
Melatonin Effects on EEG Activity During Sleep Onset in Mild-to-Moderate Alzheimer’s Disease: A Pilot Study
Abstract
There is evidence demonstrating that 5-mg of fast-release melatonin significantly reduces nocturnal sleep onset in patients with mild-to-moderate Alzheimer’s disease (AD). However, the physiological mechanism that could promote sleep installation by melatonin in patients with AD is still poorly understood. The present pilot study was designed to analyze the effects of melatonin on cortical activity during the sleep onset period (SOP) in eight mild-to-moderate AD patients treated with 5-mg of fast-release melatonin. Electroencephalographic recordings were obtained from C3-A1, C4-A2, F7-T3, F8-T4, F3-F4, and O1-O2. The relative power (RP), interhemispheric, intrahemispheric, and fronto-posterior correlations of six electroencephalographic bands were calculated and compared between two conditions: placebo and melatonin. Results show that at F7-T3, F3-F4, and C3-A1, melatonin induced an increase of the RP of the delta band. Likewise, in F7-T3, melatonin induced a decrease of the RP in the alpha1 band. Similarly, results show a lower interhemispheric correlation between the F7-T3 and F8-T4 derivations in the alpha1 band compared to the placebo condition. We conclude that the short sleep onset related to melatonin intake in AD patients was associated with a lower RP of the alpha1, a higher RP of the delta band (mainly in the left hemisphere) and a decreased interhemispheric EEG coupling in the alpha1 band. The possible role of the GABAergic neurotransmission as well as of the cascade of neurochemical events that melatonin triggers on sleep onset are discussed.
INTRODUCTION
Although difficulty in falling asleep during the night is a common symptom of Alzheimer’s disease (AD) [1–6], few studies have focused on testing pharmacological alternatives for treating sleep-related problems in AD patients [7]. One well-known treatment for sleep disturbances is melatonin (N-acetyl-5-methoxytryptamine), which plays a major role in regulating circadian rhythms. Melatonin is a hormone secreted by the pineal gland that modulates the sleep-wake cycle. Its secretion is enhanced during darkness and suppressed during day light [8]. In healthy subjects, melatonin has been shown to decrease sleep onset latency, increase total sleep time, and improve overall sleep quality [9]. Regarding neural activity, there is evidence that after melatonin administration the electroencephalographic activity (EEG) of healthy subject’s exhibits increased sleep spindles but reduced slow-wave activity [10]. In a previous study, we demonstrated that 5 mg of fast-release melatonin significantly reduced nocturnal sleep onset in patients with mild-to-moderate AD [11]. This reduction of sleep onset approaches the values reported for non-dementia geriatric patients [12]. However, the physiological mechanism through which melatonin promotes sleep installation is still only poorly understood.
The locations and pharmacology of melatonin receptors have been reviewed extensively. Two cloned receptors, MT1 and MT2, are of particular importance for rhythm physiology and pharmacology [13, 14]. Using gene knockout technology in mice and pharmacological manipulations, Liu et al. [15] found that the phase-shifting melatonin receptor in the suprachiasmatic nucleus (SCN) is MT2, while MT1 is associated with acute suppression of electrical activity in the SCN. In fact, there is evidence that MT1 receptors expressed in cell preparations [16, 17] activate Kir3 channels that underlie melatonin-induced increases in potassium conductance that, in turn, enhance GABAergic activity [18]. It has been shown that GABAergic cells in the hypothalamus, basal forebrain (BF), and the median (mPO) and ventrolateral preoptic areas (VLPO) are highly-active during sleep onset [19–22]. In addition, EEG activity observed during sleep onset has been related to GABAergic modulation in the hypothalamus, BF, mPO, and VLPO areas [23, 24].
Quantitative EEG analyses in healthy adult subjects have shown that the sleep onset period (SOP) is characterized by smooth changes during which the power of the fast frequencies decreases, while that of the low frequencies gradually increases [25–28]. Also, the degree of synchronization between EEG signals has been used to determine the functional relation between cortical areas associated with normal SOP. Morikawa et al. found that delta band synchronization between occipital and frontal regions and between central and parietal areas dropped sharply just before the alpha band disappeared during SOP [29]. Those authors further reported decreases in alpha synchronization 2-3 min before SOP, and detected significant increases in the synchronization of the fast frequencies just before the installation of stage 2 of non-rapid eye movement sleep (N-REM2) [30]. Likewise, a significant decrease in fronto-occipital and inter-frontal synchronization in the alpha band has been observed during SOP [31]. Finally, Kikuchi et al. conducted a study to examine interhemispheric EEG synchronization during the state of rest in elderly subjects, finding low EEG synchronization for the delta, theta, alpha and beta bands [32].
Considering that SOP is characterized by a specific EEG pattern in both elderly and healthy adult subjects, and that 5-mg of fast-release melatonin reduces latency to sleep onset in AD patients [11], the aim of the present study was to determine the effect of melatonin on EEG activity and the degree of EEG synchronization between different cortical areas in AD patients. We hypothesized that the facilitator effect of melatonin on SOP in AD patients will be associated with the generation of slow frequencies (delta, theta and alpha bands), together with a decrease in the activity of the fast frequencies (beta band) during SOP. Likewise, we proposed that melatonin will reduce the intrahemispheric, interhemispheric, and fronto-anterior correlation of the alpha band, while increasing the delta band correlation, compared to the placebo condition. To test these hypotheses, we evaluated the effects of melatonin during SOP on EEG spectral power and on intrahemispheric, interhemispheric, and fronto-anterior correlations in AD patients previously treated with melatonin [11].
MATERIALS AND METHODS
Participants
Eight patients with mild-to-moderate AD were recruited for this study. Their mean age was 65 years [±SE: 2.32]. Diagnoses of AD were conducted with neurological testing that clinically established the dementia syndrome. In an effort to obtain diagnoses of reversible dementia, the following tests were performed, following the suggestions of Barry et al. [33] and Amodar et al. [34]: blood count, blood chemistry, thyroid and hepatic function, simple contrasted CAT scan, and assessment of ischemic factors using the Helsinki scale. Also, an EEG study was conducted to discard epilepsy. The diagnostic criteria applied were adapted from those published by the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) Work Group [35]. Global dementia severity was classified in accordance with the Clinical Dementia Rating (CDR) scale [36], where 0 indicates no cognitive impairment and 0.5, 1, 2, and 3 indicate very mild, mild, moderate, and severe dementia, respectively. One inclusion criterion was that subjects were considered to be suffering from mild-to-moderate AD; a second was alterations in sleep onset, which were evaluated in a clinical interview designed in our laboratory and applied to the caregiver who spends the night with each subject.
