
Neonatal Neurodegeneration in Alzheimer’s Disease Transgenic Mouse Model
Abstract
Alzheimer’s disease (AD) is a progressive disorder characterized by a variety of molecular pathologies causing cortical dementia with a prominent memory deficit. Formation of the pathology, which begins decades before the diagnosis of the disease, is highly correlated with the clinical symptoms. Several proteomics studies were performed using animal models to monitor the alterations of the brain tissue proteome at different stages of AD. However, proteome changes in the brain regions of newborn transgenic mouse model have not been investigated yet. To this end, we analyzed protein expression alterations in cortex, hippocampus and cerebellum of transgenic mice carrying five familial AD mutations (5XFAD) at neonatal day-1. Our results indicate a remarkable difference in protein expression profile of newborn 5XFAD brain with region specific variations. Additionally, the proteins, which show similar expression alteration pattern in postmortem human AD brains, were determined. Bioinformatics analysis showed that the molecular alterations were mostly related to the cell morphology, cellular assembly and organization, and neuroinflammation. Moreover, morphological analysis revealed that there is an increase in neurite number of 5XFAD mouse neurons in vitro. We suggest that, molecular alterations in the AD brain exist even at birth, and perhaps the disease is silenced until older ages when the brain becomes vulnerable.
INTRODUCTION
Alzheimer’s disease (AD) is a multifactorial progressive neurodegenerative disorder that damages different brain regions, causes loss of memory and affects other brain functions. AD is neuropathologically characterized by extracellular senile plaques that are formed by the accumulation of amyloid-β (Aβ) peptides, intracellular neurofibrillary tangles (NFT) containing hyperphosphorylated microtubule-associated protein tau (MAPT), neuroinflammation, neuronal and synaptic loss. Neurodegenerative disorders, including AD, tend to be selective for specific neuron types and brain regions with specific functions. This selective degeneration also causes the clinical symptoms of the diseases. In AD, senile plaques mainly accumulate in the isocortex, then spread through the hippocampus and entorhinal cortex to other brain regions [1]. On the contrary, NFTs are first seen in the entorhinal cortex of the medial temporal lobe which is known as the most affected part in the AD brain [2]. Moreover, senile plaques spread randomly in the cortex and the hippocampus, while tangles selectively deposit in the pyramidal neurons and spread through the connections between layers [3]. Despite the different spatio-temporal patterns, both of these main pathologies are observed most intensely in the medial temporal lobe where also the atrophy caused by neuronal and synaptic loss is detected in correlation with clinical symptoms at the late stages of the disease [4, 5].
Despite many years of research, neither the mechanism of the disease is completely understood nor has a cure been found. The pathophysiological changes in AD brain appear years before the appearance of the clinical symptoms. Identifying key molecules and novel biomarkers at pre-symptomatic stage using proteomics is an invaluable way to gain insight into the pathogenesis of AD. Proteomics studies of blood and cerebrospinal fluid are important for the biomarker discoveries but not sufficient to clarify the disease mechanism. Also, brain tissue studies from AD patients are only applicable postmortem which is a late stage to examine the progression of the pathology and to find predictive or prognostic biomarkers. Hence, using transgenic AD mouse models are an advantageous way to overcome these issues.
The 5XFAD is an early-onset transgenic AD mouse model in which Aβ production increases additively and rapidly [6]. This mouse model presents intracellular Aβ depositions in cortical layer 5 and subiculum of the hippocampus within 2–4 months along with learning and memory deficits [7]. After 4 months, cognitive deficits related to frontal cortex in association with strong gliosis and amyloid plaques are identified [8]. Although 5XFAD mouse model does not present tau pathology, high amounts of toxic Aβ alters the structure and density of the synapses and causes progressive neuronal death and atrophy, where most AD mouse models lack having such characterization [9, 10].
A number of studies have examined the protein expression differences in various brain regions of transgenic AD mouse models at several time points using proteomics approach; yet newborn transgenic mouse model has not been studied [11–13]. Here, we performed a proteomics study on three brain regions—cortex, hippocampus, and cerebellum—of the newborn 5XFAD mouse model. We investigated the functional networks, which involve proteins with significantly altered expression, using Ingenuity Pathway Analysis (IPA) software. We performed primary cell culture and immunoassay experiments to validate our proteomics data. We also compared our results with the postmortem AD human brain proteome studies in order to determine the similarities between the newborn and late stage of the disease.
METHODS
Transgenic mouse model: 5XFAD
The 5XFAD mice, expressing mutant human AβPP with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial Alzheimer’s disease (FAD) mutations and two human PS1 (M146L, L286V) mutations under the Thy1 promoter, were used. (Jackson Laboratories, Bar Harbor, Maine, USA) Hemizygote transgenic mice were crossed with B6/SJL F1 breeders and extracted genomic DNA from tail biopsy of all mice were used to assess genotype by polymerase chain reaction analysis. The 5XFAD heterozygote transgenic mice (1 day-old) were used as the experimental group and non-transgenic age-matched littermates (LM) were used as the controls for the proteomics analysis. Animal care and handling was implemented according to National Institutes of Health Guidelines for the Care and Use of Laboratory Animals with approval of Local Ethics Committee of Animal Experiments of Istanbul Medipol University.
Sample preparation for LC-MS/MS analysis
Transgenic 5XFAD and LM neonatal mice (n = 3 per group) were sacrificed and cortex, hippocampus and cerebellum samples were dissected. All tissues were frozen on dry-ice and stored at –80°C. Tissues were mechanically homogenized with a pellet pestle (Sigma-Aldrich Sweden AB, Sweden) [14] in 50 mM ammonium bicarbonate and lysed via boiling at 100°C for 5 min in UPX Buffer (Expedeon, UK). The samples were centrifuged at 15,000 g at 4°C for 10 min and the supernatant were collected. Tryptic peptides were obtained according to Filter Aided Sample Preparation Protocol (FASP) [15]. Briefly, 50 μg protein extract was washed with 6M urea in the 30kDa cut-off spin column and alkylated with 10 mM iodoacetamide (IAA) in the dark for 20 min at room temperature. To digest the proteins, trypsin was added at an enzyme to substrate ratio of 1:100 and the digestion was performed overnight at 37°C. Tryptic peptides were eluted from the column and prepared at 100 ng/μl concentration.
LC-MS/MS analysis and data processing
Analysis and data processing was done according to our previously published work [16, 17]. Briefly, 200 ng of tryptic peptides were loaded on a nanoACQUITY UPLC coupled to a SYNAPT G2-Si HDMS system. The columns were equilibrated with 97% mobile phase A (0.1% formic acid in LC MS grade water (Merck)), and column temperature was set to 55°C. Peptides were separated from the trap column (Symmetry C18 5 μm, 180 μmi.d.×20 mm, Waters, Milford, MA) by gradient elution onto an analytical column (BEH C18, 1.7 μm, 75 μmi.d.×250 mm, Waters, Milford, MA) at 300 nL/min flow rate with a linear gradient from 5% to 40% mobile phase B (0.1% formic acid in hypergrade acetonitrile (Merck, NJ, USA)) over 120 min. For LC-MS/MS study, three distinct biological samples were used and each sample was analyzed in duplicate. Therefore, six separate injections were made for each biological group.
Progenensis-QI V4.0 (Nonlinear Dynamics) for proteomics software (Waters) was used for the identification and the quantification of the peptides. Glu-fib peptide with m/z 1570.6 was set as calibrant and the sample sets were normalized based on the total ion intensity. The m/z values over 50–1950 were analyzed. Carbamidomethyl-cysteine fixed modification and Acetyl N-TERM, deamidation of asparagine and glutamine, and oxidation of methionine variable modifications were set. The proteome data was searched against the reviewed mouse protein database from Uniprot (March 30, 2012, 30419). Normalization across samples were done based on total ion intensity. Similar proteins were grouped and quantitative value was given for the one with the highest score. All proteins were identified by at least 3 unique peptide sequences and identified proteins were shown in the Supplementary Table 1. The p-values and experimental ratio of proteins in two groups were calculated and filtered according to these two parameters for further experiments. To analyze the pathways related to the identified proteins, Ingenuity Pathway Analysis (IPA) and Panther Classification System 10.0 (online) were used.
