
AhRR methylation contributes to disease progression in urothelial bladder cancer
Abstract
BACKGROUND:
Bladder Cancer (BCa) is the tenth most incident malignancy worldwide. BCa is mostly attributed to environmental exposure and lifestyle, particularly tobacco smoking. The Aryl Hydrocarbon Receptor Repressor (AhRR) participates in the induction of many enzymes involved in metabolizing carcinogens, including tobacco smoke components. Additionally, studies have shown that smoking demethylates the (AhRR) gene in blood, suggesting AhRR demethylation as a specific serum smoking biomarker.
OBJECTIVE:
This study aimed to validate AhRR demethylation as a smoking biomarker in the target tissue and investigate its contribution to bladder carcinogenesis.
METHODS:
AhRR percent methylation was tested for its association with patient smoking status and oncogenic outcome indicators, particularly p53, RB1, and FGFR3 activating mutations, muscle-invasiveness, and tumor grade, in 180 BCa tissue-based DNA.
RESULTS:
Results showed significantly higher AhRR percent methylation in muscle-invasive compared to non-muscle invasive tumors (42.86% vs. 33.98%;
CONCLUSIONS:
Although AhRR methylation did not predict smoking status in BCa tumors, it may be a contributor to carcinogenesis and disease progression. Our findings constitute the basis for further research.
Abbreviations
| AhR | Aryl Hydrocarbon Receptor |
| AhRR | Aryl Hydrocarbon Receptor Repressor |
| ARNT | Aryl Hydrocarbon Receptor Nuclear |
| Translocator | |
| BCa | Urinary Bladder Cancer |
| CYP | Cytochrome P450 |
| ddPCR | Droplet Digital Polymerase Chain |
| Reaction |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| FGFR3 | Fibroblast growth factor receptor 3 |
| HG | High grade |
| LG | Low grade |
| IGFBP-5 | Insulin-like growth factor binding |
| protein-5 | |
| LOX | Lysyl Oxidase |
| MIBC | Muscle-Invasive Bladder Cancer |
| NAT1 | N-acetyltransferase 1 |
| NAT2 | N-acetyltransferase 2 |
| NMIBC | Non-Muscle Invasive Bladder Cancer |
| PCR | Polymerase Chain Reaction |
| RB1 | Retinoblastoma 1 gene |
| TP53 | Tumor protein 53 |
1.Introduction
Urinary Bladder Cancer (BCa) is the tenth most incident malignancy worldwide, accounting for 3% of all cancers, with about 200,000 deaths and 550,000 new cases reported annually on a global scale [1]. Males show a 4-fold higher BCa risk compared to females [2]. The highest incidence is encountered in industrialized countries, while a much lower incidence is observed in developing countries, with very few exceptions such as Egypt and the current case of Lebanon [3, 4]. The most common pathological type is transitional cell carcinoma (TCC), which can manifest as either non-muscle invasive bladder cancer (NMIBC) confined to the mucosa, or as a clinically aggressive muscle-invasive bladder cancer (MIBC) [5, 6]. The two different clinical outcomes have distinct histopathological characteristics, where NMIBC recurs in the form of finger-like growths that extend into the center of the bladder, known as papillary carcinoma, while MIBC tumors show flat lesions that are more likely to invade the detrusor muscle and are capable of distant metastasis [5]. At the molecular level, all studies concluded that NMIBCs and MIBCs form two broad distinct subsets, where NMIBC shows a higher prevalence of fibroblast growth factor receptor-3 (FGFR3) and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) activating mutations, while MIBC is distinguished by a higher mutation rate with copy number variations in oncogenes, particularly the tumor protein-53 (TP53), the retinoblastoma transcriptional corepressor-1 (RB1), and the E2F transcription factor-3 (E2F3). MIBC is further sub-grouped into six different molecular subtypes, including luminal-papillary (LumP), luminal-nonspecified (LumNS), luminal unstable (LumU), stroma-rich, basal/squamous (Ba/Sq), and neuroendocrine-like (NE-like), based on a recent international consensus [7].
BCa is considered an environmentally acquired malignancy from carcinogenic exposures such as arylamines and polycyclic aromatic amines, nitrosamines, water chlorination disinfection by-products, and tobacco smoke [4, 8, 9, 10]. The cell lining of the urothelial wall is exposed to these carcinogens, that are either eliminated in urine or bioactivated by drug-metabolizing enzymes [11]. Chemical carcinogenesis is thought to be particularly triggered by arylamines from tobacco smoking, which is perceived as the most significant risk factor for BCa [8, 12, 13].
Various smoking biomarkers have been examined, including cotinine levels, exhaled carbon monoxide (CO), and hemoglobin arylamine adducts. However, many of these are restricted by their short half-life, and/or relatively low specificity and sensitivity, and subsequently are of limited value as exposure biomarkers [14, 15, 16, 17]. Recently, studies reported a strong positive association between the gradual demethylation of the Aryl Hydrocarbon Receptor Repressor (AhRR) in blood and both cotinine levels and the number of cigarettes smoked daily [14, 15, 18, 19]. More than 20 different studies have elected methylation at cg05575921, a CpG residue in AhRR, as the most sensitive indicator of smoking in blood, and reported on its high accuracy as a smoking biomarker with an area under the receiver operating characteristic (ROC) curve (AUC) of 0.99 [20, 21]. Currently, the demethylation of AhRR at the cg05575921 locus is adopted as a solid biomarker for cigarette smoking in whole blood [19, 21, 22, 23, 24, 25, 26, 27]. However, AhRR methylation levels in blood may also revert to normal after reducing or ceasing smoking [28, 29], possibly due to the short lifespan of blood cells [30], and hence is restricted to marking acute exposures. In this study, we sought to validate AhRR methylation levels as a smoking biomarker in BCa tumor tissues, and we investigated its association with tumorigenic markers.
2.Methods
2.1Study population
Out of a pool of 250 histologically confirmed urothelial BCa patients diagnosed between 2013 and 2017 at two major medical centers in Beirut, Lebanon, 180 patients were sub-selected for this study. Recruited cases included Lebanese patients above the age of 50, starting with the most recently diagnosed. Exclusion criteria included non-Lebanese patients, subjects under 50 years of age, patients with additional types of cancer, and those with unavailable archival tumor tissues. Given the low BCa incidence in females, enrollment focused on male patients in order to maintain statistical power. Information on tumor grade and stage, and patients’ smoking status, was obtained from medical records. For smoking status, enrolled patients were designated as either never smokers or current smokers at the time of diagnosis or referral. Institutional Review Board approvals from the American University of Beirut and collaborating medical centers were obtained prior to conducting the study. All acquired bio-specimens and collected data were obtained as a de-identified set.