Informed consent was obtained from patients and their relatives or caregivers through the Outpatient Department. This study was approved by the Ethics Committee on Human Research of the National Institute of Psychiatry “Ramón de la Fuente Muñiz” (Project: NC093340.2) and complied with the guidelines established by the National Institutes of Health in the USA. Moreover, the work was carried out in strict accordance with The Code of Ethics of the World Medical Association (Helsinki Declaration, 1975) for experiments involving humans. It is important to note that none of the patients had received previous pharmacological treatment based on acetylcholine inhibitors or any other medication prescribed for dementia. Likewise, none had received prior pharmacological treatment for sleep disturbances or any other medication that could impact sleep.
Procedures
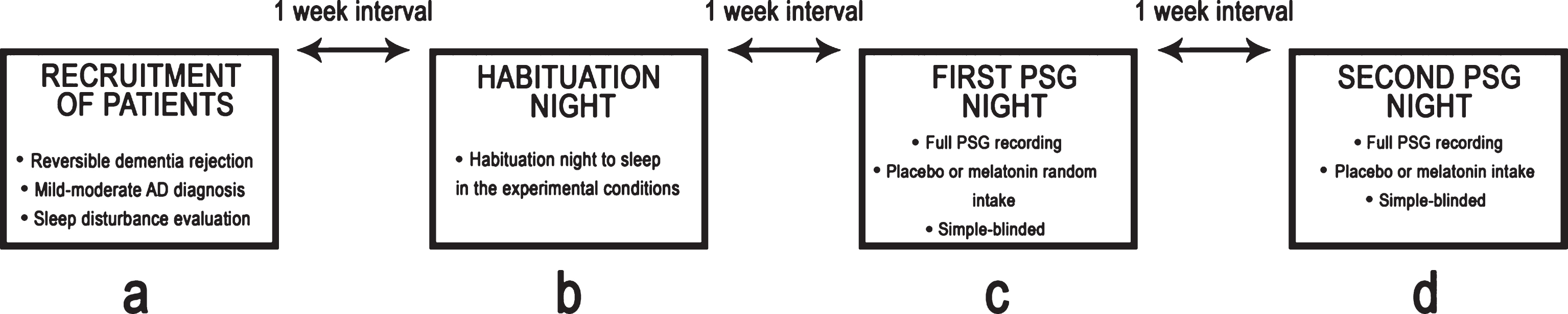
A single-blind, placebo-controlled study was conducted. A total of three nights of PSG recordings were obtained from each patient. The first night was considered a habituation session. On the second night, either an oral placebo or a 5-mg dose of fast-release melatonin (BIOQUIMED Labs) was administered, and full PSG recording was performed. On the third night, either an oral placebo or a 5-mg dose of fast-release melatonin was administered with full PSG recording. Both the melatonin and placebo were administered one hour before PSG recording began (approximately at 9:00 p.m.). The order of treatments was counterbalanced. When recording ended, and the study finalized, the patients withdrew from the laboratory. The three recordings were carried out within a one-week interval. Figure 1 illustrates our experimental design.
Fig.1
Schematic representation of the experimental design.
PSG recordings
Electrodes were placed at bipolar, F7-T3, F8-T4, F3-F4, O1-O2 and referential, C3-A1, C4-A2, derivations in accordance with the 10–20 international system for human EEG recording [37]. Two electrodes were attached superficially to both ocular canthi to obtain electrooculograms (EOG), while two additional electrodes were attached to the mentalis muscle to obtain electromyograms (EMG). All PSG recordings were performed during 9 h, from 10:00 p.m. to 7:00 a.m., in a room specially-designed for PSG studies, equipped with adequate audio and video monitoring systems. PSG signals were acquired with a Grass Model 15 polygraph and model 15A4 amplifiers (Grass-Telefactor, West Warwick, RI, USA). The sampling rate was set at 512 Hz, the high-pass at 1 Hz and the low-pass filter frequencies at 70 Hz.
Data analysis
SOP was scored, analyzed and then assessed visually in 20-s epochs. These periods consisted of transitional episodes from wakefulness to N-REM1 sleep. For the digital analyses, we chose a 6-min sample of EEG from the wake-sleep transition, the last 3 min of wakefulness, and the first 3 min of N-REM1 sleep, similar to the report of Hori’s H2-H3 states of wake-sleep transition [38]. The PSG patterns of wakefulness and N-REM1 were evaluated using international standards for visual evaluation of PSG recordings in humans [39]. Sleep latency, defined as the time that elapsed from lights off to onset of N-REM1, was also evaluated. All PSG recordings were made using GAMMA software (Grass, Co).
For digital EEG analyses, the EEG signals from the last 3 min of wakefulness and the first 3 min of N-REM1 sleep were examined off-line with CHECASEN software [40] to remove recordings altered by electrical EOG and EMG artifacts. The CHECASEN programs permits off-line, visual inspection of previously digitized electroencephalographic signals to eliminate segments contaminated by artifacts (for details about the software see: http://www.medigraphic.com/pdfs/inge/ib-2010/ib102 g.pdf). By means of the CHECASEN software, a total of 634 non-artifact-free segments of wakefulness and 610 non-artifact-free segments of N-REM1 were rejected (n = 8). Thus, a total of 180 artifact-free,1-s EEG segments (90 of wakefulness and 90 of N-REM1 sleep) from each subject and condition (placebo, melatonin) were analyzed using EEGmagic software to obtain the relative power (RP) of the EEG bands (i.e., the proportional contribution of each band expressed as a percentage of total power), and the functional synchronization between successive amplitude values of EEG segments in the EEG derivations [41]. EEGmagic first applies the Fast Fourier Transform to the EEG signals to obtain the RP values of frequencies grouped in broad bands. For purposes of the present study, the frequency bands were established as follows: delta (1–3.0 Hz), theta (4–7.0 Hz), alpha1 (8–10.0 Hz), alpha2 (11–13.0 Hz), beta1 (14–19.0 Hz), and beta2 (20–30 Hz). The parameters analyzed with EEGmagic were calculated following the steps indicated in Fig. 2. The instantaneous spectrum of the digitized signals was obtained by calculating the real and imaginary parts of the direct discrete Fourier transform using formulas 1–3 (Fig. 3a). Formulas 4–13 were applied to calculate the auto-spectra (average) and crossed spectrum (average) of the digitized signals (Fig. 3b). Later, formula 14 was applied by the EEGmagic program to calculate the Pearson product-moment correlation coefficients (r) (Fig. 3c) between bilateral regions (interhemispheric correlation: C3-A1 with C4-A2, and F7-T3 with F8-T4), between regions in the same hemisphere (intrahemispheric correlation: C3-A1 with F7-T3 and C4-A2 with F8-T4), and between fronto-posterior regions (F3-F4 with O1-O2).
