
Pre-Clinical Safety and Efficacy Evaluation of Amytrap, a Novel Therapeutic to Treat Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia. Amyloid-β (Aβ42) is implicated in AD pathogenesis. We have designed a non-immune based proprietary therapeutic, called Amytrap, a conjugate containing a retro-inverso peptide, polyethylene glycol, and human serum albumin. Amytrap not only binds Aβ42 but also prevents and dissociates aggregated Aβ42. Amytrap binds to the region in Aβ42 known to trigger its self-aggregation, thus disrupting aggregation. We have obtained proof of concept on AmyTrap in a clinically relevant mouse model, namely, AD-APPSWE/Tg2576. We synthesized and characterized Amytrap and confirmed its authenticity. Efficacy evaluations were performed on young (5 months) and old (9 months) model mice. Amytrap was injected biweekly for a period of five months. Pharmacokinetics and safety toxicology were assessed in normal mice and rats, respectively. Post treatment, younger mice showed significant improvements in cognition and Aβ42 levels in plasma, brain, and cerebrospinal fluid, while older mice showed less significant benefits. Immunohistochemistry of brain sections showed similar differences between young and old mice. They all had diminished size and number of plaques in the brain of Amytrap-treated mice. Further, treated mice did not develop antibodies to Amytrap, suggesting Amytrap is non-immunogenic. Safety toxicological studies in rats showed that Amytrap was well tolerated and therefore safe (even at 50 X the efficacy dose). Stability tests showed Amytrap is stable at 4°C for up to one year. Efficacy and safety features make Amytrap a promising candidate for treating or modulating AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia affecting 25 million people worldwide. The incidence of AD and associated costs are predicted to double in the next 20 years due to increasing life expectancy [1–3]. Accumulation of amyloid-β (Aβ42) in the brain is consistently observed in AD and is believed to cause the accumulation and conversion of tau into its toxic state [4]. Tau in its toxic state results in damage and destruction of synapses that mediate memory and cognition [4, 5]. Furthermore, accumulation of Aβ42 leads to the deposition of insoluble toxic Aβ42 plaques in the brain. Since Aβ42 is not only pathogenic by itself but also triggers tau pathology, it is an important target for therapy.
Extensive efforts were devoted to developing therapeutic interventions targeting Aβ42 but with little or no success [6]. Earlier attempts to target Aβ42 included development of inhibitors of β-secretase or γ-secretase but have been unsuccessful because of inherent medicinal chemistry problems or undesirable side effects [7, 8]. Direct immunization with Aβ42 or passive immunization with anti-Aβ42 antibodies is one of the most advanced approaches. However, the first clinical trial of immunization with pre-aggregated Aβ42 (AN1792) was stopped due to adverse effects involving detrimental T-cell mediated brain inflammation [9]. Passive transfer with anti-Aβ42 antibodies showed no significant improvement in symptoms and exhibited immunological adverse reactions in AD patients [10]. Based on the foregoing results, the validity of targeting Aβ42 for treating AD has been questioned. However, data gathered in these trials call for critical examination of the Aβ42 theory before a conclusion rejecting the relevancy of Aβ42 as a target can be drawn. An important observation favoring Aβ42 is the promising efficacy of antibodies. In trials with three different Aβ42 antibodies (solanezumab, crenezumab, and aducanumab) [11–13], post-hoc analysis suggested a slowing of cognitive decline in mild AD subjects. Especially interesting, a clinical study with Aducanumab (Biogen) showed a cognitive benefit with a significant reduction in Aβ42 burden in the brain in patients with AD. This development endorses Aβ42 as a viable target for developing AD treatments. Therefore, the Aβ42 hypothesis is currently being revisited by researchers with a focus on anti-Aβ42 antibodies. This shift in treatment paradigm toward targeting Aβ42 in early stages could turn out to be very beneficial to AD therapy. Recently, a phase II AD trial once nearly consigned to the heap of disappointing attempts against the disease has re-emerged with new positive results, showing that an anti-Aβ42 beta protofibril antibody can slow clinical symptom decline, and reduce the accumulation of plaque in the brain [14]. An alternative disease-modifying therapy for AD involves the search for candidates that can prevent Aβ42 aggregation or disassociate preformed Aβ42 aggregates. One such approach would be to screen or develop peptides that prevent Aβ42 aggregation or disassociate preformed Aβ42 aggregates as detailed [15].
We had previously selected a peptide, FFVLK [16], and used it as a prototype to test our concept. Eventually, we tested other known peptide molecules including RGTWEGKW [17] and QSHYRHISPAQV [18] in AD therapy using our approach. In the present study, we attempt to test one of those peptides in vitro and in vivo. This peptide is the active pharmacological ingredient in our drug conjugate, namely ‘Amytrap’. We chose to study this peptide based on encouraging preliminary experimental data (unpublished). As native peptides are less stable in circulation, we synthesized tetrameric retro-inverso (RI) form (WKGEWTGR) which was then pegylated and conjugated to human serum albumin (HSA) to enhance its bioavailability. Retro-inversion is considered an attractive approach for drug and vaccine design by providing modified peptides with prolonged half-life in vivo [19, 20]. Aβ42 has a motif GXXXG that has been shown to be responsible for the self-oligomerization and aggregation [17]. Amytrap peptide has a high affinity to bind the GXXXG region and thus blocks oligomerization and aggregation (Fig. 1). By inhibiting this critical motif we believe Amytrap will act as a sink and shift the homeostasis away from the brain.
Fig.1
Aβ sequence and the binding sites. The diagram shows the binding regions of the anti-Aβ antibodies (red) and the Amytrap-1 (yellow) in Aβ. The region (the circled amino acids) where the AmyTrap-1 peptide binds has been shown to promote hair-pin formation of toxic Aβ [17].
![Aβ sequence and the binding sites. The diagram shows the binding regions of the anti-Aβ antibodies (red) and the Amytrap-1 (yellow) in Aβ. The region (the circled amino acids) where the AmyTrap-1 peptide binds has been shown to promote hair-pin formation of toxic Aβ [17].](https://content.iospress.com:443/media/adr/2019/3-1/adr-3-1-adr190107/adr-3-adr190107-g001.jpg)
We demonstrate the effective binding of immobilized RI-peptide to Aβ42 in vitro and the efficacy of Amytrap conjugate in vivo in the clinically relevant model mouse of AD-APPSWE/Tg2576. We evaluated its safety in normal rats and demonstrate that Amytrap conjugate was safe even at 50x the efficacy dose. Overall, the results show reduction in Aβ42 levels and significant improvement in memory tasks in younger mice.
MATERIALS AND METHODS
Peptide synthesis
The D-amino acid containing tetramer RI-peptide WKGEWTGR representing the native sequence RGTWEGKW was synthesized at LifeTein (NJ, USA). Amytrap monomers were linked into a fork-like structure by β-Alanine and Lysine. Molecular weight (MW) and purity of 95% was confirmed by LC-MS and HPLC. The synthetic peptide was collected and analyzed by mass spectrometry and the intensity versus m/z (mass-to-charge ratio) was plotted to calculate the mass of the injected RI-peptide. The plot indicated that the mass of the peptide injected was 4796.3, which is the anticipated molecular mass of the RI-peptide. To check the purity, 5 μl (one mg) of peptide solution was injected into a 4.6 X 250 mm, KLM PS/DVB column, preequilibrated with 0.1% of Trifluoro acetic acid (TFA) and 90% of acetonitrile (buffer A). The peptide was eluted with a step gradient of 35% of buffer A and 65% of buffer B (0.1% TFA in water) for 25 min at a flow rate of 1.0 ml/min. Purity was determined by HPLC where a major peak with 95% purity was eluted with a retention time 13.38 min (data not shown). Five hundred milligrams of peptide was successfully prepared and tested for purity and function as described below.
Confirmation of purity and functional studies
Purity of the RI tetramer peptide was analyzed by SDS-PAGE. RI-tetramer peptide was resolved on a 4–12% SDS-PAGE gel which was then stained with Coomassie Brilliant Blue (CBB). A single band at 5 kDa corresponding to that of tetramer RI-peptide was observed indicating that the synthesized peptide was at least 95% pure. ELISA was performed to confirm that the RI tetramer peptide was able to bind a biotinylated Aβ42 (bio-Aβ42) following a previously utilized protocol [16].
Preparing albumin conjugate
To prepare the RI-peptide-PEG-HSA conjugate (Amytrap), we followed the ‘Click’ chemistry approach [21, 22] using these three stages: 1) Conjugation of RI-peptide with Dibenzocyclooctyne-maleimide (DBCO-Maleimide) linker to form RI-DBCO; 2) Conjugation of HSA protein with azide-(PEG)400-NHS linker to form HSA-(PEG)400-azide intermediate; and 3) Conjugation of RI-peptide-DBCO with HSA-(PEG)5K-azide to form the final HSA-(PEG) 5K-Amytrap product.
In the first reaction, 2 mg of RI-peptide was dissolved in 100 μL phosphate-buffered saline (PBS) at pH 7.4 and reacted with 0.1 mg Tris (2-carboxyethyl) phosphine (TCEP) for 10 min at room temperature (RT). One milligram DBCO-Maleimide was added and incubated at 4°C overnight (ratio of RI: Linker is 1 : 5). The reaction mixture was dialyzed overnight in PBS at 4°C. An aliquot was analyzed on HPLC. In the second step, HSA and azide-PEG4-NHS linker 1 : 10 (molar ratio) was added to PBS buffer and incubated for 2 h at RT and 4°C O/N to form HSA-PEG4-azide intermediate. The unreacted linkers were removed using extensive dialysis using a 1 kDa MW cut-off tubing (MW of the linker is 472 Da). The dialysis was carried out in PBS buffer O/N at 4°C using tubing with MW cut-off at 8 14 kDa to obtain azide-PEG4-HSA. HSA-PEG4-azide (10 mg/mL) was reacted with RI-peptide-DBCO (2.85 mg/mL, molar ratio of HSA-PEG4-azido: RI-peptide-DBCO=1 : 4) in PBS at pH 7.4. The reaction was allowed for 8 h at RT and O/N at 4°C to obtain Amytrap conjugate (HSA-PEG4-RI-peptide). The final conjugate was dialyzed against ddH2O using a 30 kDa MW-cutoff tubing. Earlier, an antibody was raised in a rabbit and tested in ELISA to have a high titer for the tetramer peptide (unpublished data). The finished Amytrap conjugate was further characterized by SDS-PAGE (4–12%) followed by CBB staining. Western blotting on the conjugate utilizing the anti-tetramer antibody confirmed that conjugation was successful. Briefly, 1 μg aliquots of HSA, and Amytrap conjugate were resolved on an SDS-PAGE gel (150 V, 1 h) and then transferred at 90 V to polyvinylidene difluoride (PVDF) membranes. The membrane was blocked in 5% nonfat dry milk in TBST (25 mM Tris-HCl pH 8.0, 125 mM NaCl, and 0.1% Tween-20) overnight at 4°C. The membrane was probed with the rabbit anti-tetramer antibody (1 : 5,000) for 1 h at RT. The membrane was washed 3 times with TBST and incubated with a horse radish peroxidase (HRP) labeled goat anti-rabbit antibody (1 : 10,000) for 1 h. The membrane was washed three times and proceeded further for enhanced chemiluminescence (ECL) and image development as per manufacturer’s instructions.
RI-peptide binding to Aβ42 by ELISA
We tested Aβ42 binding activity of the tetramer RI-peptide by ELISA. Microplate wells were coated with 100 ng of the tetramer RI-peptide in carbonate buffer pH 9.6 for 1 h at 37°C. Wells were blocked with 3% gelatin in Tris-buffered saline (TBS) for 1 h at 37°C. To the bound RI-peptide different concentrations (1, 5, and 10 ng) of biotinylated Aβ42 (AnaSpec, CA, USA) was added in triplicate and incubated for 1 h at 37°C. Plates were washed 3 times with TBST and incubated with streptavidin-HRP (1 : 5000) (KPL, Gaithersburg, MD) for 1 h at 37°C. After washing the wells 3 times with TBST, 100 μl HRP substrate solution (SureBlue, KPL, Gaithersburg, MD, USA) was added and incubated for 1 min at RT. One hundred μl of 1 N HCl was added to stop the reaction and the absorbance was read at 450 nm.
Cell cytotoxicity assay
We have established a neuronal cell-based apoptosis assay to test any compound or drug in vitro. We use SH-SY5Y neuroblastoma cell line (Sigma Aldrich, Cat. No. 94030304) for this assay as it closely resembles in vivo conditions. Utilizing this assay, we examined whether tetramer RI-peptide could inhibit and or reverse Aβ42-induced toxicity. SH-SY5Y cells were grown and sub-cultured in Eagle’s Minimal Essential Medium (EMEM) at 37°C in a humidified 5% CO2 incubator. 2,600 cells/well were seeded in 100 μL of EMEM in a 96-well plate and incubated overnight at 37°C in a humidified, 5% CO2 incubator. In another plate, a column of 8-wells was seeded with cells for “Zero Hour” reading. Next day, EMEM was replaced with 100 μL of fresh medium containing different concentrations of RI Tetramer peptide with 10 μM of Aβ42 was added in duplicate. Cells in the “Zero Hour” plate were mixed with 100 μL of CellTiterGlo (CTG) reagent from Promega (Catalog No. G7573) and read in a luminometer (Biotek, FLx800) for “Zero Hour” reading. After 72-h incubation with the compounds, cell viability was measured in a luminometer after the addition of 100 μL /well CTG assay reagent. “Zero Hour” reading was subtracted from all values, readings of untreated cells were taken as 100% growth. Percent inhibition was calculated with reference to untreated cells using SoftMax software.
Pharmacokinetics of RI-peptide, Amytrap-PEG, and RI-peptide-PEG-HSA conjugate
To establish bioavailability and determine dosing periods of the RI-peptide, pharmacokinetics (PK) was performed in Balb/c mice at Pocono Rabbit Farms and Laboratory (PA). Mice (male and female) were dosed with a single injection (200 μg/ 140 μl) of RI-peptide, RI-peptide-PEG conjugate, or RI-peptide-PEG-HSA subcutaneously. Prior to injection, a plasma sample was taken to account for pre-injection levels. Plasma samples were taken from mice post injection at the time intervals mentioned (0, 1, 2, 4, 8, or 24 h or 0, 2, 5, 9, 12, or 16 days) and tested for RI-peptide levels using ELISA. Plasma protein concentration was estimated by BCA and 10 and 100 μg/well was plated on a 96-well Grenier flat bottom plate for 1 h at 37°C then blocked with 3% gelatin for 1 h at 37°C. Wells were probed with rabbit anti-RI-peptide antibody for 1 h at 37°C, then washed with TBST. The wells were treated with goat anti-rabbit HRP antibody for 1 h at 37°C. Wells were washed 8 times with TBST before 100 μl of HRP substrate solution (LumiGLO®, KPL, Gaithesburg, MD) was added to each well and read on a luminometer.