2.2DNA extraction
Multiple sections of 5
2.3AhRR DNA methylation testing
AhRR methylation levels at the cg05575921 locus in tumorigenic DNA was determined by bisulfite conversion, pre-amplification by Polymerase Chain Reaction (PCR), and DNA methylation measurement by Droplet Digital Polymerase Chain Reaction (ddPCR).
2.3.1Bisulfite conversion of extracted DNA
Complete bisulfite conversion and cleanup of the extracted DNA for methylation analysis were performed in a 96-well setup, using the EpiTect 96 Bisulfite Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions for sodium bisulfite conversion of unmethylated cytosines in DNA isolated from FFPE tissue samples using a centrifuge. The samples were divided into 96-well plates for bisulfite conversion. For each sample, DNA was dissolved in a bisulfite mix and RNase-free water, in a total volume of 140
2.3.2Pre-amplification by PCR
After bisulfite conversion, the AhRR region in the bisulfite converted DNA was amplified in 96-well plates, according to the manufacturer’s instructions using a Smoke Signature Assay Kit (IBI Scientific, Iowa). For each sample, 3–5
2.3.3DNA methylation assessment by ddPCR
Prior to performing ddPCR, an initial dilution step was performed to achieve an optimal number of independent strand templates (
2.3.4Tumors’ molecular markers mutation detection
PCR and restriction digestion with optimized conditions were used to detect mutations in the tumors for the following molecular markers: TP53 at Exon 4-Codon 72 (rs1042522) and at Exon 7-Codon 248 (rs121912651), RB1 non-sense mutation (rs137853293) at Exon 23, and FGFR3 somatic activating mutations at Exon 7, Codon 248 (C248; rs121913482), and Codon 249 (C249; rs121913483). Designed PCR reactions, primers, annealing temperatures, and restriction enzymes were previously described [31].
2.4Statistical analysis
Descriptive statistics were used to summarize the characteristics of the tumors. Boxplot and histogram were generated to check the normality of the continuous variables in the study sample. Univariate analysis consisted of frequency and percentage distributions for the different categorical variables in the study. Means, standard deviations (SDs), 95% Confidence Intervals (CIs), and ranges were computed for the continuous variable. Associations between AhRR methylation and each of the variables (smoking status, tumor grade, invasiveness, TP53, FGFR3 C248, and FGFR3 C249) were then tested using the Student’s
3.Results
During AhRR methylation assessment, 11 samples were discarded due to lack of any methylated or unmethylated droplet clusters in ddPCR results. A sample was also rejected due to a difference of more than 30% methylation between duplicates [23]. Therefore, our results are based on a final sample size of 168 patients.
3.1Study population characteristics
The majority of tumors (150) were high grade (HG), while only 7 were low grade (LG); 68 samples were muscle-invasive, while 99 were non-muscle-invasive (Table 1). In addition, 74.4% of the patients were current smokers, while 20.2% reported they were never smokers. Frequencies of TP53, FGFR3, and RB1 genotypes were as follows: 47.6% showed the TP53 C72 wild-type genotype, while 49.3% presented with at least one mutation (Table 2); 95.8% of samples had the FGFR3 C248 wild type genotype, while only 11.9% had the FGFR3 C249 wild type; mutations in TP53 C248 and RB1 E23 were almost absent (Table 2). AhRR methylation ranged between 0 and 99.65% with a mean AhRR methylation of 37.46
Table 1
Study population characteristics in the total sample (
| Characteristic |
| Frequency (%) |
|---|---|---|
| Tumor grade | ||
| High grade | 150 | 89.2 |
| Low grade | 7 | 4.1 |
| Unknown | 11 | 6.5 |
| Invasiveness | ||
| Invasive | 68 | 40.4 |
| Non-invasive | 99 | 58.9 |
| Unknown | 1 | 0.5 |
| Smoking status | ||
| Current smoker | 125 | 74.4 |
| Never smoker | 34 | 20.2 |
| Unknown | 9 | 5.35 |
Table 2
Frequency of TP53, FGFR3, RB1, NAT1, and NAT2 genotypes in the total sample (
| SNP/mutation | Undetermined | ||||||
|---|---|---|---|---|---|---|---|
| TP53 C72 | 80 | (47.6%) | 34 | (20.2%) | 49 | (29.1%) | 5 (2.9%) |
| TP53 C248 | 161 | (95.8%) | 1 | (0.6%) | 2 | (1.2%) | 4 (2.4%) |
| FGFR3 E7 C248 | 161 | (95.8%) | 1 | (0.6%) | 6 | (3.5%) | 0 (0.0%) |
| FGFR3 E7 C249 | 20 | (11.9%) | 2 | (1.2%) | 146 | (86.9%) | 0 (0.0%) |
| RB1 E23 | 138 | (82.1%) | 0 | (0.0%) | 0 | (0.0%) | 30 (17.9%) |
| NAT1 G | 95 | (56.5%) | 1 | (0.6%) | 72 | (42.8%) | 0 (0.0%) |
| NAT2 G | 119 | (70.8%) | 5 | (2.9%) | 41 | (24.4%) | 3 (1.7%) |
Figure 1.