Cell culture experiments
Three-day old 5XFAD mice and their non-transgenic littermates were anesthetized and euthanized by cervical transection. The cortex, hippocampus and cerebellum were dissected under a stereomicroscope in cold Leibovitz’s L-15 modification medium (L15, Wicent Inc.). The cortex samples were transferred into Hibernate A medium (Hib A) (Life Technologies, Gaithersburg, MD, USA); the hippocampus and cerebellum samples were transferred into L-15 medium, both containing 2 mM Glutamax-I (Invitrogen), 100 U penicillin, 100 mg streptomycin, 250 ng/mL amphotericin B (Sigma-Aldrich), and 100 U/mL papain (Sigma-Aldrich). They were all agitated with a custom-made agitator vibrating at 50 Hz at +4°C for 45 min. After agitation, the cortex samples were washed in Hank’s buffered salt solution (Sigma-Aldrich) three times, and triturated in Hib A for 15 min by gently and repeatedly pipetting through the tips of narrowing bores (from 2 mm diameter down). The cell suspension was spun at 300 g for 3 min, the supernatant was discarded and the pellet was resuspended in PNBM medium supplemented with 2% NSF-1 (PNBM-NSF, Lonza, Walkersville, MD, USA) containing 2 mM Glutamax-I, 100U penicillin, 100 mg streptomycin and 250 ng/mL amphotericin B. Similar to preparation for cortex, after agitation, hippocampus and cerebellum were triturated for 15 min by gently and repeatedly pipetting through the tips of narrowing bores (from 2 mm diameter down). The cell suspension was spun at 300 g for 3 min, supernatant was discarded and the pellet was resuspended in L-15 and spun at 300 g for 3 min. The supernatant was discarded and the pellet was resuspended in PNBM-NSF. The final cell suspensions of cortex, hippocampus and cerebellum were seeded on 35-mm glass-bottomed petri dishes, which had been previously covered with poly-D lysine (1.8 lg/cm2, 3 h at room temperature). The dishes were left in the incubator (37°C, 5% CO2), for 2 h to let the neurons attach to the bottom. The dishes were gently washed with PBS to remove unattached cells and remaining debris, filled with PNBM-NSF and returned to the incubator. After 48-hours, the cells were fixed with 4% (v/v) paraformaldehyde in PBS for 15 min at room temperature, followed by two washes in PBS. Multiple 20X pictures of cell cultures were acquired at various locations using a Zeiss Axio Observer.A1 inverted microscope with a Hoffman modulation coupled to a CCD camera (Zeiss Axiocam 503 Color) after fixation. Somas and neurites, which were branched from the last nodes, were counted. Neurite per neuron numbers were calculated.
Immunocytochemistry
Fixated cells were permeabilized and blocked with PBS containing 0.1% Triton X-100, 3% bovine serum albumin and 1% goat serum (Sigma-Aldrich) for 45 min at room temperature. Cells were incubated with rabbit anti-Tubulin IgG (1:500, Sigma-Aldrich) primary antibody overnight at 4°C. Cells were then washed two times with PBS and incubated with goat anti-rabbit (1:1000, Alexa Fluor®594) secondary antibody for 3 h. Labeled cells were then washed in 0.02% Tween-20 in PBS and incubated in 1:1000 DAPI in PBS. After final wash with 0.02% Tween-20 in PBS, immuno-stained cells were observed under Zeiss Confocal microscope.
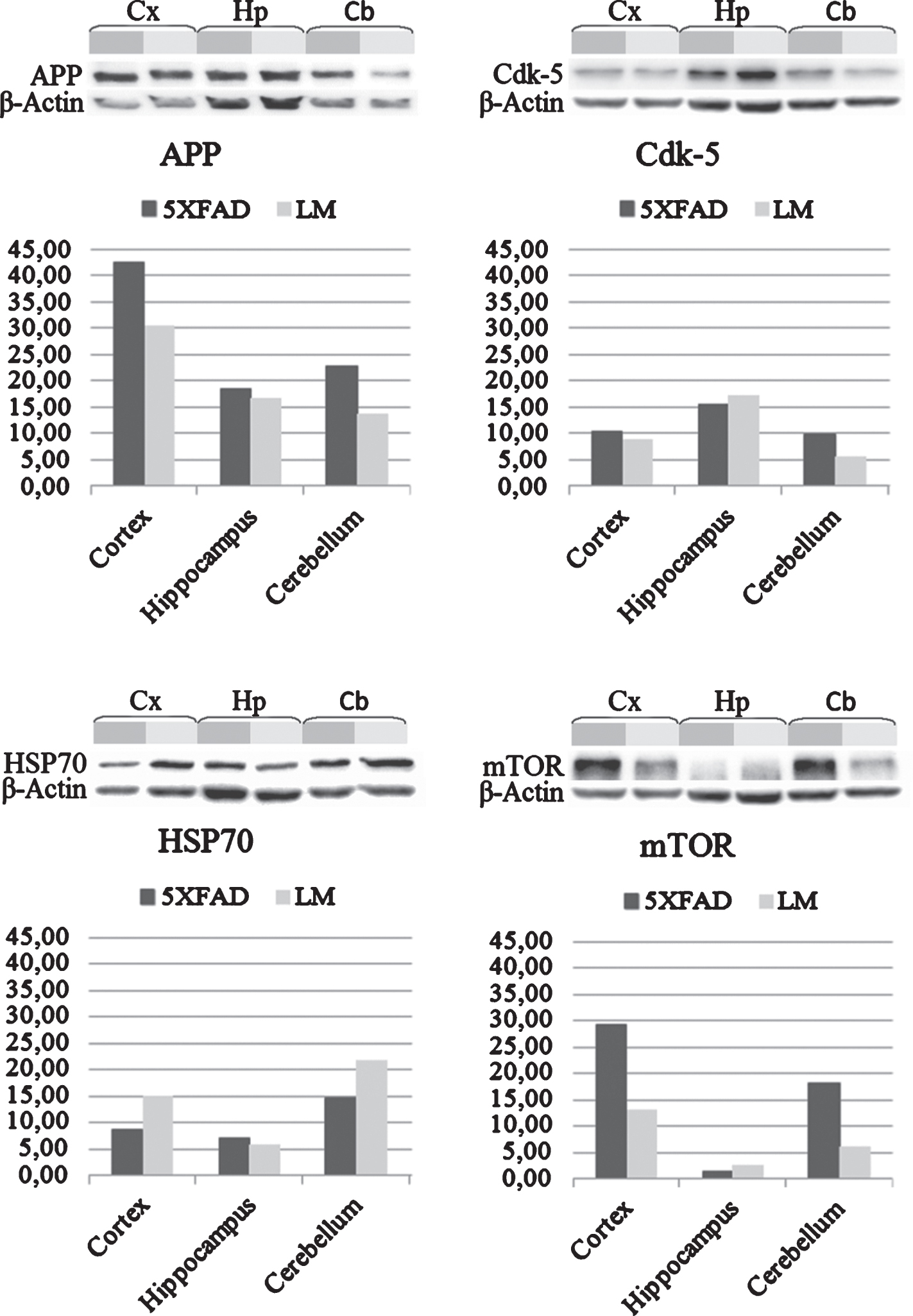
Western blot analysis
Proteins were extracted from the cortex, hippocampus and cerebellum as described above (see Sample preparation for LC-MS/MS analysis section). 20 μg of proteins from each tissue lysate were resolved by 4–20% Mini-PROTEAN TGX precast gels (BioRad) and transferred to a PVDF membrane (BioRad). The membrane was first blocked in the blocking buffer (5% nonfat dry milk, 0.1% Tween-20; in PBS) for 1 h at room temperature and then incubated with anti-Aβ antibody (1:500; 4G8; Signet, USA), anti-HSP70 antibody (1:200; Santa Cruz Biotechnology, USA), anti-mTOR antibody (1:250; Santa Cruz Biotechnology, USA), and anti-Cdk-5 antibody (1:200; Santa Cruz Biotechnology, USA) in blocking buffer for overnight at 4°C. After washing, the membrane was incubated with an HRP-conjugated goat anti-mouse secondary antibody (1:2000; Santa Cruz Biotechnology, USA) for 1 h at room temperature and detected by chemiluminescence (Bio-Rad). Equal protein loading was confirmed by stripping the blot and re-probing with a β-actin antibody (1:1000; Santa Cruz Biotechnology, USA). Western blots were quantified by densitometry using the ImageJ software.
Immunohistochemistry
Sagittal brain sections (18 μm) of newborn 5XFAD and LM mice were fixated via 4% PFA. Sections were blocked with pre-diluted horse serum provided in avidin/biotin complex (ABC) kit (Vector Laboratories, Burlingame, CA) for 10 min. Sections were incubated in 1:30 goat polyclonal antibody against IL-4 (Santa Cruz Biotechnology, USA) overnight at 4°C. After washing, the sections were incubated with biotinylated horse anti-mouse Ig/rabbit Ig secondary antibody for 15 min. Amplification of secondary antibody signal was achieved through incubation in ABC complex for 5 min. For color development, the vector diaminobenzidine (DAB) peroxidase kit (Vector Laboratories) was used. Sections were mounted onto slides and cover-slipped. Image acquisition was performed using Zeiss AxioZoom V16 microscope. Multiple pictures from cortex, hippocampus and cerebellum of 5XFAD and LM brain sections were taken with 10X, 40X, 63X objectives to be analyzed.