Analysis of Amytrap conjugate stability
Analysis of stability was performed by assessing long-term storage at three temperatures (RT, 4°C, and -20°C) and 4 time points (3, 6, 9, and 12 months). Amytrap conjugate powder was pre-weighed into microcentrifuge tubes and initial weights were marked. A ratio of RI tetramer peptide to Amytrap conjugate powder was previously determined to be 1 : 6.4. At the selected timepoint, powders at each temperature were dissolved with filtered distilled water to 10 mg/ml. Protein estimation was performed by bicinchoninic acid assay and western blotting was performed as described in the section “Preparing Albumin conjugate.” Band images were captured on x-ray film. The MW of the bands was established and marked by matching the film and PVDF membrane. The film was scanned to a computer and analyzed with ImageJ software.
Efficacy evaluation in 5-month-old APPSWE 2576 AD model mice
The efficacy of Amytrap conjugate was tested in an AD mouse model (APPSWE 2576 mouse model, human amyloid-β precursor protein expresser) [23] available with Taconic Biosciences (Rennselaer, NY). The mouse expresses high concentrations of mutant Aβ42, develops significant Aβ42 plaques, and displays memory deficits. Therefore, it serves as a model to study drugs designed to treat AD. Thirty-six 4-month-old female mice were purchased and housed at Taconic Biosciences for aging. At 5 months of age, mice were divided into three groups and received treatment as shown in Table 1. Injections were subcutaneously given biweekly for 20 weeks (10 doses in total). The animals were tested for cognitive improvement by Y-maze test before the dosing period and before sacrificing ten days after the last injection. Blood plasma, cerebrospinal fluid (CSF), and brains from these animals were collected to estimate Aβ42 burden by immunostaining.
Table 1
Efficacy studies performed in 5-month-old AD mice. This table outlines the treatment for designated groups of APPSWE/Tg2576 female mice. Mice received saline or The Amytrap conjugate every 15 days for 5 months
| Mouse strain: APPSWE/Tg2576 (36 females) | Study Duration: 5 months | ||
| Mouse Group | Dose Group | Number of Mice | Peptide Dose |
| Group 1 | Control | N=12 | 0 μg |
| Group 2 | [FFVLK]4 | N=12 | 100 μg |
| Group 3 | Amytrap | N=12 | 100 μg |
Behavioral analysis
Spontaneous Y-Maze test was performed as described [23]. Briefly, mice were individually placed in the center of a Y maze, with all three arms (arm length is 38 cm, and arm width is 7.5 cm) available for exploration for a period of 10 min. The percentage of triads in which all three arms were recorded as an alternation to estimate short-term memory of the last arms entered. Alternations were counted by a trained technician. Consecutive entries into the three different arms (i.e., ABC, ACB, BCA, BAC, CAB, or CBA), and percent alternation (number of alterations * 100 / number of arm visits – 2) was used as an index of working memory performance. After the test, data was transferred from hard copy to Excel spreadsheet. Following the test, each mouse was returned to its home cage, and the arena was cleaned. Mice that made less than 5 arm entries were excluded from the analysis.
Measurement of Aβ42 in brain tissue from mice
AD model mice brain tissue was homogenized utilizing a protocol as described [24, 25]. Briefly, frozen brains were homogenized followed by sonication in a 10-fold volume of TBS (pH 7.6) containing 2% SDS and protease inhibitors (complete protease inhibitor cocktail, Boehringer Mannheim, Mannheim, Germany) followed by centrifugation at 100,000 x g for 1 h at 4°C. The supernatant was removed and the pellet sonicated with 70% formic acid and centrifuged at 100,000 x g for 20 min at RT. Extracts were neutralized by dilution (1 : 20) into 1 M Tris phosphate buffer (pH 11). Supernatants were vacuum-dried and solubilized in TBS. Extracts were quantified for brain Aβ42 in pMols/gram brain [25].
Immunohistochemistry of brain sections to assess Aβ42 burden
Brain plaques were detected and analyzed utilizing a protocol as described [25]. Serial sections (5 μm) were cut sagittally through the hemisphere embedded in paraffin with rotary microtome. Every 3rd section of each series was mounted on Superfrost Plus slide and processed with a monoclonal mouse anti-human Aβ42 antibody (G48, Covance, WI). Sections were deparaffinized in xylene, rinsed in graded ethanol, and then rehydrated to distilled water. Endogenous peroxidase activity was inactivated with hydrogen peroxidase and then antigen retrieval was performed as described in the manufacturer’s literature. Sections were incubated in 0.01 M phosphate buffered saline containing normal blocking serum, BSA (Jackson ImmunoResearch Labs, West Grove, PA), Triton X-100, and the specific antibody (1 : 200) o/n at 4°C. Immunoreaction products were visualized through avidin-biotin complex Vectastin elite ABC kit (Vector Lab., Burlingame, CA) and 3’, 3. – diaminobenzidine as a chromogen. Sections were rinsed in distilled water and counterstained with hematoxylin. Sections were prepared for analysis as described in the methods [25] Immunohistology services were performed by FD Neurotechnologies (Maryland) and pathology assistance was provided by Vertinary Histology Consultants (CT).
Efficacy evaluation in 9-month-old APPSWE 2576 AD model mice
The efficacy of Amytrap conjugate was further tested in 9-month-old APPSWE 2576 mice to evaluate dosing concentrations. Thirty-two 3-month-old female mice were purchased and housed at Taconic Biosciences for aging. At 9 months of age, mice were divided into three groups and received treatment as shown in Table 2. This entire efficacy experiment was performed at our CRO, Brains Online (CA) which was recently acquired by Charles River Laboratories. Injections were subcutaneously given biweekly for 20 weeks (10 doses in total). The animals were tested for cognitive improvement by novel object recognition (NOR) test and Y-maze test before sacrificing ten days after the last injection. Blood plasma, CSF, and brains from these animals were collected to estimate Aβ42 burden by immunostaining as performed in the 5-month-old AD model mice study.
Table 2
Efficacy studies performed in 9-month-old AD mice. This table outlines the treatment for designated groups of APPSWE/Tg2576 female mice. Mice received saline or The Amytrap conjugate every 15 days for 5 months
| Mouse strain: APPSWE/Tg2576 (32 females) | Study Duration: 5 months | ||||
| Mouse Group | Dose Group | Number of Mice | Treatment (mg/kg bodyweight) | Conjugate injected | Peptide equivalent |
| Group 1 | Control | N=10 | Saline | 0 μg | 0 μg |
| Group 2 | Low Dose | N=11 | 6.45 | 128 μg | 20 μg |
| Group 3 | High Dose | N=11 | 16.1 | 320 μg | 50 μg |
Measurement of Aβ40 and Aβ42 in plasma and in CSF from mice
Aβ40 and Aβ42 from plasma and CSF were measured with the SensoLyte® ELISA kit as per manufacturer’s instructions (AnaSpec, CA) [26].
Assessment of immunogenicity in mouse plasma by ELISA
ELISA was performed as described in the section “RI-peptide binding to Aβ42 by ELISA,” 10 ng/well tetramer peptide was coated onto a 96-well microtiter plate. Triplicate wells were probed with either positive control antibody or with individual mouse plasma (1 : 10,000). Absorbance obtained by individual groups are expressed as mean±SE from triplicate experiments.
Behavioral analysis
Along with Y-maze test which was performed as stated in the 5-month-old AD model mice study, the NOR test was performed to assess the ability of mice to recognize a familiar over a novel object within a stipulated time frame (10 min) [27]. Experiments were carried out in a plastic chamber (16 in X 16 in X 14 in) with opaque floors and walls, designated as the experimental arena. The arenas were placed in a dark experimental room illuminated with consistent, dim light in the arenas. Each day, at the end of each trial, mice were returned to their home cage and the chambers were cleaned with 70% ethanol. Mice were allowed to explore the empty area for 10 min on Day 1. On Day 2, mice were placed into the same arena with two identical objects (object A) and allowed to explore the objects for 10 min. On Day 3, mice were, once again, placed in the same arena as the previous days, but one of the objects from Day 2 was replaced with a new object (object B). The time spent exploring each object was recorded and analyzed. Exploration was defined as touching the object or directing nose towards the object at a distance less than 2 cm. A minimum exploration criterion was used such that only animals with a total object exploration time greater than 5 s were included. Animals were excluded from analysis if they climbed on top of the objects.
Immunohistochemistry of brain sections to assess Aβ42 burden
Immunohistochemistry of brain sections and analyses was performed by our CRO, FD NeuroTechnologies (Columbia, MD). Mice were anesthetized and perfused transcardially with saline followed by the fixative, phosphate-buffered 4% paraformaldehyde. Harvested brains were fixed in the fixative for 24 h at 4°C and cryoprotected in 0.1 M phosphate buffer (pH 7.4) containing 20% sucrose for 72 h at 4°C. Then, brains were rapidly frozen in isopentane pre-cooled to –75°C with dry ice and stored at –80°C before sectioning. Serial cryostat sections (40 μm) were cut coronally through the entire cerebrum, approximately from Bregma 3.20 mm to –5.20 mm [28]. Every 1st, 2nd, 3rd, 4th, 5th, 6th, and 7th sections of each series of 7 sections (interval: 280 μm) were collected separately (30 sections per set per brain). All sections were stored free-floating in FD sections storage solution at –20°C before further processing. The sections of the 1st set were processed for anti-Aβ42 antibody (BioLegend, San Diego, CA) immunohistochemistry. Briefly, after inactivating endogenous peroxidase activity with hydrogen peroxidase, sections were incubated separately with avidin and biotin solutions (Vector Lab, Burlington, ON, Canada) to block nonspecific binding of endogenous biotin, biotin-binding protein and lectins, followed by incubation of sections in PBS containing mouse IgG blocking reagent (PK-2200, Vector Lab). Subsequently, sections were incubated free-floating in PBS containing Triton X-100 (Sigma, St. Louis, MO), M.O.M.™ protein concentrate (PK-2200, Vector Lab), and a monoclonal mouse anti-6E10 antibody (1 : 5000) for 17 h at 4°C. The immunoreaction product was visualized according to the avidin-biotin complex method with Vector® M.O.M.™ Immunodetection Kit (PK-2200 Kit) as per the manufacturer’s instructions from Vector Lab, Burlington, ON, Canada. This was followed by incubation of the sections for 2 min in acetate-imidazole buffer containing nickel sulfate, 3,3′-diaminobenzidine tetrahydrochloride (Sigma), and H2O2. All steps were carried out at RT except indicated, and each step was followed by washes in PBS. After thorough rinses in distilled water, all sections were mounted on gelatin-coated microscope slides, dehydrated in ethanol, cleared in xylene, and coverslipped in Permount® (Fisher Scientific, Fair Lawn, NJ).
Quantitative analysis of 6E10-immunostained Aβ42 plaques was performed in cerebral cortices and hippocampi of 32 mouse left hemispheres. Brain sections were prepared, 10 (for hippocampus, interval: 280 μm) or 15 (for cerebral cortex, interval: 560 μm) equally spaced sections were chosen. For each section, the number of 6E10-immunostained Aβ42 plaques was counted, and surface area of each plaque was measured with a bright-field microscope equipped with a digital camera and Olympus CellSens Dimension imaging software [29]. The total number obtained from each region of each brain was used for statistical analysis. For statistical analysis, the mean numbers, including both the number of Aβ42 plaques and the surface area of Aβ42 plaques from all animal groups were analyzed with Student’s t-test and an analysis of variance (ANOVA) by using PSPP software. In addition, images were taken under an Olympus microscope (BX43) with a 10X/0.4 or a 40X/0.95 objective from representative mouse brains to show Aβ42 plaques in various cortical areas and the hippocampus.
Safety toxicological analyses in rats
A total of 24 Sprague Dawley (SD) rats (age, sex, and weight matched) were divided into four groups of six rats each. The first group served as the control while the remaining three groups were administered subcutaneously 0.64 mg (low dose), 6.4 mg (medium dose), and 32 mg (high dose) of Amytrap conjugate (RI-peptide-PEG-HSA, 0.1 mg, 1 mg, and 5 mg peptide equivalent) per animal in 400 μl of sterile filtered water. Animals received four injections two weeks apart. All animals were allowed free access to food and water. Weekly measurements of food intake and body weights were recorded. Blood samples were harvested at five time points 0, 15, 30, 45, and 55 days. The animals were sacrificed on day 55. Blood, livers, kidneys, brains, and hearts were collected for histopathological analyses. Biochemical analysis of plasma proteins was performed to detect if changes occurred in protein levels. Plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), total proteins, albumin, total bilirubin, urea and creatinine were measured from blood samples taken on day 0 and 55 due to practical reasons of cost. The animal experiment was performed by our CRO, Pocono Rabbit Farm and Laboratory, and the protein and enzyme analyses were performed at Comparative Pathology Laboratory (UC Davis, CA).
Histopathology
Liver, kidney, brain, and heart were collected, fixed in formalin and embedded in paraffin. Sections (5 microns) were stained with hematoxylin and eosin (H&E) and examined by light microscopy. Histopathology results were documented in a summary from a histopathologist from Comparative Pathology Laboratory, (UC Davis, CA).
RESULTS
Synthesis and functional characterization of RI-peptide
The newly synthesized RI-peptide was analyzed for purity by SDS-PAGE and tested for functional binding of Aβ42 by ELISA. A method we previously utilized with a Bio-Aβ42 demonstrated concentration dependent binding by the RI-peptide (Fig. 2). We assessed the RI-peptide for toxicity and tolerance in neuroblastoma cell line (SH-SY5Y) to ensure that the tetrameric peptide was safe. The results show that the RI-peptide was not toxic up to a concentration of 20 μM (Fig. 3). The test also assessed the ability of tetramer RI-peptide to reverse Aβ42-induced toxicity. Varying concentrations of RI-peptide were incubated in the presence of a fixed concentration of Aβ42. The results are illustrated in Figure 2. RI tetramer peptide up to 20 μM concentration significantly countered the toxic effects of Aβ42 as evidenced by healthy cell growth.
Fig.2
Binding of Aβ42 by Amytrap. RI tetramer peptide binding to Aβ42 by ELISA. 100 ng of RI tetramer peptide was coated and probed with 0, 1, 5, or 10 ng/well of biotinylated Aβ42. Values are expressed mean absorbance±SD is plotted.