Fig.2
Method used to calculate the relative power spectra and correlation: (a) the analog signals A and B are digitized and then used to obtain the instantaneous spectra of each signal; (b) subsequently, the autospectra (SAA and SBB) and crossed spectrum (SAB) are calculated. The cross-correlation function is obtained with the Fourier Inverse Transform (F-1) of the crossed spectrum.
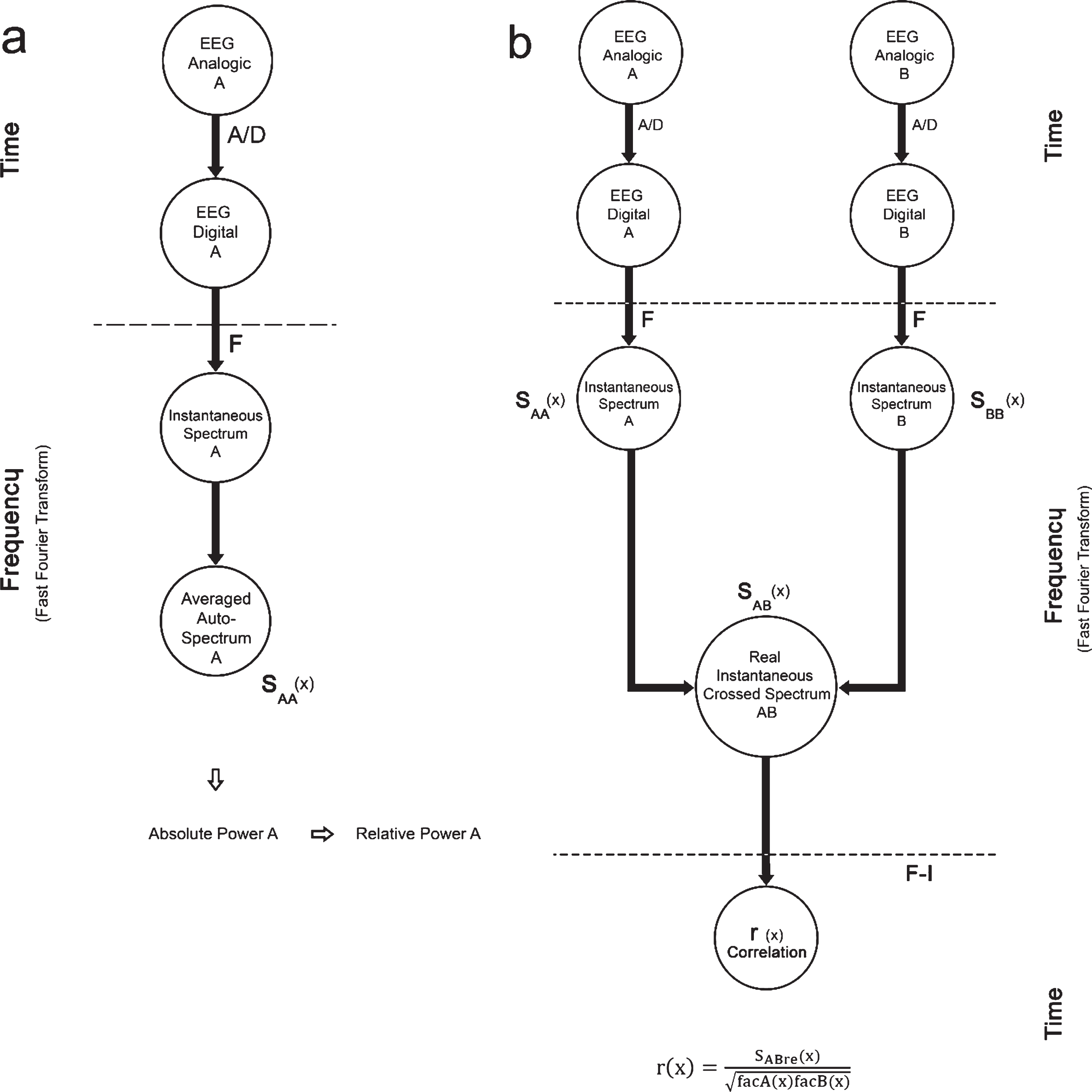
Fig.3
Formulas used to calculate the power spectra (a, b,) and the electroencephalographic correlation (c).

Statistical analyses
Sleep latencies under the placebo and melatonin conditions were assessed using two correlated-groups Student t tests with significance set at p≤0.05. For RP and r analyses, the EEG bands were also assessed using correlated-groups Student t tests with significance set at p≤0.05. For statistical purposes, r values were transformed to Fisher’s z-scores and RP values were transformed to natural logarithms.
RESULTS
Sociodemographic and sleep data
The sociodemographic information and sleep characteristics of the eight patients with mild-to-moderate AD are shown in Table 1. Their mean age was 65.62 years (SE±1.01) and mean education level was 9.37 years (SE±1.05). Only one was living alone at the time of study. Diagnoses determined that 50% of these patients had mild AD and that the other 50% had moderate AD. The most common sleep disturbance reported was insomnia. The sleep latency of the patients in the placebo condition had a mean of 34.75 min (SE±8.54), but after melatonin administration this decreased significantly (mean = 15.25 min, SE±2.10) (t = 2.21, df = 14, p = 0.04).