Lastly, Nissl staining were done for neighbor sections as control. Stained sagittal brain section images were obtained by Zeiss Confocal microscope.
Statistical and bioinformatics analysis
Statistical analyses of nanoLC-ESI-MS/MS were performed by the two-tailed Student’s t-test. Proteins identified by label-free LC-MS/MS were analyzed by the Ingenuity Pathway Analysis Tool (IPA, Ingenuity Systems, Redwood City, CA) for biological functions, associated diseases and biological networks. Right-tailed Fisher’s exact test was used to determine the p-value, which indicates that the probability of biological functions, canonical pathways and diseases associated with the networks are because of chance alone. One-tailed Student’s t-test was used for the statistical analysis of neurite numbers of cell culture and western blot experiments.
RESULTS
In order to analyze the protein expression differences in the cortex, hippocampus and cerebellum of 5XFAD compared to their non-transgenic littermates (LM), mass spectrometry based proteomics methods were used following the dissection of the brain regions. Charge state deconvolution and deisotoping were done with Progenesis QI-P and 1598 proteins were identified and 325 proteins were observed with statistically significant changes (fold change>1.5 and p < 0.05) across different brain regions of neonatal 5XFAD mouse model (Supplementary Table 1). A total of 243, 132, and 80 of these proteins belong to cortex, hippocampus and cerebellum respectively. Out of 243 proteins, 138 were specific to the cortex while 68 proteins were also altered in the hippocampus and 17 proteins were also altered in the cerebellum. Hippocampus and cerebellum shared 5 proteins. Additionally, 20 proteins were observed to be altered within all regions (Fig. 1A). Figure 2 shows the list of common 20 proteins within all three regions and fold change of protein level in these regions of 5XFAD mice compared to LM. Notably, almost all overlapping proteins were regulated in the same direction, except one protein was upregulated in the cortex while downregulated in the hippocampus (Thada-Thyroid adenoma-associated protein) and one protein upregulated in the cerebellum while downregulated in the cortex (Neil2-Endonuclease 8-like2). Among the top ten significantly altered proteins in each region, CD44, ATP-dependent 6-phosphofructokinase (PFKL) and (Fragile X mental retardation protein 1) FMR1 in cortex and hippocampus (Serine protease inhibitor A3G) SERPINA3G in cortex and cerebellum, Protein SOGA3 in hippocampus and cerebellum were overlapping (Fig. 1B).
Fig.1
Overview of protein expression profiles in cortex, hippocampus and cerebellum of newborn 5XFAD mice. A) Number of overlapping and non-overlapping proteins in three brain regions. B) Top ten altered proteins in three brain regions listed by rank of fold change. Cut-off for FC>1.5 and p < 0.05 comparing 5XFAD with LM. (↑:upregulated, ↓: downregulated).
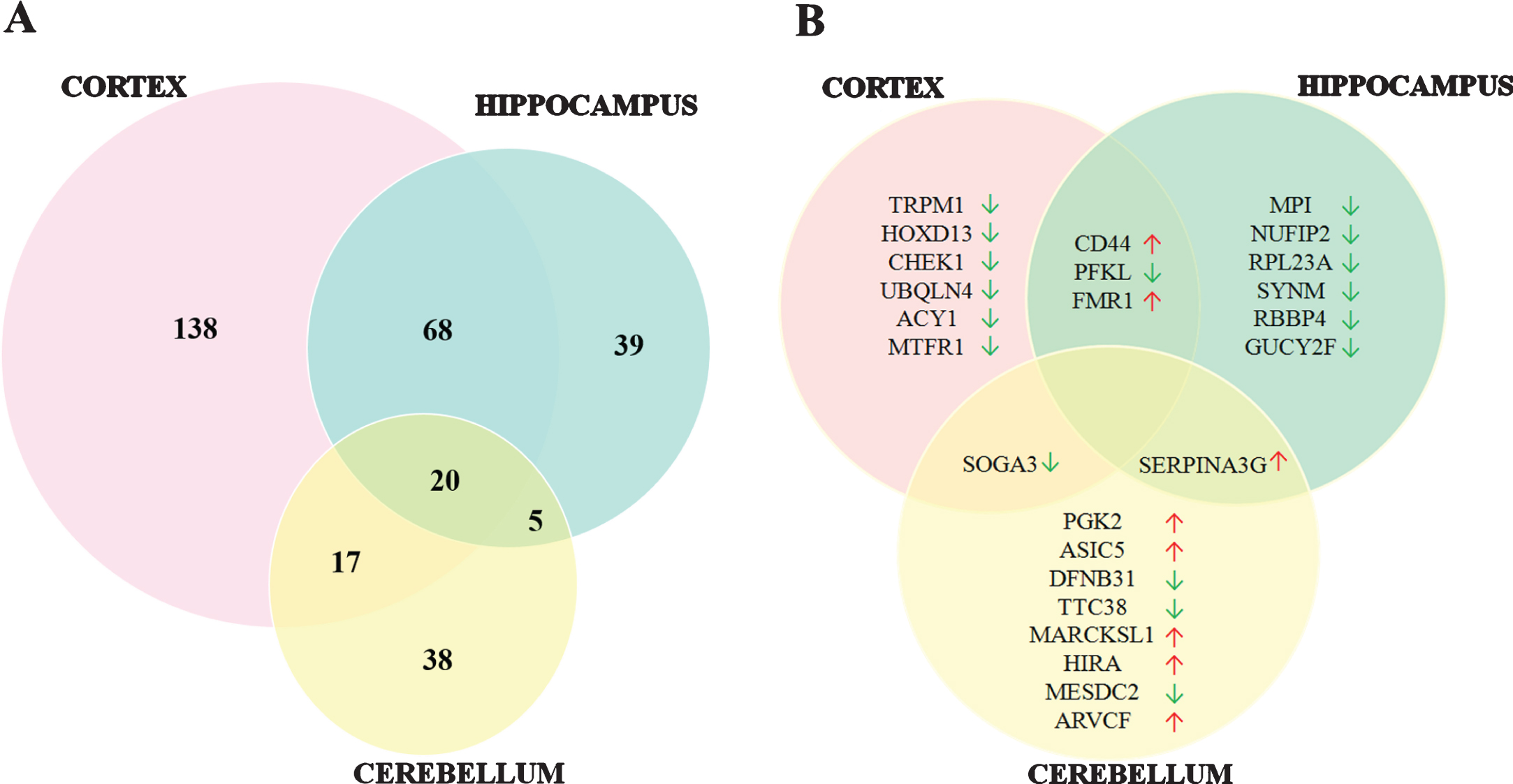
Fig.2
Co-regulated proteins between cortex, hippocampus and cerebellum in 5XFAD mice compared to control. Proteins that were elevated and diminished compared to control animals. (Cut-off for FC>1.5 and p < 0.05).
