
Effects of Thioflavin T and GSK-3 Inhibition on Lifespan and Motility in a Caenorhabditis elegans Model of Tauopathy
Abstract
The nematode Caenorhabditis elegans (C. elegans) is a powerful model organism to study lifespan and aging, protein aggregation, and neurodegeneration, as well as to carry out drug screenings. The C. elegans strain aex-3/T337 expresses human pathogenic V337M mutant tau under a pan-neuronal promoter and presents uncoordinated locomotion, accumulation of phosphorylated insoluble tau, and shortened lifespan. Herein we have used this strain to assay two compounds that could affect tau aggregation and/or phosphorylation, and looked for phenotypic changes in their lifespan and motility. The first compound is Thioflavin T (ThT), a member of the tetracycline family with protein antiaggregant properties, yet to be tested in a tauopathy model. The second is a novel small molecule, NP103, a highly selective inhibitor of glycogen synthase kinase-3 (GSK-3), the main kinase contributing to pathogenic tau hyperphosphorylation. Importantly, we find that ThT extends lifespan of aex-3/T337 worms as it does with control N2 animals, showing both strains similar locomotion features under this treatment. By contrast, NP103 improves the paralysis phenotype of aex-3/T337 mutants but not their lifespan. Our results show that both treatments present beneficial effects for this model of tauopathy and encourage pursuing further investigations on their therapeutic potential for AD and other tauopathies.
INTRODUCTION
Aging is associated with a gradual decline of physiological and cognitive functions [1], and is a major risk factor for various neurodegenerative diseases, including Alzheimer’s, Huntington’s, and Parkinson’s diseases [2]. Although aging is a complex phenomenon, failure to maintain proper protein homeostasis plays a major role [3, 4]. Dysfunctional protein homeostasis results in protein misfolding and aggregation, being these two hallmarks characteristic of aging and age-related diseases [5]. Flavonoid Thioflavin T (ThT) belongs to the tetracycline family and is an amyloid-binding dye widely used for neuropathological analysis able to inhibit aggregation of pre-formed amyloid fibrils in vitro [6–8]. ThT has also been shown to promote longevity in the nematode Caenorhabditis elegans (C. elegans) and to significantly suppress amyloid-β (Aβ) aggregation-mediated paralysis [9].
Tau is the major neuronal microtubule-associated protein and its activity is regulated by its phosphorylation state [10]. Aberrant phosphorylation and aggregation of tau have been linked to axonal transport problems, synaptic malfunction and neurodegeneration [11] and are found in several neurodegenerative diseases collectively called tauopathies. Glycogen synthase kinase-3 (GSK-3) is the main kinase contributing to tau hyperphosphorylation, being hyperactivated in Alzheimer’s disease (AD) [12, 13]. Tau aggregates are amyloid in nature [14] and form neurofibrillary tangles that together with Aβ plaques are the two major histopathological hallmarks of AD [12, 13]. During the last years there is a growing interest in studying tau pathology in neurodegeneration as well as tau as a therapeutic target [15, 16]. In particular, studies have focused in tau aggregation, hyperphosphorylation, and other post-translational modifications [10, 17, 18].
The nematode worm C. elegans has become intensely studied due to its experimental advantages to investigate neuronal cell degeneration and death. Particularly, its short lifespan has made C. elegans to emerge as a powerful model organism for the study of aging [19–21]. Moreover, it is also an important model to study protein aggregation in aging as well as the molecular mechanisms of drug action and disease pathogenesis [22, 23]. C. elegans has been used as a model of various age-associated neurodegenerative diseases, including AD [24], Parkinson’s [25, 26] and Huntington’s disease [27]. Studying human tau mutants in C. elegans has the advantage of avoiding interference with endogenous tau, since the only tau homologue in the worm, PTL-1, is only expressed in a small subset of neurons [28, 29]. The C. elegans strain aex-3/T337 expresses full-length human tau bearing the pathogenic mutation V337M by using the aex-3 pan-neuronal promoter and shows sufficient similarities to human disease to give confidence that they are relevant to the toxic mechanism of human tauopathies [30, 31]. This line presents progressively worsening uncoordinated locomotion, accumulation of phosphorylated insoluble human tau, and shortened lifespan.
The structure of amyloidogenic intermediates, such as those formed by Aβ and tau, is highly conserved [32], suggesting that compounds blocking their formation independently of the protein they contain could be useful for several protein aggregation disorders. In fact, ThT and other tetracyclines can disaggregate Aβ oligomeric forms and other misfolded proteins [33–36]. However, to our knowledge there are no previous studies on the effects of these compounds on tau neurotoxicity, even though ThT binds tau aggregates and is also widely used as dye to stain tau amyloid fibrils [37–39]. The use of therapeutic compounds for aging-associated neurodegenerative diseases in C. elegans has been recently reviewed, but studies of ThT effects on tauopathy models are not documented [23].
Because ThT was successfully used to improve the phenotype of a C. elegans model of Aβ pathology [9], herein we have assayed this drug in the C. elegans model of tauopathy aex-3/T337, and analyzed whether it could improve the phenotype associated to tau toxicity. Importantly, we find that ThT extends the lifespan of aex-3/T337 worms. In addition, since GSK-3 is the major kinase driving tau pathologic hyperphosphorylation, we have also tested a novel and highly specific GSK-3 inhibitor, NP103. This novel compound improves significantly aex-3/T337 worms’ paralysis phenotype but shows no effects on the extension of their lifespan. Our results indicate that although both treatments can modulate tau-neurotoxic uncoordinated phenotype, ThT renders more beneficial effects associated to aging for this model of tauopathy.
MATERIALS AND METHODS
C. elegans strains
N2 (Bristol) was used as wild-type C. elegans [40]. Line aex-3/T337 [myo-2:GFP, aex-3:tau-V337M] carries chromosomally integrated transgenes encoding GFP and the 1N4R isoform of human tau carrying the V337M FTDP-17 mutation with expression driven by the pan-neuronal promoter aex-3. This strain is GFP-positive and shows a pronounced uncoordinated phenotype [30].