Table 1
Sociodemographic and sleep characteristics of eight patients with mild-to-moderate Alzheimer’s disease
| Age (y) | Education (y) | Living arrangements | CDR evaluation | Sleep disturbances | Sleep latency (min) | ||
| Placebo | Melatonin* | ||||||
| Patient1 | 65 | 9 | Wife and others | Moderate | Insomnia/sleep bruxism | 45.5 | 15.0 |
| Patient2 | 61 | 15 | Wife only | Mild | Insomnia/periodic limb movement | 11.5 | 13.0 |
| Patient3 | 65 | 10 | Other relatives | Moderate | Insomnia | 37.0 | 14.0 |
| Patient4 | 69 | 6 | Alone | Moderate | Insomnia/parasomnia | 80.0 | 9.0 |
| Patient5 | 69 | 7 | Others | Mild | Insomnia/sleep bruxism | 42.0 | 8.5 |
| Patient6 | 63 | 12 | Others | Mild | Insomnia | 16.5 | 27.0 |
| Patient7 | 68 | 7 | Wife and others | Moderate | Insomnia/periodic limb movement | 4.0 | 16.0 |
| Patient8 | 65 | 9 | Wife and others | Mild | Insomnia | 41.5 | 19.5 |
| Mean = 65,62 | Mean = 9.37 | Mean = 34.75 | Mean = 15.25 | ||||
| SE±1.017 | SE±1.05 | SE±8.54 | SE±2.10 | ||||
| *(t = 2.21, df = 14, p = 0.04) | |||||||
Relative power
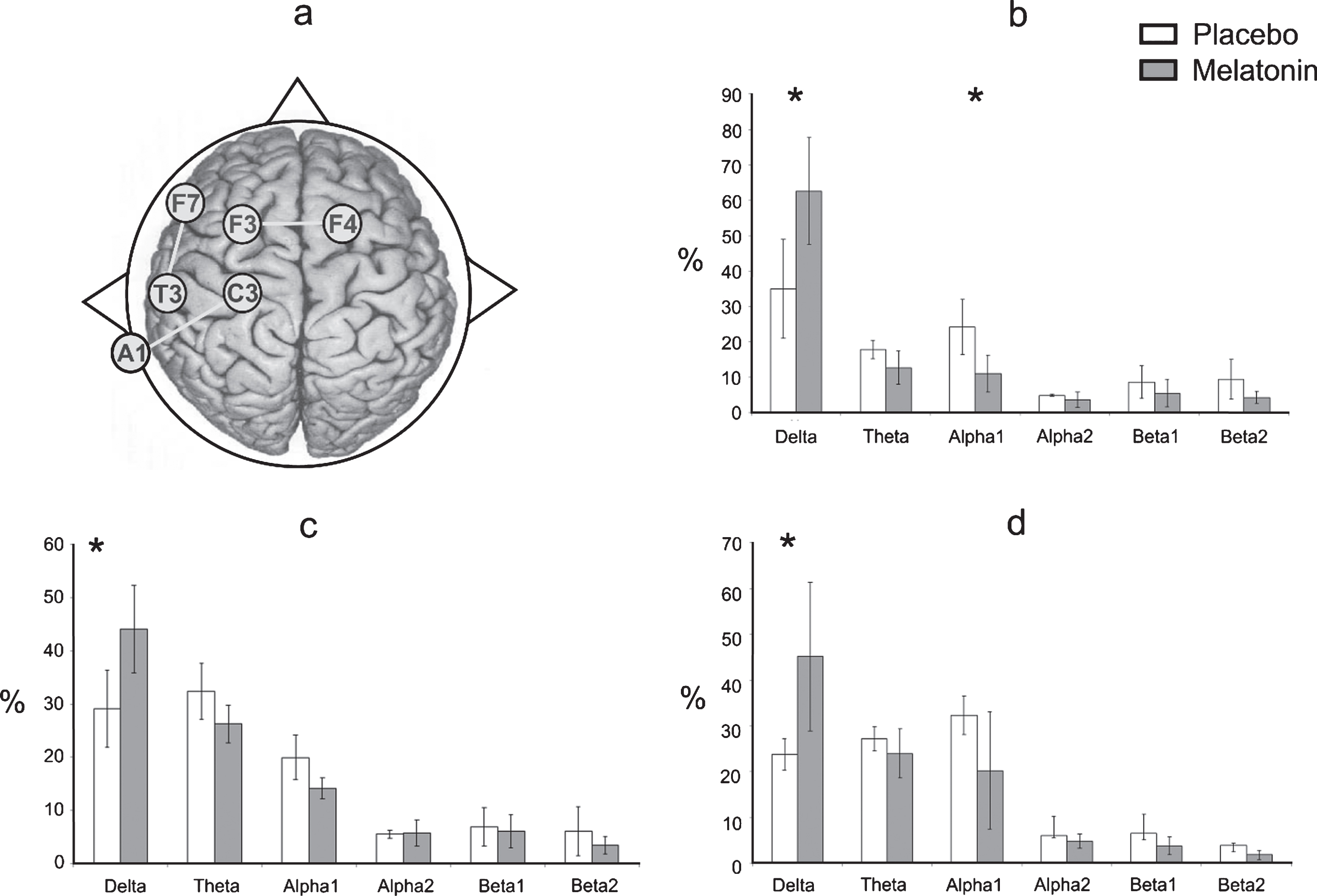
After melatonin administration, a significant increase of RP in delta (t = –5.49, df = 7, p = 0.0009) with a decrease in alpha1 (t = 5.048, df = 7, p = 0.0014) were observed at F7-T3 compared to the placebo group (Fig. 4b). Likewise, after melatonin administration, a significant increase of RP in delta (t = –2.54, df = 7, p = 0.03) was observed at F3-F4 compared to the placebo group (Fig. 4c). At C3-A1, an important increase of RP in delta (t = –2.495, df = 7, p = 0.04) was obtained compared to the placebo group (Fig. 4d). No significant differences were observed at the O1-O2 derivations, nor were significant changes in the RP of the EEG bands seen after melatonin administration in the right hemisphere compared to the placebo group.
Fig.4
a) This diagram represents the EEG derivations in which significant differences were obtained. Relative power (in %) of the EEG bands recorded at F7-T3 (b), F3-F4 (c) and C3-A1 (d) of the EEG derivations during the wake-sleep transition in the placebo (white bars) and melatonin (gray bars) conditions. Results are shown as mean±2SE. Asterisks indicate significant differences with *p≤0.05.