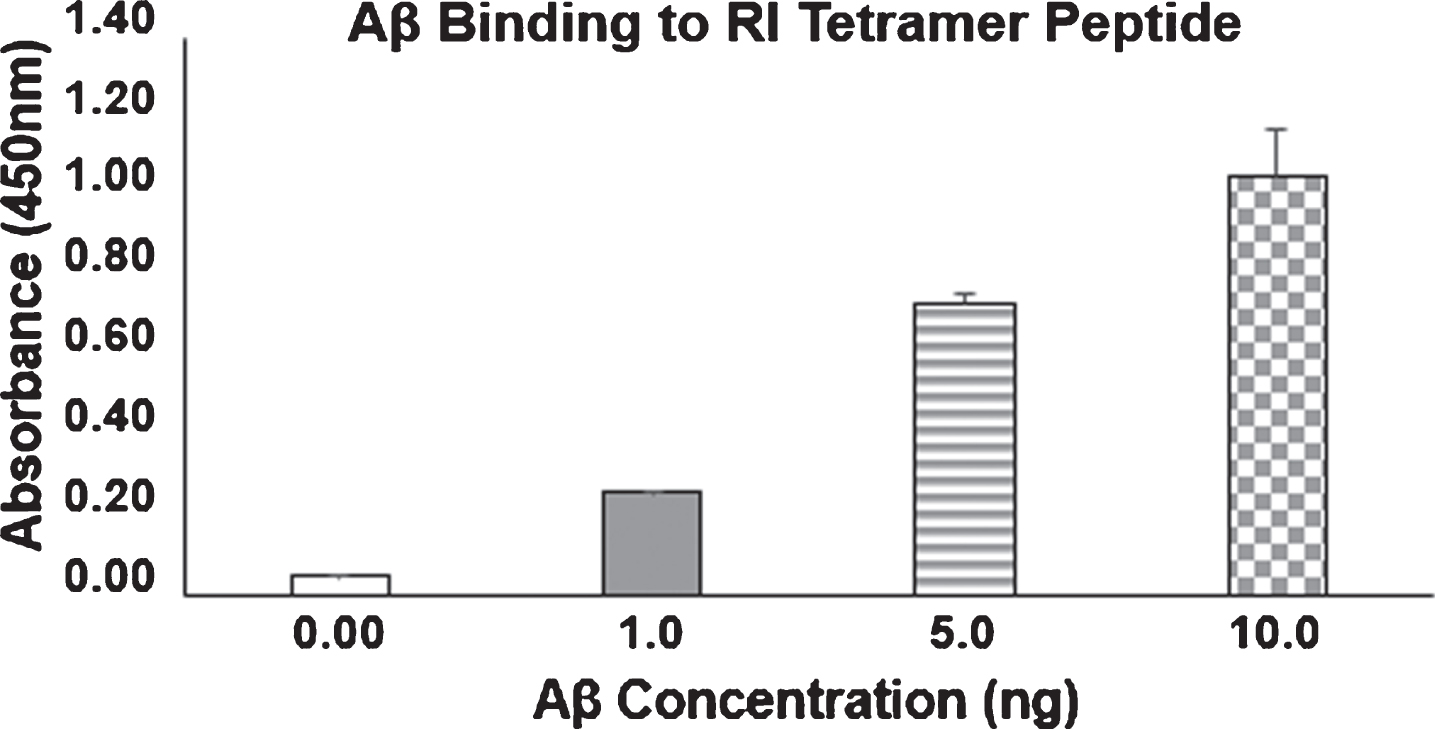
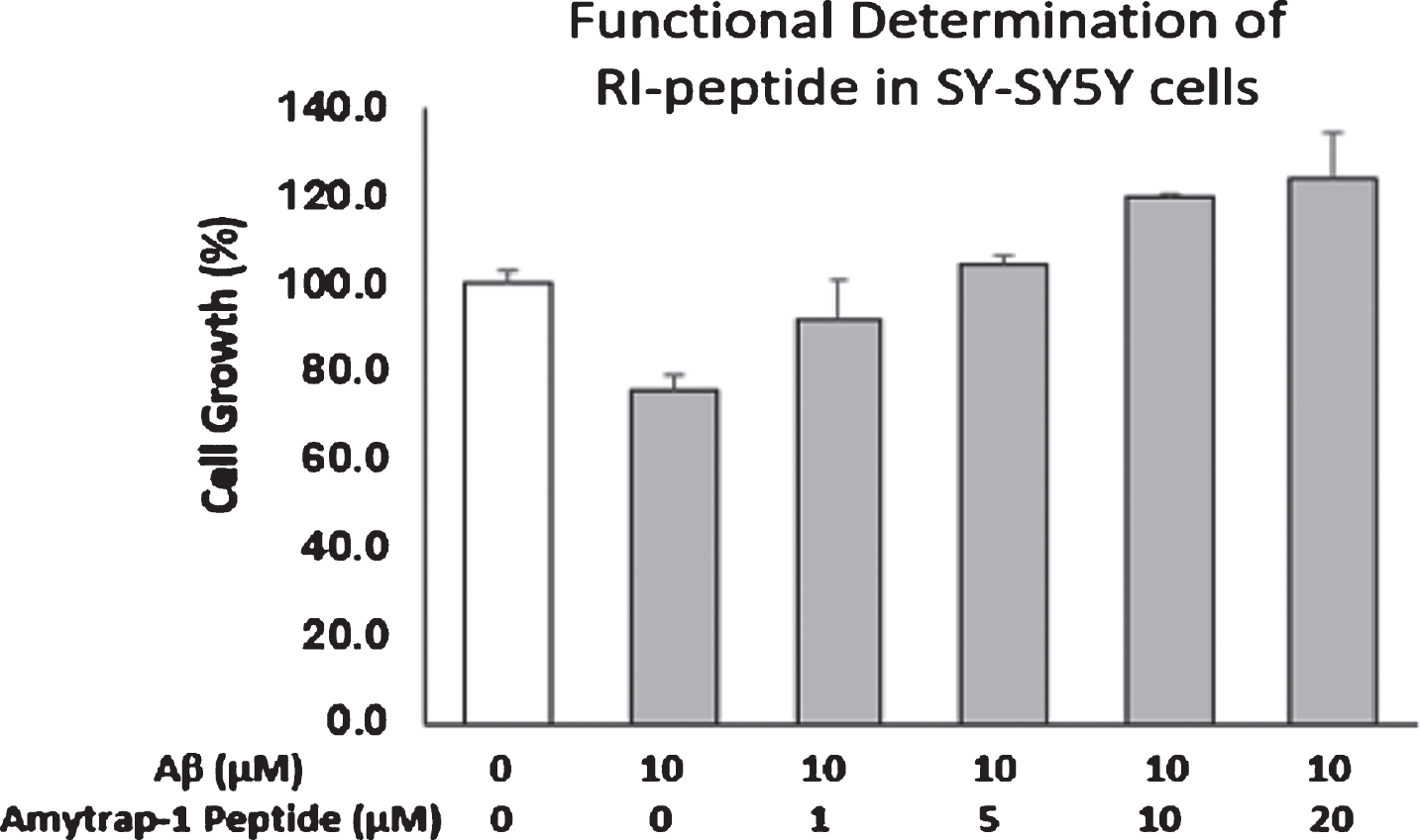
Fig.3
Reversal of cellular cytotoxicity by RI-peptide. SH-SY5Y cells were incubated with 10 μM Aβ42 in the presence of different concentrations of Amytrap peptide in duplicates for 72 h. Cell growth was measured with CellTiterGlo and read in a luminometer. Percentage of cell growth was compared with untreated cells (unfilled bar, 100%). Values are expressed as Mean±SD.
Synthesis and characterization of Amytrap conjugate
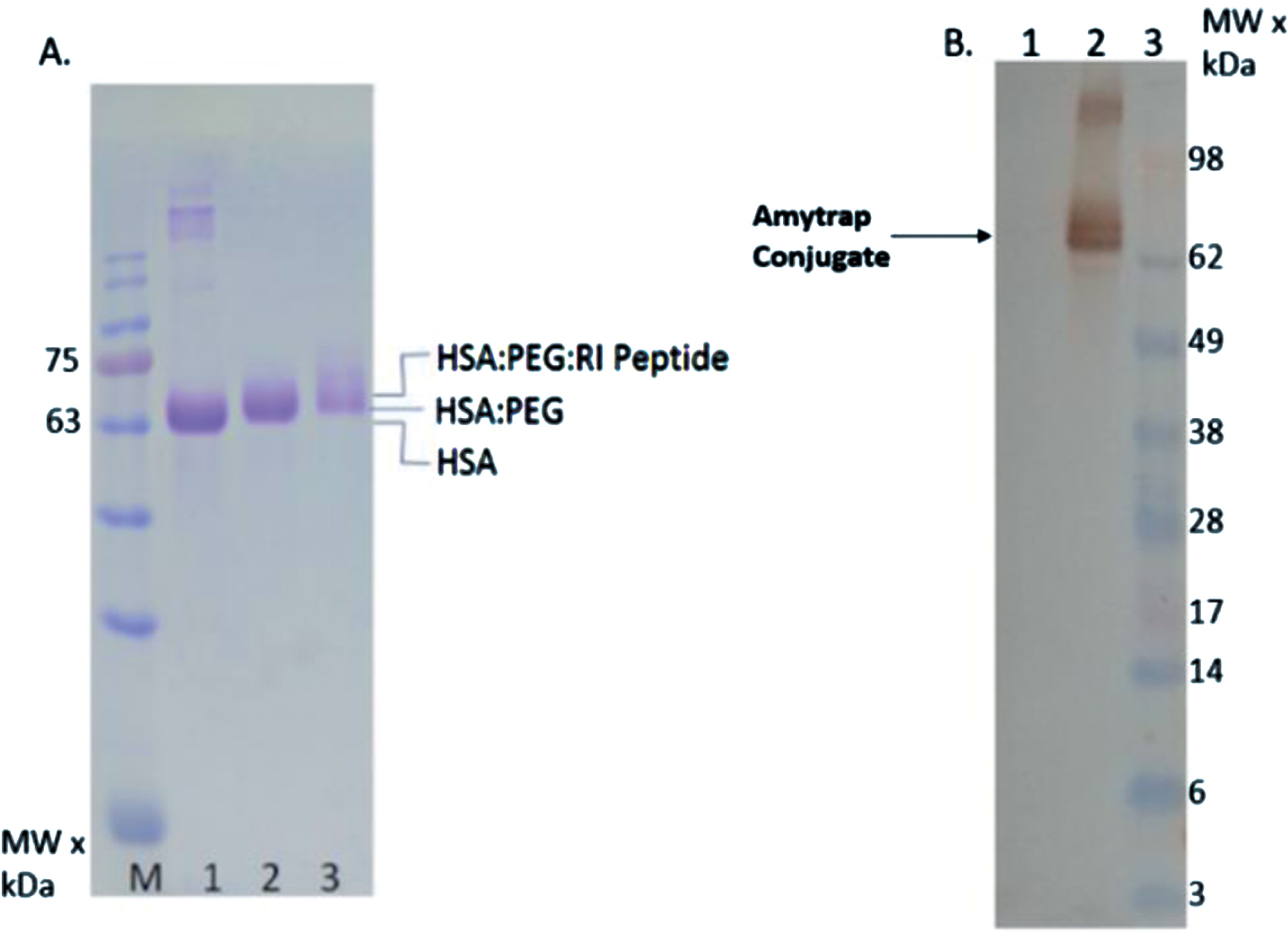
Click chemistry was followed as described in the methods to conjugate the RI-peptide to PEG and human serum albumin (HSA) and was purified by HPLC. In order to verify the click chemistry product, SDS-PAGE followed by Coomassie staining was performed with an aliquot of the Amytrap-conjugate and the intermediary products. As anticipated, a shift in MW matching the expected size was observed with the HSA-PEG-RI-Peptide relative to HSA or HSA-PEG, indicating successful formation of the Amytrap conjugate (Fig. 4A). The HSA band was observed on Coomassie stained gel at the right MW (>62 kDa, Fig. 4A, lane 1). In addition, western blotting was performed utilizing an anti-RI-peptide antibody which confirmed the shifted conjugate band contained the RI-peptide. HSA, used as negative control, did not show reactivity to the antibody (Fig. 4b, lane 1). Thus, the results proved beyond doubt the conjugation process was successful.
Fig.4
Conjugation of Amytrap peptide to albumin. A) Analysis of Amytrap conjugate by SDS-PAGE, Lane M, MW marker, lane 1, HSA, lane 2, HSA-PEG, lane 3 Amytrap conjugate (HSA-PEG-RI-peptide). B) Analysis of the conjugate by western blot. The blot was probed with anti-RI-peptide antibody followed by peroxidase labeled antibody. Lane 1, HSA, lane 2, Amytrap conjugate, lane 3, MW marker.
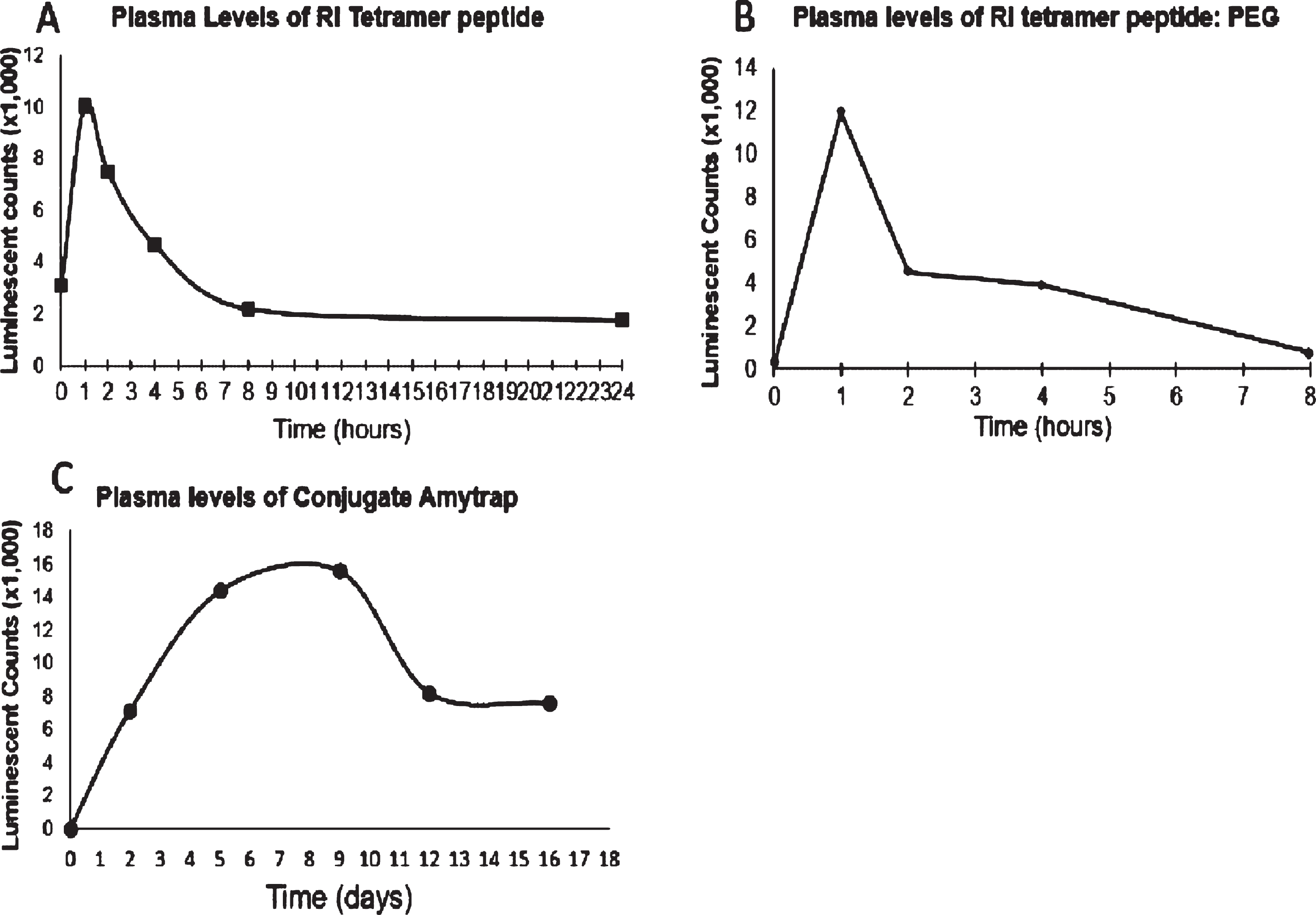
Bioavailability of Amytrap conjugates
To establish dosing parameters and determine the availability of Amytrap conjugate, Balb/c mice were injected subcutaneously with a single dose (200 μg) each of RI-peptide, RI-peptide-PEG, or Amytrap conjugate. We tested the presence of the RI-peptide alone, RI-peptide-PEG, or Amytrap conjugate (RI-peptide-PEG-HSA) in Balb/c mice (Fig. 5). This was important to determine the dosing interval of injections for efficacy and safety studies. RI-peptide could be detected up to 4 h and the RI-peptide-PEG could be detected up to 8 h (Fig. 5a, b). The complete Amytrap conjugate could be detected until 12 days (Fig. 5c). The experiment was repeated and the data was reproducible. From these results we determined the bioavailability of Amytrap conjugate to be 12–14 days as well as the injection duration of every 15 days in the efficacy and safety studies. Preliminary studies performed in live-imaging Balb/C mice utilizing fluorescent Amytrap peptide showed that Amytrap did not cross the blood-brain-barrier (unpublished). Our original hypothesis that Amytrap exerts its Aβ42 sequestration via peripheral suction was thus validated.
