dollé F , Tavitian B ((2011) ) Reduced PBR/TSPO expression after minocycline treatment in a rat model of focal cerebral ischemia: A PET study using [18 F] DPA-714. Mol Imaging Biol 13: , 10–15. |
[346] | Cheng S , Hou J , Zhang C , Xu C , Wang L , Zou X , Chen G ((2015) ) Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci Rep 5: , 10535. |
[347] | Kraus RL , Pasieczny R , Lariosa-Willingham K , Turner MS , Jiang A , Trauger JW ((2005) ) Antioxidant properties of minocycline: Neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem 94: , 819–827. |
[348] | Choi Y , Kim HS , Shin KY , Kim EM , Kim M , Kim HS , Chang KA ((2007) ) Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology 32: , 2393–2404. |
[349] | Sinha N , Dua D ((2015) ) Lycopene: Most potent antioxidant with endless benefits. Int J Pharm Bio Sci 6: , 838–846. |
[350] | Rao AV , Ali A ((2007) ) Biologically active phytochemicals in human health: Lycopene. Int J Food Prop 10: , 279–288. |
[351] | Chauhan K , Sharma S , Agarwal N , Chauhan B ((2011) ) Lycopene of tomato fame: Its role in health and disease. Int J Pharm Sci Rev Res 10: , 99–115. |
[352] | Devaraj S , Mathur S , Basu A , Aung HH , Vasu VT , Meyers S , Jialal I ((2008) ) A dose-response study on the effects of purified lycopene supplementation on biomarkers of oxidative stress. J Am Coll Nutr 27: , 267–273. |
[353] | Trumbo PR ((2005) ) Are there adverse effects of lycopene exposure? J Nutr 135: , 2060S–2061S. |
[354] | Sachdeva AK , Chopra K ((2015) ) Lycopene abrogates Aβ (1–42)-mediated neuroinflammatory cascade in an experimental model of Alzheimer’s disease. J Nutr Biochem 26: , 736–744. |
[355] | Prakash A , Kumar A ((2014) ) Implicating the role of lycopene in restoration of mitochondrial enzymes and BDNF levels in β-amyloid induced Alzheimer’s disease. Eur J Pharmacol 741: , 104–111. |
[356] | Bala R , Khanna D , Mehan S , Kalra S ((2015) ) Experimental evidence for the potential of lycopene in the management of scopolamine induced amnesia. RSC Adv 5: , 72881–72892. |
[357] | Kumar P , Kalonia H , Kumar A ((2010) ) Protective effect of lycopene against 3-nitropropionic acid-induced Huntington’s-like symptoms in rats. Alzheimers Dement 6: , S186. |
[358] | Weaver BA ((2014) ) How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 25: , 2677–2681. |
[359] | Kumar S , Mahdi H , Bryant C , Shah JP , Garg G , Munkarah A ((2010) ) Clinical trials and progress with paclitaxel in ovarian cancer. Int J Womens Health 2: , 411–427. |
[360] | Bishop JF , Dewar J , Toner GC , Tattersall MH , Olver IN , Ackland S , Kennedy I , Goldstein D , Gurney H , Walpole E , Levi J ((1997) ) Paclitaxel as first-line treatment for metastatic breast cancer. The Taxol Investigational Trials Group, Australia and New Zealand. Oncology (Williston Park) 11: , 19–23. |
[361] | Brambilla L , Romanelli A , Bellinvia M , Ferrucci S , Vinci M , Boneschi V , Tedeschi L ((2008) ) Weekly paclitaxel for advanced aggressive classic Kaposi sarcoma: Experience in 17 cases. Br J Dermatol 158: , 1339–1344. |
[362] | Shemesh OA , Spira ME ((2011) ) Rescue of neurons from undergoing hallmark tau-induced Alzheimer’s disease cell pathologies by the antimitotic drug paclitaxel. Neurobiol Dis 43: , 163–175. |
[363] | Appleby BS , Nacopoulos D , Milano N , Zhong K , Cummings JL ((2013) ) A review: Treatment of Alzheimer’s disease discovered in repurposed agents. Dement Geriatr Cogn Disord 35: , 1–22. |
[364] | Hung SY , Fu WM ((2017) ) Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci 24: , 47. |