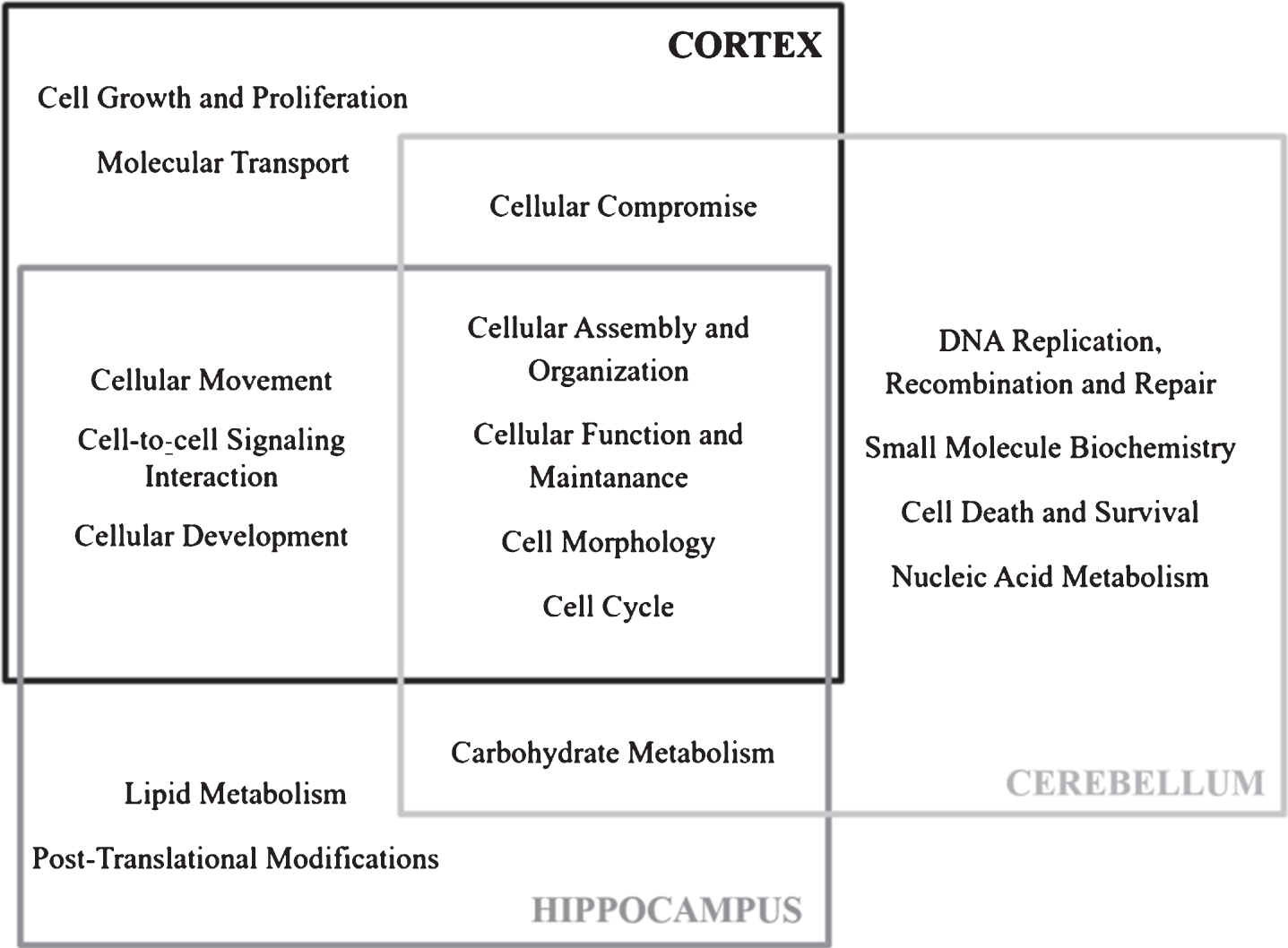
According to IPA analysis of significantly altered proteins, the related canonical pathways were Ephrin B Signaling, Clathrin-mediated Endocytosis Signaling, Integrin Signaling for the cortex, Netrin Signaling, Mevalonate Pathway, Rho Family GTPases signaling for the hippocampus and 2-ketoglutrate Dehydrogenase Complex, AMPK Signaling and BRCA1 in DNA Damage Response pathways for the cerebellum. In addition to that, we analyzed 20 co-regulated proteins using IPA. IL-4 was found to be the top upstream regulator (Supplementary Table 2) while the top network of these 20 proteins indicated a relation with inflammation network (Supplementary Figure 1). All the pathways stated above were expected to influence molecular and cellular functions such as; cellular assembly and organization, cellular function and maintenance, cell morphology, and cell cycle according to IPA (Fig. 3).
Fig.3
Molecular and cellular functions associated to significantly altered proteins in newborn 5XFAD cortex, hippocampus and cerebellum according to IPA.
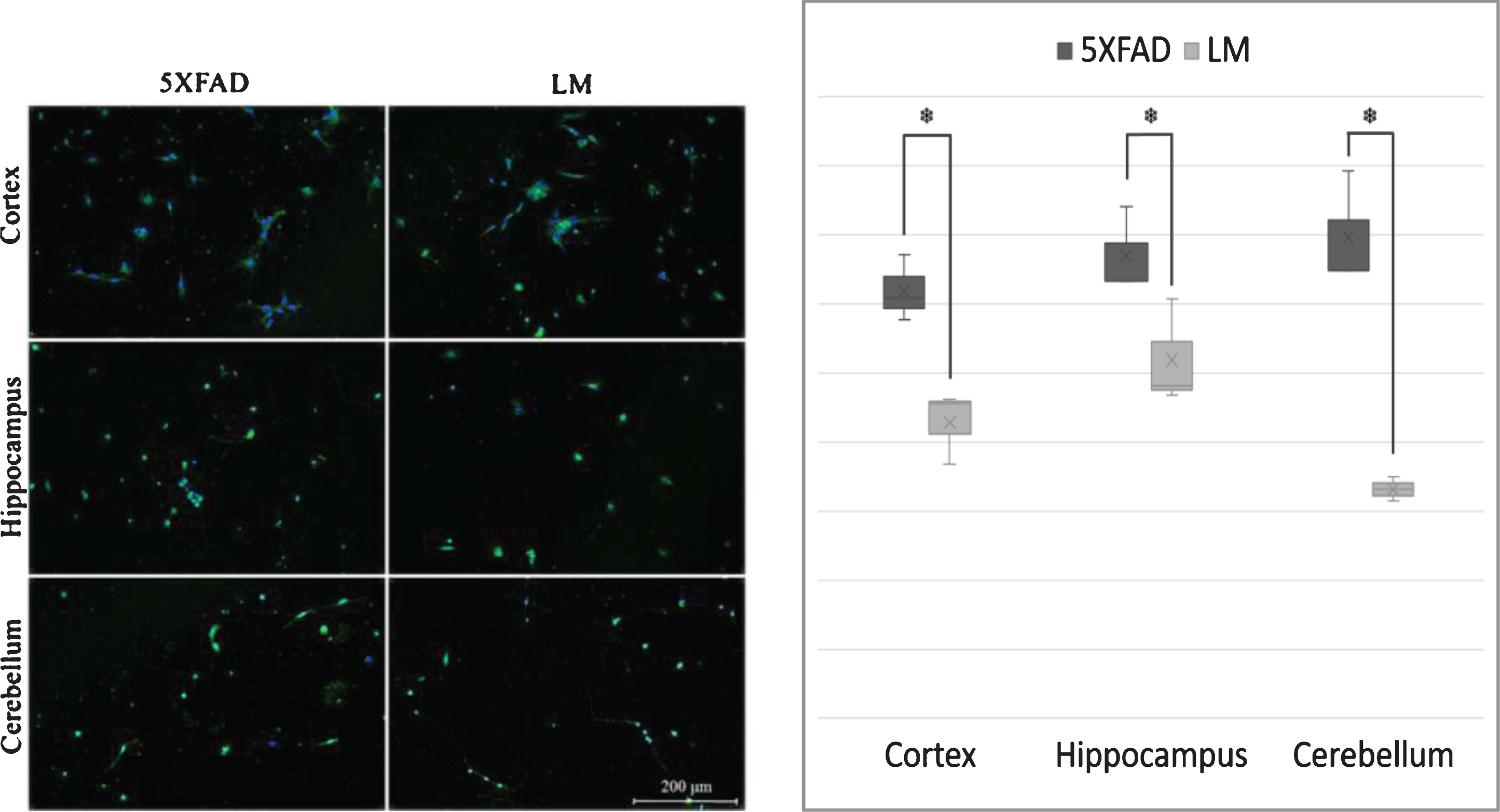
We used in vitro techniques to determine the possible morphological alterations that IPA suggested. Primary cells of cortex, hippocampus and cerebellum of 3 day-old 5XFAD mice and their non-transgenic littermates were cultured and immunofluorescence stained with β-III Tubulin to detect cytoskeletal morphology changes. Obtained images were analyzed and neurite per neuron ratios were calculated for each brain region. Results indicated an increase in neurite number of the newborn 5XFAD cortex, hippocampus, and cerebellum (Fig. 4).
Fig.4
In vitro Morphology Analysis. (A) Cell culture images and (B) quantified number of neurite per neuron values of newborn cortex, hippocampus and cerebellum. (Tubulin = green, DAPI = blue; n = 3; *p < 0.05).