Maintenance and treatment of nematode strains
C. elegans strains were propagated at 20°C on solid sterile Nematode Growth Medium (NGM) seeded with 100 μl spots of E.Coli (OP50) for food on p60 plates. They were transferred to a fresh plate every 3-4 days. Synchronized animals were prepared by bleaching adult worms. Briefly, worms were collected with sterile water from a p60 plate. After washing all the bacteria from the supernatant they were lysed with hypochlorite solution for a few minutes. Then they were washed and centrifuged under a sucrose gradient. Finally, eggs were allowed to grow overnight in M9 medium and hatchlings were seeded on fresh NGM plates. The worms from L4 larvae stage (3 days old) were fed with the vehicle, Thioflavin T, or the GSK-3β inhibitor (NP103) at different concentrations as specified in figure legends and every single day they were transferred to a new fresh plate containing the compound.
Lifespan assay and paralysis analysis
Lifespan assays were performed as described in [9]. Briefly, twenty synchronized L4 worms were transferred to a fresh p35 plate containing 4 ml of agar in the presence or absence of 100 μL of the corresponding compound at different concentrations. Both plates and compounds were prepared under sterile conditions and spreading the compound over the surface of the plate. We considered that the compound had an even distribution throughout the 4 ml of agar. At this point, someone different from the scorer coded the plates so that the scoring was blind to experimental conditions. Plates were allowed to dry at room temperature for 1 h and then 60 μL of a concentrated suspension of E.Coli OP50 were spotted to form a circular layer in the center of the plate. All lifespan experiments were performed at 20°C. Every day animals were transferred to fresh plates prepared the day before and next day nematodes were scored as alive, dead or lost. We followed the same criteria established in [9] to determine if a worm was dead or not. Survival curves were plotted using Prism® 5.0 (GraphPad Software Inc, La Jolla, CA). At the moment of the passage, motility was visualized by the trails left by the worms crawling on the surface of the new plates by an observer blind to the plates codes. In the case of aex-3/T337, paralysis was analyzed by a scorer blind to experimental conditions assigning different values to worms with normal wild type-like movement (value 3), slow movement (value 2), or paralyzed (value 1), following a method to measure Aβ paralysis [41]. Paralyzed animals were distinguished from dead by testing movement by prodding the body with the worm picker. The percentage of aex-3/T337 worms scored with different values of movement along time of treatment with vehicle or NP103 was determined each day when passing the animals to a fresh plate.
Reagents, enzymes, and substrates
[α-33P]-ATP (1000–3000 Ci/mmol) was obtained from Amersham Biosciences (cat n° AH9968). The following human recombinant enzymes were purchased from Upstate: GSK-3β (cat n° 14–306), GSK-3β (cat n° 14–492), MAPK1/ERK1 (cat n° 14–439), CDK1 (cat n° 14–450), and PKA (cat n° 14–440), CK2 (cat n° 14–197). Human Recombinant Cdk5/p35 was purchased from Invitrogen (cat n° PV3000). The following enzyme substrates were purchased from Upstate: myelin basic protein (cat n° 13–104), histone H1 (cat n° 14–155), and Kemptide (cat n° 20–199). The phospho-glycogen synthase kinase peptide-2 (GS2) was purchased from Tocris (cat n° 1352). The proprietary compound NP103 was provided directly by the company Noscira S.A. Unless otherwise stated, all other reagents were of the highest purity available from Sigma (St Louis, MO).
Enzymatic assays
Enzymatic assays were performed essentially as described in [42]. Briefly, the activity of GSK-3β (both wild-type and C199A versions) was measured with the FRET-based Z’-LYTE™ technology from Invitrogen using the so-called “Ser/Thr-9” peptide, a fluorescein- and coumarin-double-labeled 11-mer based on the sequence of the human glycogen synthase I containing Ser-641, as peptide substrate. The assay was carried out at 25°C according to the manufacturer’s instructions in a final volume of 10 μL in 384-well low-volume round bottom black plates (Corning, Corning, NY) with 50 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM EGTA, and 0.01% Brij-35 as assay buffer. Enzyme concentration ranged from 2 to 5 nM as determined by active-site titration with the well-known inhibitor CT99021. Unless otherwise stated, ATP and peptide concentrations were 12.5 μM and 2 μM, respectively, corresponding to their respective previously determined Km values. Typically, the assays were run during 1 h in the presence or absence of compounds in a final DMSO concentration of 1% and samples were processed according to the manufacturer’s instructions. When concentration responses of the compounds were evaluated, their potencies were determined as the negative logarithm of the IC50 (i.e., the pIC50); as the experiments were carried out by performing serial dilutions, the range of concentrations was evenly distributed in a logarithmic rather than a linear scale. pIC50s were calculated by fitting the resulting data to Equation 1, using the nonlinear regression function of Prism® 5.0 (GraphPad Software Inc, La Jolla, CA).
ATP competition
Recombinant human GSK-3β activity was assayed in vitro at 0.5 mU/μL in the absence or presence of NP103 (5 and 25 μM) and increasing concentrations of ATP (from 3.125 to 100 μM) with [α33P]-ATP (0.025 μCi/μL). GS2 peptide (62.5 μM) was used as substrate. Reaction was carried out in a final volume of 20 μL for 30 min at 30°C. After that, 15 μL aliquots were spotted onto P81 phosphocellulose papers, washed four times in 1% phosphoric acid and counted in a Trilux Microbeta scintillation counter.
Statistical analysis
Data were analyzed and plotted by using the Graph Pad Prism® software (GraphPad Software Inc, La Jolla, CA) and the IC50 was calculated from the average of the logarithmic values obtained. The results are expressed as mean SEM. Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison test (selected pairs) where p < 0.05 was considered significant. A Chi-square test was used to compare movement differences of aex-3/T337 under the indicated treatment.
