
Combined HPV 16 E2 and L1 methylation predict response to treatment with cidofovir and imiquimod in patients with vulval intraepithelial neoplasia
Abstract
BACKGROUND:
Topical cidofovir and imiquimod can effectively treat approximately 55% of patients with vulval intraepithelial neoplasia (VIN), thus avoiding the need for surgery. Human papillomavirus (HPV)
OBJECTIVE:
This work aimed to determine if the applicability and predictive power of the
METHODS:
HPV E2 methylation and S5 classifier score were measured in fresh tissue samples collected pre-treatment from 132 patients with biopsy-proven VIN grade 3 who participated in a multicentre clinical trial and were randomised to treatment with cidofovir or imiquimod.
RESULTS:
Combining HPV16
CONCLUSIONS:
Combined HPV
1.Introduction
Vulval intraepithelial neoplasia (VIN) is a chronic condition of vulval skin and diagnosis is confirmed histologically by the identification of cellular changes associated with a premalignant state. VIN is commonly caused by human papillomavirus (HPV), which is present in around 76% of cases [1]. VIN can be very distressing for patients and often takes a long time to diagnose. If untreated, VIN may progress to vulval cancer.
Surgical excision has historically been the gold standard of care for patients with VIN but is associated with significant morbidity [2, 3]. With an increasingly younger population of women being affected by VIN, and recurrence rates in the region of 40–60% [4, 5], optimised alternative treatments are urgently needed. In a recent randomised trial (RT3VIN), topical treatment with imiquimod (an immune response modulator) versus cidofovir (a cytotoxic cytosine analogue specially formulated for topical treatment) both demonstrated complete response rates of 46% [6]. Both these treatments are challenging for patients, causing pain, irritation and ulceration, leading to premature discontinuation of treatment in up to 26% of patients. However, in patients who respond, recurrence rates appear to be lower than the recurrence rates seen in patients treated surgically and were lower with cidofovir compared to imiquimod (6% vs 28% at 18 months) [7]. The ability to identify patients more likely to respond to these treatments prior to their commencement could help to motivate the patient to persevere with the treatment and reassure the clinician that the right treatment is being offered.
Previous research demonstrated that HPV 16
The potential of HPV
The S5 classifier is a DNA methylation panel including both human and HPV DNA sequences that has been validated as a triage test in the detection of CIN and cervical cancer [11]. Additionally, it and has been shown to have high potential as a prognostic biomarker to accurately identify progressive cervical intraepithelial neoplasia 2 (CIN 2) [12]. The S5 classifier targets the late (L1 and/or L2) viral regions of HPV 16, 18, 31 and 33 as well as host gene EPB41L3. This assay has several potential advantages: it is not reliant on HPV positivity to generate a result (due to inclusion of the EPB41L3 host gene), it detects methylation in three additional HPV genotypes and targets a region (
The aims of this work were to:
• determine whether or not the HPV 16
• determine whether or not the S5 classifier or any of its constituent parts could predict response to treatment
• explore combining the S5 classifier or any of its constituent parts with HPV 16
2.Materials and methods
2.1Patients, samples and DNA extraction
This study utilised a subset of bioresources from the RT3VIN clinical trial which included women who were age 16 years or older and had biopsy-proven VIN grade 3 (including visible perianal disease not extending into the anal canal) within the past 3 months. Baseline translational research samples were available in addition to the clinical outcome data (
Consent for sample collection and their use in research was obtained from patients when they were recruited into the RT3VIN trial and was approved by the UK Multi Research Ethics Committee (08/NIR03/82).
2.2HPV detection
A type-specific PCR targeting the HPV 16
2.3Methylation analysis in the Cardiff University HPV research laboratory (CU HPV Lab)
HPV DNA methylation testing was restricted to HPV16 positive (
2.4Methylation analysis in Queen Mary’s University of London laboratory (QMUL Lab)
All 132 baseline samples were tested for the components of the S5 classifier as well as HPV16
2.5Statistical methods
Statistical analyses were conducted using STATA/SE v16 according to a pre-specified analysis plan. The dataset used in this analysis included all patients from the RT3VIN trial who had both a pre-treatment tissue sample and a post-treatment histological assessment response available.