Fig.5
Mouse PK analysis. Balb/C mice received a single injection of (A) RI tetramer, (B) Pegylated RI-peptide, and (C) Amytrap Conjugate at 200 μg. Plasma levels of (A) RI tetramer peptide and (B) Pegylated RI-peptide at specified time points (0, 1, 2, 4, 8, and 24 h) after injection of the compound were analyzed. C) Amytrap conjugate was injected and plasma levels at 0, 2, 5, 9, 12, and 16 days were analyzed. Values are expressed as luminescent counts (x1000).
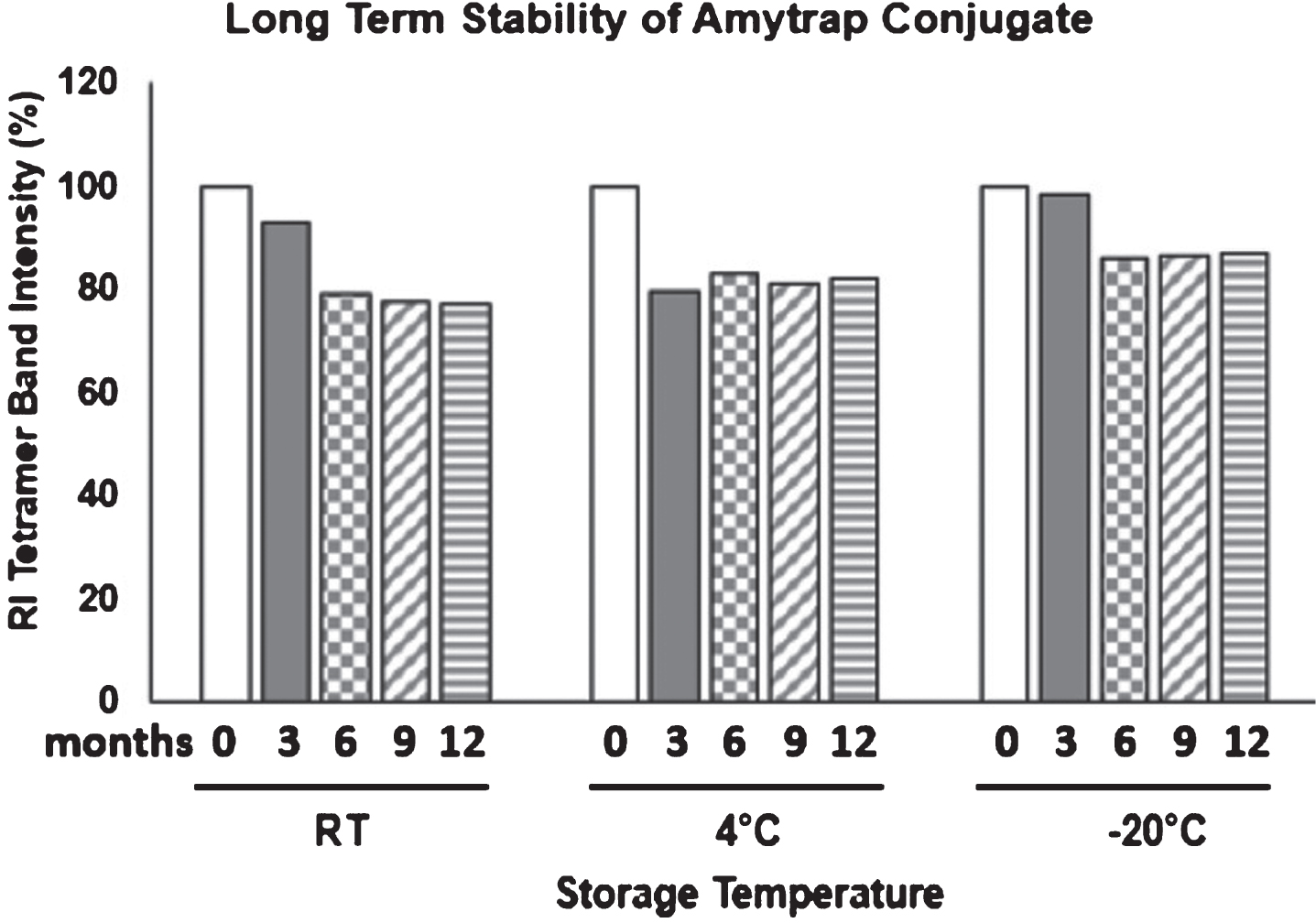
Amytrap conjugate stability on storage
To assess the long-term stability of the Amytrap conjugate, a 1-year stability test was performed. Amytrap conjugate powder was pre-weighed into 12 microcentrifuge tubes and stored at RT, 4°C, and –20°C for 4 timepoints. At specified time points, Amytrap conjugate powder was dissolved in filtered distilled water and analyzed by western blotting. The intensity of the band from the 0-month sample was taken as 100% and the relative values from other samples were plotted (Fig. 6). Amytrap stored at RT and –20°C was stable for the first three months but the stability decreased after. At 4°C, Amytrap conjugate decreased in stability at 3 months but was able to maintain its stability up through 12 months.
Fig.6
Long-term stability of Amytrap conjugate. Stability of Amytrap conjugate at RT, 4°C and -20°C was tested at 3, 6, 9, and 12 months as represented by tetramer band intensity on immunoblot. Samples were dissolved and analyzed by SDS-page. 3-month samples were taken as 100%.
Efficacy evaluation of Amytrap in 5-month-old mice
Efficacy evaluation of the Amytrap conjugate was performed in 5-month-old APPSWE/Tg2576 mice. Three groups of mice (n = 12) as described in the methods section were used. Control mice received saline, while the test groups received 100 μg doses of Amytrap or [FFVLK]4, respectively. Injections were given subcutaneously, biweekly for 5 months. Blood was collected before and at the end of the study. Y-maze tests were performed before dosing and 10 days after the last injection.
Reduction of brain amyloid
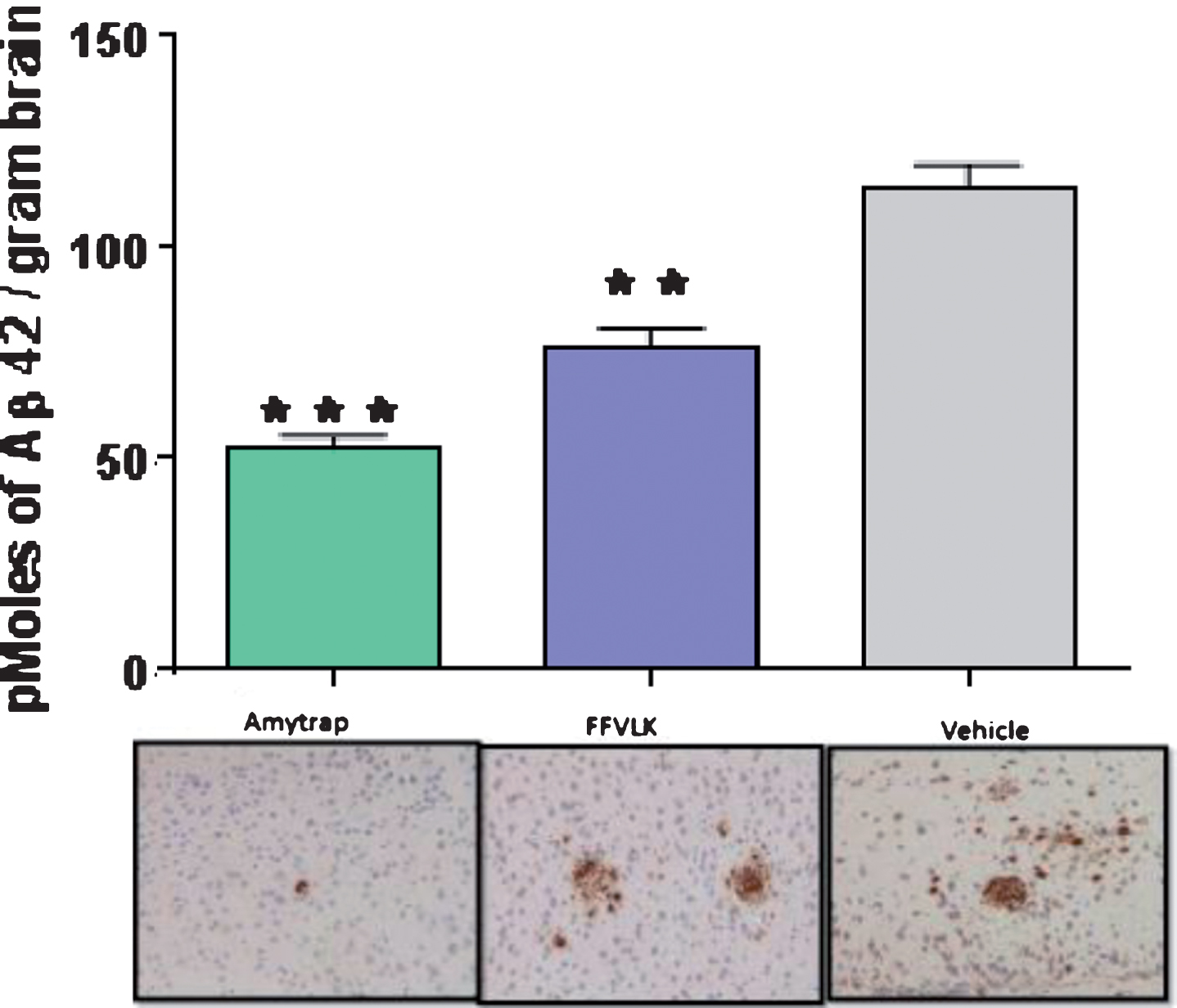
Brain Aβ42 levels were analyzed from homogenate extracts from APPSWE 2576 AD model mice from control and treated groups to evaluate Amytrap against FFVLK. Brain homogenates from mice treated with FFVLK or Amytrap had significantly lower levels of Aβ42 plaque formation in comparison with control mice (Fig. 7). Brain homogenates from Amytrap treated mice showed significantly greater reduction in Aβ42 when compared with FFVLK as well. Representative images of brain sections show this reduction as well.
Fig.7
Brain amyloid in 5-month-old AD model mice. Aβ42 content in brain extracts by ELISA (histogram) and brain sections (lower panel) by immunohistochemistry from mice treated 5 months (n = 12). Values are expressed as Mean±SD and statistical significance was calculated by the students t-test; ***p < 0.001 and **p < 0.05
Cognitive changes as evaluated by Y-maze spontaneous alternation
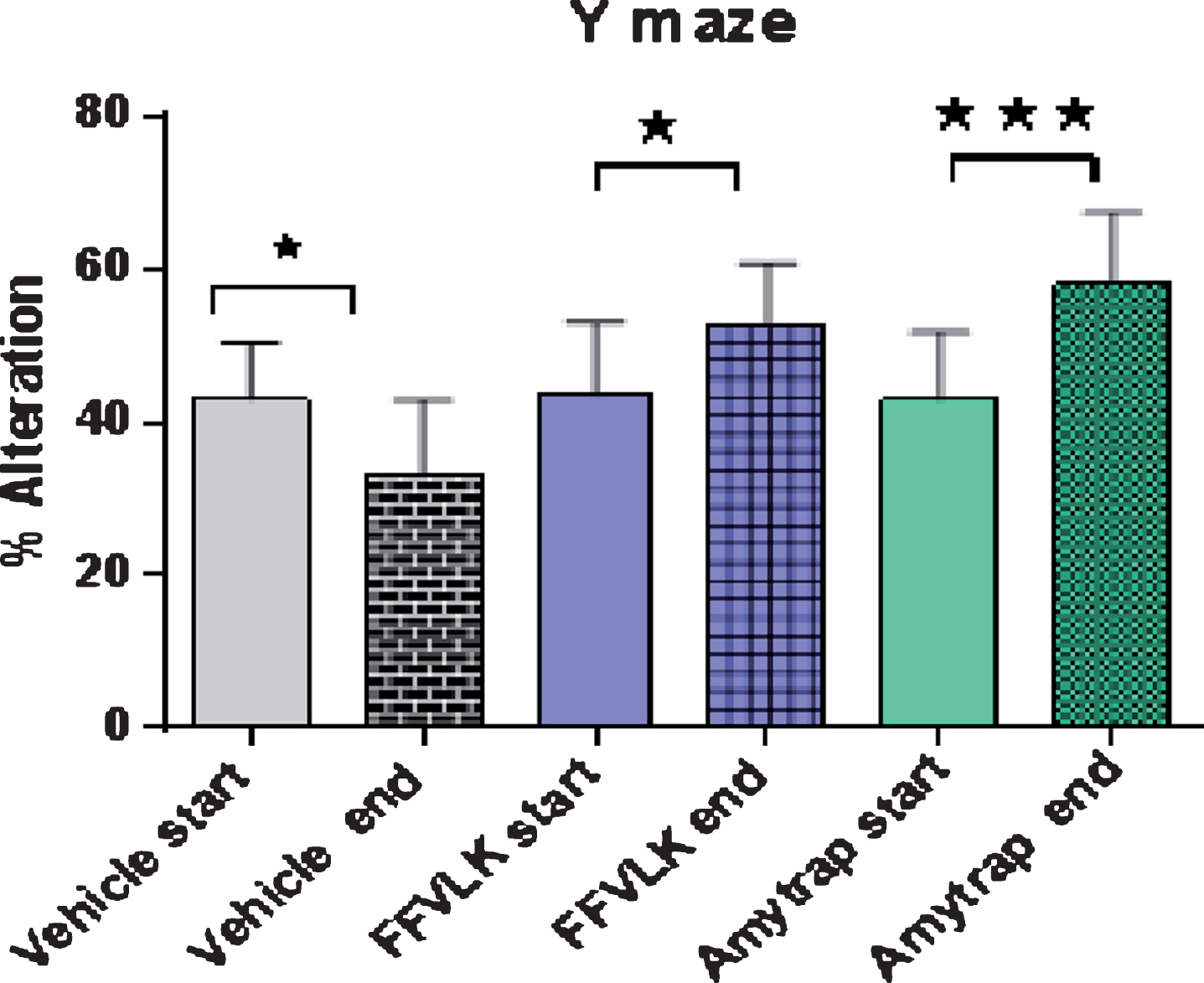
Experimental mice were assessed by spontaneous alternation in the Y-maze. Before the treatment phase of the study, a baseline analysis was performed for comparative purposes. AD model mice that were in the control group were evaluated and found to have a lower number of % alternations in comparison to the results from the start of the study (Fig. 8). Mice from both of the treatment groups [FFVLK]4 and Amytrap showed improved % alternation results after treatment. The mice that were treated with Amytrap showed even greater signs of improvement in cognitive function in comparison to mice treated with [FFVLK]4.