RESULTS
The anti-aggregant compound Thioflavin T extends the lifespan of the C. elegans model of tauopathy aex-3/T337
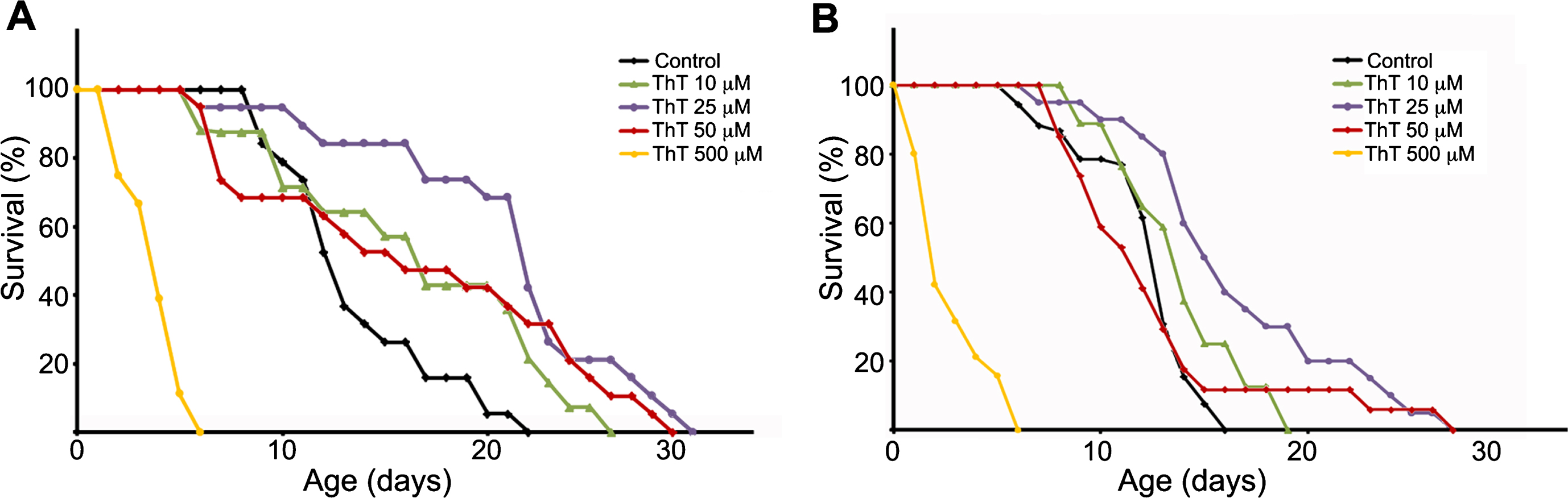
To investigate if ThT could have any beneficial effects on tauopathy models, we selected the C. elegans model of tauopathy aex-3/T337 [30], and tested different doses of ThT. In agreement with previous observations [30], we could confirm that these worms presented an uncoordinated phenotype and a shorter lifespan compared with wild type N2 strain (not shown). Before analyzing the effects of ThT in aex-3/T337 animals, we initially carried out control lifespan experiments using wild-type N2 worms treated with different concentrations of this drug. The treatment was started in synchronized worms when they reached the L4 stage, and was maintained at 20°C until the experiment was finished. Every day animals were transferred to fresh plates prepared the day before. First, we performed ThT dose-response curves (10 μM to 500 μM) to establish the optimal concentration rendering the maximum lifespan extension (Fig. 1A). Note that the highest ThT dose tested (500 μM) was toxic (Fig. 1A). However, lifespan of N2 worms treated with 10, 25, or 50 μM ThT was significantly increased when compared to those non-treated (Fig. 1A). Next, we performed ThT dose-response experiments using the aex-3/T337 strain and obtained similar results (Fig. 1B). While 500 μM ThT resulted equally toxic as to N2 control animals, there was a clear improvement of lifespan in aex-3/T337 worms exposed to lower doses of ThT (Fig. 1B). Visual observation after 16 days of treatment revealed that aex-3/T337 worms exposed to ThT presented a phenotype reminiscent of that of wild type animals, with improved locomotion (not shown).
Fig.1
Thioflavin T (ThT) extends lifespan of a C. elegans model of tauopathy. Dose–response Kaplan–Meier survival curve of synchronized populations of wild-type (N2) worms (A) or mutant aex-3/T337 worms (B), exposed to 0 μM (control) to 500 μM ThT at 20°C. These data correspond to one representative experiment out of three independent assays (n = 20 worms per experiment and dose).
A novel selective GSK-3 inhibitor improves the uncoordinated phenotype of aex-3/T337 strain without extending their lifespan
As GSK-3β is widely accepted as the major kinase responsible for tau hyperphosphorylation (reviewed in [43]), we studied whether its inhibition could contribute to lifespan extension and/or improvement in locomotion in aex-3/T337 worms. For that purpose, we used a novel GSK-3β inhibitor, NP103, a synthetic small molecule derived from the marine compound Oroidin developed by Noscira S.A. [44, 45]. NP103 inhibits in vitro recombinant human GSK-3β and GSK-3α with similar IC50s, of 0.56 μM and 0.29 μM, respectively (Fig. 2A, B). The compound shows a good solubility up to over 500 μM (slightly lower at very basic pH), a logP of 1.61, membrane permeability and is stable in biological fluids. Preliminary data in mice show NP103 having a very good (>70%) oral bioavailability and readily crossing the blood-brain barrier (20–25%).
Fig.2
Characterization of NP103 compound as GSK-3 selective inhibitor. A, B) NP103 inhibits GSK-3β and GSK-3α in vitro. Data represent the mean values + SD of the percentage of enzymatic activity. Each data point was assayed in duplicates. IC50 values were calculated from the concentration-response curves. C) Effect of different ATP concentrations on GSK-3β activity. A double-reciprocal Lineweaver-Burk plotting of the enzyme kinetics in the presence of two different concentrations of NP103 is shown. Each point represents the mean value of two independent experiments. No competition with substrate was observed (not shown). D) NP103 kinase inhibition selectivity profile. A comparison among external and in-house obtained results in selected kinases.