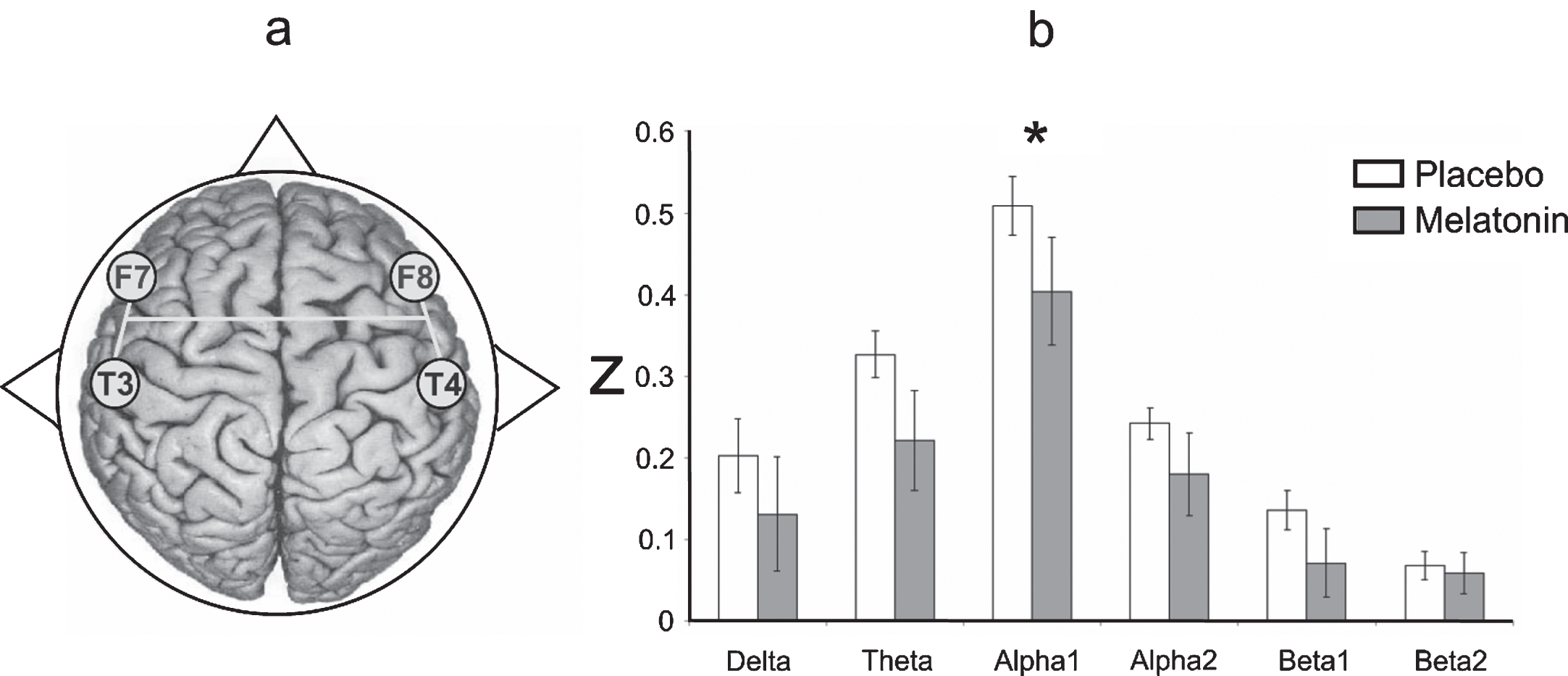
Interhemispheric correlation
As Fig. 5 shows, the degree of EEG coupling between the F7-T3 and F8-T4 derivations was lower in the alpha1 band in patients treated with melatonin (t = 2.39, df = 7, p = 0.04) than in those who received the placebo. No differences were found when the intrahemispheric and fronto-posterior correlations between the placebo and melatonin conditions were compared.
Fig.5
a) Representative diagram of the EEG derivations in which significant differences were obtained. b) Interhemispheric correlation (in z-values) of the EEG bands recorded between F7-T3 and F8-T4 during the wake-sleep transition after administering placebo (white bars) and melatonin (gray bars). Results are shown as mean±2SE. Asterisks indicate significant differences with *p≤0.05.
DISCUSSION
To the best of our knowledge, this is the first study to analyze the effect of melatonin on EEG activity and its relation to the installation of sleep in AD patients. In general, melatonin administration induced EEGs similar to those observed in healthy subjects in our AD patients during SOP [25–27,42–44]; i.e., a significant increase of RP in the slow frequencies (mainly delta) with a decrease of RP in the alpha band. However, there is evidence that the most representative change in SOP in healthy subjects is the predominance of slow-wave activity in the prefrontal cortex [45]. In this regard, our results showed a significant increase in the delta band at C3-A1 and F7-T3 during SOP that was related to melatonin administration. Likewise, we observed an increase in the RP of the delta band after melatonin administration at F3-F4. These results suggest that the predominance of slow waves in the prefrontal cortex is necessary for an optimal transition from wakefulness to sleep, and that this can be facilitated by administering melatonin to AD patients.
In addition to EEG power, our study analyzed the degree of correlation between the EEG signals from different cortical areas, with the result that characteristic degrees of cortical correlation in the different conditions were found. The degree of interhemispheric correlation between the F7-T3 and F8-T4 derivations in the alpha1 band was lower after melatonin administration than in the placebo condition. This result supports our hypothesis and concurs with various other studies which have reported that EEG synchronization during SOP in healthy subjects is associated with decreased correlation in the alpha band [28, 30–32].
There is evidence that the extension of synchronizing signals from associative prefrontal to posterior areas plays a role in the wake-to-sleep transition [46]. Topographic maps of coherence in delta-to-theta band activity have demonstrated that the synchronous component at the anterior-central areas of the scalp appears to correspond to increasing power [47]. Likewise, a prevalence of an occipital-to-frontal information flow in the delta, theta and alpha bands during the pre-sleep period, and a prevalence of a frontal-to-parieto-occipital information flow in all bands during SOP have been demonstrated in healthy subjects [48]. However, we did not observe a higher degree of EEG coupling between the F3-F4 and O1-O2 derivations after melatonin administration. Although the precise mechanism underlying these coherence abnormalities is unclear, it has been suggested that as the inter-neuronal distance for information propagation increases coherence is lowered, since AD shows diffuse and widespread cerebral degenerations [49, 50]. These studies suggest that a decrease in EEG correlation is associated with the presence of periventricular white matter lesions, which can cause functional disconnections between brain regions. Since neuroimaging studies have shown that periventricular white matter lesions, or leukoaraiosis, are observed more frequently in AD patients than in control subjects [51, 52], changes such as these may be another factor that reduces functional connectivity in AD patients.