Fig.8
Y-maze % alternation in 5-month-old AD model mice. Changes in percent alteration in Y maze task performed by 5-month-old mice following treatment with vehicle, FFVLK or Amytrap at 100 μg monthly for 5 months. Start and End in the figure equate to Before treatment and After treatment, respectively. Values are expressed as Mean±SD (n = 12). *p < 0.05, ***p < 0.001.
Efficacy evaluation of Amytrap in 9-month-old mice
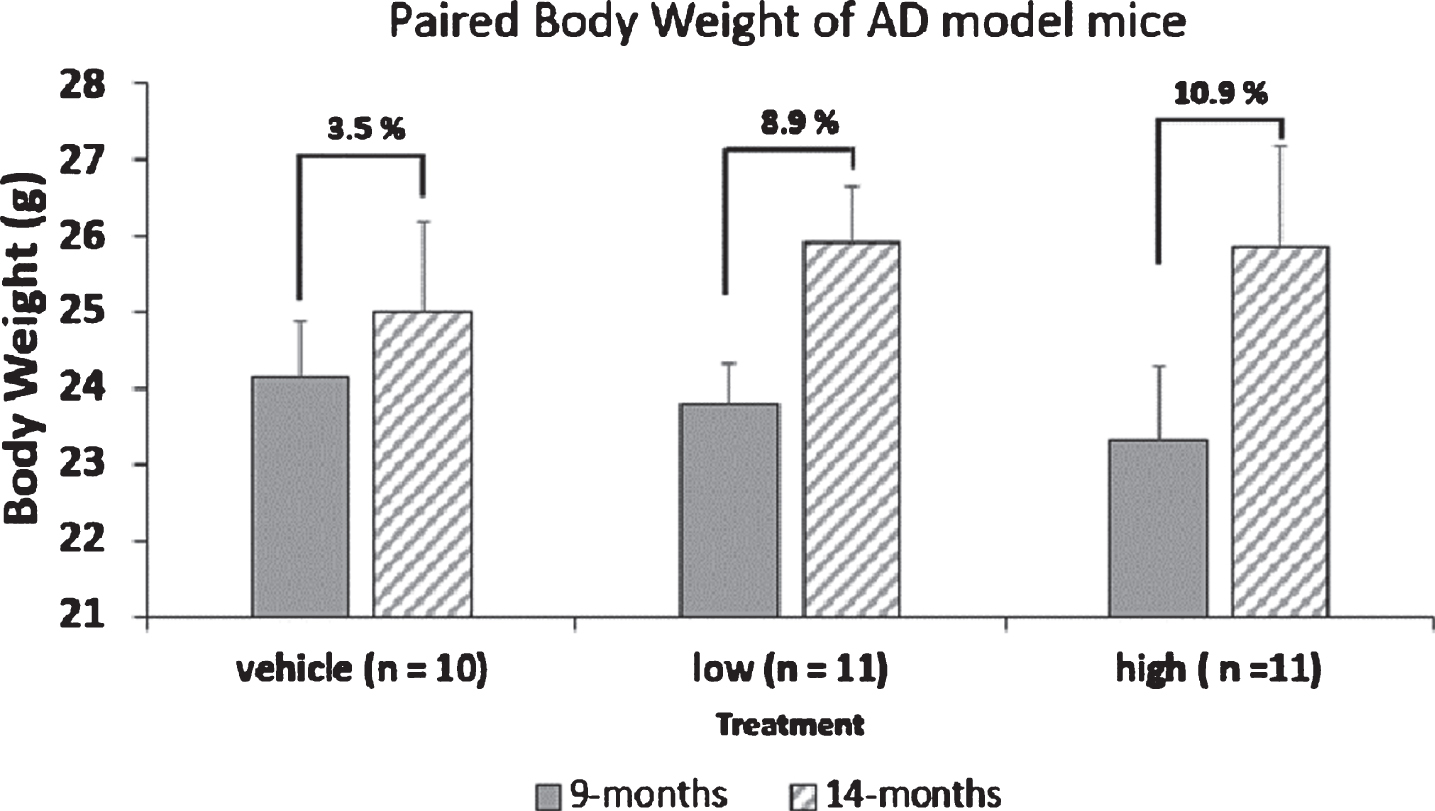
Evaluation of Amytrap to recover cognitive faculties in 9-month-old AD model mice was performed. Three groups of mice (n = 10/group) as described in the methods section were used. Control mice received saline, while the test groups received 6.45 and 16.1 mg/kg body weight of Amytrap. Injections were given subcutaneously, biweekly for 5 months. Body weight, food intake, and other routine monitoring was performed. No change in food intake was found, however, mice treated with Amytrap (low and high dose) had gained more body weight than control mice (Fig. 9). Blood was collected before and at the end of the study. Y-maze test and Novel object recognition tests were performed 10 days after the last injection.
Fig.9
Paired body weight of AD model mice. Mice were weighed at the start and throughout the study. Individual weights were obtained in grams and the average weight for mice at 9 months of age and 14 months of age (start and end) of the study were calculated. The values given represent body weight as Mean±SEM.
Measurement of Aβ40 and Aβ42 levels in plasma, CSF and immunohistochemistry
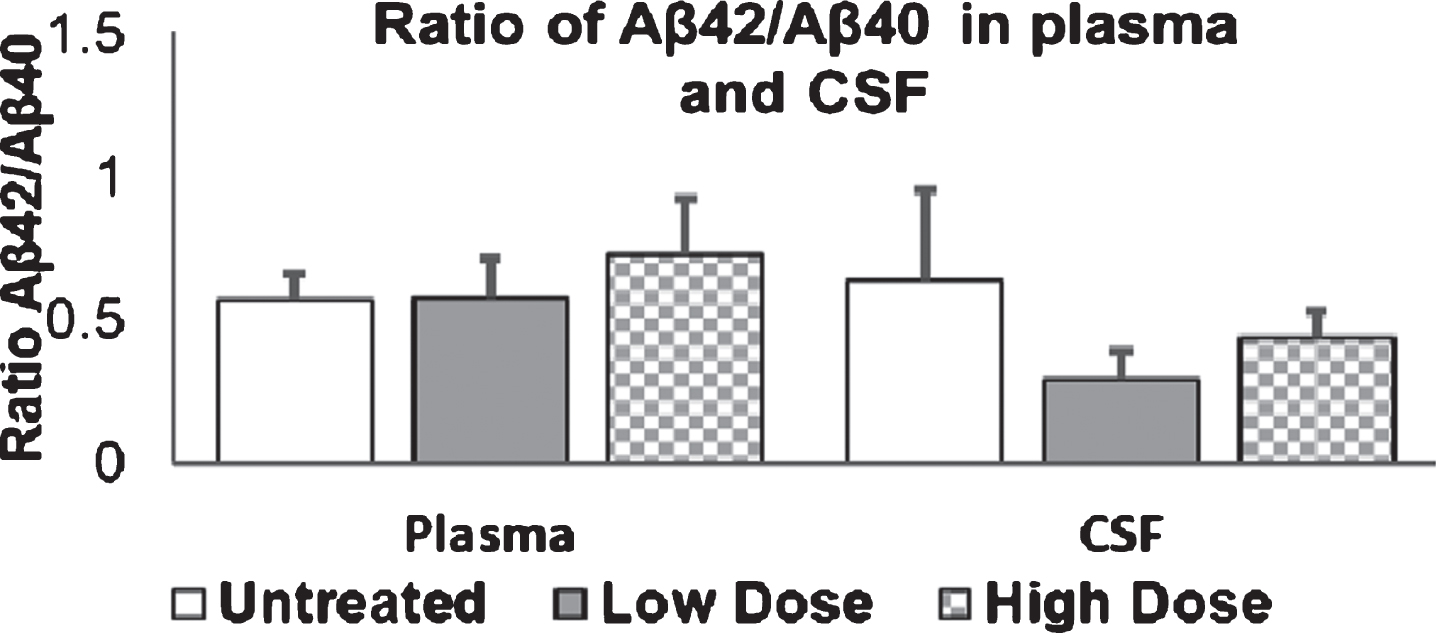
Aβ40 and Aβ42 levels were measured in plasma and CSF. Results are presented in Figs. 10 and 11. Amytrap injection induced a reduction in Aβ40 and Aβ42 levels in plasma of treated mice compared to untreated controls. Although there was a concentration dependent decrease of Aβ40 in the plasma, the reduction in levels of Aβ42 in the low dose group was evident, but higher dose did not further decrease the levels (Fig. 10). A significant difference can be seen when the low dose and high dose were compared with the control group. CSF concentrations of Aβ40 were found to gradually increase with increasing doses of Amytrap conjugate. Aβ42 levels in CSF were found to be on average 4, 2, or 3 ng/ml for the control, low, or high dose, respectively. CSF levels of Aβ42 was found to decrease with low and high doses, while Aβ40 does appear to be elevated in concentration (Fig. 11). In addition, the ratio of Aβ42 to Aβ40 was calculated to see if Amytrap treatment alters it (Fig. 12). Plasma ratios of Aβ42 to Aβ40 were found to be slightly elevated in mice that received high dose of Amytrap but did not change with low dose when compared to the control. The ratio was almost significantly reduced in the low dose whereas less significant reduction was observed in the high dose.
Fig.10
Aβ40 and Aβ42 levels in plasma of AD mice. 9-month-old mice were injected with Amytrap conjugate (6.45 or 16.1 mg/kg; Low or High dose) or saline (control) biweekly for 5 months. Plasma amyloid is expressed in pg/mL. Values are expressed as Mean±SE. Significance of Aβ40 or Aβ42 was determined using Student’s t-test *p < 0.05, **p < 0.01. Experiments were repeated in triplicate.
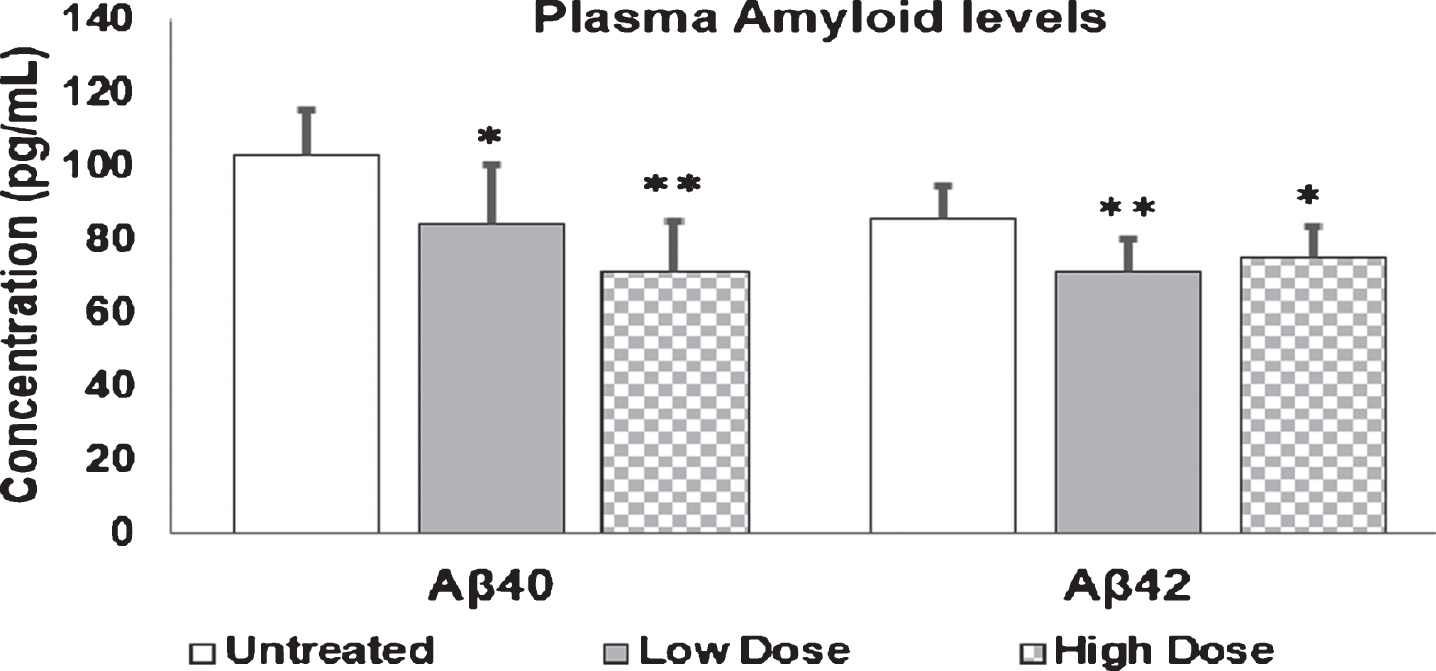
Fig.11
Aβ40 and Aβ42 levels in mice CSF. 9-month-old mice were injected with Amytrap conjugate (6.45 or 16.1 mg/kg; Low or High dose) or saline (control) biweekly for 5 months. CSF amyloid levels are expressed in ng/mL. Values are expressed as Mean±SE. Experiments were repeated in triplicate. Vehicle/control (n = 10), Low dose or high dose (n = 11).