[365] | Das V , Miller JH ((2012) ) Microtubule stabilization by peloruside A and paclitaxel rescues degenerating neurons from okadaic acid-induced tau phosphorylation. Eur J Neurosci 35: , 1705–1717. |
[366] | Zhang B , Maiti A , Shively S , Lakhani F , McDonald-Jones G , Bruce J , Toleikis PM ((2005) ) Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A 102: , 227–231. |
[367] | Makani V , Zhang B , Han H , Yao Y , Lassalas P , Lou K , Smith AB ((2016) ) Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta Neuropathol Commun 4: , 106. |
[368] | Brunden KR , Gardner NM , James MJ , Yao Y , Trojanowski JQ , Lee VMY , Smith III A B ((2013) ) MT-stabilizer, dictyostatin, exhibits prolonged brain retention and activity: Potential therapeutic implications. ACS Med Chem Lett 4: , 886–889. |
[369] | Javidnia M , Kurd-Misto BT , Mouss CE ((2017) ) An update on clinical trials targeting human tauopathies. Clin Trials Degener Dis 2: , 66. |
[370] | Pinheiro L , Faustino C ((2018) ) Therapeutic strategies targeting tau protein: Implications for Alzheimer’s disease. Alzheimer’s Disease & Treatment. MedDocs Publishers LLC. |
[371] | Cortes J , Baselga J ((2007) ) Targeting the microtubules in breast cancer beyond taxanes: The epothilones. Oncologist 12: , 271–280. |
[372] | Isbrucker RA , Cummins J , Pomponi SA , Longley RE , Wright AE ((2003) ) Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem Pharmacol 66: , 75–82. |
[373] | Abidi A ((2013) ) Cabazitaxel: A novel taxane for metastatic castration-resistant prostate cancer-current implications and future prospects. J Pharmacol Pharmacother 4: , 230–237. |
[374] | Semiond D , Sidhu SS , Bissery MC , Vrignaud P ((2013) ) Can taxanes provide benefit in patients with CNS tumors and in pediatric patients with tumors? An update on the preclinical development of cabazitaxel. Cancer Chemother Pharmacol 72: , 515–528. |
[375] | Cisternino S , Bourasset F , Archimbaud Y , Sémiond D , Sanderink G , Scherrmann JM ((2003) ) Nonlinear accumulation in the brain of the new taxoid TXD258 following saturation of P-glycoprotein at the blood–brain barrier in mice and rats. Br J Pharmacol 138: , 1367–1375. |
[376] | Manley PE , Trippett T , Smith AA , Macy ME , Leary SE , Boklan J , Devin J ((2018) ) A phase 1/2 dose-finding, safety, and activity study of cabazitaxel in pediatric patients with refractory solid tumors including tumors of the central nervous system. Pediatr Blood Cancer 65: , e27217. |
[377] | Fitzgerald DP , Emerson DL , Qian Y , Anwar T , Liewehr DJ , Steinberg SM , Steeg PS ((2012) ) TPI-287, a new taxane family member, reduces the brain metastatic colonization of breast cancer cells. Mol Cancer Ther 11: , 1959–1967. |
[378] | US National Library of Medicine ((2018) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02133846. ClinicalTrials.gov Identifier: NCT02133846, Posted on May 8, 2014, Last updated May 3, 2018. |
[379] | Tan CC , Zhang XY , Tan L , Yu JT ((2018) ) Tauopathies: Mechanisms and therapeutic strategies. J Alzheimers Dis 61: , 487–508. |
[380] | Ivashko-Pachima Y , Maor-Nof M , Gozes I ((2019) ) NAP (davunetide) preferential interaction with dynamic 3-repeat Tau explains differential protection in selected tauopathies. PloS One 14: , e0213666. |
[381] | Vulih-Shultzman I , Pinhasov A , Mandel S , Grigoriadis N , Touloumi O , Pittel Z , Gozes I ((2007) ) Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther 323: , 438–449. |
[382] | US National Library of Medicine ((2019) ) ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01056965. ClinicalTrials.gov Identifier: NCT01056965, Posted on January 26, 2010, Last updated April 5, 2019. |