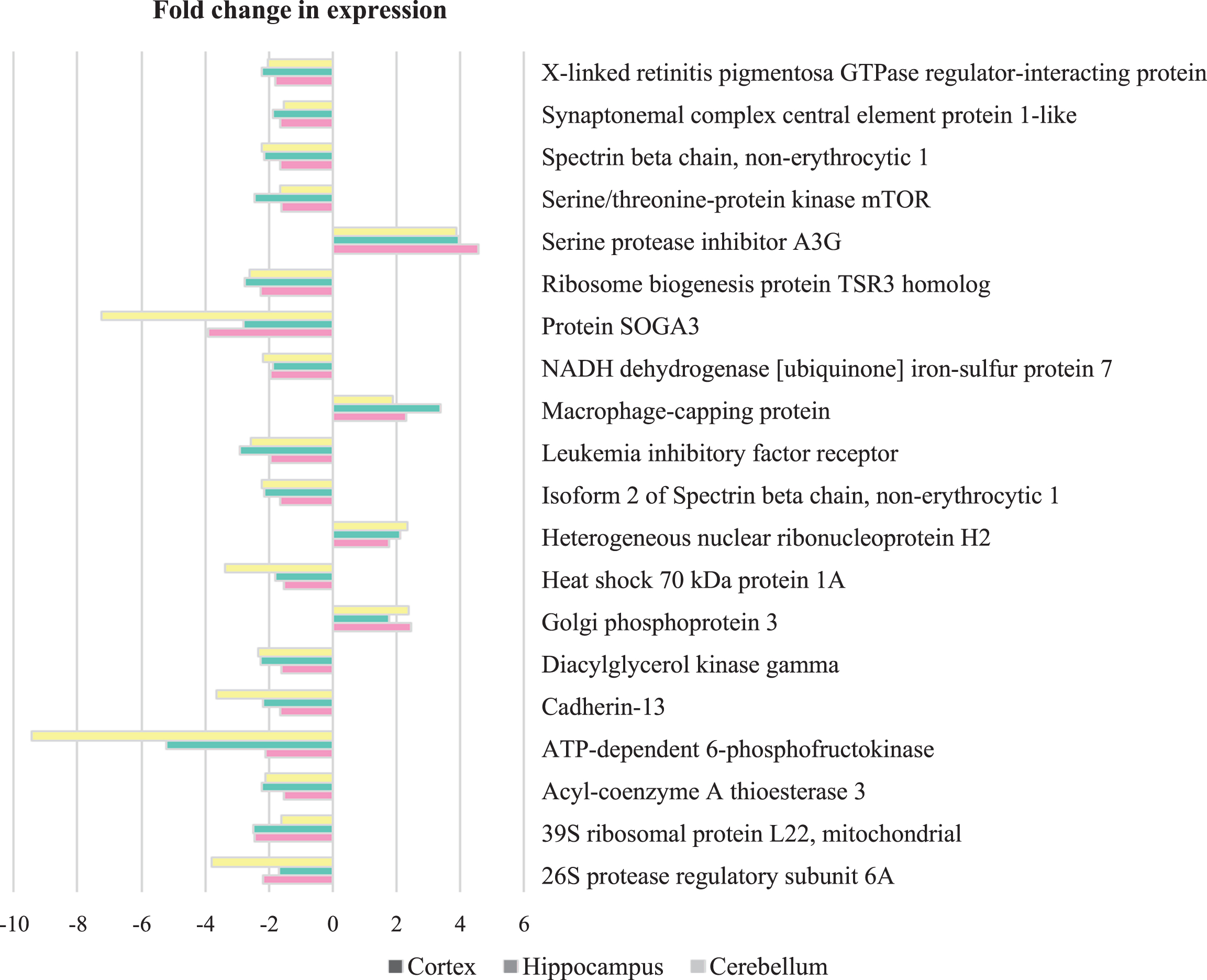
Several studies suggest that elevated levels of AβPP may enhance the neurite outgrowth and branching before it becomes neurotoxic. Therefore, we performed a western blot analysis to detect the expression change in AβPP of 5XFAD mice compared to LM mice. Results showed an increase in the 5XFAD cortex, hippocampus and cerebellum (Fig. 5). Also, we analyzed the expression change of three proteins—Cyclin-dependent-like kinase 5 (Cdk-5), Heat shock protein 70 (HSP70) and Serine/threonine-protein kinase mTOR—which are known to be altered in AD brain, by western blot to validate our proteomics results. Expression of Cdk-5 was found to be increased in cortex and cerebellum of 5XFAD whereas decreased in the hippocampus. Expression of HSP70 was decreased in cortex and cerebellum as indicated by proteomics data. On the other hand, expression of mTOR in cortex and cerebellum of 5XFAD mouse was increased as opposed to proteomics data (Fig. 5).
Fig.5
Validations with western blot. Western blot analysis of cortex, hippocampus and cerebellum shows expression change of AβPP, Cdk-5, HSP70, and mTOR proteins in newborn 5XFAD mice compared to LM.
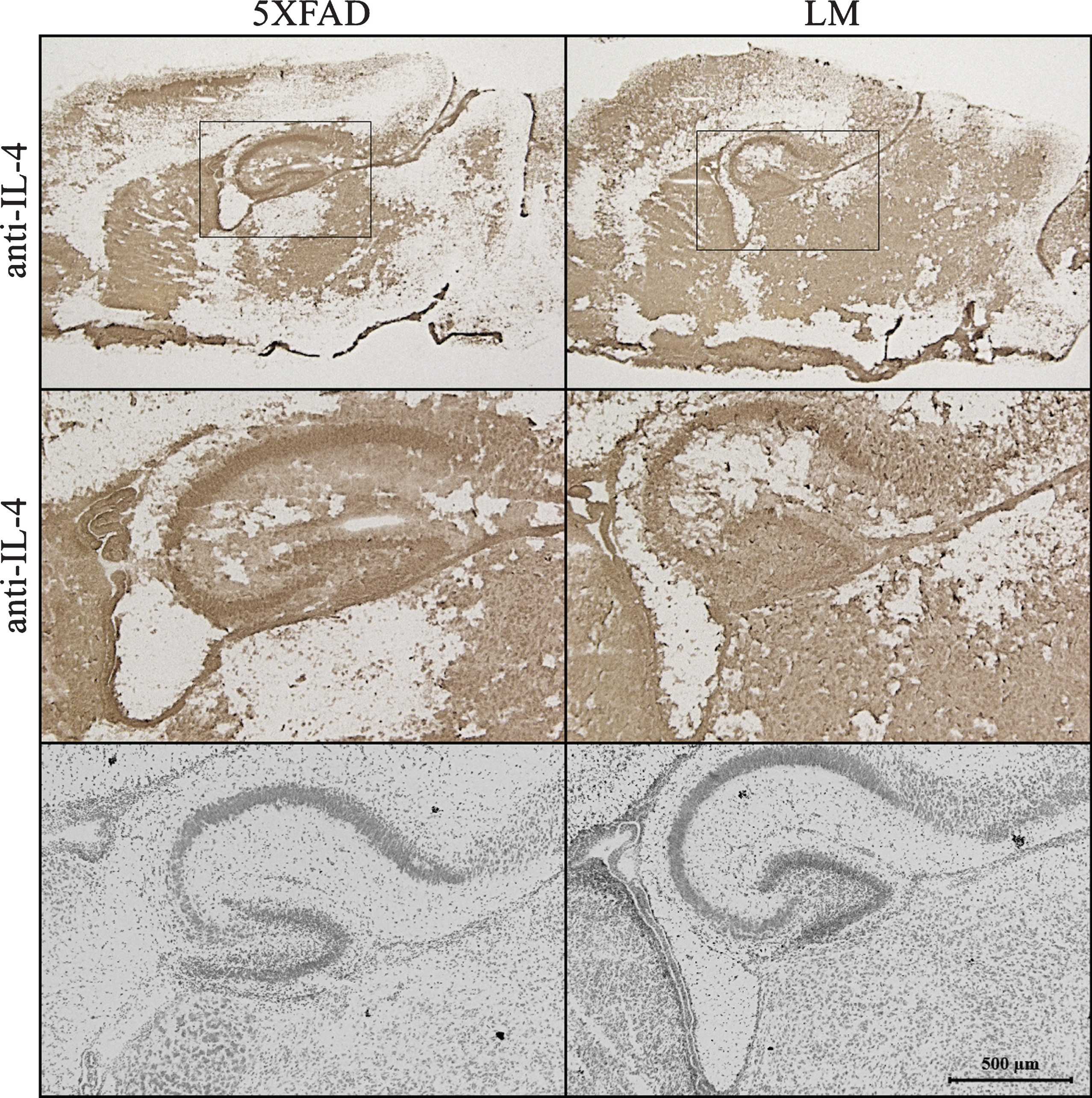
Expression of IL-4 protein, which was suggested by IPA to be an upstream regulator of the co-regulated proteins, was validated using immunohistochemistry (Fig. 6). The result indicated an increase in IL-4 expression in CA1 area of the 5XFAD hippocampus.
Fig.6
Immunostaining. IL-4 staining of newborn sagittal brain sections of 5XFAD and LM mice. (Top: IL-4 staining of whole brain, Middle: IL-4 staining of hippocampus, Bottom: Nissl Staining of hippocampus).
Among the proteins with a modulated expression, a bibliographic search was carried out to identify those reported in the previous proteomics studies of postmortem human AD brains (Table 1). We found that 10 proteins from cortex, 2 proteins from hippocampus, and 1 protein from cerebellum presented in the previous studies were also altered in our study of newborn 5XFAD mice in the same direction.