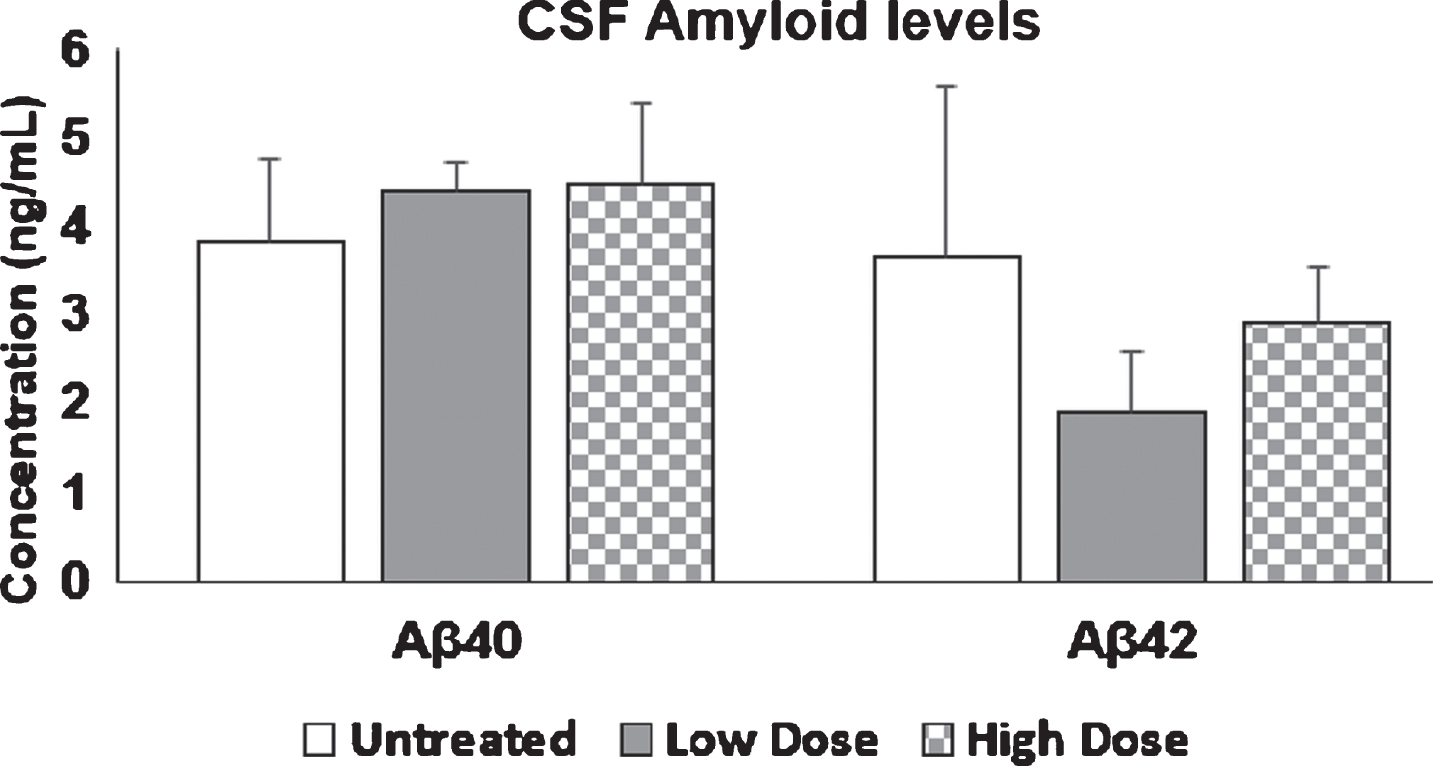
Fig.12
Ratio of Aβ42/Aβ40 in plasma and CSF. 9-month old mice were injected with Amytrap conjugate (6.45 or 16.1 mg/kg; Low or High dose) or saline (control) biweekly for 5 months. Ratios were taken for both Plasma and CSF Aβ42/Aβ40. Values are expressed as Mean±SE. Significance was determined using Student’s t-test. Vehicle/control (n = 10), Low dose or high dose (n = 11).
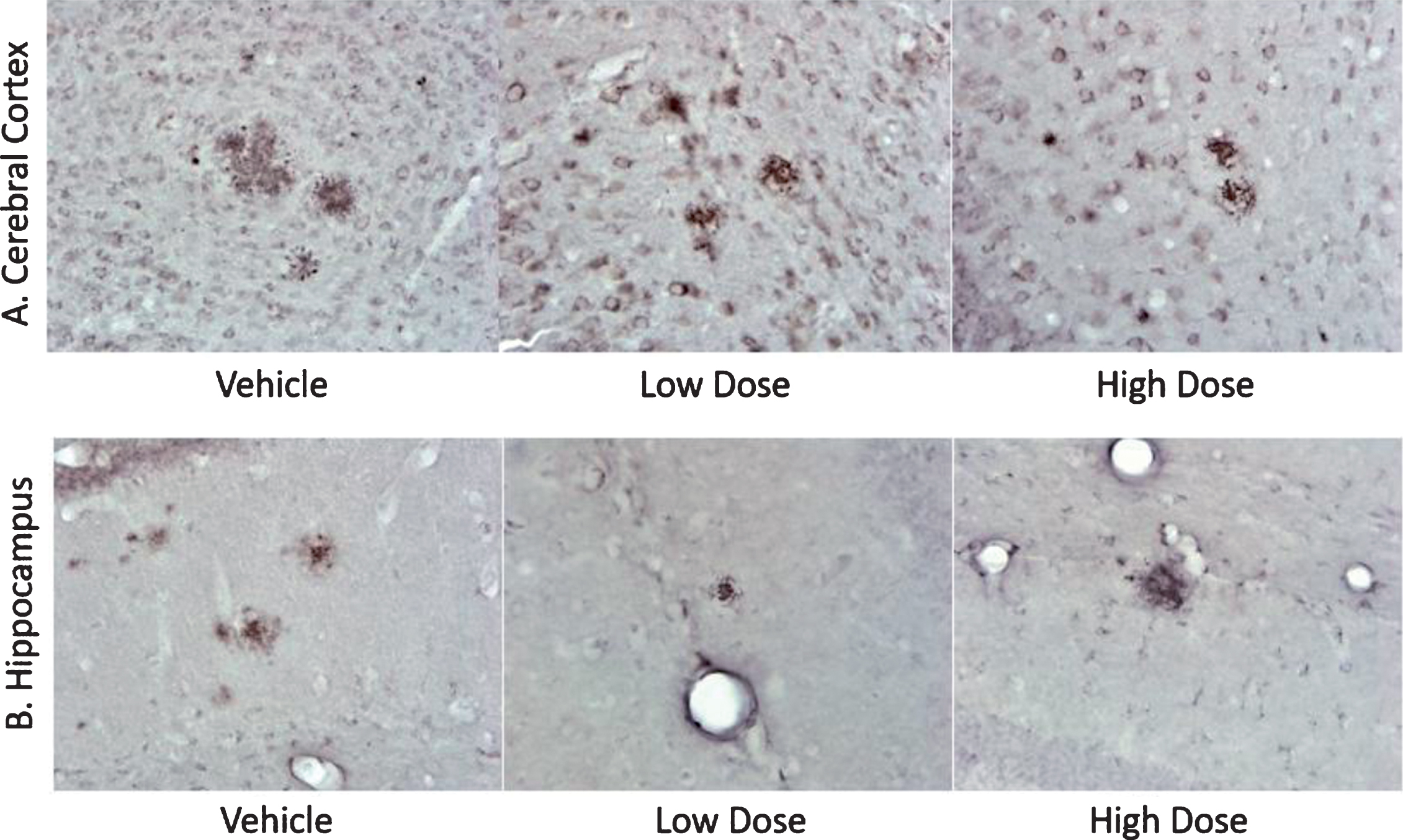
Immunochemical analyses showed that the number of plaques in the cerebral cortex and hippocampi of mice upon treatment with Amytrap were reduced in the low dose and high dose but neither appear to be statistically significant (Table 3). Images of cerebral cortex and hippocampi reflect these results (Fig. 13).
Table 3
Analysis of Amytrap’s effect on reducing amyloid plaques. Mice brain sections (cerebral cortices) or hippocampi were analyzed for the number of amyloid plaques and plaque sizes. Significance was calculated utilizing both ANOVA and Student’s t-test. Values are expressed as Mean±SD
| Mouse Group | Cerebral Cortices Number of Plaques | Cerebral Cortices Plaque surface area (μm2) | Hippocampi Number of Plaques | Hippocampi Plaque surface area (μm2) |
| Control | 196.3±53.3 | 104386.6±27175.7 | 37.4±25.0 | 18948.1±10262.5 |
| Low Dose | 140.7±30.9 | 89632.2±20023.9 | 17.8±6.9 | 10946.3±3506.1 |
| High Dose | 196.6±65.0 | 105441.0±32915.2 | 28.9±14.3 | 17990.7±6235.4 |
Fig.13
Histology sections of AD mice from efficacy study. Immunohistology sections of cerebral cortices (A) and hippocampi (B) from experimental mice as indicated representing amyloid plaques (dark red spots, at 40x magnification). The cumulative data representing total number of plaques and the surface area are presented in Table 2.
Behavioral evaluation by Y-Maze spontaneous alternation
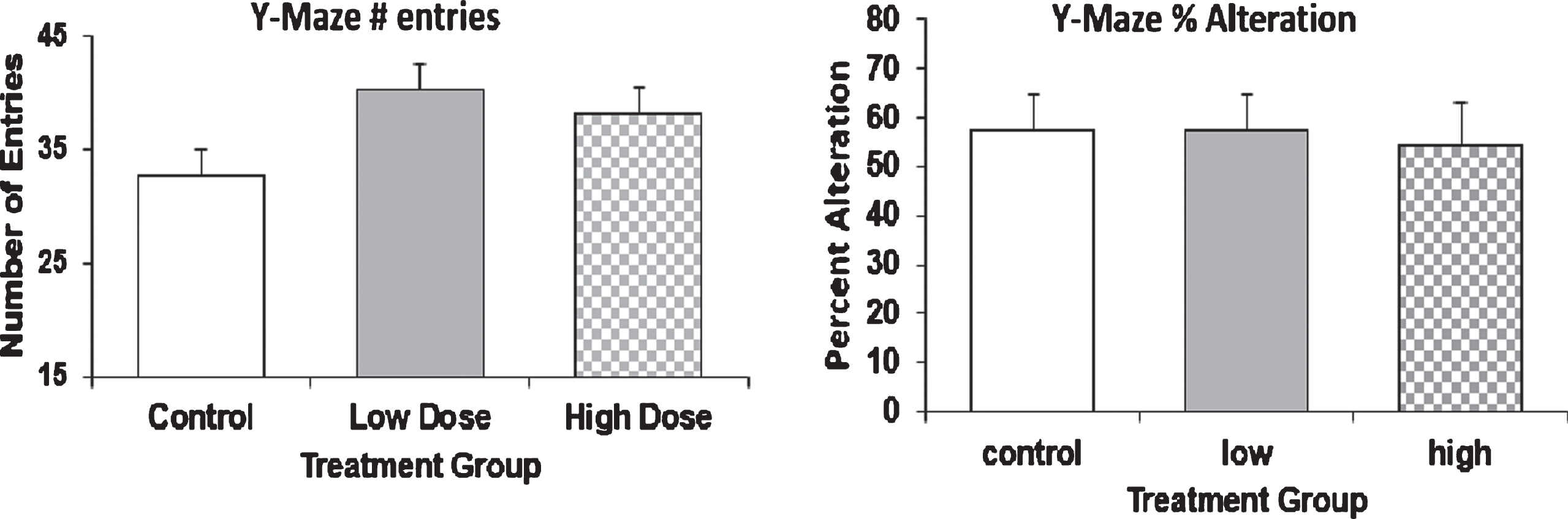
Experimental mice were assessed by entry and spontaneous alternation in the Y-maze. One untreated animal was excluded from the total number of entries data and one animal treated with high dose was excluded from the percent alternation data, as these data points were identified as significant outliers using Grubb’s Outlier Test. No significant differences between treatment groups were found in either parameter (One-Way ANOVA). All groups displayed low percent alternation (54–57%, Fig. 14) in Y-maze task and no differences were found between each group in the number of entries.
Fig.14
Behavioral analysis of AD mice by Y-Maze. 9-month old mice were injected with Amytrap conjugate (6.45 or 16.1 mg/kg; Low or High dose) or saline (control) biweekly for 5 months. Behavioral analysis was tested through Y-Maze. The number of entries (left) and percent alteration (right) in the Y-Maze. Values are expressed as Mean±SD.