Boxplot (a) and histogram (b) showing AhRR percent methylation distribution across the total sample (
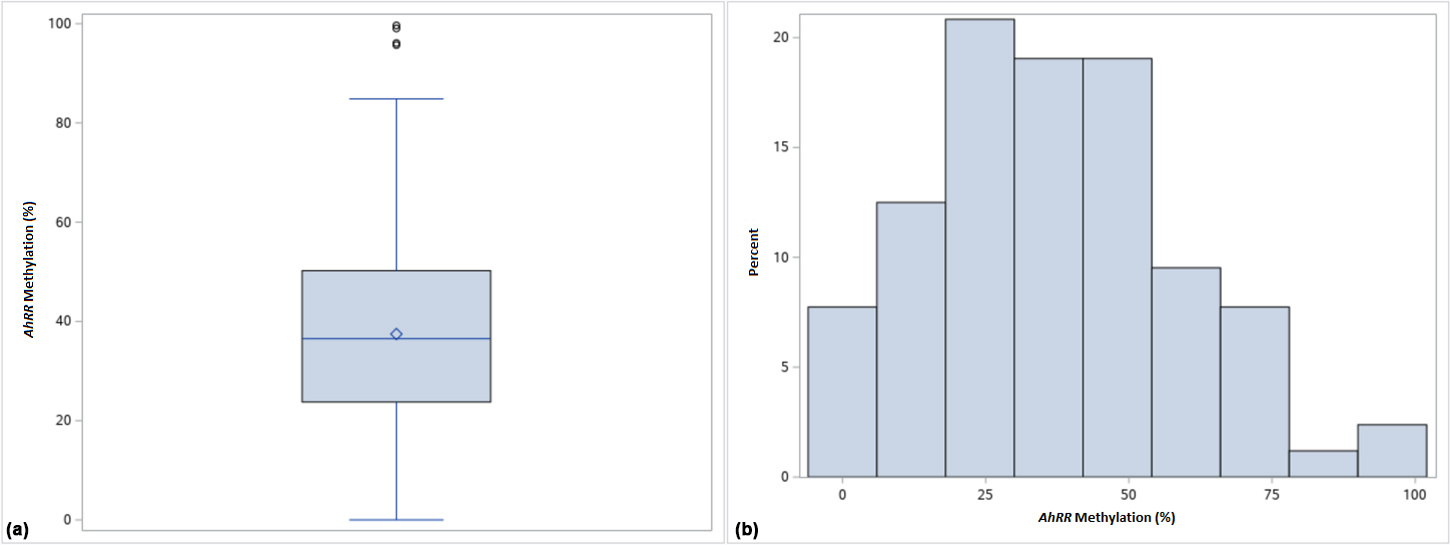
3.2Bivariate analysis
In the bivariate analysis, results showed that never smokers had a higher mean AhRR percent methylation of 40.16%
Table 3
AhRR % methylation across smoking status and tumorigenic indicators in the total sample (
| Variable | Mean AhRR % methylation (95% CI) | |
|---|---|---|
| Smoking status ( | ||
| Current smoker | 36.88 (32.89–40.86) | 0.456 |
| Never smoker | 40.16 (32.04–48.28) | |
| Tumor grade ( | ||
| High grade | 37.00 (33.42–40.58) | 0.223 |
| Low grade | 26.59 (10.80–42.38) | |
| Invasiveness ( | ||
| Invasive | 42.86 (37.05–48.63) | 0.011 |
| Non-invasive | 33.98 (29.92–38.04) | |
| TP53 C72 ( | ||
| Mutant | 39.69 (34.72–44.66) | 0.114 |
| Wild-type | 34.28 (29.71–38.85) | |
| FGFR3 E7 C248 ( | ||
| Mutant | 28.11 (19.38–36.85) | 0.036 |
| Wild-type | 37.87 (34.36–41.38) | |
| FGFR3 E7 C249 ( | ||
| Mutant | 37.30 (33.71–40.90) | 0.799 |
| Wild-type | 38.66 (27.81–49.51) |
On the other hand, results showed that muscle-invasive tumors (42.86%
Table 4
AhRR % methylation across smoking status and tumorigenic indicators in high grade (HG) tumors (
| Variable | Mean AhRR % methylation (95% CI) | |
|---|---|---|
| Smoking status ( | 36.87 (33.14–40.61) | |
| Current smoker | 40.15 (31.77–48.52) | 0.345 |
| Never smoker | 35.89 (31.68–40.11) | |
| Invasiveness ( | 37.00 (33.42–40.58) | |
| Invasive | 42.09 (35.97–48.22) | 0.018 |
| Non-invasive | 33.40 (29.15–37.66) | |
| TP53 C72 ( | 36.50 (32.93–40.06) | |
| Mutant | 38.97 (33.47–44.47) | 0.174 |
| Wild-type | 34.06 (29.44–38.68) | |
| FGFR3 E7 C248 ( | 37.00 (33.42–40.58) | |
| Mutant | 28.11 (19.38–36.85) | 0.044 |
| Wild-type | 37.43 (33.70–41.16) | |
| FGFR3 E7 C249 ( | 37.00 (33.42–40.58) | |
| Mutant | 36.74 (32.90–40.58) | 0.721 |
| Wild-type | 38.66 (27.80–49.51) |
3.3Stratified analysis
Further analysis using Wilcoxon Rank Sum test with stratification by muscle-invasiveness showed that in muscle-invasive tumors, FGFR3 C248 genotype was not significantly associated with AhRR percent methylation (
Table 5
AhRR % methylation association with FGFR3 Codons 248 and 249, stratified by invasiveness using Wilcoxon Rank Sum test
| FGFR3 C248 | FGFR3 C249 | |||
| Invasive | Non- invasive | Invasive | Non- invasive | |
| Mutant | ||||
| | 1 | 6 | 60 | 87 |
| Mean % AhRR | 21.60 | 29.20 | 44.39 | 32.66 |
| Wild-type | ||||
| | 67 | 93 | 8 | 12 |
| Mean % AhRR | 43.17 | 34.29 | 31.35 | 43.53 |
| 0.231 | 0.491 | 0.159 | 0.125 | |
3.4Gene-gene interactions
We had previously examined polymorphisms of N-acetylation metabolic enzymes and its association with key MIBC and NMIBC tumor biomarkers in the target population as described [31]. In the current study, interaction between AhRR percent methylation and previously detected NAT1 G
4.Discussion
Unlike studies that report strong associations between smoking and AhRR demethylation in blood [19, 21, 23, 27], our study found no significant associations between AhRR methylation and smoking status in the BCa tissue. Therefore, our findings could suggest that AhRR methylation in bladder cancer tissue is not a marker of smoking. However, smoking could be acting on different AhRR CpG-sites in the tumor. For instance, one study on lung cancer reports that current smokers exhibited considerably lower AhRR methylation levels at the cg21161138 loci in lung tissues compared to never smokers [32]. Thus, assessing tumor-based methylation levels at different AhRR CpG-sites may provide further insight into relationships between tobacco smoking and AhRR methylation in BCa tumor tissues. It is important to note here that very few studies have examined the association between smoking status and AhRR methylation in tissue, and none in bladder cancer. In one of these studies, the analysis of buccal epithelium and placenta did not identify a smoking-associated methylation difference at any AhRR locus [33]. In another study, investigating a genome-wide analysis of smoking-related methylation in adipose tissue, AhRR was found to be hypomethylated and upregulated in current smokers [34].