Table 1
Proteins that significantly altered in newborn 5XFAD mouse model in comparison with those previously reported in proteomics studies of postmortem AD brain between 2002–2016
| Accession No | Protein | Newborn 5XFAD | postmortem AD |
| Cortex | |||
| P03995 | Glial fibrillary acidic protein* | ↓ | [65–67] |
| ↑↓ | |||
| P17182 | Alpha enolase | ↓ | [65, 68] |
| ↑↓ | |||
| P51807 | Dynein light chain | ↑ | [65] |
| ↑ | |||
| P62141 | Serine/threonine-protein phosphatase PP1-beta catalytic subunit | ↓ | [69] |
| ↓ | |||
| Q80Z24 | Neuronal growth regulator 1* | ↓ | [69] |
| ↓ | |||
| Q8BHZ0 | Protein FAM49A | ↑ | [69] |
| ↑ | |||
| Q99MB2 | Mitochondrial fission regulator 1 | ↓ | [69] |
| ↓ | |||
| Q9JKK7 | Tropomodulin-2* | ↑ | [69] |
| ↑ | |||
| Q9QZM0 | Ubiquilin-2* | ↓ | [67] |
| ↓ | |||
| Q9WUK2 | Eukaryotic translation initiation factor 4H | ↑ | [69] |
| ↑ | |||
| Hippocampus | |||
| P14824 | Annexin A6* | ↑ | [53] |
| ↑ | |||
| P62204 | Calmodulin | ↑ | [68] |
| ↑ | |||
| Cerebellum | |||
| Q9QYR6 | Microtubule-associated protein 1A* | ↑ | [53] |
| ↑ |
(↑: upregulated, ↓: downregulated) (*Cell morphology, cellular assembly and organization, cellular function and maintenance, cellular growth and proliferation related proteins).
DISCUSSION
Although AD is an age-related neurodegenerative disorder that demonstrates symptoms at mid-60 s for the late-onset form [18] which represents more than 95% of the AD cases, alterations of the brain are known to start decades before the symptoms. These alterations selectively affect the particular functional regions of the brain, such as cortex and hippocampus, instead of dominating the whole brain. Previous proteomics studies of transgenic mouse models include different time-points varying between 1 month to 12 months [11, 19–26]. Here, we carried out a proteomics study with newborn 5XFAD mice focusing on three brain regions; cortex, hippocampus and cerebellum. Much to our surprise, a noteworthy change in protein expression profile was determined, especially considering the other different pre-symptomatic time point studies in the literature [11, 19].
Neurodegenerative disorders have symptoms reflecting the functional area that is damaged by pathology. In AD, the earliest symptom is memory loss, which indicates hippocampus and entorhinal cortex defect. In the next stage of the disease, carrying out the cognitive tasks such as reading, recognizing objects and decision making become much harder according to spreading of the disease through the cortex. At the final stage, widespread brain atrophy causes loss of language skills, basic motor skills and personality changes [27–31]. Even, schizophrenia is detected to be linked with cerebellum damage in some cases. Our data exhibit region specific alterations in protein expression pattern (Fig. 1). The protein expression alterations were found mainly localized in the cortex and the hippocampus, which are known to be the most affected regions in AD, while cerebellum displays fewer alterations. Though the top canonical pathways related to cortex, hippocampus and cerebellum differed, cellular and molecular functions, that were expected to change in these regions, showed similarity (Fig. 3). This result suggests that increased Aβ expression of the mouse model caused by mutation, leads to similar functional alterations even though alterations occur through different proteins.
Among the top ten cellular functions, obtained by IPA, we determined that four functions, cellular assembly and organization, cellular function and maintenance, cell morphology and cell cycle, were affected in all three brain regions (Fig. 3). According to our current knowledge, cortex and hippocampus are primarily effected brain regions in AD and they interact with each other directly by neural connections [1, 2]. Thus, it is not a surprise that they shared more functional alterations as well as protein expression patterns. However, cerebellum, the least and latest affected brain region, showed the minimum protein expression change in newborn AD brain.
Several in vitro studies showed that low concentrations of aggregated Aβ, Aβ fibrils including Aβ40 and Aβ42, and soluble AβPP induce the neurite outgrowth and branching [32–34] even though Aβ is the main toxic element of senile plaque pathology of AD. AβPP was also identified as an independently operating growth cone cell adhesion molecule that affects axon guidance in adult and developing brains [35]. Besides the fact that it is a critical factor for neural circuit development, its expression is increased in reactive glia after neuronal damage [36]. Our IPA analysis, suggesting that the proteome alteration in 5XFAD mice is related to cell morphology, cellular movement and, cellular assembly and organization, led us to perform a primary cell culture experiment to investigate morphological and organizational differences in newborn 5XFAD mice compared to LM mice. Our analysis revealed an increase in neurite number in cultured cells from newborn 5XFAD cortex, hippocampus and cerebellum (Fig. 4). We predicted that this increase may be the result of the rapid, but not high enough to be toxic, production of AβPP and Aβ. Western blot analysis verified the increase in AβPP expression in all three brain regions (Fig. 5). These results indicate that even at an early time point as neonatal day-1, molecular shift in 5XFAD mouse brain causes observable and significant alterations in cellular structure.
Tau pathology has not been reported for 5XFAD mouse model yet and we did not perform any experiments to investigate tauopathy in newborn mice, however, some of the proteins we identified demonstrate a relation with tauopathy. Heat shock 70 kDa protein, which was one of 20 co-regulated proteins (Fig. 2), is a molecular chaperon that prevents aggregate formation in cells, suppress AD conditions and protects neurons in various animal models of AD [37–39]. HSP70 might promote tau binding to microtubules and implicate blocking tau aggregation or directly prevent tau aggregation, resulting in prevention of tauopathy [40–42]. Downregulation of HSP70, we detected in 5XFAD cortex, hippocampus and cerebellum, might favor the possible tauopathy. Cyclin-dependent kinase 5 is another important protein for tauopathy and AD. Cdk5 plays an important role in neuronal development, synaptic plasticity, cognition and neuronal survivor [43–46]. Hyperactivation of Cdk5 under neurotoxic or stress conditions, as seen in AD, leads to hyperphosphorylation of several cytoskeletal components such as tau and neurofilaments, thus disturbances in cytoskeletal organization and loss of dendritic spines [47–50]. Our results show elevated expression of Cdk5 (Fig. 5) which was previously reported in the brain and CSF of AD patients [51, 52]. We also detected elevated levels of Microtubule-Associated Protein MAP1A, parallel to previous postmortem AD studies [53], which might be an afford of neurons to compensate the loss of one functional member of MAP family; tau [54].
Neuroinflammation is an important factor for AD progress. The glial activation starts as a reaction to Aβ toxicity and neural damage becomes harmful in time [55]. IL-4 is an anti-inflammatory cytokine that suppresses pro-inflammatory cytokine production to neutralize neuroinflammatory process in general and in AD [56–59]. Several in vitro and in vivo studies showed exogenous IL-4 reduces the accumulation of Aβ [59–61] while endogenous IL-4 leads to neurodegeneration in hippocampal neurons [62]. Our IPA analysis points out the top upstream regulator of the common proteins within all three regions is IL-4 (Supplementary Table 2). In addition, when the pathway of inflammation-related proteins in literature and co-regulated proteins of our data are combined, the total pathway shows these two groups of proteins are connected directly or indirectly while they do not share any protein (Supplementary Figure 1). This might be an indicator of activated neuroinflammation reaction of the brain against increased amount of Aβ in newborn 5XFAD. Therefore, we investigated the expression change of IL-4 by immunochemistry which showed an increase in IL-4 levels in CA1 area of 5XFAD hippocampus compared to LM hippocampus (Fig. 6). Since the previous data is inconclusive, it is hard to say whether this increase is a neuroprotective activation in response to Aβ elevation or a part of neurotoxic events. Nevertheless, we may conclude there is a disturbance in balance between pro- and anti-inflammatory cytokines.
We also compared our data to the proteomic studies which were previously performed with postmortem human brain tissue of AD patients (Table 1). Both in the literature and in our study, we determined 13 proteins that showed expression change in the same direction. Overall, 6 of these proteins were stated as related to cellular functions such as; cellular assembly and organization, cellular function and maintenance, and cell morphology via IPA. Another important aspect of these results is that, the postmortem human brain studies we used for comparison were performed on sporadic AD cases; nevertheless, the results are still considerably similar to our transgenic mouse model study results. This similarity supports the reliability of 5XFAD transgenic mouse model. It is understandable to detect a protein expression change caused by the overexpression of APP and PSEN genes in such familial mouse model, however, observing the alterations in cellular extend is intriguing. On the other hand, conducting a pre-symptomatic stage study with sporadic AD cases is not possible, and clinical and pathophysiological features of the familial and sporadic AD are known to be similar except small differences like severity and age of on-set [63, 64].