Behavioral assessment by novel object recognition
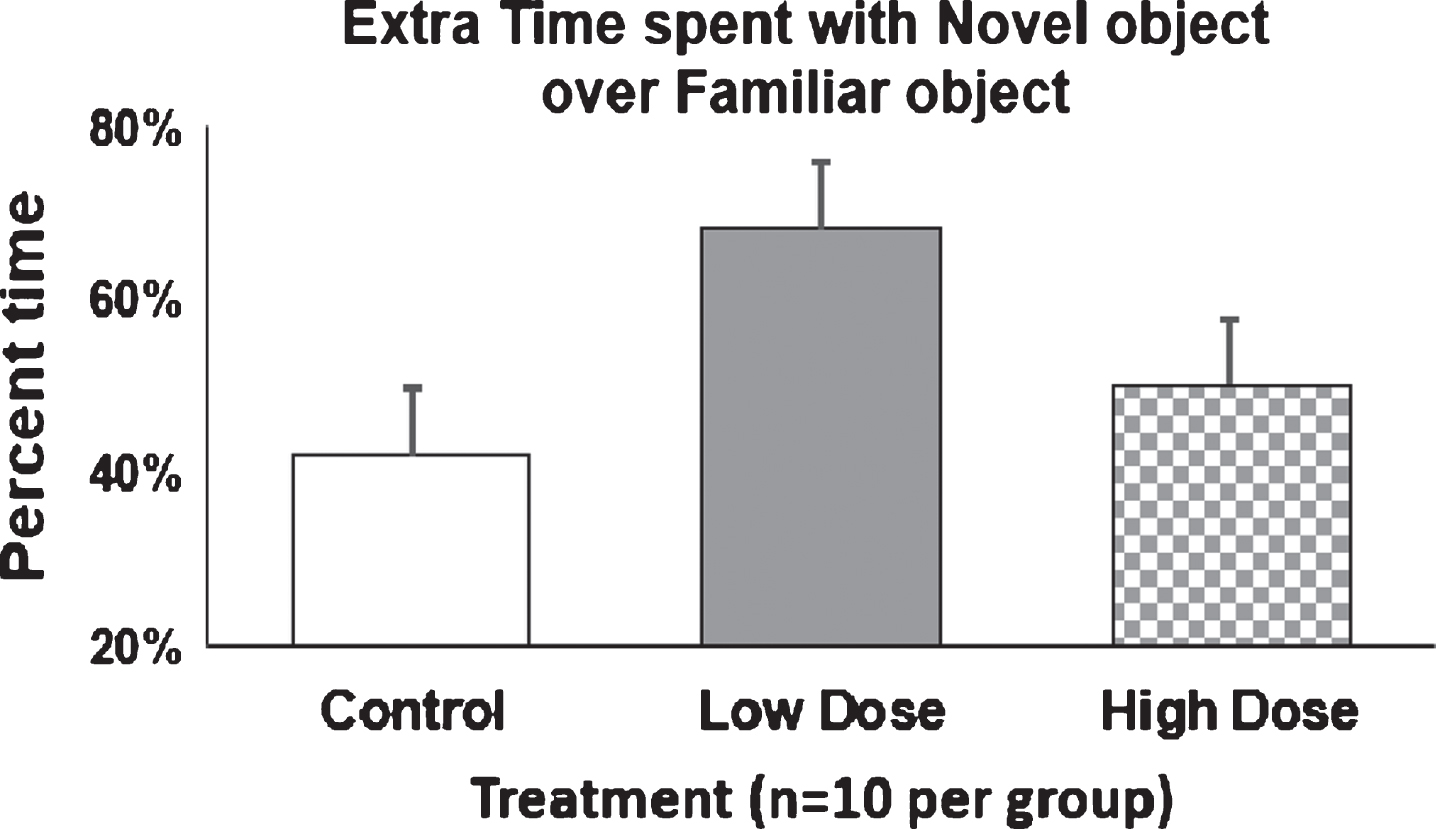
The 10-min NOR task was performed as an additional behavioral assessment to detect if Amytrap conjugate treatment led to cognitive improvements. Prior to data analysis, animals were excluded according to the predetermined exclusion criteria as described in the methods. Based on these criteria, animals were excluded as follows: Untreated n = 1; Low dose n = 3; high dose n = 1. No significant outliers were identified using Grubb’s Outlier Test. A two-way ANOVA was calculated and showed that treatment did affect how animals were exploring the objects, but there was no significant interaction between object exploration and treatment. Furthermore, none of the groups showed a significant preference for exploring the novel object compared to the familiar object (Sidak’s multiple comparisons post-hoc test). In addition to the previous data, the extra time that animals spent exploring the novel object versus familiar object was calculated. Untreated mice spent 42% more time with the novel object, low dose spent 68% more time, and high dose spent 52% more time with the novel object (Fig. 15), suggesting a statistically insignificant but noticeable improvement in cognitive behavior.
Safety toxicology
Fig.15
Extra time spent with novel object by AD mice. 9-month old mice were injected with Amytrap conjugate (6.45 or 16.1 mg/kg; Low or High dose) or saline (control) biweekly for 5 months. The percent difference in the extra time spent with the novel object over familiar object was calculated for each group and expressed as Mean±SD. Two mice each from low and high dose were found to be major outliers and removed from analyses. Vehicle/control (n = 10) and low and high dose (n = 10).
Safety toxicology in rats was performed to determine if Amytrap exhibits any toxicity. A total of 24 SD rats were divided into four groups (control, low dose, medium dose, and high dose) of six rats each and administered Amytrap as described in the methods. Amytrap conjugate did not cause physical abnormalities or death in any dose group during the study. In addition, Amytrap did not show any impact on bodyweight or food-intake in these animals. After 55 days the total protein, albumin, AST, ALT, creatinine, BUN, ALP, and total bilirubin content in serum were quantified to monitor the effect of Amytrap on liver and kidney function. None of the marker levels in each dosing group was statistically significant (ANOVA) compared to untreated controls (Table 4). Brain, liver, kidney, and heart tissue sections collected from rats were stained by H&E and examined for any histopathological changes. No significant or adverse effects between treatment groups or in comparison with control rats were found. In kidneys, mild glomerulopathy was present in multiple rats but not found to be statistically significant or relatively severe. One rat in the high dose group was observed to have a focal cardiac scar that was incidental but was not significant (Table 5). Thus, Amytrap conjugate was not found to be toxic in the Sprague Dawley rats even at 50 times the efficacy dose (high dose).
Table 4
Toxicology evaluation parameters in rats (n = 6 per group). Rat plasma was analyzed for the stated parameters in the left most column. Significance was analyzed by ANOVA for each group. Values are expressed as Mean±SD
| Group (Mean±SD) | Untreated | Low Dose | Medium Dose | High Dose |
| Alanine Transaminase U/L | 67.72±8.8 | 70.18±12.7 | 76.13±11.9 | 72.63±7.8 |
| Albumin g/dL | 4.19±0.2 | 4.24±0.2 | 4.22±0.3 | 4.01±0.2 |
| Alkaline Phosphatase U/L | 189.60±25.2 | 185.57±43.5 | 170.45±34.4 | 170.85±20.1 |
| Aspartate Transaminase U/L | 169.88±18.7 | 162.17±10.2 | 164.17±22.1 | 136.98±9.8 |
| Blood Urea Nitrogen mg/dL | 17.42±1.8 | 16.18±1.9 | 17.28±1.5 | 17.02±1.7 |
| Creatinine mg/dL | 0.31±0.0 | 0.33±0.0 | 0.32±0.0 | 0.33±0.0 |
| Total Bilirubin mg/dL | 0.05±0.0 | 0.04±0.0 | 0.04±0.0 | 0.04±0.0 |
| Total Protein g/dL | 6.28±0.2 | 6.16±0.2 | 6.15±0.4 | 6.13±0.2 |
Table 5
Rat tissue toxicology. Rat brains, livers, hearts and kidneys were analyzed for any toxic damage
| Group | Brain | Liver | Heart | Kidney | MPGN grade |
| Control | NSF | NSF | NSF | NSF | 0 |
| Control | NSF | NSF | NSF | NSF | 0 |
| Control | NSF | NSF | NSF | Mild MPGN | 2 |
| Control | NSF | NSF | NSF | NSF | 0 |
| Control | NSF | NSF | NSF | NSF | 0 |
| Control | NSF | NSF | NSF | NSF | 0 |
| Low | NSF | NSF | NSF | NSF | 0 |
| Low | NSF | NSF | NSF | Mild MPGN | 2 |
| Low | NSF | NSF | NSF | NSF | 0 |
| Low | NSF | NSF | NSF | NSF | 0 |
| Low | NSF | NSF | NSF | NSF | 0 |
| Low | NSF | NSF | NSF | NSF | 0 |
| Medium | NSF | NSF | NSF | Minimal MPGN | 1 |
| Medium | NSF | NSF | NSF | Minimal MPGN | 1 |
| Medium | NSF | NSF | NSF | Minimal MPGN | 1 |
| Medium | NSF | NSF | NSF | Minimal MPGN | 1 |
| Medium | NSF | NSF | NSF | NSF | 0 |
| Medium | NSF | NSF | NSF | NSF | 0 |
| High | NSF | NSF | NSF | Minimal MPGN | 1 |
| High | NSF | NSF | Focal scar | Mild MPGN | 2 |
| High | NSF | NSF | NSF | NSF | 0 |
| High | NSF | NSF | NSF | NSF | 0 |
| High | NSF | NSF | NSF | NSF | 0 |
| High | NSF | NSF | NSF | NSF | 0 |
Histological findings: See results for individual tissues examined in the table above. NSF, No significant histologic changes were observed. MPGN, multifocal membranoproliferative glomerulonephritis with protein casts. Grade 0 - no lesions. Grade 1 - minimal multifocal; Grade 2 - mild multifocal. Scar, cardiomyocyte dropout with interstitial fibrosis and reactive inflammation.
Assessment of immunogenicity by Amytrap
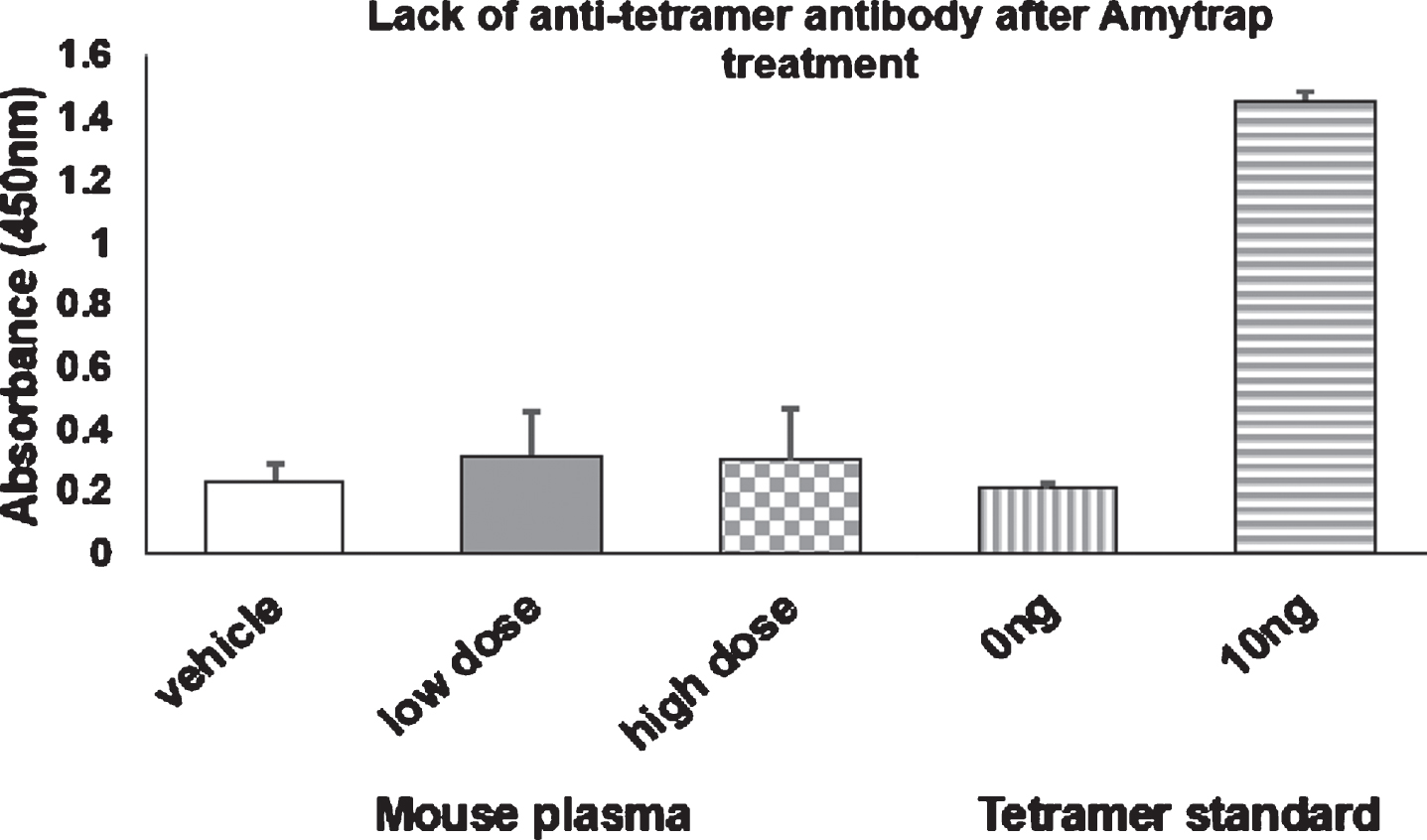
In order to assess the possible immunogenicity against the injected Amytrap conjugate, plasma harvested from the experimental mice was analyzed for the presence of anti-tetramer antibodies by ELISA. A pure anti-tetramer antibody was used as a positive control for binding of the Amytrap tetramer peptide. Mice treated with the saline vehicle were found to have absorbances similar to the wells treated with 0 ng of tetramer. Both the low dose and high dose treated mice have slightly elevated levels but were not found to be significantly different from vehicle treated mice. The results indicate no antibodies were detectable in mice sera treated with Amytrap conjugate thus confirming no immunogenicity was induced by Amytrap (Fig. 16).
Fig.16
Immunogenicity of Amytrap peptide. 9-month-old mice were injected with Amytrap conjugate (6.45 or 16.1 mg/kg; Low or High dose) biweekly for 5 months. Amytrap peptide was coated into each well at 10 ng/well. Measurement of anti-RI-peptide antibody in plasma in comparison with a standardized anti-RI-peptide antibody. Absorbance values are expressed as Mean±SE of absorbance. Each mouse plasma was taken in triplicate from vehicle (n = 10), low dose (n = 11), and high dose (n = 11) groups. Experiments were performed in triplicate.
DISCUSSION
Extensive efforts were devoted to developing therapeutic interventions to treat AD. Aβ42 or tau protein have been the most studied targets [4, 5]. Recombinant Technologies has been pioneering the development of non-immune, peptide-based therapeutics to treat AD for over a decade [16, 25, 30, 31]. Initially our research led to the development of a prototype RI-peptide (with seq from 16–23 of Aβ peptide) which was utilized to obtain proof of concept data in vitro followed by in vivo studies in APPSWE/Tg2576 mice [16]. The present work is an extension of this earlier work with the identification and characterization of a patented Amytrap peptide (RI-peptide) conjugate, HSA-PEG-RI-peptide, designed to specifically target, bind, and sequester Aβ42. Multiple studies have been recently performed supporting the efficiency of D-peptides as therapeutic compounds to treat AD [32]. RI-peptides have been shown to be very selective for their target and are thus less likely to cross-react with other proteins [33].
Concentration dependent binding of Aβ42 by the RI-peptide confirmed the authenticity of the synthesized peptide in vitro on microplates and in cell culture system. These studies found that the RI-peptide was nontoxic in cells at 20 μM concentrations and it was able to reverse Aβ42-induced toxicity. This observation confirmed Aβ42 target engagement by the synthesized RI-peptide earlier in our own study [25] and agreed with similar studies [34]. Conjugation of this peptide to both PEG and HSA was successfully completed to form the HSA-PEG-RI-peptide conjugate. Previous studies have utilized PEG and HSA to increase peptide bioavailability and stability [35, 36] in circulation. Further, albumin is a biological carrier and is well known for its protective role in binding Aβ42 and inhibiting its oligomerization [37].
Bioavailability of the Amytrap conjugate and its components was established in order to confirm downstream dosing parameters. The period of time that Amytrap conjugate circulated was considerably longer than its intermediates (Fig. 4). While the intermediate products peaked within a few hours they could not be detected beyond 24 hours, Amytrap conjugate was available in circulation and could be detected until 14 days post injection. As we had anticipated the bio availability was improved by the conjugation of PEG and HSA. These observations are supported by similar studies where conjugation of peptides to PEG and HSA improved bioavailability [24].