On the other hand, AhRR methylation levels in blood may have reverted to normal after ceasing or even reducing smoking. In one epigenome-wide association study in European peripheral-blood DNA, the impact of cigarette smoking on DNA methylation was found to be partially but not completely reversible, starting three months after smoking cessation [35]. In another genome-wide methylation profiling study, also conducted in European populations, two homogeneous classes of smoking biomarkers were reported in blood: reversible markers whose methylation reverts to normal within decades after smoking cessation, and persistent markers whose methylation levels remain even 35 years after smoking cessation [36]. However, no studies have reported a reverting effect in tissues, which preclude us from making such assumptions in the context of our study. At the same time, our target group did not include former smoking patients. As a conclusion, future studies should examine these observed tissue-specific smoking-related epigenetic effects, and attempt to benefit from advancements in urine-tumor DNA methylation assessment, which is emerging as a much more promising approach for early diagnosis, residual tumor detection, and surveillance in BCa, as compared to routine detection methods [37, 38].
In this study, we found that muscle-invasiveness is significantly associated with higher AhRR percent methylation, hence a higher repression of its transcription. This is supported in the literature, where AhRR is suggested to play a key role in tumor suppression and the fact that the expression of AhRR is downregulated in various cancer tissues [39, 40, 41, 42]. AhRR repression may contribute to aggressive tumorigenic phenotype, including increased migration and invasiveness, and reduced apoptosis in cancer cells [41]. Moreover, our findings are consistent with recent studies suggesting that the Aryl Hydrocarbon Receptor (AhR) is associated with higher BCa progression [43]. AhRR contribution to disease progression may be occurring via AhR repression, leading to muscle-invasiveness of flat lesions. Interestingly, a recent study reports a significantly higher expression of AhR in NMIBC tumors compared to MIBC and normal tissue in mice models [44]. Therefore, this mechanism is worthy of further investigation. Alternatively, increased AhRR expression may result in advancing chronic inflammation, a known risk factor of BCa that may be promoting carcinogenesis [45, 46, 47]. At the same time, it is important to note that AhRR normal levels of expression may vary between different tissues given the possibility of tissue-specific gene expression and regulation [46, 48, 49]. AhR induces expression of AhRR in a tissue-specific manner, which in turn inhibits AhR-induced expression of certain genes through different mechanisms [50, 51]. Those genes include common predictors across all targets including the insulin-like growth factor binding protein-5 (IFGBp-5), the Lysyl Oxidase (LOX), and the Cytochrome P450 CYP1B1 which are thought to contribute to malignant transformation in different types of tissues [52, 53, 54, 55, 56]. Throughout these common predictor genes, AhR mediates gene expression in many pathways involved in development and progression of cancer; this includes the matrix metalloproteinase-9 (MMP-9) through the c-Jun-dependent pathway, the ERK-FAK-Rac-1 pathway, as well as the Snail superfamily transcriptional repressor-1 (Snail-1), the TWIST family of basic helix-loop-helix transcription factors Twist1 and Twist2, and vimentin in the E-cadherin-related pathways [57]. Variation in expression of many of these genes have been linked to BCa [58, 59, 60], which further highlights the role of the AhR pathway in bladder carcinogenesis.
In addition, we found that mutant FGFR3 C248 genotype is significantly associated with lower AhRR methylation. This is another evidence arguing for a role for AhRR methylation in carcinogenesis of the bladder, particularly that this FGFR3 activating mutation is one of the drivers of malignancy in several human tissues including the bladder [31, 61, 62]. FGFR3 C248 (rs121913482) is a missense mutation where a cytosine in the 5’-GCG-3’ nucleotide sequence is substituted with a thymine, subsequently resulting in a 5’-GTG-3’ sequence [63]. Knowing that the AhR-ARNT (AhR Nuclear Translocator) transcription complex has affinity for the 5’-GTG-3’ sequence [64, 65], we can postulate that an increase in expression of AhR may cause overexpression of FGFR3 when mutated, and that AhRR may be acting through an unknown regulatory pathway to suppress tumors induced by overexpression of AhR and FGFR3. This may explain the observed association between the lower AhRR methylation and FGFR3 mutation. However, this requires further research, particularly examining the expression of AhR, ARNT, and other related components of this particular pathway.
Nonetheless, when we stratified by muscle-invasiveness, the significant association was not maintained between FGFR3 C248 and AhRR methylation levels. This may be due to lack of power, given that only one tumor with mutant FGFR3 C248 was within the invasive subgroup. At the same time, no association was found between tumor grade and AhRR methylation, although HG tumors showed a notably higher mean AhRR percent methylation compared to LG tumors, which is in agreement with some studies in the literature [41]. Observed results may also be related to the fact that the majority of our samples were HG (89.2%). A larger sample is needed to is needed to re-examine those associations.
Several limitations may have affected the results of this study. We relied on patients’ self-reported smoking status, which may be biased by misreporting or underreporting. In fact, research has shown that self-reported smoking status may be inaccurate, especially since smoking could be a socially unacceptable behavior in certain contexts [66, 67]. However, this does not apply much to the Lebanese context, where smoking is highly prevalent [68, 69]. The ability of contacting surviving patients to obtain blood samples to validate smoking status through assessing AhRR methylation or cotinine levels in blood DNA was not possible due to the de-identified nature of the data acquired. Similarly, data on smoking extent or cessation, and the types of tobacco (cigarettes vs. waterpipe) were not obtained from the sampled patients, and the association between AhRR methylation and extent of smoking and dosage was not possible due to lack of detailed smoking information. Furthermore, data on other potential occupational and environmental exposures was unavailable. Another limitation is that DNA was extracted from archival tumor samples, which may have affected DNA integrity, resulting in potential degradation of AhRR methylation sites. This would explain the lack of ddPCR clusters in 11 samples in this study. Nevertheless, ddPCR has shown better accuracy in assessing DNA methylation in archival FFPE tumors, when compared to other methods of DNA methylation assessment [70, 71]. Another limitation is the focus on tumors from one gender only. Due to the low BCa incidence in females, enrollment focused solely on male patients to maintain statistical power, hence restricting the possibility of stratifying the analysis by gender. A much larger sample size is needed to represent female patients in future studies. Despite the above-mentioned limitations, our study has several strengths. First, we targeted a population with a high BCa incidence and a high tobacco smoking prevalence, which provides power to the reported associations. Second, the study is original in both evaluating AhRR methylation as a smoking marker in the target tissue (BCa), and in relation to specific tumorigenic outcome indicators in order to get an insight into the mechanisms of carcinogenesis. Moreover, samples were acquired from two medical centers situated in different parts of the capital city, attracting people from various rural and urban regions of the country, which may have potential influence on the representativeness of the sample given the centralized nature of tertiary treatment in the country. Patients from different regions in Lebanon seek complex tertiary treatment, such as that for cancer, in large and equipped hospitals in the capital city. Thus, the examined total pool of samples has better representativeness of the target population, ensuring genetic diversity. On the other hand, despite relying on FFPE tumors as mentioned above, where prolonged formalin fixation could have caused DNA crosslinking and breakages in nucleotide sequence, we were able to optimize the extraction procedure to obtain a high quality and a sufficient yield of starting material [72]. Another strength of this study is that AhRR percent methylation was measured using ddPCR, a highly sensitive technique that partitions DNA strands into thousands of droplets that are assessed individually, hence providing a highly accurate assessment of percent methylation. The validity of the data obtained was further enhanced when we ran samples in duplicates and reported the average percent methylation for each patient, while discarding samples with outlying results.