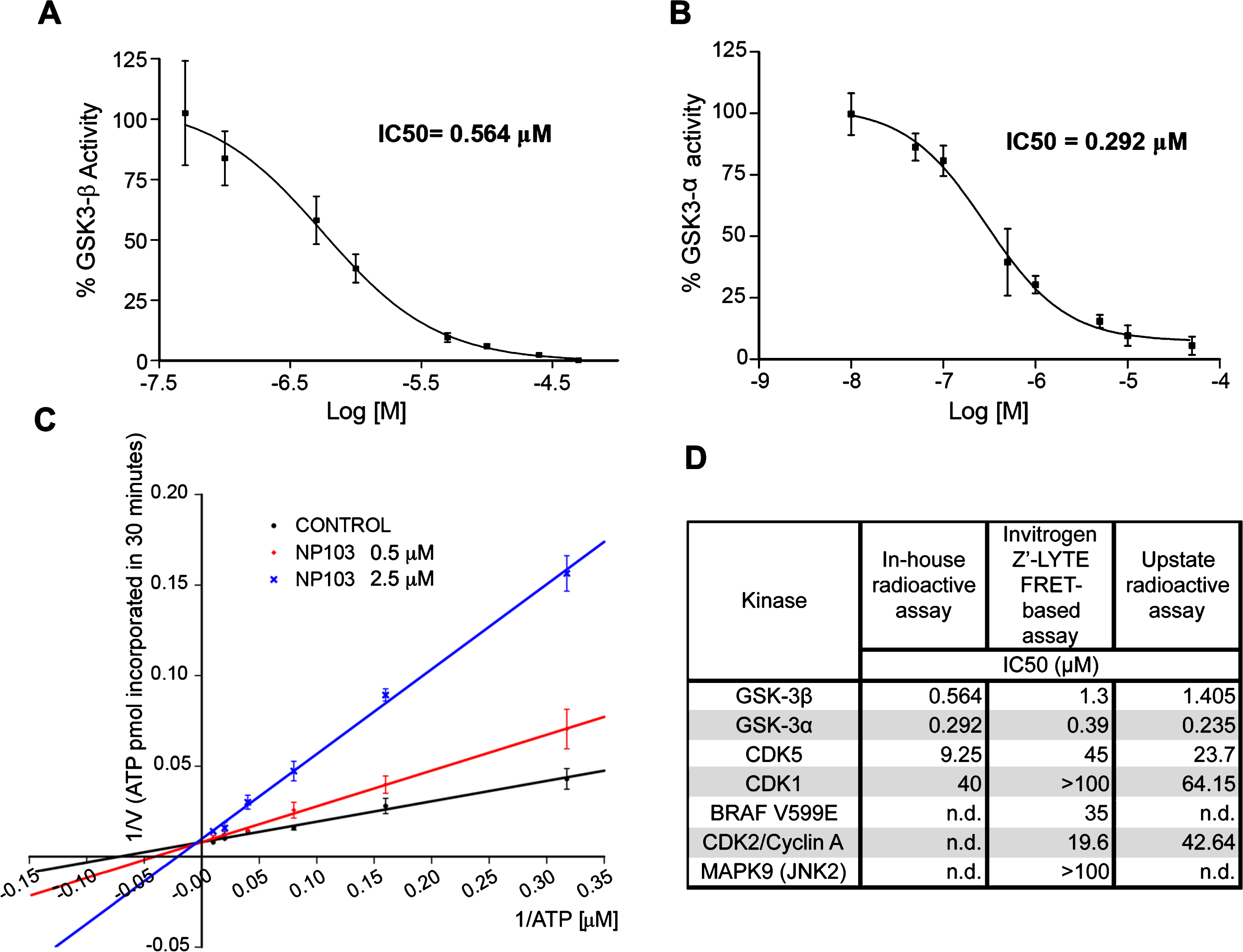
In addition, increasing ATP concentrations can overcome GSK-3β inhibition by NP103, thus indicating that this compound acts as an ATP-competitive inhibitor (Fig. 2C). Further in silico docking studies confirmed the binding to the catalytic active site of GSK-3β. To assess compound’s selectivity profile, its inhibitory properties were evaluated against a panel of 231 different protein kinases by an external CRO. Only a few protein kinases showed significant inhibition and thus were selected for performing detailed curve-response studies and IC50 calculation. As shown in (Fig. 2D), NP103 is a highly selective inhibitor for both GSK-3 isoforms. We also know that NP103 decreases tau protein phosphorylation in cell-based assays at the epitope Ser396 (a site specifically phosphorylated by GSK-3 in cells) without exerting cytotoxic effects (not shown) [44]. More recently, NP103 has been shown to inhibit tau phosphorylation in vivo after intracerebral administration in a mouse model of epilepsy [46]. Together, these results make NP103 an especially interesting molecule, since blocking the hyperphosphorylation step of tau may be a prime target at which to halt the pathogenic cascade.
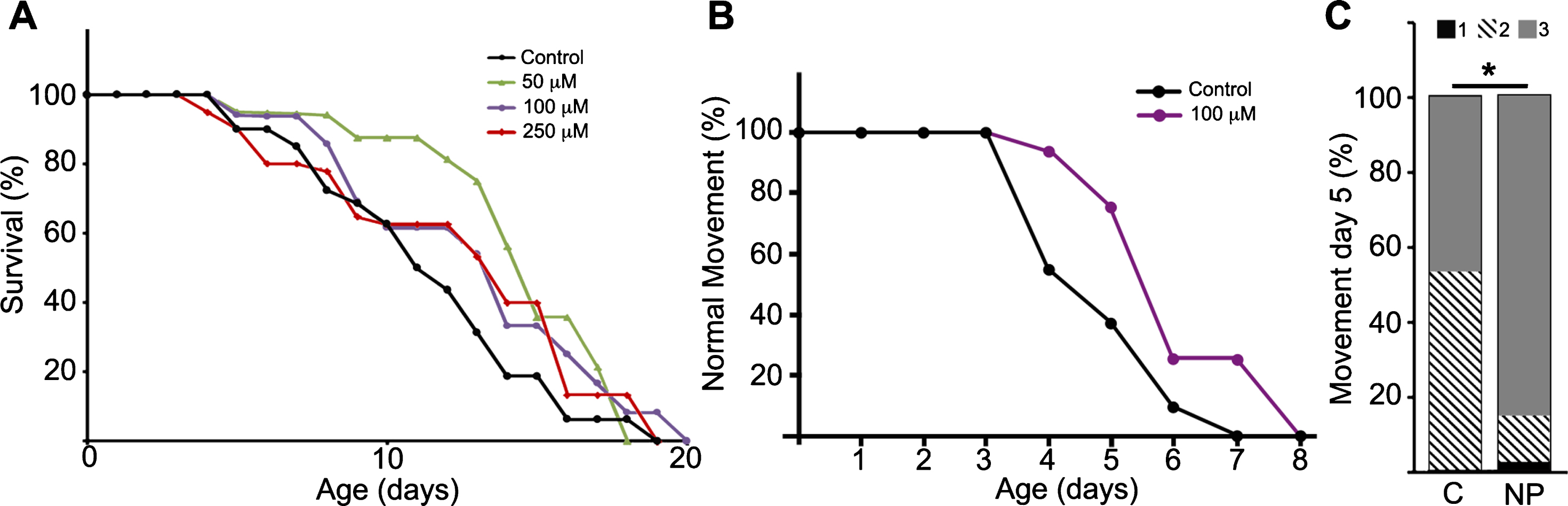
C. elegans GSK-3 and human GSK-3α and GSK3β show a high degree of homology and conservation of sequences crucial for kinase activity: the ATP binding site, the proton acceptor site and the activation loop at the catalytic domain. Given that NP103 is an ATP-competitive inhibitor, it is highly predictable this compound would be able to equally inhibit ATP-binding in the orthologues. Thus, we performed lifespan assays in the pathogenic tau aex-3/T337 transgenic strain using NP103 at different doses. Figure 3A shows that NP103 did not present toxicity at any concentration tested and produced no major changes on lifespan extension of aex-3/T337 worms. In addition, none of the doses used showed effects on wild type N2 worms or was able to prevent the appearance of the paralysis phenotype that occurs in aex-3/T337 animals since approximately day 9. However, 100 μM NP103 raised the percentage of animals showing normal locomotion along time of treatment, being this increase statistically significant at day 5 (Fig. 3B-D). This result indicates that GSK-3 inhibition can improve the quality of life of aex-3/T337 animals without extending their lifespan.