As mentioned above, there is evidence to support the idea that melatonin induces sleep by modulating GABAA receptors [53–56]. Indeed, study data have shown that GABAergic cells of the hypothalamus, BF, mPO, and VLPO in the hypothalamic area are highly-active during sleep onset [19–22]. Moreover, it has been demonstrated that EEG activity observed during sleep onset is related to GABAergic activity in these structures [24, 57–65]. Hence, it is probable that the beneficial effect of melatonin (i.e., decreased sleep latency) on sleep onset in AD patients could be associated with a functioning of the GABAergic system that was sufficient to generate an EEG pattern similar to those manifested by healthy subjects during normal sleep onset. AD is associated with a broad loss of synapse density and the continuous degeneration of cholinergic and glutamatergic pathways [66]. Although the occurrence of disruptions of excitatory pathways is widely-recognized, inhibitory GABAergic pathways are generally thought to be well-preserved in AD [67, 68].
The present study found a reduction of sleep onset latencies, noting that they approached the normal time previously reported for geriatric non-dementia patients [12]. In fact, the effect of rapid installation of sleep after melatonin administration in non-dementia geriatric patients has been documented previously through observations which found that 3 mg of this indolamine facilitate the rapid installation of non-REM sleep [69], as was observed in our study. These results could suggest that, even in patients with mild-to-moderate AD, the brain structures involved in sleep control and regulation (such as the suprachiasmatic nucleus, thalamic-cortical circuits and the brain stem), are capable of reacting to the cascade of events that melatonin triggers to facilitate sleep installation. Our results show that in three patients, SOP was unexpectedly prolonged after melatonin as compared to placebo. This unforeseen finding suggests that in some patients with mild-to-moderate AD, the brain structures involved in sleep control and regulation may be affected by AD neurodegeneration.
Study limitations
It is important to note that the present work has some methodological limitations intrinsic to a pilot study, such as the lack of a control group, because we used a pretest/post-test design for a single group. This type of design permits inferring causal relations between the independent and dependent variables, though with some restrictions [70]. The small sample size (n = 8) is another limitation because there is evidence that small reduced samples undermine internal and external validity, and so increase the likelihood of type I error, which decreases the power of the study. It is important to note that, however, that although the sample is small with only eight drug-naive, mild-to-moderate AD patients, our findings do provide new and important data for understanding sleep disturbances and their associated treatments in cases of AD, since our subjects had not been treated previously with any neurological or psychiatric medication. Another limitation of our study is the lack of correction of multiple comparisons. Due to the small “n” we decided to illustrate all differences without such correction, though this could increase the level of speculation. Likewise, the use of measures of EEG correlations instead of coherence may be a methodological limitation, because coherence measures the co-variation between two signals as a function of frequency, taking into account both amplitude and phase changes between the signals involved, while correlation measures the co-variation between the signals as a function of time, considering only the phase relationship between the signals that are being analyzed, without considering amplitude.
We conclude that the short sleep onset related to melatonin intake in AD patients was associated with a lower RP of the alpha1 band and a higher RP of the delta band during the sleep onset period, mainly in the left hemisphere. These changes in EEG power were accompanied by a decreased degree of interhemispheric EEG coupling in the alpha1 band after melatonin administration. Our findings thus suggest that melatonin may have reduced sleep latency in these AD patients, probably due to an increase in the activity of GABAA receptors, mainly in the hypothalamus and basal forebrain. It is probable that structures like the suprachiasmatic nucleus, thalamic-cortical circuits and structures of the brain stem related to sleep control and regulation–even in mild-to-moderate AD–are capable of reacting to the cascade of events that melatonin triggers on SOP.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This study forms part of the postdoctoral fellowship of PhD. Manuel Alejandro Cruz-Aguilar, who thanks CONACyT, the Universidad de Guadalajara, and the Instituto Nacional de Psiquiatría “Ramón de la Fuente Muñiz”, for the support and facilities provided. The placebo and melatonin were donated by BIOQUIMED Laboratory. This work was partially funded by CONACyT Project 46593-M, led by Gloria Benítez-King.
REFERENCES
[1] | Cole MG , Dendukuri N ((2003) ) Risk factors for depression among elderly community subjects: A systematic review and meta-analysis. Am J Psychiatry 160: , 1147–1156. |
[2] | Hidalgo JL , Gras CB , Garcia YD , Lapeira JT , Del Campo del Campo JM , Verdejo MA ((2007) ) Functional status in the elderly with insomnia. Qual Life Res 16: , 279–286. |
[3] | Stone KL , Ancoli-Israel S , Blackwell T , Ensrud KE , Cauley JA , Redline S , Hillier TA , Schneider J , Claman D , Cummings SR ((2008) ) Actigraphy-measured sleep characteristics and risk of falls in older women. Arch Intern Med 168: , 1768–1775. |
[4] | Oosterman JM , Van Someren EJ , Vogels RL , Van Harten B , Scherder EJ ((2009) ) Fragmentation of the rest-activity rhythm correlates with age-related cognitive deficits. J Sleep Res 18: , 129–135. |