5.Conclusions
In summary, our study shows that muscle-invasiveness is associated with higher AhRR methylation levels, while oncogenic FGFR3 C248 activating mutation is associated with lower AhRR methylation levels. These findings are novel, may help improve knowledge of BCa mechanisms of carcinogenesis in both MIBC and NMIBC, and can make the basis for potential methods of treatment targeting AhRR. Future studies should validate exposure to tobacco smoke by assessing AhRR methylation in blood, accounting for smoking cessation or reduction, as well as more comprehensively at multiple loci in BCa tumors, and the surrounding non-cancerous bladder tissues. In addition, future studies should investigate the mechanisms of bladder carcinogenesis by examining all the components of the AhR pathway in a large sample size that allows further stratification by gender and tumor outcome indicators, and that accounts for potential occupational and environmental BCa risk factors.
Ethics approval
This research was approved by the Institutional Review Board (IRB) of the American University of Beirut prior to collecting samples and data.
Authors’ contributions
Conception: H.R.D.
Interpretation or analysis of data: H.R.D., M.K, N.W.H., and K.A.
Preparation of the manuscript: N.W.H., and H.R.D.
Revision for important intellectual content: N.W.H., H.R.D., M.K., M.E.J., M.A.M., K.A., and R.R.H.
Supervision: H.R.D., and M.K.
Availability of data and materials
The datasets used in the current study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare that they have no competing interests.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-220002.
Acknowledgments
This research is funded by the American University of Beirut Research Board (URB) Award #103785.
References
[1] | J. Ferlay, M. Ervik, F. Lam, M. Colombet, L. Mery, M. Piñeros, A. Znaor, I. Soerjomataram and F. Bray, Global Cancer Observatory: Cancer Today, Lyon, France: International Agency for Research on Cancer, International Agency for Research on Cancer, Lyon, France, (2018) . |
[2] | P.A. Geavlete, D. Georgescu, R. Mulţescu, M. Drăguţescu, M. Jecu and B. Geavlete, Endoscopic Treatment of Bladder Tumors, Elsevier, (2016) , 83–203. |
[3] | The Lancet, GLOBOCAN 2018: counting the toll of cancer, The Lancet 392: ((2018) ), 985. |
[4] | N.A. Lakkis, S.M. Adib, G.N. Hamadeh, R.T. El-Jarrah and M.H. Osman, Bladder cancer in lebanon: Incidence and comparison to regional and western countries, Cancer Control 25: ((2018) ), 1–7. |
[5] | D.J. McConkey and W. Choi, Molecular subtypes of bladder cancer, Curr Oncol Rep 20: ((2018) ), 77. |
[6] | M.J. Al-Husseini, A. Kunbaz, A.M. Saad, J.V. Santos, S. Salahia, M. Iqbal and F. Alahdab, Trends in the incidence and mortality of transitional cell carcinoma of the bladder for the last four decades in the USA: A SEER-based analysis, BMC Cancer 19: ((2019) ). |
[7] | A. Kamoun, A. De Reyniès, Y. Allory, G. Sjödahl, A.G. Robertson, R. Seiler, K.A. Hoadley, C.S. Groeneveld, H. Al-Ahmadie, W. Choi, M.A.A. Castro, J. Fontugne, P. Eriksson, Q. Mo, J. Kardos, A. Zlotta, A. Hartmann, C.P. Dinney, J. Bellmunt, T. Powles, N. Malats, K.S. Chan, W.Y. Kim, D.J. McConkey, P.C. Black, L. Dyrskjøt, M. Höglund, S.P. Lerner, F.X. Real, F. Radvanyi, M. Aine, H. Al-Ahmadie, Y. Allory, J. Bellmunt, I. Bernard-Pierrot, P.C. Black, M.A.A. Castro, K.S. Chan, W. Choi, B. Czerniak, C.P. Dinney, L. Dyrskjøt, P. Eriksson, J. Fontugne, E.A. Gibb, C.S. Groeneveld, A. Hartmann, K.A. Hoadley, M. Höglund, A. Kamoun, J. Kardos, J. Kim, W.Y. Kim, D.J. Kwiatkowski, T. Lebret, S.P. Lerner, F. Liedberg, N. Malats, D.J. McConkey, Q. Mo, T. Powles, F. Radvanyi, F.X. Real, A. De Reyniès, A.G. Robertson, A. Siefker-Radtke, N. Sirab, R. Seiler, G. Sjödahl, A. Taber, J. Weinstein and A. Zlotta, A consensus molecular classification of muscle-invasive bladder cancer, European Urology 77: ((2020) ), 420–433. |
[8] | M. Burger, J.W. Catto, G. Dalbagni, H.B. Grossman, H. Herr, P. Karakiewicz, W. Kassouf, L.A. Kiemeney, C. La Vecchia, S. Shariat and Y. Lotan, Epidemiology and risk factors of urothelial bladder cancer, Eur Urol 63: ((2013) ), 234–241. |
[9] | M.G.K. Cumberbatch, I. Jubber, P.C. Black, F. Esperto, J.D. Figueroa, A.M. Kamat, L. Kiemeney, Y. Lotan, K. Pang, D.T. Silverman, A. Znaor and J.W.F. Catto, Epidemiology of bladder cancer: A systematic review and contemporary update of risk factors in 2018, Eur Urol 74: ((2018) ), 784–795. |
[10] | A.A. Deb, C.E. Okechukwu, S. Emara and S.A. Abbas, Occupational exposure as risk factor for kidney and bladder cancer: A systematic review and meta-analysis, Urology & Nephrology Open Access Journal 7: ((2019) ), 143–151. |
[11] | S. Rendic and F.P. Guengerich, Contributions of human enzymes in carcinogen metabolism, Chemical Research in Toxicology 25: ((2012) ), 1316–1383. |