Other studies focusing on proteome profile changes in the brain tissue of AD mice during the progression of the disease showed that the number of alterations between the proteome of AD and healthy brains increase as an age-dependent manner. Surprisingly, our data of newborn AD mouse model displays more similarities with postmortem human AD proteomes than other progression-stage proteome studies. One of the possible explanations of how these similarities in the molecular and morphological alterations do not cause disease pathology and symptoms for decades, may be the activation of both detrimental and protective mechanisms via the altered proteins which were discovered in the current study. Thereby, it may be assumed that the disease falls into a silenced period. However, through the course of time, the burden of the cell increases because of increasing DNA damage, oxidative stress, mitochondrial dysfunction, protein aggregates, and defects in proteolysis system accompanied by aging the disease may override.
Finally, based on our present data and previous studies, unrevealing the early molecular events is crucial to get a better understanding of AD pathogenesis. Further investigation is necessary to figure out whether neonatal period represents a favorable model for old age. Our findings suggest a radical new aspect of the genesis of neurodegeneration where we show that at very early stages there is evidence to support the initiation of altered molecular pathways which might lead to cognitive impairment.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/ADR-170049.
REFERENCES
[1] | Thal DR , Rub U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[2] | Bobinski M , de Leon MJ , Convit A , De Santi S , Wegiel J , Tarshish CY , Saint Louis LA , Wisniewski HM ((1999) ) MRI of entorhinal cortex in mild Alzheimer’s disease. Lancet 353: , 38–40. |
[3] | Pearson RC , Esiri MM , Hiorns RW , Wilcock GK , Powell TP ((1985) ) Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci U S A 82: , 4531–4534. |
[4] | de Leon MJ , George AE , Stylopoulos LA , Smith G , Miller DC ((1989) ) Early marker for Alzheimer’s disease: The atrophic hippocampus. Lancet 2: , 672–673. |
[5] | Hyman BT , Van Hoesen GW , Damasio AR , Barnes CL ((1984) ) Alzheimer’s disease: Cell-specific pathology isolates the hippocampal formation. Science 225: , 1168–1170. |
[6] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26: , 10129–10140. |
[7] | Girard SD , Jacquet M , Baranger K , Migliorati M , Escoffier G , Bernard A , Khrestchatisky M , Feron F , Rivera S , Roman FS , Marchetti E ((2014) ) Onset of hippocampus-dependent memory impairments in 5XFAD transgenic mouse model of Alzheimer’s disease. Hippocampus 24: , 762–772. |
[8] | Girard SD , Baranger K , Gauthier C , Jacquet M , Bernard A , Escoffier G , Marchetti E , Khrestchatisky M , Rivera S , Roman FS ((2013) ) Evidence for early cognitive impairment related to frontal cortex in the 5XFAD mouse model of Alzheimer’s disease. J Alzheimers Dis 33: , 781–796. |
[9] | Eimer WA , Vassar R ((2013) ) Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8: , 2. |
[10] | Jawhar S , Trawicka A , Jenneckens C , Bayer TA , Wirths O ((2012) ) Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease.196 e. Neurobiol Aging 33: , 129–140. |
[11] | Lv J , Ma S , Zhang X , Zheng L , Ma Y , Zhao X , Lai W , Shen H , Wang Q , Ji J ((2014) ) Quantitative proteomics reveals that PEA15 regulates astroglial Abeta phagocytosis in an Alzheimer’s disease mouse model. J Proteomics 110: , 45–58. |
[12] | Manavalan A , Mishra M , Sze SK , Heese K ((2013) ) Brain-site-specific proteome changes induced by neuronal P60TRP expression. Neurosignals 21: , 129–149. |
[13] | Martin B , Brenneman R , Becker KG , Gucek M , Cole RN , Maudsley S ((2008) ) iTRAQ analysis of complex proteome alterations in 3xTgAD Alzheimer’s mice: Understanding the interface between physiology and disease. PLoS One 3: , e2750. |
[14] | O’Neil SE , Sitkauskiene B , Babusyte A , Krisiukeniene A , Stravinskaite-Bieksiene K , Sakalauskas R , Sihlbom C , Ekerljung L , Carlsohn E , Lotvall J ((2011) ) Network analysis of quantitative proteomics on asthmatic bronchi: Effects of inhaled glucocorticoid treatment. Respir Res 12: , 124. |
[15] | Wisniewski JR , Zougman A , Nagaraj N , Mann M ((2009) ) Universal sample preparation method for proteome analysis. Nat Methods 6: , 359–362. |
[16] | Beker MC , Caglayan B , Yalcin E , Caglayan AB , Turkseven S , Gurel B , Kelestemur T , Sertel E , Sahin Z , Kutlu S , Kilic U , Baykal AT , Kilic E ((2018) ) Time-of-day dependent neuronal injury after ischemic stroke: Implication of circadian clock transcriptional factor Bmal1 and survival kinase AKT. Mol Neurodegener 55: , 2565–2576. |
[17] | Demircan T , Keskin I , Dumlu SN , Aytürk N , Avşaroğlu ME , Akgün E , Öztürk G , Baykal AT ((2017) ) Detailed tail proteomic analysis of axolotl (Ambystoma mexicanum) using an mRNA-seq reference database. Proteomics 17: . doi: 10.1002/pmic.201600338 |
[18] | Prince M , Bryce R , Albanese E , Wimo A , Ribeiro W , Ferri CP ((2013) ) The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement 9: , 63–75 e62. |
[19] | Hartl D , Rohe M , Mao L , Staufenbiel M , Zabel C , Klose J ((2008) ) Impairment of adolescent hippocampal plasticity in a mouse model for Alzheimer’s disease precedes disease phenotype. PLoS One 3: , e2759. |
[20] | Sun Y , Rong X , Lu W , Peng Y , Li J , Xu S , Wang L , Wang X ((2015) ) Translational study of Alzheimer’s disease (AD) biomarkers from brain tissues in AbetaPP/PS1 mice and serum of AD patients. J Alzheimers Dis 45: , 269–282. |
[21] | Yang H , Wittnam JL , Zubarev RA , Bayer TA ((2013) ) Shotgun brain proteomics reveals early molecular signature in presymptomatic mouse model of Alzheimer’s disease. J Alzheimers Dis 37: , 297–308. |
[22] | Chang SH , Jung IS , Han GY , Kim NH , Kim HJ , Kim CW ((2013) ) Proteomic profiling of brain cortex tissues in a Tau transgenic mouse model of Alzheimer’s disease. Biochem Biophys Res Commun 430: , 670–675. |
[23] | Walls KC , Coskun P , Gallegos-Perez JL , Zadourian N , Freude K , Rasool S , Blurton-Jones M , Green KN , LaFerla FM ((2012) ) Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J Biol Chem 287: , 30317–30327. |
[24] | Chou JL , Shenoy DV , Thomas N , Choudhary PK , Laferla FM , Goodman SR , Breen GA ((2011) ) Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J Proteomics 74: , 466–479. |
[25] | Rhein V , Song X , Wiesner A , Ittner LM , Baysang G , Meier F , Ozmen L , Bluethmann H , Drose S , Brandt U , Savaskan E , Czech C , Gotz J , Eckert A ((2009) ) Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A 106: , 20057–20062. |
[26] | Shin SJ , Lee SE , Boo JH , Kim M , Yoon YD , Kim SI , Mook-Jung I ((2004) ) Profiling proteins related to amyloid deposited brain of Tg2576 mice. Proteomics 4: , 3359–3368. |
[27] | Arnold SE , Hyman BT , Flory J , Damasio AR , Van Hoesen GW ((1991) ) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex 1: , 103–116. |
[28] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[29] | DeKosky ST , Scheff SW ((1990) ) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27: , 457–464. |
[30] | DeKosky ST , Scheff SW , Styren SD ((1996) ) Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration 5: , 417–421. |
[31] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[32] | Susen K , Blochl A ((2005) ) Low concentrations of aggregated beta-amyloid induce neurite formation via the neurotrophin receptor p75. J Mol Med (Berl) 83: , 720–735. |
[33] | Wujek JR , Dority MD , Frederickson RC , Brunden KR ((1996) ) Deposits of A beta fibrils are not toxic to cortical and hippocampal neurons in vitro. Neurobiol Aging 17: , 107–113. |