Fig.3
NP103 shows minor effects on lifespan extension of pathogenic tau aex-3/T337 transgenic strain but improves their paralysis phenotype. A) Dose–response Kaplan–Meier survival curve of synchronized mutant aex-3/T337 worms exposed to different doses (0–250 μM) of NP103. B) Curve representing the percentage of aex-3/T337 worms non-treated (black line) or NP103 100 μM treated (purple line) showing wild type-like movement along time. One representative experiment out of three independent assays are shown in (A) and (B); (n = 20 worms per experiment and dose). C) Statistical analysis of aex-3/T337 worms motility after 5 days of treatment with vehicle (control, C) or 100 μM NP103 (NP). Motility was scored assigning different values to worms with normal wild type-like movement (value 3), slow movement (value 2) or paralyzed (value 1). Data show means±s.e.m of three independent experiments (n = 20 worms per experiment). Chi-square test was performed (*p < 0.05).
DISCUSSION
Herein we have explored new benefits of ThT and GSK-3 inhibition in the C. elegans model of tauopathy aex-3/T337 that expresses the pathogenic mutant form of human tau V337M [30, 31]. We demonstrate that ThT extends lifespan in these transgenic worms, improving their locomotion. By contrast, inhibition of GSK-3 improves the paralysis phenotype of aex-3/T337 worms with minor effects on their lifespan.
In C. elegans, tetracycline and its analogues, including ThT, can reduce Aβ oligomer and amyloid fibrillar deposition because they can interact directly in vivo with different Aβ assemblies and reverse their structures to non-amyloidogenic forms. In this way, these compounds protect transgenic C. elegans strains from the paralysis phenotype of human Aβ associated toxicity. For instance, ThT can ameliorate detrimental effects during development of the Aβ aggregate-related pathology in C. elegans [9]. Notably, thioflavins bind aggregates containing different hyperphosphorylated forms of tau [37–39, 47]. However, ThT effects on tau aggregates are largely unknown, although ThT can retard significantly tau and phospho-tau aggregation in vitro [37]. The lowest concentration of ThT used for anti-tau aggregation in these in vitro assays was 30 μM. Here we detect effects of ThT on N2 worms and aex-3/T337 mutants lifespan at 10, 25, and 50 μM doses. Together these observations could indicate that low concentrations of ThT may be likely affecting different processes in the worms but not tau aggregation. We cannot however exclude that higher ThT concentrations could present combined effects both modifying tau aggregation properties and other tau-independent cellular functions.
In this regard, according to Alavez and collaborators [9], amyloid-binding compounds such as ThT, could act as a stress response mimetic and extend lifespan and influence protein aggregation through improved global protein homeostasis by affecting chaperones and protein misfolding cascades. They demonstrate that the pro-longevity effect of ThT depends on the transcription factors heat shock factor-1 (HSF-1) and the stress resistance and longevity Nrf2-like factor SKN-1 [9]. HSF-1 is induced by the accumulation of damaged proteins that controls expression of the ubiquitination/proteasome pathway for protein degradation preventing their aggregation [48, 49].
In the tauopathy model used in our study, ThT could be activating the ubiquitination/proteasome pathway by inducing HSF-1. This step could promote the elimination of tau accumulation, as one way of tau degradation can be proteasome-dependent [50, 51]. A parallel induction of HSF-1 and SNK-1 transcription programs, including small heat-shock protein and stress response encoding genes that in turn promote longevity [9, 52, 53], could be also acting this way in ThT-treated aex-3/T337 worms. In this context, a very recent publication demonstrates that molecular chaperones confer protection against tau aggregation [54]. An additional recent work shows that hydralazine, an activator of SKN-1 signaling, extends healthy lifespan in wild type and in a tauopathy model of C. elegans derived from aex-3/T337 [55]. Together these data support the findings we report here regarding ThT effects on aex-3/T337 worms’ longevity, and suggest the participation of similar mechanisms and signaling cascades. Further research using this and other tauopathy models will establish the precise molecular mechanisms involved in the reduction of tau neurotoxicity by ThT.
Treatments with ThT or the GSK-3 inhibitor NP103 reduce the paralysis of aex-3/T337 animals, indicating decreased neurotoxicity. This beneficial effect could be due to the induction of autophagy-lysosomal functions also involved the degradation of tau aggregates [50, 51]. It is well known that ThT mediated-protection requires this pathway [9] and that GSK-3 inhibition triggers autophagy [56, 57]. Whether ThT could act on autophagy-lysosome system through the inhibition of GSK-3, linking the actions of both components to a common cellular process dedicated to eliminate aggregated proteins would be an interesting issue to explore in the future.
In AD and other tauopathies GSK-3 hyperactivation is frequently accompanied by an imbalance of tau phosphatases, such as PP2A, that contribute to maintain tau hyperphosphorylated [58]. Therefore, although GSK-3 inhibition would avoid the novo phosphorylation of unphosphorylated target sites, residues that were already phosphorylated could remain in that state if deficiencies in phosphatases activities occur. This could contribute to observe mild effects of GSK-3 inhibitors, like the ones obtained here. In addition to GSK-3, other kinases such as Cdk5 may contribute to aex-3/T337 tau hyperphosphorylation that will be resistant to the use of GSK-3 inhibitors [59–61].