Table 1
Distribution of DNA methylation factors in baseline samples
| Cidofovir | Imiquimod | |||||||
| Randomised | 89 | 91 | ||||||
| with response endpoint | 72 (80.9) | 71 (78.0) | ||||||
| and baseline sample | 65 (73.0) | 67 (73.6) | ||||||
| Analysis dataset | 65 | 67 | ||||||
| Unifocal VIN | 32 (49.2) | 33 (49.3) | ||||||
| Multifocal VIN | 33 (50.8) | 34 (50.8) | ||||||
| First VIN | 35 (53.9) | 36 (53.7) | ||||||
| Recurrent VIN | 30 (46.2) | 31 (46.3) | ||||||
| HPV+ve | 59 (90.1) | 55 (82.1) | ||||||
| HPV16+ve | 52 (80.0) | 54 (80.6) | ||||||
| % methylation | % methylation | |||||||
| EPB41L3 | 65 | (100) | 5.3 | (3.5–11.9) | 67 | (100) | 4.7 | (3.1–7.8) |
| HPV 16 | 45 | (69.2) | 62.5 | (22.0–92.8) | 47 | (70.1) | 51.91 | (20.0–92.7) |
| HPV 16 | 40 | (61.5) | 11.7 | (9.1–82.4) | 38 | (56.7) | 18.2 | (9.8–67.8) |
| HPV 18 | 1 | (1.5) | 32.6 | 0 | (0) | – | ||
| HPV 31 | 0 | (0) | – | 1 | (1.5) | 22.64 | ||
| HPV 33 | 8 | (12.3) | 43.7 | (13.8–75.5) | 5 | (7.5) | 46.0 | (41.1–64.6) |
| S5 | 65 | (100) | 10.9 | (7.3–18.0) | 67 | (100) | 10.5 | (6.1–14.9) |
| HPV 16 | 35 | (53.8) | 4.5 | (3.4–42.5) | 38 | (56.7) | 7.8 | (4.0–81.6) |
| HPV 16 | 30 | (46.2) | 4.3 | (2.2–35.6) | 34 | (50.7) | 3.4 | (2.4–33.6) |
HPV 16
S5
The HPV 16 E2 methylation results were compared between laboratories using a scatter and Bland-Altman plot but all other analyses used the HPV 16 E2 methylation results from the QMUL Lab as it was thought to better represent a routine NHS clinical laboratory.
HPV positivity was defined as any PapilloCheck HPV genotype or HPV16
For each treatment arm, Wilcoxon rank sum tests were used to compare the distribution of each methylation marker, and chi-square tests to compare the proportion with HPV 16/any HPV positivity, between the group who responded to treatment and the group who did not respond to treatment. Multiple comparisons were accounted using the Benjamini-Hochberg procedure [16] by setting the false discovery rate (Q) at 0.1 and calculating each individual
In order to examine the validity of the pragmatic imputation of missing methylation values as zero, we conducted post-hoc sensitivity analyses to examine the consistency of findings across subgroups of patients: those with missing methylation and no relevant HPV genotype detected (thought to be most likely to be true zeros) and those with missing methylation but relevant genotype detected (thought to potentially be explained by viral integration and therefore potentially spurious zeros).