[383] | Javitt DC , Buchanan RW , Keefe RS , Kern R , McMahon RP , Green MF , Jarskog F ((2012) ) Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr Res 136: , 25–31. |
[384] | Matsuoka Y , Jouroukhin Y , Gray AJ , Ma L , Hirata-Fukae C , Li HF , Arancio O ((2008) ) A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. J Pharmacol Exp Ther 325: , 146–153. |
[385] | Morimoto BH , Schmechel D , Hirman J , Blackwell A , Keith J , Gold M , AL-108-211 Study Investigators ((2013) ) A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Dis 5: , 325–339. |
[386] | Anand K , Sabbagh M ((2015) ) Early investigational drugs targeting tau protein for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs 24: , 1355–1360. |
[387] | Gozes I , Iram T , Maryanovsky E , Arviv C , Rozenberg L , Schirer Y , Furman-Assaf S ((2014) ) Novel tubulin and tau neuroprotective fragments sharing structural similarities with the drug candidate NAP (Davuentide). J Alzheimers Dis 40: , 23–36. |
[388] | Vollmer LL , Jiménez M , Camarco DP , Zhu W , Daghestani HN , Balachandran R , Day BW ((2011) ) A simplified synthesis of novel dictyostatin analogues with in vitro activity against epothilone B–resistant cells and antiangiogenic activity in zebrafish embryos. Mol Cancer Ther 10: , 994–1006. |
[389] | Kovalevich J , Cornec AS , Yao Y , James M , Crowe A , Lee VMY , Brunden KR ((2016) ) Characterization of brain-penetrant pyrimidine-containing molecules with differential microtubule-stabilizing activities developed as potential therapeutic agents for Alzheimer’s disease and related tauopathies. J Pharmacol Exp Ther 357: , 432–450. |
[390] | Zhang B , Yao Y , Cornec AS , Oukoloff K , James MJ , Koivula P , Brunden KR ((2018) ) A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model. Mol Neurodegener 13: , 59. |
[391] | Dent EW ((2017) ) Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol Biol Cell 28: , 1–8. |
[392] | Wang-Gillam A , Arnold SM , Bukowski RM , Rothenberg ML , Cooper W , Wang KK , Lockhart AC ((2012) ) A phase I dose escalation study of TTI-237 in patients with advanced malignant solid tumors. Invest New Drugs 30: , 266–272. |
[393] | Habener LJ , Hooper JN , Carroll AR ((2016) ) Chemical and biological aspects of marine sponges from the family Mycalidae. Planta Med 82: , 816–831. |
[394] | Singh AJ , Xu CX , Xu X , West LM , Wilmes A , Chan A , Ghosh AK ((2009) ) Peloruside B, a potent antitumor macrolide from the New Zealand marine sponge Mycale hentscheli: Isolation, structure, total synthesis, and bioactivity. J Org Chem 75: , 2–10. |
[395] | West LM , Northcote PT , Battershill CN ((2000) ) Peloruside A: A potent cytotoxic macrolide isolated from the New Zealand marine sponge Mycale sp. J Org Chem 65: , 445–449. |
[396] | Das V , Miller JH ((2012) ) Microtubule stabilization by peloruside A and paclitaxel rescues degenerating neurons from okadaic acid-induced tau phosphorylation. Eur J Neurosci 35: , 1705–1717. |
[397] | Parésys L , Hoffmann K , Froger N , Bianchi M , Villey I , Baulieu EE , Fuchs E ((2015) ) Effects of the synthetic neurosteroid: 3β-methoxypregnenolone (MAP4343) on behavioral and physiological alterations provoked by chronic psychosocial stress in tree shrews. Int J Neuropsychopharmacol 19: , pyv119. |
[398] | Kramp VP ((2012) ) List of drugs in development for neurodegenerative diseases: Update October 2011. Neurodegener Dis 9: , 210–283. |
[399] | Murakami K , Fellous A , Baulieu EE , Robel P ((2000) ) Pregnenolone binds to microtubule-associated protein 2 and stimulates microtubule assembly. Proc Natl Acad Sci U S A 97: , 3579–3584. |