[12] | F.H. van Osch, S.H. Jochems, F.J. van Schooten, R.T. Bryan and M.P. Zeegers, Quantified relations between exposure to tobacco smoking and bladder cancer risk: A meta-analysis of 89 observational studies, Int J Epidemiol 45: ((2016) ), 857–870. |
[13] | S. Kispert, J. Marentette and J. McHowat, Cigarette smoking promotes bladder cancer via increased platelet-activating factor, Physiol Rep 7: ((2019) ), e13981. |
[14] | N.L. Benowitz, Biomarkers of environmental tobacco smoke exposure, Environmental Health Perspectives 107: ((1999) ), 349–355. |
[15] | C.M. Chang, S.H. Edwards, A. Arab, A.Y. Del Valle-Pinero, L. Yang and D.K. Hatsukami, Biomarkers of tobacco exposure: Summary of an FDA-sponsored public workshop, Cancer Epidemiology Biomarkers & Prevention 26: ((2017) ), 291–302. |
[16] | A. Florescu, R. Ferrence, T. Einarson, P. Selby, O. Soldin and G. Koren, Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: Focus on developmental toxicology, Therapeutic Drug Monitoring 31: ((2009) ), 14–30. |
[17] | J. Hukkanen, P.J. Iii and N.L. Benowitz, Metabolism and disposition kinetics of nicotine, Pharmacological reviews 57: ((2005) ), 79–115. |
[18] | Y. Zhang, I. Florath, K.-U. Saum and H. Brenner, Self-reported smoking, serum cotinine, and blood DNA methylation, Environmental Research 146: ((2016) ), 395–403. |
[19] | A.M. Andersen, R.A. Philibert, F.X. Gibbons, R.L. Simons and J. Long, Accuracy and utility of an epigenetic biomarker for smoking in populations with varying rates of false self-report, Am J Med Genet B Neuropsychiatr Genet 174: ((2017) ), 641–650. |
[20] | R. Philibert, N. Hollenbeck, E. Andersen, T. Osborn, M. Gerrard, F.X. Gibbons and K. Wang, A quantitative epigenetic approach for the assessment of cigarette consumption, Frontiers in Psychology 6: ((2015) ). |
[21] | R. Philibert, M. Dogan, S.R.H. Beach, J.A. Mills and J.D. Long, AHRR methylation predicts smoking status and smoking intensity in both saliva and blood DNA, Am J Med Genet B Neuropsychiatr Genet, (2019) . |
[22] | T. Mikeska and J.M. Craig, DNA methylation biomarkers: Cancer and beyond, Genes 5: ((2014) ), 821–864. |
[23] | S.E. Bojesen, N. Timpson, C. Relton, G. Davey Smith and B.G. Nordestgaard, AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality, Thorax 72: ((2017) ), 646–653. |
[24] | R.A. Philibert, S.R. Beach, M. Lei and G.H. Brody, Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking, Clin Epigenetics 5: ((2013) ), 1–8. |
[25] | N.S. Shenker, P.M. Ueland, S. Polidoro, K.V. Veldhoven, F. Ricceri, R. Brown, J.M. Flanagan and P. Vineis, DNA methylation as a long-term biomarker of exposure to tobacco smoke, Epidemiology 24: ((2013) ), 712–716. |
[26] | S. Zeilinger, B. Kuhnel, N. Klopp, H. Baurecht, A. Kleinschmidt, C. Gieger, S. Weidinger, E. Lattka, J. Adamski, A. Peters, K. Strauch, M. Waldenberger and T. Illig, Tobacco smoking leads to extensive genome-wide changes in DNA methylation, PLoS One 8: ((2013) ), e63812. |
[27] | K. Dawes, A. Andersen, E. Papworth, B. Hundley, N. Hutchens, H. El Manawy, A. Becker, L. Sampson, W. Philibert, F.X. Gibbons, M. Gerrard and R. Philibert, Refinement of cg05575921 demethylation response in nascent smoking, Clinical Epigenetics 12: ((2020) ). |
[28] | R. Philibert, N. Hollenbeck, E. Andersen, S. McElroy, S. Wilson, K. Vercande, S.R.H. Beach, T. Osborn, M. Gerrard, F.X. Gibbons and K. Wang, Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation, Frontiers in Psychiatry 7: ((2016) ), 55–55. |
[29] | R. Philibert, J.A. Mills, J.D. Long, S.E. Salisbury, A. Comellas, A. Gerke, K. Dawes, M. Vander Weg and E.A. Hoffman, The reversion of cg05575921 methylation in smoking cessation: A potential tool for incentivizing healthy aging, Genes 11: ((2020) ), 1415. |
[30] | S. Manik, L.M. Saini and N. Vadera, Counting and classification of white blood cell using Artificial Neural Network (ANN), IEEE. |
[31] | M. El Kawak, H.R. Dhaini, M.E. Jabbour, M.A. Moussa, K. El Asmar and M. Aoun, Slow N-acetylation as a possible contributor to bladder carcinogenesis, Mol Carcinog 59: ((2020) ), 1017–1027. |
[32] | M. De Vries, D.A. Van Der Plaat, I. Nedeljkovic, R.N. Verkaik-Schakel, W. Kooistra, N. Amin, C.M. Van Duijn, C.-A. Brandsma, C.C. Van Diemen, J.M. Vonk and H. Marike Boezen, From blood to lung tissue: Effect of cigarette smoke on DNA methylation and lung function, Respiratory Research 19: ((2018) ). |
[33] | B. Novakovic, J. Ryan, N. Pereira, B. Boughton, J.M. Craig and R. Saffery, Postnatal stability, tissue, and time specific effects of<i>AHRR</i>methylation change in response to maternal smoking in pregnancy, Epigenetics 9: ((2014) ), 377–386. |
[34] | P.-C. Tsai, C.A. Glastonbury, M.N. Eliot, S. Bollepalli, I. Yet, J.E. Castillo-Fernandez, E. Carnero-Montoro, T. Hardiman, T.C. Martin, A. Vickers, M. Mangino, K. Ward, K.H. Pietiläinen, P. Deloukas, T.D. Spector, A. Viñuela, E.B. Loucks, M. Ollikainen, K.T. Kelsey, K.S. Small and J.T. Bell, Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health, Clinical Epigenetics 10: ((2018) ). |