Efficacy of Amytrap was evaluated in the clinically relevant APPSWE/Tg2576. Five-month-old mice treated with Amytrap showed significant reduction in brain Aβ42 level by quantitative measurement and by immunohistochemical analysis of brain tissue. These observations are in agreement with a previous study utilizing a different RI peptide [34]. However, in 9-month-old mice, Amytrap produced less significant benefits. While Amytrap was able to significantly reduce both Aβ42 and Aβ40 from the plasma, the same reduction was not observed in the CSF in 9-month-old mice. Amytrap is capable of depleting Aβ42, but it seems to be more indicative of an early treatment. Overall, younger mice showed more pronounced efficacy than older mice. These observations reflect the outcome from clinical trials wherein a drug targeting Aβ42 has been shown to be more efficacious in early or mild cases of AD than in moderate and severe cases [38]. It is clear that Amytrap does not enter the brain based on live imaging experiments performed on mice (unpublished observation). We believe that Amytrap acts like a sink by sequestering Aβ42 from the circulation via peripheral action and follows the path of HSA through the liver clearance.
Immunogenicity against the injected Amytrap conjugate was not observed as no antibodies to the tetramer peptide could be detected. The lack of antibody formation was not surprising as D-peptides are generally less toxic and have been found to have a lower probability of eliciting an immune response because of weaker binding to MHC II molecules [39]. Further, the lack of immune response makes Amytrap superior to most failed antibodies (which showed damaging immunologic side effects) to treat AD. Further, Amytrap is cost effective and easy to transport and administer.
The cognitive patterns of 5-month-old Amytrap-treated mice revealed improved behavior in the treated groups (Fig. 8). This was comparable to our positive control peptide [FFVLK]4 [16]. The observations from the 5-month-old mice study are also in agreement with similar studies [40, 41]. However, 9-month-old Amytrap treated mice revealed no significant improvement when compared to control mice although a pattern in the right direction could be observed. This performance was less favorable than what was seen in the 5-month-old mice study again agreeing with the pattern observed in human trials. Thus, the observation is consistent with human clinical trials wherein patients with longer duration of disease responded less favorably to Aβ42 targeted therapies compared to those with recent disease onset [38]. An alternate behavioral assessment test, 10-min NOR task was performed to determine any improvement in the 9-month-old mice. No significant difference was found in the length of time mice spent with the novel object over the familiar. However, the treated mice spent extra time exploring the novel object (Fig. 14) which may indicate an improvement in cognitive behavior. Again, the starting age of the mice in this study may have impacted the performance of Amytrap conjugate, thus leading to the lower benefit observed.
Immunohistochemistry was performed to evaluate brain Aβ42 plaques. In 5-month-old mice, Amytrap reduced the number and size of plaques within the brain (Fig. 7). This reduction was found to be even greater than the reduction by the positive control [FFVLK]4. In studies utilizing an RI-peptide to treat AD, a similar reduction in soluble Aβ42 plaque formation was observed [34]. However, in 9-month-old mice, the number and size of plaques in the cerebral cortex and hippocampi of mice upon treatment with Amytrap were reduced in the low dose (Table 2) but was not statistically significant. It is worth noting that the trend is positive in older mice too. A trend was observed that suggests that as the dose was increased the number of mice that did not develop Aβ42 plaques increased. The high dose had a lesser effect on plaque size and number. It could mean that the older mice need to be treated with Amytrap for a longer duration. Further, as discussed before the starting age of the mice appear to be critical. We also would like to speculate that it is possible that the high dose aggregates Amytrap-bound Aβ42 due to steric hindrance. This argument is based on earlier studies while designing the peptide. We synthesized monomer, tetramer, pentamer, hexamer, octamer, or decamer versions of the RI-peptide and allowed them to bind Aβ42 on ELISA. The results indicated that the tetrameric version had the best binding qualities with the best binding constant (Kd = 0.07×10-2 μM). The peptides containing more than 4 copies exhibited suboptimal binding of Aβ42, which could be due to steric hindrance. We feel that a similar hindrance could be attributable for the lack of significant reduction of Aβ42 levels in the high dose.
Amytrap toxicity was tested in SD rats. Blood chemistry was analyzed for alterations in a panel of parameters. Amytrap appeared to be nontoxic, even after 50X the efficacy dose. Necropsy results demonstrated that no significant or adverse pathology were observed upon exposure to Amytrap treatment at all doses tested. It is worth mentioning that these observations are consistent with other D-peptides, where no adverse effects were reported [39, 42]. Stability of the conjugate was analyzed through a 1-year stability study to establish temperature and light conditions with quarterly evaluations. While the RT storage was sufficient for the first 3 months, cooler storage temperatures appear to maintain the conjugates stability after 3 months. These observations are in agreement with other reports that have found D-peptide based therapeutics to have greater stability [35, 36, 39].
Overall, the outcome of these studies warrants further investigation into the application of Amytrap as a disease modulator or a combination therapeutic to treat AD. Amytrap was shown to be efficacious in young mice, which may mean that it is required for earlier treatment. Revisiting dosing in older mice may be necessary as Amytrap is moved forward towards human clinical trials.
CONFLICT OF INTEREST
We declare that there is no actual and potential conflict of interest on this study.
Ethics Approval and Consent to Participate
The animal studies were approved by Brains Online (CA) and Poconos Rabbit Farms and Laboratory (PA).
ACKNOWLEDGMENTS
Partial salary support to Dr. Pazhani Sundaram and Omkar Gandbhir was provided by a small business innovative research phase 2 grant # R44 AG0 50336, from National Institute on Aging, National Institutes of Health, USA. Dr. P.S., the principal investigator and corresponding author thanks NIH for the grant award. The authors would like to acknowledge and thank Drs. Madan Anant, Ranjini Sundaram, and Bijan Almassian for their technical support and advise with these studies.
REFERENCES
[1] | Alzheimer’s Association ((2006) ) Early onset dementia: A national challenge, a future crisis. Alzheimer’s Association, Washington, DC, https://www.alz.org/media/Documents/early-younger-onset-full-r.pdf |
[2] | Wilson RS , Weir DR , Leurgans SE , Evans DA , Hebert LE , Langa KM , Plassman BL , Small BJ , Bennett DA ((2011) ) Sources of variability in estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement 7: , 74–79. |
[3] | Alzheimer’s Association ((2012) ) 2012 Alzheimer’s disease facts and figures. Alzheimers Dement 8: , 131–168. |
[4] | Bloom GS ((2014) ) Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: , 505–508. |
[5] | Hong S , Beja-Glasser VF , Nfonovim BM , Frouin A , Li S , Ramakrishnan S , Merry KM , Shi Q , Rosenthal A , Barres BA , Lemere CA ((2016) ) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: , 712–716. |
[6] | Becker RE , Greig NH , Giacobini E , Schneider LS , Ferruci L ((2014) ) A new roadmap for drug development for Alzheimer’s disease. Nat Rev Drug Discov 13: , 156. |
[7] | Panza F , Solfrizzi V , Frisardi V , Capurso C , D’introno A , Colacicco AM , Vendemiale G , Capurso A , Imbimbo BP ((2009) ) Disease-modifying approach to the treatment of Alzheimer’s disease: From alpha-secretase activators to gamma-secretase inhibitors and modulators. Drugs Aging 26: , 537–555. |
[8] | Crump CJ , Johnson DS , Li YM ((2013) ) Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry 52: , 3197–3216. |
[9] | Zotova E , Bharambe V , Cheaveau M , Morgan W , Holmes C , Harris S , Neal JW , Love S , Nicoll JA , Boche D ((2013) ) Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain 136: , 2677–2696. |
[10] | Salloway S , Sperling R , Fox NC , Blennow K , Klunk W , Raskind M , Sabbagh M , Honig LS , Porsteinsson AP , Ferris S , Reichert M ((2014) ) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 322–333. |
[11] | Doody RS , Thomas RG , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , Raman R , Sun X , Aisen PS , Siemers E ((2014) ) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 311–321. |
[12] | Cummings JL , Cohen S , van Dyck CH , Brody M , Curtis C , Cho W , Ward M , Friesenhahn M , Rabe C , Brunstein F , Quartino A ((2018) ) ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 10: , 212. |
[13] | Chiao P , Bedell BJ , Avants B , Zijdenbos AP , Grand’Maison M , O’Neill P , O’Gorman J , Chen T , Koeppe R ((2018) ) Impact of reference/target region selection on amyloid PET standard uptake value ratios in the phase 1b PRIME study of aducanumab. J Nucl Med 18: , 118. |
[14] | Kresge N ((2018) ) Biogen surges after positive results in Alzheimer’s Trial. Bloomberg https://www.bloomberg.com/news/articles/2018-07-06/eisai-surges-after-positive-results-in-alzheimer-s-drug-trial?in_source=video_page. |
[15] | Goyal D , Shuaib S , Mann S , Goyal B ((2017) ) Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Aβ) aggregation: Potential therapeutics of Alzheimer’s disease. ACS Comb Sci 19: , 55–80. |
[16] | Sundaram RK , Kasinathan C , Stein S , Sundaram P ((2008) ) Detoxification depot for amyloid-β peptides. Curr Alzheimer Res 5: , 26–32. |
[17] | Sato T , Kienlen-Campard P , Ahmed M , Liu W , Li H , Elliott JI , Aimoto S , Constantinescu SN , Octave JN , Smith SO ((2006) ) Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry 45: , 5503–5516. |
[18] | Wiesehan K , Buder K , Linke RP , Patt S , Stoldt M , Unger E , Schmitt B , Bucci E , Willbold D ((2003) ) Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1-42 by mirror image phage display. Chembiochem 4: , 748–753. |
[19] | Chorev M , Goodman M ((1995) ) Recent developments in retro peptides and proteins—an ongoing topochemical exploration. Trends Biotechnol 13: , 438–445. |
[20] | Dintzis HM , Symer DE , Dintzis RZ , Zawadzke LE , Berg JM ((1993) ) A comon of the immunogenicity of a pair of enantiomeric proteins. Proteins 16: , 306–308 paris. |
[21] | Li H , Aneja R , Chaiken I ((2013) ) Click chemistry in peptide-based drug design. Molecules 18: , 9797–9817. |
[22] | Slagle CJ , Thamm DH , Randall EK , Borden MA ((2018) ) Click conjugation of cloaked peptide ligands to microbubbles. Bioconjug Chem 29: , 1534–1543. |
[23] | Hsiao K , Chapman P , Nilsen S , Eckman C , Harigaya Y , Younkin S , Yang F , Cole G ((1996) ) Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science 274: , 99–102. |
[24] | Kawarabayashi T , Younkin LH , Saido TC , Shoji M , Ashe KH , Younkin SG ((2001) ) Age dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21: , 372–381. |
[25] | Sundaram RK , Kasinathan C , Stein S , Sundaram P ((2012) ) Novel detox gel depot sequesters β-amyloid peptides in a mouse model of Alzheimer’s disease. Int J Pept Res Ther 18: , 99–106. |
[26] | Yang H , Hou T , Wang W , Luo Y , Yan F , Jia J ((2018) ) The effect of chronic cerebral hypoperfusion on amyloid-β metabolism in a transgenic mouse model of Alzheimer’s disease (PS1V97L). J Alzheimers Dis 62: , 1609–1621. |
[27] | Bevins RA , Besheer J ((2006) ) Object recognition in rats and mice: A one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1: , 1306–1311. |
[28] | Franklin KB , Paxinos G ((2007) ) The mouse brain in stereotaxic coordinates. Academic Press, San Diego. |
[29] | Gregori M , Taylor M , Salvati E , Re F , Mancini S , Balducci C , Forloni G , Zambelli V , Sesana S , Michael M , Michail C ((2017) ) Retro-inverso peptide inhibitor nanoparticles as potent inhibitors of aggregation of the Alzheimer’s Aβ peptide. Nanomedicine 13: , 723–732. |
[30] | Sundaram RK , Nambiar AK , Sundaram P ((2014) ) Patents on potential drugs to treat Alzheimer’s disease: Special emphasis on small peptides. Recent Pat CNS Drug Discov 9: , 71–75. |
[31] | Gandbhir O , Sundaram P ((2019) ) ‘Amytrapper’, a novel immobilized sepharose API matrix, removes amyloid-β from circulation. J Alzheimers Dis Rep 3: , 19–29. |
[32] | Klein AN , Ziehm T , van Groen T , Kadish I , Elfgen A , Tusche M , Thomaier M , Reiss K , Brener O , Gremer L , Kutzsche J ((2017) ) Optimization of D-peptides for Aβ monomer binding specificity enhances their potential to eliminate toxic Aβ oligomers. ACS Chem Neurosci 8: , 1889–1900. |
[33] | Cerchietti LC , Yang SN , Shaknovich R , Hatzi K , Polo JM , Chadburn A , Dowdy SF , Melnick A ((2009) ) A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 113: , 3397–3405. |
[34] | Parthsarathy V , McClean PL , Hölscher C , Taylor M , Tinker C , Jones G , Kolosov O , Salvati E , Gregori M , Masserini M , Allsop D ((2013) ) A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease.10. PLoS One 8: , 1371. |
[35] | Feng J , Zhao C , Wang L , Qu L , Zhu H , Yang Z , An G , Tian H , Shou C ((2018) ) Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics 8: , 2094. |
[36] | Di L ((2015) ) Strategic approaches to optimizing peptide ADME properties. AAPS J 17: , 134–143. |
[37] | Boada M , Ramos-Fernández E , Guivernau B , Munoz FJ , Costa M , Ortiz AM , Jorquera JI , Nunez L , Torres M , Paez A ((2016) ) Treatment of Alzheimer disease using combination therapy with plasma exchange and haemapheresis with albumin and intous immunoglobul: Rationale and treatment aroach of the AMBAR (Alzheimer Management By Albumin Replacement) study. Neurología 31: , 473–481. |
[38] | Sevigny J , Chiao P , Bussière T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J ((2016) ) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[39] | Hervé M , Maillére B , Mourier G , Texier C , Leroy S , Ménez A ((1997) ) On the immunogenic properties of retro-inverso peptides. Total retro-inversion of T-cell epitopes causes a loss of binding to MHC II molecules. Mol Immunol 34: , 157–163. |
[40] | Fernandez-Martos CM , Atkinson RA , Chuah MI , King AE , Vickers JC ((2017) ) Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimers Dement 3: , 92–106. |
[41] | Sigurdsson EM , Scholtzova H , Mehta PD , Frangione B , Wisniewski T ((2001) ) Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer’s disease-associated pathology in transgenic mice. Am J Pathol 159: , 439–447. |
[42] | Zhang M , Zhang M , Wang J , Cai Q , Zhao R , Yu Y , Tai H , Zhang X , Xu C ((2018) ) Retro-inverso follicle-stimulating hormone peptide-mediated polyethylenimine complexes for targeted ovarian cancer gene therapy. Drug Deliv 25: , 995–1003. |