[5] | Allen SR , Seiler WO , Stahelin HB , Spiegel R ((1987) ) Seventy-two hourpolygraphic and behavioral recordings of wakefulness and sleep in a hospital geriatric unit: Comparison between demented and nondemented patients. Sleep 10: , 143–159 |
[6] | Pollak CP , Perlick D ((1991) ) Sleep problems and institutionalization of the elderly. J Geriatr Psychiatry Neurol 4: , 204–210. |
[7] | McCleery J , Cohen DA , Sharpley AL ((2016) ) Pharmacotherapies for sleep disturbances in dementia. Cochrane Database Syst Rev 11: , CD009178. |
[8] | Claustrat B , Brun J , Chazot G ((2005) ) The basic physiology and pathophysiology of melatonin. Sleep Med Rev 9: , 11–24. |
[9] | Ferracioli-Oda E , Qawasmi A , Bloch MH ((2013) ) Meta-analysis: Melatonin for the treatment of primary sleep disorders. PLoS One 8: , e63773. |
[10] | Dijk DJ , Roth C , Landolt HP , Werth E , Aeppli M , Achermann P , Borbély AA ((1995) ) Melatonin effect on daytime sleep in men: Suppression of EEG low frequency activity and enhancement of spindle frequency activity. Neurosci Lett 201: , 13–16. |
[11] | Cruz-Aguilar MA , Ramirez-Salado I , Cruz-Ulloa C , Benitez-King G ((2013) ) Melatonin effects on macro sleep architecture in Alzheimer’s disease patients. Salud Mental 36: , 243–249. |
[12] | Reinoso-Suárez F ((1999) ) The neurobiology of slow wave sleep. An R Acad Nac Med 116: , 209–226. |
[13] | Masana MI , Dubocovich ML ((2001) ) Melatonin receptor signaling: Finding the path through the dark. Sci STKE 2001: , pe39-pe39. |
[14] | Reppert SM , Weaver DR , Cassone VM , Godson C , Kolakowski LF ((1995) ) Melatonin receptors are for the birds: Molecular analysis of two receptor subtypes differentially expressed in chick brain. Neuron 15: , 1003–1015. |
[15] | Liu C , Weaver DR , Ji X , Shearman LP , Pieschl RL , Gribkoff VK , Reppert SM ((1997) ) Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron 19: , 91–102. |
[16] | Stehle J , Vanecek J , Vollrath L ((1989) ) Effects of melatonin on spontaneous electrical activity of neurons in rat suprachiasmatic nuclei: An in vitro iontophoretic study. J Neural Transm 78: , 173–177. |
[17] | Jiang ZG , Nelson CS , Allen CN ((1995) ) Melatonin activates an outward current and inhibits Ih in rat suprachiasmatic nucleus neurons. Brain Res 687: , 125–132. |
[18] | Nelson CS , Marino JL , Allen CN ((1996) ) Melatonin receptors activate heteromeric G-protein coupled Kir3 Channels. Neuroreport 7: , 717–720. |
[19] | Saper CB , Scammell TE , Lu J ((2005) ) Hypothalamic regulation of sleep and circadian rhythms. Nature 437: , 1257–1263. |
[20] | Llinás RR , Steriade M ((2006) ) Bursting of thalamic neurons and states of vigilance. J Neurophysiol 95: , 3297–3308. |
[21] | Datta S , MacLean RR ((2007) ) Neurobiological mechanisms for the regulation of mammalian sleep-wake behavior: Reinterpretation of historical evidence and inclusion of contemporary cellular and molecular evidence. Neurosci Biobehav Rev 31: , 775–824. |
[22] | Mignot E ((2008) ) Why we sleep: The temporal organization of recovery. PLoS Biol 6: , e106. |
[23] | Manns ID , Alonso A , Jones BA ((2000) ) Discharge properties of juxtacellularly labeled and immunohistochemically identified cholinergic basal forebrain neurons recorded in association with the electroencephalogram in anesthetized rats. J Neurosci 20: , 1505–1518. |
[24] | Lee MG , Manns ID , Alonso A , Jones BE ((2004) ) Sleep-wake related discharge properties of basal forebrain neurons recorded with micropipettes in head-fixed rats. J Neurophysiol 92: , 1182–1198. |
[25] | Merica H , Fortune RD ((2004) ) State transitions between wake and sleep, and within the ultradian cycle, with focus on the link to neuronal activity. Sleep Med Rev 8: , 473–485. |
[26] | De Gennaro L , Vecchio F , Ferrara M , Curcio G , Rossini PM , Babiloni C ((2004) ) Changes in fronto-posterior functional coupling during sleep onset in humans. J Sleep Res 13: , 209–217. |
[27] | Hori T ((1985) ) Spatiotemporal changes of EEG activity during waking-sleeping transition period. Int J Neurosci 27: , 101–114. |
[28] | De Gennaro L , Ferrara M , Curcio G , Cristiani R ((2001) ) Anteroposterior EEG changes during the wakefulness-sleep transition. Clin Neurophysiol 112: , 1901–1911. |
[29] | Morikawa T , Hayashi M , Hori T ((1989) ) Coherence analysis of the EEG in hypnagogic period. In Memoirs of the Faculty of Integrated Arts and Sciences III Fluctuations Hiroshima Univ (Japanese; English abstract), vol. 13: , pp. 81–89. |
[30] | Hori T , Hayashi M , Morikawa T ((1990) ) Topography and coherence analysis of the hypnagogic EEG. In Sleep ’90, Pontenagel Press, Bochum, pp. 10–12. |
[31] | Cantero JL , Atienza M , Salas RM , Gómez CM ((1999) ) Alpha EEG coherence in different brain states: An electrophysiological index of the arousal level in human subjects. Neurosci Lett 271: , 167–170. |
[32] | Kikuchi M , Wada Y , Koshino Y , Nanbu Y , Hashimoto T ((2000) ) Effect of normal aging upon interhemispheric EEG coherence: Analysis during rest and photic stimulation. Clin Electroenceph 31: , 170–174. |
[33] | Barry PP , Moskowitz MA ((1988) ) The diagnosis of reversible dementia in the elderly: A critical review. Arch Intern Med 148: , 1914–1918. |
[34] | Chari D , Ali R , Gupta R ((2017) ) Reversible dementia in elderly: Really uncommon? J Geriatr Ment Health 2: , 30–37. |
[35] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34: , 939–944. |
[36] | Hughes C , Berg L , Danziger WL , Coben LA , Martin RL ((1982) ) A new clinical scale for the staging of dementia. Br J Psychiatry 140: , 566–572. |
[37] | Jasper HH ((1958) ) The ten–twenty electrode system of the International Federation. Electroencephalogr Clin Neurophysiol 10: , 367–380. |
[38] | Hori T , Hayashi M , Morikawa T ((1994) ) Topographic EEG changes and the hypnagogic experience. In Sleep Onset: Normal and Abnormal Processes, Ogilvie RD, Harsh JR, eds. American Psychological Association, Washington DC, US, pp. 237–253. |