[35] | L.G. Tsaprouni, T.-P. Yang, J. Bell, K.J. Dick, S. Kanoni, J. Nisbet, A. Viñuela, E. Grundberg, C.P. Nelson, E. Meduri, A. Buil, F. Cambien, C. Hengstenberg, J. Erdmann, H. Schunkert, A.H. Goodall, W.H. Ouwehand, E. Dermitzakis, T.D. Spector, N.J. Samani and P. Deloukas, Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation, Epigenetics 9: ((2014) ), 1382–1396. |
[36] | F. Guida, T.M. Sandanger, R. Castagné, G. Campanella, S. Polidoro, D. Palli, V. Krogh, R. Tumino, C. Sacerdote, S. Panico, G. Severi, S.A. Kyrtopoulos, P. Georgiadis, R.C.H. Vermeulen, E. Lund, P. Vineis and M. Chadeau-Hyam, Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation, Human Molecular Genetics 24: ((2015) ), 2349–2359. |
[37] | X. Chen, J. Zhang, W. Ruan, M. Huang, C. Wang, H. Wang, Z. Jiang, S. Wang, Z. Liu, C. Liu, W. Tan, J. Yang, J. Chen, Z. Chen, X. Li, X. Zhang, P. Xu, L. Chen, R. Xie, Q. Zhou, S. Xu, D.L. Irwin, J.-B. Fan, J. Huang and T. Lin, Urine DNA methylation assay enables early detection and recurrence monitoring for bladder cancer, Journal of Clinical Investigation 130: ((2020) ), 6278–6289. |
[38] | W. Chung, J. Bondaruk, J. Jelinek, Y. Lotan, S. Liang, B. Czerniak and J.-P.J. Issa, Detection of bladder cancer using novel DNA methylation biomarkers in urine sediments, Cancer Epidemiology Biomarkers & Prevention 20: ((2011) ), 1483–1491. |
[39] | P. Kumar, M. Yadav, K. Verma, R. Dixit, J. Singh, S.K. Tiwary, G. Narayan and V.K. Dixit, Expression analysis of aryl hydrocarbon receptor repressor (AHRR) gene in gallbladder cancer, Saudi Journal of Gastroenterology: Official Journal of the Saudi Gastroenterology Association 27: ((2021) ), 54–59. |
[40] | S. Vacher, P. Castagnet, W. Chemlali, F. Lallemand, D. Meseure, M. Pocard, I. Bieche and M. Perrot-Applanat, High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism, PLOS ONE 13: ((2018) ), e0190619. |
[41] | E. Zudaire, N. Cuesta, V. Murty, K. Woodson, L. Adams, N. Gonzalez, A. Martínez, G. Narayan, I. Kirsch, W. Franklin, F. Hirsch, M. Birrer and F. Cuttitta, The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers, Journal of Clinical Investigation, (2008) . |
[42] | C.F.A. Vogel and T. Haarmann-Stemmann, The aryl hydrocarbon receptor repressor – more than a simple feedback inhibitor of AhR signaling: Clues for its role in inflammation and cancer, Curr Opin Toxicol 2: ((2017) ), 109–119. |
[43] | L.H.G. Matheus, S.V. Dalmazzo, R.B.O. Brito, L.A. Pereira, R.J. De Almeida, C.P. Camacho and H. Dellê, 1-Methyl-D-tryptophan activates aryl hydrocarbon receptor, a pathway associated with bladder cancer progression, BMC Cancer 20: ((2020) ). |
[44] | J. Yu, Y. Lu, S. Muto, H. Ide and S. Horie, The dual function of aryl hydrocarbon receptor in bladder carcinogenesis, Anticancer Research 40: ((2020) ), 1345–1357. |
[45] | K.M. Jordahl, A.I. Phipps, T.W. Randolph, H.A. Tindle, S. Liu, L.F. Tinker, K.T. Kelsey, E. White and P. Bhatti, Differential DNA methylation in blood as a mediator of the association between cigarette smoking and bladder cancer risk among postmenopausal women, Epigenetics 14: ((2019) ), 1065–1073. |
[46] | M. Wan, B.D. Bennett, G.S. Pittman, M.R. Campbell, L.M. Reynolds, D.K. Porter, C.L. Crowl, X. Wang, D. Su, N.A. Englert, I.J. Thompson, Y. Liu and D.A. Bell, Identification of smoking-associated differentially methylated regions using reduced representation bisulfite sequencing and cell type-specific enhancer activation and gene expression, Environ Health Perspect 126: ((2018) ), 047015. |
[47] | D.-D. Zhang, W.-T. Wang, J. Xiong, X.-M. Xie, S.-S. Cui, Z.-G. Zhao, M.J. Li, Z.-Q. Zhang, D.-L. Hao, X. Zhao, Y.-J. Li, J. Wang, H.-Z. Chen, X. Lv and D.-P. Liu, Long noncoding RNA LINC00305 promotes inflammation by activating the AHRR-NF-κB pathway in human monocytes, Scientific Reports 7: ((2017) ), 46204. |
[48] | A.R. Sonawane, J. Platig, M. Fagny, C.-Y. Chen, J.N. Paulson, C.M. Lopes-Ramos, D.L. Demeo, J. Quackenbush, K. Glass and M.L. Kuijjer, Understanding tissue-specific gene regulation, Cell Reports 21: ((2017) ), 1077–1088. |
[49] | O. Brandstätter, O. Schanz, J. Vorac, J. König, T. Mori, T. Maruyama, M. Korkowski, T. Haarmann-Stemmann, D. Von Smolinski, J.L. Schultze, J. Abel, C. Esser, H. Takeyama, H. Weighardt and I. Förster, Balancing intestinal and systemic inflammation through cell type-specific expression of the aryl hydrocarbon receptor repressor, Scientific Reports 6: ((2016) ), 26091. |
[50] | E.J. Dougherty and R.S. Pollenz, ARNT: A Key bHLH/PAS Regulatory Protein Across Multiple Pathways, Elsevier, (2010) , 231–252. |
[51] | J. Yamamoto, K. Ihara, H. Nakayama, S. Hikino, K. Satoh, N. Kubo, T. Iida, Y. Fujii and T. Hara, Characteristic expression of aryl hydrocarbon receptor repressor gene in human tissues: Organ-specific distribution and variable induction patterns in mononuclear cells, Life Sciences 74: ((2004) ), 1039–1049. |