It is also important to take into account that GSK-3 is part of regulatory networks where other non-tau proteins are phosphorylated. Thus, it is difficult to determine whether the effects seen here could be directly caused by reduced tau phosphorylation or by some other effect on another pathway that indirectly influences tau-mediated uncoordinated phenotype and lifespan shortening. In this sense, it is well established that HSF1 and SKN-1 activities are repressed by GSK-3 [62–65]. These two longevity factors might be repressed in pathologies accompanied by GSK-3 hyperactivity, such as tauopathies and AD, favoring tau hyperphosphorylation and accumulation. Therefore, it would be expected that GSK-3 inhibition should result in the restoration of the activities of these two transcription factors and their pro-longevity programs. Indeed, it has been reported recently that the use of lithium extends Drosophila lifespan through inhibition of GSK-3 and activation of Nrf2-dependent hormesis [66]. However, our data show that GSK-3 inhibition does not significantly extend lifespan in the model of tauopathy studied here. One possible explanation would be that lithium could modulate GSK-3 and additional signaling pathways and autophagy-lysosomal degradation, regulating longevity factors [65, 67, 68]. More selective and specific inhibitors of GSK-3, such as NP103, would not show these lithium-combined effects.
Tetracyclines cross the blood brain barrier and are already used in clinical practice for central nervous system injury, offering the advantage of a safe toxicological profile and well-characterized pharmacological properties [69–71]. Based on in vitro studies and animal models, some drug development efforts have been made in recent years to assess the therapeutic potential of tetracyclines and derivatives in various disorders [72]. A search of public databases (clinicaltrials.gov; drugbank.ca) shows at least three clinical trials undertaken to assess possible clinical benefits of tetracyclines in the treatment of AD (minocycline; NCT01463384) or transthyretin amyloidosis (doxycycline; NCT01677286).
The therapeutic effect of several GSK-3 inhibitors, such as Lithium and Tideglusib, has been evaluated in various clinical studies for the treatment of AD and other tauopathies (see review [73]). Some Lithium studies reported mild effects [74–76] while Tideglusib study showed it to be safe and a considerable improvement in cognitive function measurements [77] and reduced the progression of brain atrophy in patients with supranuclear progressive palsy [78]. Therefore, there is an interest and thus encourages continuing further clinical trials with these compounds as treatment options.
It would be worth examining any synergistic effects of ThT and NP103 on tau pathology using combined treatments as well as to investigate whether ThT and NP103 show therapeutic potential in studies using other C. elegans models and mouse models of tauopathies, including AD. If proven effective in preclinical assays, next step would be to promote their use for clinical testing in an effort to curb the progression of neurotoxic symptoms in AD and other tauopathies.
CONFLICT OF INTEREST
M.M. is a former employee of Noscira S.A. The other authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by grants SAF2014-52737-P and SAF2017-88885-R to T.I., SAF2016-78603-R to M.M. from Ministerio de Ciencia, Innovación y Universidades (Spain). It was also funded by B2017/BMD-3700 (NEUROMETAB-CM) from Comunidad de Madrid to T.I., and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED, Instituto de Salud Carlos III, Spain) to T.I. and M.M. A.G.M was funded by a contract from CIBERNED. The cost of this publication has been paid in part by FEDER (European Funds for Regional Development) funds. We are grateful to Professor B.C. Kraemer (Geriatrics Research Education and Clinical Center, Seattle Veterans Affairs Puget Sound Health Care System and University of Washington, Seattle, Washington, USA) for kindly providing aex-3/T337 strain and Professor Giovanni Lesa for his generous scientific advice and help for the experimental use of C. elegans. We also thank members of Noscira’s R&D department and Dr. J. Jurado and Dr. Yuri Chiodo for their help and support, and members of our laboratories for constructive suggestions.
REFERENCES
[1] | Bektas A , Schurman SH , Sen R , Ferrucci L ((2018) ) Aging, inflammation and the environment. Exp Gerontol 105: , 10–18. |
[2] | Wyss-Coray T ((2016) ) Ageing, neurodegeneration and brain rejuvenation. Nature 539: , 180–186. |
[3] | Koga H , Kaushik S , Cuervo AM ((2011) ) Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev 10: , 205–215. |
[4] | Klaips CL , Jayaraj GG , Hartl FU ((2018) ) Pathways of cellular proteostasis in aging and disease. J Cell Biol 217: , 51–63. |
[5] | Sweeney P , Park H , Baumann M , Dunlop J , Frydman J , Kopito R , McCampbell A , Leblanc G , Venkateswaran A , Nurmi A , Hodgson R ((2017) ) Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl Neurodegener 6: , 6. |
[6] | Forloni G , Colombo L , Girola L , Tagliavini F , Salmona M ((2001) ) Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett 487: , 404–407. |
[7] | Airoldi C , Colombo L , Manzoni C , Sironi E , Natalello A , Doglia SM , Forloni G , Tagliavini F , Del Favero E , Cantu L , Nicotra F , Salmona M ((2011) ) Tetracycline prevents Abeta oligomer toxicity through an atypical supramolecular interaction. Org Biomol Chem 9: , 463–472. |
[8] | Xue C , Lin TY , Chang D , Guo Z ((2017) ) Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R Soc Open Sci 4: , 160696. |
[9] | Alavez S , Vantipalli MC , Zucker DJ , Klang IM , Lithgow GJ ((2011) ) Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472: , 226–229. |
[10] | Medina M , Hernandez F , Avila J ((2016) ) New features about tau function and dysfunction. Biomolecules 6: , pii: E21. |
[11] | Kneynsberg A , Combs B , Christensen K , Morfini G , Kanaan NM ((2017) ) Axonal degeneration in tauopathies: Disease relevance and underlying mechanisms. Front Neurosci 11: , 572. |