Table 2
Distribution of DNA methylation factors in responders and non-responders by trial arm
| Cidofovir ( | Imiquimod ( | |||||||||||
| No response ( | Response ( | Rank sum result | (i/m) Q | No response ( | Response ( | Rank sum result | (i/m) Q | |||||
| HPV 16 | n (%) | n (%) | n (%) | n (%) | ||||||||
| Negative | 5 | (38.5) | 8 | (61.5) | 0.071 | 3 | (23.1) | 10 | (76.9) | 0.029 | ||
| Positive | 25 | (48.1) | 27 | (51.9) | 24 | (44.4) | 30 | (55.6) | ||||
| Any HPV | n (%) | n (%) | n (%) | n (%) | ||||||||
| Negative | 2 | (50.0) | 2 | (50.0) | 0.1 | 0 | (0.0) | 4 | (100.0) | 0.021 | ||
| Positive | 28 | (45.9) | 33 | (54.1) | 26 | (42.9) | 36 | (57.1) | ||||
| Methylation |
Median (IQR) |
Median (IQR) |
Median (IQR) |
Median (IQR) | ||||||||
| HPV16 | 2.7 | (0–3.7) | 2.7 | (0–15.7) | 0.057 | 3.2 | (0–81.6) | 2.2 | (0–6.6) | 0.043 | ||
| S5 | 9.8 | (7.4–16.3) | 13.4 | (6.7–18.2) | 0.050 | 13.4 | (8.9–17.7) | 2.2 | (9.3–13.4) | 0.014 | ||
| EPB41L3 | 5.1 | (3.4–11.2) | 5.7 | (3.5–13.7) | 0.064 | 4.8 | (2.7–10.4) | 4.7 | (3.2–5.9) | 0.093 | ||
| HPV 16 | 21.2 | (0–85.6) | 28.8 | (0–92.1) | 0.079 | 36.0 | (14.4–93.4) | 12.2 | (0–55.7) | 0.007 | ||
| HPV 16 | 8.1 | (0–10.6) | 8.1 | (0–62.2) | 0.086 | 8.9 | (0–67.8) | 3.2 | (0–15.0) | 0.036 | ||
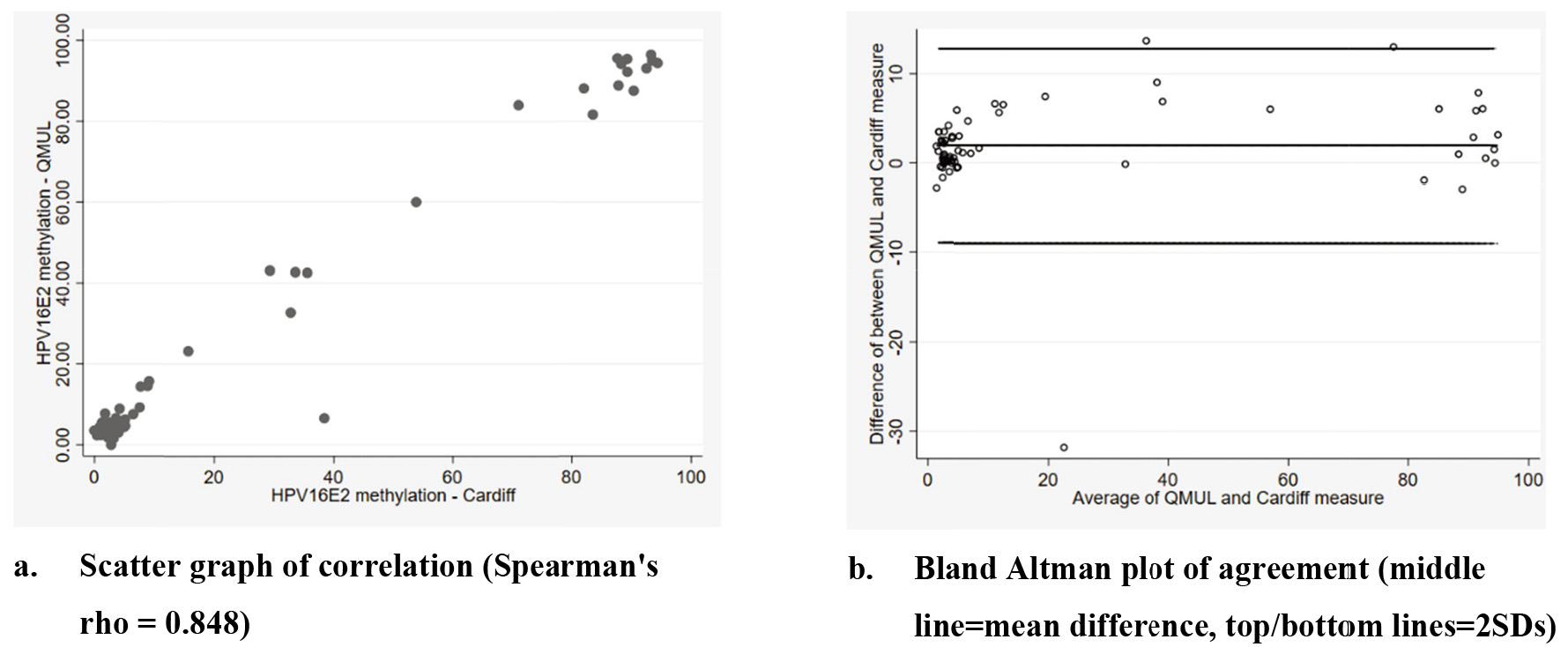
Figure 1.
HPV16E2 methylation detection by laboratory.
3.Results
3.1Distribution of DNA methylation markers by treatment group
Table 1 shows the distribution of pre-treatment DNA methylation biomarkers tested in the two treatment groups. Of all the patients randomised into the RT3VIN trial, 65/89 (73.0%) of patients in the cidofovir arm, and 67/91 (73.6%) in the imiquimod arm, had both a baseline sample and response assessment available; these 132 samples were used for the current study. HPV positivity and methylation assay results were reasonably well balanced between treatment arms. It can be seen that HPV 18, 31 and 33 positivity were relatively low in this cohort and that therefore the S5 score is largely dependent upon EPB41L3, HPV 16 L1 and L2.
Figure 2.
Distribution of methylation levels in responders and non-responders by trial arm.