[39] | Rechtschaffen A , Kales A ((1968) ) A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. Los Angeles: Brain Information Service/Brain Research Institute University of California. |
[40] | Guevara MA , Hernández-González M , Sanz-Martin A ((2010) ) CHECASEN: Programa para revisar señales EEG fuera de línea. Rev Mex Ing Biom 31: , 135–141. |
[41] | Guevara MA , Hernández-González M ((2009) ) EEGmagic: Programa para analizar señales electroencefalográficas. Rev Mex Ing Biom 30: , 41–53. |
[42] | Badia P , Wright Jr , Kenneth P , Wauquier A ((1994) ) Fluctuations in single-hertz EEG activity during the transition to sleep. In Sleep Onset: Normal and Abnormal Processes, Ogilvie RD, Harsh JR, eds. American Psychological Association, Washington DC, US, pp. 201–218. |
[43] | Wright KP Jr , Badia P , Wauquier A ((1995) ) Topographical and temporal patterns of brain activity during the transition from wakefulness to sleep. Sleep 18: , 880–889. |
[44] | De Gennaro L , Ferrara M , Bertini M ((2001) ) The boundary between wakefulness and sleep: Quantitative electroencephalographic changes during the sleep onset period. Neuroscience 107: , 1–11. |
[45] | Marzano C , Moroni F , Gorgoni M , Nobili L , Ferrara M , De Gennaro L ((2013) ) How we fall asleep: Regional and temporal differences in electroencephalographic synchronization at sleep onset. Sleep Med 14: , 1112–1122. |
[46] | De Gennaro L , Vecchio F , Ferrara M , Curcio G , Rossini PM , Babiloni C ((2005) ) Antero-posterior functional coupling at sleep onset: Changes as a function of increased sleep pressure. Brain Res Bull 65: , 133–140. |
[47] | Tanaka H , Hayashi M , Hori T ((2000) ) Topographical characteristics of slow wave activities during the transition from wakefulness to sleep. Clin Neurophysiol 111: , 417–427. |
[48] | De Gennaro L , Vecchio F , Ferrara M , Curcio G , Rossini PM , Babiloni C ((2004) ) Changes in fronto-posterior functional coupling at sleep onset in humans. J Sleep Res 13: , 209–217. |
[49] | Besthorn C , Forstl H , Geiger-Kabisch C , Sattel H , Gasser T , Schreiter-Gasser U ((1994) ) EEG coherence in Alzheimer disease. Electroencephalogr Clin Neurophysiol 90: , 242–245. |
[50] | Leuchter AF , Dunkin JJ , Lufkin RB , Anzai Y , Cook IA , Newton TF ((1994) ) Effect of white matter disease on functional connections in the aging brain. J Neurol Neurosurg Psychiatry 57: , 1347–1354. |
[51] | Meyer JS , Kawamura J , Terayama Y ((1992) ) White matter lesions in the elderly. J Neurol Sci 110: , 1–7. |
[52] | Kawamura J , Meyer JS , Terayama Y , Weathers S ((1992) ) Leukb-Araiosis and cerebral hypoperfusion compared in elderly normal and Alzheimer’s dementia. J Am Geriatr Soc 40: , 375–380. |
[53] | Wang F , Jing-Cai LI , Chun-Fu WU , Jing-Yu Y , Feng XU , Fei P ((2002) ) Hypnotic activity of melatonin: Involvement of semicarbazide hydrochloride, blocker of synthetic enzyme for GABA. Acta Pharmacol Sin 23: , 860–864 . |
[54] | Xu F , Li JC , Ma KC , Wang M ((1995) ) Effects of melatonin on hypothalamic gamma-aminobutyric acid, aspartic acid, glutamic acid, beta-endorphin and serotonin levels in male mice. Biol Signals 4: , 225–231. |
[55] | Golombek DA , Pévet P , Cardinali DP ((1996) ) Melatonin effects on behavior: Possible mediation by the central GABAergic system. Neurosci Biobehav Rev 20: , 403–412. |
[56] | Acufla-Castroviejo D , Escames G , Macks M , Muñóz-Hoyos A , Molina-Carballo A , Arauzo M , Montes R , Vives F ((1995) ) Minireview: Cell protective role of melatonin in the brain. J Pineal Res 19: , 57–63. |
[57] | Glotzbach SF , Heller HC ((1984) ) Changes in the thermal characteristics of hypothalamic neurons during sleep and wakefulness. Brain Res 309: , 17–26. |
[58] | Kaitin KI ((1984) ) Preoptic area unit activity during sleep and wakefulness in the cat. Exp Neurol 83: , 347–357. |
[59] | Findlay ALR , Hayward JN ((1969) ) Spontaneous activity of single neurons in the hypothalamus of rabbits during sleep and waking. J Physiol 201: , 237–258. |
[60] | Lincoln DW ((1969) ) Correlation of unit activity in the hypothalamus with EEG patterns associated with the sleep cycle. Exp Neurol 24: , 1–18. |
[61] | McGinty D , Szymusiak R ((1990) ) Keeping cool: A hypothesis about mechanisms and functions of slow wave sleep. Trends Neurosci 13: , 480–487. |
[62] | Koyama Y , Hayaishi O ((1994) ) Firing of neurons in the preoptic/anterior hypothalamic areas in rat: Its possible involvement in slow wave sleep and paradoxical sleep. Neurosci Res 19: , 31–38. |
[63] | Alam MN , McGinty DJ , Szymusiak R ((1995) ) Neuronal discharge of preoptic/anterior hypothalamic thermosensitive neurons: Relation to NREM sleep. Am J Physiol 269: , R1240–R1249. |
[64] | Szymusiak R , Alam N , Steininger TL , McGinty D ((1998) ) Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res 803: , 178–188. |
[65] | Suntsova N , Szymusiak R , Alam MN , Guzman-Marin R , McGinty D ((2002) ) Sleep-waking discharge patterns of median preoptic nucleus neurons in rats. J Physiol 543: , 665–677. |
[66] | Selkoe DJ ((2002) ) Alzheimer’s disease is a synaptic failure. Science 298: , 789–791. |
[67] | Rissman RA , Mobley WC ((2011) ) Implications for treatment: GABAA receptors in aging, Down syndrome and Alzheimer’s disease. J Neurochem 117: , 613–622. |
[68] | Luchetti S , Huitinga I , Swaab DF ((2011) ) Neurosteroid and GABA-A receptor alterations in Alzheimer’s disease, Parkinson’s disease and multiple sclerosis. Neuroscience 19: , 6–21. |
[69] | Cardinali DP , Brusco LI , Liberczu C , Furio AM ((2002) ) The use of melatonin in Alzheimer’s disease. Neuro Endocrinol Lett 23: , 20–23. |
[70] | Campbell DT , Stanley JC ((1996) ) Experimental and quasi-experimental designs for research. In Handbook of research on teaching, Gage NL, ed. Houhton Mifflin, Boston, pp. 171–246. |