[12] | Hernandez F , Avila J ((2008) ) Tau aggregates and tau pathology. J Alzheimers Dis 14: , 449–452. |
[13] | Spillantini MG , Goedert M ((2013) ) Tau pathology and neurodegeneration. Lancet Neurol 12: , 609–622. |
[14] | Barghorn S , Davies P , Mandelkow E ((2004) ) Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 43: , 1694–1703. |
[15] | Mudher A , Colin M , Dujardin S , Medina M , Dewachter I , Naini SMA , Mandelkow EM , Mandelkow E , Buee L , Goedert M , Brion JP ((2017) ) What is the evidence that tau pathology spreads through prion-like propagation?. Acta Neuropathol Commun 5: , 99. |
[16] | Sotiropoulos I , Galas MC , Silva JM , Skoulakis E , Wegmann S , Maina MB , Blum D , Sayas CL , Mandelkow EM , Mandelkow E , Spillantini MG , Sousa N , Avila J , Medina M , Mudher A , Buee L ((2017) ) Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun 5: , 91. |
[17] | Wischik CM , Harrington CR , Storey JM ((2014) ) Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88: , 529–539. |
[18] | Simic G , Babic Leko M , Wray S , Harrington C , Delalle I , Jovanov-Milosevic N , Bazadona D , Buee L , de Silva R , Di Giovanni G , Wischik C , Hof PR ((2016) ) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: , 6. |
[19] | Beck CD , Rankin CH ((1993) ) Effects of aging on habituation in the nematode Caenorhabditis elegans. Behav Processes 28: , 145–163. |
[20] | Murakami S , Murakami H ((2005) ) The effects of aging and oxidative stress on learning behavior in C. elegans. Neurobiol Aging 26: , 899–905. |
[21] | Tissenbaum HA ((2015) ) Using C. elegans for aging research. Invertebr Reprod Dev 59: , 59–63. |
[22] | David DC , Ollikainen N , Trinidad JC , Cary MP , Burlingame AL , Kenyon C ((2010) ) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8: , e1000450. |
[23] | Chen X , Barclay JW , Burgoyne RD , Morgan A ((2015) ) Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem Cent J 9: , 65. |
[24] | Alexander AG , Marfil V , Li C ((2014) ) Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front Genet 5: , 279. |
[25] | Nass R , Hall DH , Miller DM 3rd , Blakely RD ((2002) ) Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99: , 3264–3269. |
[26] | Martinez BA , Caldwell KA , Caldwell GA ((2017) ) C. elegans as a model system to accelerate discovery for Parkinson disease. Curr Opin Genet Dev 44: , 102–109. |
[27] | Faber PW , Alter JR , MacDonald ME , Hart AC ((1999) ) Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci U S A 96: , 179–184. |
[28] | Goedert M , Baur CP , Ahringer J , Jakes R , Hasegawa M , Spillantini MG , Smith MJ , Hill F ((1996) ) PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J Cell Sci 109: , 2661–2672. |
[29] | Gordon P , Hingula L , Krasny ML , Swienckowski JL , Pokrywka NJ , Raley-Susman KM ((2008) ) The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev Genes Evol 218: , 541–551. |
[30] | Kraemer BC , Zhang B , Leverenz JB , Thomas JH , Trojanowski JQ , Schellenberg GD ((2003) ) Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A 100: , 9980–9985. |
[31] | Kraemer BC , Burgess JK , Chen JH , Thomas JH , Schellenberg GD ((2006) ) Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet 15: , 1483–1496. |
[32] | Kayed R , Head E , Thompson JL , McIntire TM , Milton SC , Cotman CW , Glabe CG ((2003) ) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: , 486–489. |
[33] | Thomas M , Le WD , Jankovic J ((2003) ) Minocycline and other tetracycline derivatives: A neuroprotective strategy in Parkinson’s disease and Huntington’s disease. Clin Neuropharmacol 26: , 18–23. |
[34] | Malmo C , Vilasi S , Iannuzzi C , Tacchi S , Cametti C , Irace G , Sirangelo I ((2006) ) Tetracycline inhibits W7FW14F apomyoglobin fibril extension and keeps the amyloid protein in a pre-fibrillar, highly cytotoxic state. FASEB J 20: , 346–347. |
[35] | Sirangelo I , Irace G ((2010) ) Inhibition of aggregate formation as therapeutic target in protein misfolding diseases: Effect of tetracycline and trehalose. Expert Opin Ther Targets 14: , 1311–1321. |
[36] | Giorgetti S , Raimondi S , Pagano K , Relini A , Bucciantini M , Corazza A , Fogolari F , Codutti L , Salmona M , Mangione P , Colombo L , De Luigi A , Porcari R , Gliozzi A , Stefani M , Esposito G , Bellotti V , Stoppini M ((2011) ) Effect of tetracyclines on the dynamics of formation and destructuration of beta2-microglobulin amyloid fibrils. J Biol Chem 286: , 2121–2131. |
[37] | Sui D , Liu M , Kuo MH ((2015) ) In vitro aggregation assays using hyperphosphorylated tau protein. J Vis Exp 95: , e51537. |
[38] | Shammas SL , Garcia GA , Kumar S , Kjaergaard M , Horrocks MH , Shivji N , Mandelkow E , Knowles TP , Mandelkow E , Klenerman D ((2015) ) A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun 6: , 7025. |
[39] | Pickhardt M , Neumann T , Schwizer D , Callaway K , Vendruscolo M , Schenk D , St George-Hyslop P , Mandelkow EM , Dobson CM , McConlogue L , Mandelkow E , Toth G ((2015) ) Identification of small molecule inhibitors of tau aggregation by targeting monomeric tau as a potential therapeutic approach for tauopathies. Curr Alzheimer Res 12: , 814–828. |
[40] | Brenner S ((1974) ) The genetics of Caenorhabditis elegans. Genetics 77: , 71–94. |
[41] | Dostal V , Link CD ((2010) ) Assaying beta-amyloid toxicity using a transgenic C. elegans model. J Vis Exp 44: , pii: 2252. |
[42] | Dominguez JM , Fuertes A , Orozco L , del Monte-Millan M , Delgado E , Medina M ((2012) ) Evidence for irreversible inhibition of glycogen synthase kinase-3 beta by tideglusib. J Biol Chem 287: , 893–904. |
[43] | Medina M , Avila J ((2014) ) New insights into the role of glycogen synthase kinase-3 in Alzheimer’s disease. Expert Opin Ther Targets 18: , 69–77. |
[44] | Medina M , Martín-Aparicio E , Pérez-Puerto MJ , Fuertes A , Alonso D , Alonso M , Castro A , Navarro-Rico ML , Orozco-Muñoz L , Monte-Millán M , Martínez A ((2008) ) P2-402: A novel, highly specific glycogen synthase kinase-3 inhibitor of marine origin. Alzheimers Dement 4: , T491. |