[52] | V.P. Androutsopoulos, I. Spyrou, A. Ploumidis, A.E. Papalampros, M. Kyriakakis, D. Delakas, D.A. Spandidos and A.M. Tsatsakis, Expression profile of CYP1A1 and CYP1B1 enzymes in colon and bladder tumors, PLoS ONE 8: ((2013) ), e82487. |
[53] | Y.M. Al-Saraireh, F.O.F.O. Alshammari, A.M.M. Youssef, S. Al-Sarayreh, G.H. Almuhaisen, N. Alnawaiseh, J.M. Al Shuneigat and H.M. Alrawashdeh, Profiling of CYP4Z1 and CYP1B1 expression in bladder cancers, Scientific Reports 11: ((2021) ). |
[54] | H.A. Basma, L.H. Kobeissi, M.E. Jabbour, M.A. Moussa and H.R. Dhaini, Original article CYP2E1 and NQO1 genotypes and bladder cancer risk in a Lebanese population, International Journal of Molecular Epidemiology and Genetics 4: ((2013) ), 207–217. |
[55] | X. Yang, S. Solomon, L.R. Fraser, A.F. Trombino, D. Liu, G.E. Sonenshein, E.V. Hestermann and D.H. Sherr, Constitutive regulation ofCYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue, Journal of Cellular Biochemistry 104: ((2008) ), 402–417. |
[56] | B.-J. Breitkreutz, C. Stark and M. Tyers, Osprey: A network visualization system, Genome Biology 4: ((2003) ), R22. |
[57] | Z. Wang, M. Snyder, J.E. Kenison, K. Yang, B. Lara, E. Lydell, K. Bennani, O. Novikov, A. Federico, S. Monti and D.H. Sherr, How the AHR became important in cancer: The role of chronically active AHR in cancer aggression, Int J Mol Sci 22: ((2020) ). |
[58] | F.-C. Zeng, S. Cen, Z.-Y. Tang and X.-L. Kang, Elevated matrix metalloproteinase-9 expression may contribute to the pathogenesis of bladder cancer, Oncology Letters 11: ((2016) ), 2213–2222. |
[59] | Q. Yu, K. Zhang, X. Wang, X. Liu and Z. Zhang, Expression of transcription factors snail, slug, and twist in human bladder carcinoma, Journal of Experimental & Clinical Cancer Research 29: ((2010) ), 119. |
[60] | J. Lobo, S. Monteiro-Reis, C. Guimarães-Teixeira, P. Lopes, I. Carneiro, C. Jerónimo and R. Henrique, Practicability of clinical application of bladder cancer molecular classification and additional value of epithelial-to-mesenchymal transition: prognostic value of vimentin expression, Journal of Translational Medicine 18: ((2020) ). |
[61] | M. Foth, N.F.B. Ismail, J.S.C. Kung, D. Tomlinson, M.A. Knowles, P. Eriksson, G. Sjödahl, J.M. Salmond, O.J. Sansom and T. Iwata, FGFR3 mutation increases bladder tumourigenesis by suppressing acute inflammation, The Journal of Pathology 246: ((2018) ), 331–343. |
[62] | E.K. Duperret, S.J. Oh, A. McNeal, S.M. Prouty and T.W. Ridky, Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors, Cell Cycle 13: ((2014) ), 1551–1559. |
[63] | S.T. Sherry, dbSNP: The NCBI database of genetic variation, Nucleic Acids Research 29: ((2001) ), 308–311. |
[64] | H.I. Swanson, W.K. Chan and C.A. Bradfield, DNA Binding Specificities and Pairing Rules of the Ah Receptor, ARNT, and SIM Proteins, Journal of Biological Chemistry 270: ((1995) ), 26292–26302. |
[65] | S.G. Bacsi, S. Reisz-Porszasz and O. Hankinson, Orientation of the heterodimeric aryl hydrocarbon (dioxin) receptor complex on its asymmetric DNA recognition sequence, Molecular Pharmacology 47: ((1995) ), 432–438. |
[66] | S.C. Gorber, S. Schofield-Hurwitz, J. Hardt, G. Levasseur and M. Tremblay, The accuracy of self-reported smoking: A systematic review of the relationship between self-reported and cotinine-assessed smoking status, Nicotine & Tobacco Research 11: ((2009) ), 12–24. |
[67] | N.A. Morales, M.A. Romano, K. Michael Cummings, J.R. Marshall, A.J. Hyland, A. Hutson and G.W. Warren, Accuracy of self-reported tobacco use in newly diagnosed cancer patients, Cancer Causes & Control 24: ((2013) ), 1223–1230. |
[68] | R.T. Nakkash, J. Khalil and R.A. Afifi, The rise in narghile (shisha, hookah) waterpipe tobacco smoking: A qualitative study of perceptions of smokers and non smokers, BMC Public Health 11: ((2011) ), 315. |
[69] | N. Bejjani, C. El Bcheraoui and S.M. Adib, The social context of tobacco products use among adolescents in Lebanon (MedSPAD-Lebanon), Journal of Epidemiology and Global Health 2: ((2012) ), 15. |
[70] | L. Van Wesenbeeck, L. Janssens, H. Meeuws, O. Lagatie and L. Stuyver, Droplet digital PCR is an accurate method to assess methylation status on FFPE samples, Epigenetics 13: ((2018) ), 207–213. |
[71] | D. Lissa, T. Ishigame, R. Noro, M.J. Tucker, V. Bliskovsky, S. Shema, J.A. Beck, E.D. Bowman, C.C. Harris and A.I. Robles, HOXA9 methylation and blood vessel invasion in FFPE tissues for prognostic stratification of stage I lung adenocarcinoma patients, Lung Cancer 122: ((2018) ), 151–159. |
[72] | A.K. Sarnecka, D. Nawrat, M. Piwowar, J. Ligȩza, J. Swadźba and P. Wójcik, DNA extraction from FFPE tissue samples – a comparison of three procedures, Współczesna Onkologia 23: ((2019) ), 52–58. |