[34] | Milward EA , Papadopoulos R , Fuller SJ , Moir RD , Small D , Beyreuther K , Masters CL ((1992) ) The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron 9: , 129–137. |
[35] | Sosa LJ , Bergman J , Estrada-Bernal A , Glorioso TJ , Kittelson JM , Pfenninger KH ((2013) ) Amyloid precursor protein is an autonomous growth cone adhesion molecule engaged in contact guidance. PLoS One 8: , e64521. |
[36] | Siman R , Card JP , Nelson RB , Davis LG ((1989) ) Expression of beta-amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 3: , 275–285. |
[37] | Evans CG , Wisen S , Gestwicki JE ((2006) ) Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. J Biol Chem 281: , 33182–33191. |
[38] | Muchowski PJ , Wacker JL ((2005) ) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6: , 11–22. |
[39] | Bobkova NV , Garbuz DG , Nesterova I , Medvinskaya N , Samokhin A , Alexandrova I , Yashin V , Karpov V , Kukharsky MS , Ninkina NN , Smirnov AA , Nudler E , Evgen’ev M ((2014) ) Therapeutic effect of exogenous hsp70 in mouse models of Alzheimer’s disease. J Alzheimers Dis 38: , 425–435. |
[40] | Dou F , Netzer WJ , Tanemura K , Li F , Hartl FU , Takashima A , Gouras GK , Greengard P , Xu H ((2003) ) Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A 100: , 721–726. |
[41] | Patterson KR , Ward SM , Combs B , Voss K , Kanaan NM , Morfini G , Brady ST , Gamblin TC , Binder LI ((2011) ) Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 50: , 10300–10310. |
[42] | Jinwal UK , O’Leary 3rd JC , Borysov SI , Jones JR , Li Q , Koren 3rd J , Abisambra JF , Vestal GD , Lawson LY , Johnson AG , Blair LJ , Jin Y , Miyata Y , Gestwicki JE , Dickey CA ((2010) ) Hsc70 rapidly engages tau after microtubule destabilization. J Biol Chem 285: , 16798–16805. |
[43] | McLinden KA , Trunova S , Giniger E ((2012) ) At the fulcrum in health and disease: Cdk5 and the balancing acts of neuronal structure and physiology. Brain Disord Ther 2012: , 001. |
[44] | Nikolic M , Dudek H , Kwon YT , Ramos YF , Tsai LH ((1996) ) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10: , 816–825. |
[45] | Fischer A , Sananbenesi F , Schrick C , Spiess J , Radulovic J ((2002) ) Cyclin-dependent kinase 5 is required for associative learning. J Neurosci 22: , 3700–3707. |
[46] | Tan TC , Valova VA , Malladi CS , Graham ME , Berven LA , Jupp OJ , Hansra G , McClure SJ , Sarcevic B , Boadle RA , Larsen MR , Cousin MA , Robinson PJ ((2003) ) Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol 5: , 701–710. |
[47] | Qu J , Nakamura T , Cao G , Holland EA , McKercher SR , Lipton SA ((2011) ) S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A 108: , 14330–14335. |
[48] | Ahlijanian MK , Barrezueta NX , Williams RD , Jakowski A , Kowsz KP , McCarthy S , Coskran T , Carlo A , Seymour PA , Burkhardt JE , Nelson RB , McNeish JD ((2000) ) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A 97: , 2910–2915. |
[49] | Lee MS , Kwon YT , Li M , Peng J , Friedlander RM , Tsai LH ((2000) ) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: , 360–364. |
[50] | Noble W , Olm V , Takata K , Casey E , Mary O , Meyerson J , Gaynor K , LaFrancois J , Wang L , Kondo T , Davies P , Burns M , Veeranna, Nixon R , Dickson D , Matsuoka Y , Ahlijanian M , Lau LF , Duff K ((2003) ) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38: , 555–565. |
[51] | Sadleir KR , Vassar R ((2012) ) Cdk5 protein inhibition and Abeta42 increase BACE1 protein level in primary neurons by a post-transcriptional mechanism: Implications of CDK5 as a therapeutic target for Alzheimer disease. J Biol Chem 287: , 7224–7235. |
[52] | Olah Z , Kalman J , Toth ME , Zvara A , Santha M , Ivitz E , Janka Z , Pakaski M ((2015) ) Proteomic analysis of cerebrospinal fluid in Alzheimer’s disease: Wanted dead or alive. J Alzheimers Dis 44: , 1303–1312. |
[53] | Manavalan A , Mishra M , Feng L , Sze SK , Akatsu H , Heese K ((2013) ) Brain site-specific proteome changes in aging-related dementia. Exp Mol Med 45: , e39. |
[54] | Iqbal K , Alonso Adel C , Chen S , Chohan MO , El-Akkad E , Gong CX , Khatoon S , Li B , Liu F , Rahman A , Tanimukai H , Grundke-Iqbal I ((2005) ) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: , 198–210. |
[55] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WS , Hampel H , Hull M , Landreth G , Lue L , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[56] | Furlan R , Bergami A , Lang R , Brambilla E , Franciotta D , Martinelli V , Comi G , Panina P , Martino G ((2000) ) Interferon-beta treatment in multiple sclerosis patients decreases the number of circulating T cells producing interferon-gamma and interleukin-4. J Neuroimmunol 111: , 86–92. |
[57] | Chao CC , Molitor TW , Hu S ((1993) ) Neuroprotective role of IL-4 against activated microglia. J Immunol 151: , 1473–1481. |
[58] | Ledeboer A , Breve JJ , Poole S , Tilders FJ , Van Dam AM ((2000) ) Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 30: , 134–142. |
[59] | Paludan SR , Lovmand J , Ellermann-Eriksen S , Mogensen SC ((1997) ) Effect of IL-4 and IL-13 on IFN-gamma-induced production of nitric oxide in mouse macrophages infected with herpes simplex virus type 2. FEBS Lett 414: , 61–64. |
[60] | Kawahara K , Suenobu M , Yoshida A , Koga K , Hyodo A , Ohtsuka H , Kuniyasu A , Tamamaki N , Sugimoto Y , Nakayama H ((2012) ) Intracerebral microinjection of interleukin-4/interleukin-13 reduces beta-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 207: , 243–260. |
[61] | Latta CH , Sudduth TL , Weekman EM , Brothers HM , Abner EL , Popa GJ , Mendenhall MD , Gonzalez-Oregon F , Braun K , Wilcock DM ((2015) ) Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-beta using BV2 microglial cells and APP/PS1 transgenic mice. J Neuroinflammation 12: , 41. |
[62] | Park KW , Baik HH , Jin BK ((2008) ) Interleukin-4-induced oxidative stress via microglial NADPH oxidase contributes to the death of hippocampal neurons in vivo. Curr Aging Sci 1: , 192–201. |
[63] | Lippa CF , Saunders AM , Smith TW , Swearer JM , Drachman DA , Ghetti B , Nee L , Pulaski-Salo D , Dickson D , Robitaille Y , Bergeron C , Crain B , Benson MD , Farlow M , Hyman BT , George-Hyslop SP , Roses AD , Pollen DA ((1996) ) Familial and sporadic Alzheimer’s disease: Neuropathology cannot exclude a final common pathway. Neurology 46: , 406–412. |
[64] | Swearer JM , O’Donnell BF , Drachman DA , Woodward BM ((1992) ) Neuropsychological features of familial Alzheimer’s disease. Ann Neurol 32: , 687–694. |
[65] | Musunuri S , Kultima K , Richard BC , Ingelsson M , Lannfelt L , Bergquist J , Shevchenko G ((2015) ) Micellar extraction possesses a new advantage for the analysis of Alzheimer’s disease brain proteome. Anal Bioanal Chem 407: , 1041–1057. |
[66] | Tsuji T , Shiozaki A , Kohno R , Yoshizato K , Shimohama S ((2002) ) Proteomic profiling and neurodegeneration in Alzheimer’s disease. Neurochem Res 27: , 1245–1253. |
[67] | Minjarez B , Calderon-Gonzalez KG , Rustarazo ML , Herrera-Aguirre ME , Labra-Barrios ML , Rincon-Limas DE , Del Pino MM , Mena R , Luna-Arias JP ((2016) ) Identification of proteins that are differentially expressed in brains with Alzheimer’s disease using iTRAQ labeling and tandem mass spectrometry. J Proteomics 139: , 103–121. |
[68] | Zahid S , Oellerich M , Asif AR , Ahmed N ((2014) ) Differential expression of proteins in brain regions of Alzheimer’s disease patients. Neurochem Res 39: , 208–215. |
[69] | Andreev VP , Petyuk VA , Brewer HM , Karpievitch YV , Xie F , Clarke J , Camp D , Smith RD , Lieberman AP , Albin RL , Nawaz Z , El Hokayem J , Myers AJ ((2012) ) Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J Proteome Res 11: , 3053–3067. |