[45] | Kramer T , Schmidt B , Lo Monte F ((2012) ) Small-molecule inhibitors of GSK-3: Structural insights and their application to Alzheimer’s disease models. Int J Alzheimers Dis 2012: , 381029. |
[46] | Engel T , Gómez-Sintes R , Alves M , Jiménez-Mateos E , Fernández-Nogales M , Sanz-Rodríguez A , Morgan J , Beamer E , Rodríguez-Matellán A , Dunleavy M , Sano T , Ávila J , Medina M , Hernández F , Lucas JJ , Henshall DC ((2018) ) Bi-directional genetic modulation of GSK-3β exacerbates hippocampal neuropathology in experimental status epilepticus. Cell Death Dis 9: , 969. |
[47] | Santa-Maria I , Perez M , Hernandez F , Avila J , Moreno FJ ((2006) ) Characteristics of the binding of thioflavin S to tau paired helical filaments. J Alzheimers Dis 9: , 279–285. |
[48] | Pirkkala L , Nykanen P , Sistonen L ((2001) ) Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 15: , 1118–1131. |
[49] | Li J , Labbadia J , Morimoto RI ((2017) ) Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol 27: , 895–905. |
[50] | Wang Y , Mandelkow E ((2012) ) Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans 40: , 644–652. |
[51] | Lee MJ , Lee JH , Rubinsztein DC ((2013) ) Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 105: , 49–59. |
[52] | Hsu AL , Murphy CT , Kenyon C ((2003) ) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300: , 1142–1145. |
[53] | Morley JF , Morimoto RI ((2004) ) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15: , 657–664. |
[54] | Mok SA , Condello C , Freilich R , Gillies A , Arhar T , Oroz J , Kadavath H , Julien O , Assimon VA , Rauch JN , Dunyak BM , Lee J , Tsai FTF , Wilson MR , Zweckstetter M , Dickey CA , Gestwicki JE ((2018) ) Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat Struct Mol Biol 25: , 384–393. |
[55] | Dehghan E , Zhang Y , Saremi B , Yadavali S , Hakimi A , Dehghani M , Goodarzi M , Tu X , Robertson S , Lin R , Chudhuri A , Mirzaei H ((2017) ) Hydralazine induces stress resistance and extends C. elegans lifespan by activating the NRF2/SKN-1 signalling pathway. Nat Commun 8: , 2223. |
[56] | Sarkar S , Floto RA , Berger Z , Imarisio S , Cordenier A , Pasco M , Cook LJ , Rubinsztein DC ((2005) ) Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170: , 1101–1111. |
[57] | Parr C , Carzaniga R , Gentleman SM , Van Leuven F , Walter J , Sastre M ((2012) ) Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Mol Cell Biol 32: , 4410–4418. |
[58] | Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2005) ) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22: , 1942–1950. |
[59] | Dolan PJ , Johnson GV ((2010) ) The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Devel 13: , 595–603. |
[60] | Medina M , Garrido JJ , Wandosell FG ((2011) ) Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front Mol Neurosci 4: , 24. |
[61] | Lee S , Hall GF , Shea TB ((2011) ) Potentiation of tau aggregation by cdk5 and GSK3beta. J Alzheimers Dis 26: , 355–364. |
[62] | Chu B , Soncin F , Price BD , Stevenson MA , Calderwood SK ((1996) ) Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J Biol Chem 271: , 30847–30857. |
[63] | Wang X , Grammatikakis N , Siganou A , Calderwood SK ((2003) ) Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol Cell Biol 23: , 6013–6026. |
[64] | An JH , Vranas K , Lucke M , Inoue H , Hisamoto N , Matsumoto K , Blackwell TK ((2005) ) Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proc Natl Acad Sci U S A 102: , 16275–16280. |
[65] | Blackwell TK , Steinbaugh MJ , Hourihan JM , Ewald CY , Isik M ((2015) ) SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic Biol Med 88: , 290–301. |
[66] | Castillo-Quan JI , Li L , Kinghorn KJ , Ivanov DK , Tain LS , Slack C , Kerr F , Nespital T , Thornton J , Hardy J , Bjedov I , Partridge L ((2016) ) Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep 15: , 638–650. |
[67] | Freland L , Beaulieu JM ((2012) ) Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci 5: , 14. |
[68] | Kim E , Sakata K , Liao FF ((2017) ) Bidirectional interplay of HSF1 degradation and UPR activation promotes tau hyperphosphorylation. PLoS Genet 13: , e1006849. |
[69] | Choi KJ , Son HJ , Kim SH ((2007) ) Ionic treatment for removal of sulfonamide and tetracycline classes of antibiotic. Sci Total Environ 387: , 247–256. |
[70] | Forloni G , Salmona M , Marcon G , Tagliavini F ((2009) ) Tetracyclines and prion infectivity. Infect Disord Drug Targets 9: , 23–30. |
[71] | Noble W , Garwood C , Stephenson J , Kinsey AM , Hanger DP , Anderton BH ((2009) ) Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J 23: , 739–750. |
[72] | Stoilova T , Colombo L , Forloni G , Tagliavini F , Salmona M ((2013) ) A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J Med Chem 56: , 5987–6006. |
[73] | Pandey MK , DeGrado TR ((2016) ) Glycogen synthase kinase-3 (GSK-3)-targeted therapy and imaging. Theranostics 6: , 571–593. |
[74] | Forlenza OV , Diniz BS , Radanovic M , Santos FS , Talib LL , Gattaz WF ((2011) ) Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: Randomised controlled trial. Br J Psychiatry 198: , 351–356. |
[75] | Hampel H , Ewers M , Burger K , Annas P , Mortberg A , Bogstedt A , Frolich L , Schroder J , Schonknecht P , Riepe MW , Kraft I , Gasser T , Leyhe T , Moller HJ , Kurz A , Basun H ((2009) ) Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 70: , 922–931. |
[76] | Macdonald A , Briggs K , Poppe M , Higgins A , Velayudhan L , Lovestone S ((2008) ) A feasibility and tolerability study of lithium in Alzheimer’s disease. Int J Geriatr Psychiatry 23: , 704–711. |
[77] | del Ser T , Steinwachs KC , Gertz HJ , Andres MV , Gomez-Carrillo B , Medina M , Vericat JA , Redondo P , Fleet D , Leon T ((2013) ) Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J Alzheimers Dis 33: , 205–215. |
[78] | Hoglinger GU , Huppertz HJ , Wagenpfeil S , Andres MV , Belloch V , Leon T , Del Ser T ((2014) ) Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord 29: , 479–487. |