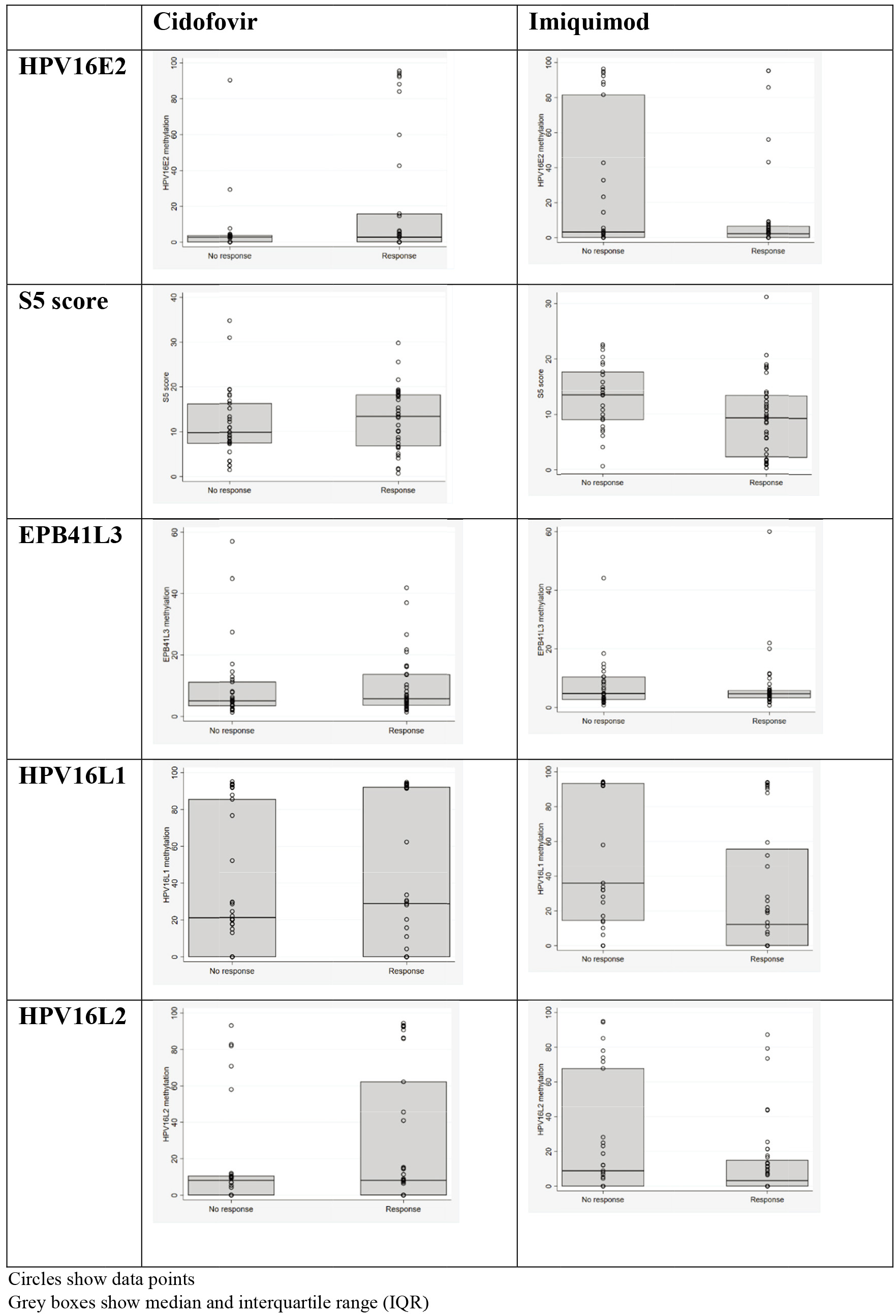
Table 3
Response rates in methylation biomarker defined subgroups of the treatment groups
|
| Cidofovir | Imiquimod |
| |||
|---|---|---|---|---|---|---|
| HPV 16 | 67 | 16/33 | (48.5) | 26/34 | (76.5) | 0.018 |
|
HPV16L1 | 26 | 8/13 | (61.5) | 10/13 | (76.9) | 0.395 |
|
HPV16L1 | 14 | 4/7 | (57.1) | 7/7 | (100.0) | 0.051 |
|
HPV16L1 | 27 | 4/13 | (30.8) | 9/14 | (64.3) | 0.082 |
| HPV 16 | 65 | 19/32 | (59.4) | 14/33 | (42.4) | 0.172 |
| HPV 16 | 85 | 21/46 | (45.7) | 24/39 | (61.5) | 0.144 |
|
HPV16E2 | 26 | 8/13 | (61.5) | 10/13 | (76.9) | 0.395 |
|
HPV16E2 | 33 | 9/17 | (52.9) | 9/16 | (56.3) | 0.849 |
|
HPV16E2 | 26 | 4/16 | (25.0) | 5/10 | (50.0) | 0.192 |
| HPV 16 | 47 | 14/19 | (73.7) | 16/28 | (57.1) | 0.247 |
| HPV 16 | 76 | 21/36 | (58.3) | 21/40 | (52.5) | 0.610 |
| HPV 16 | 56 | 14/29 | (48.2) | 19/27 | (70.4) | 0.093 |
| HPV 16 | 36 | 12/15 | (80.0) | 9/21 | (42.9) | 0.026 |
| HPV 16 | 96 | 23/50 | (46.0) | 31/46 | (67.4) | 0.035 |
HPV 16
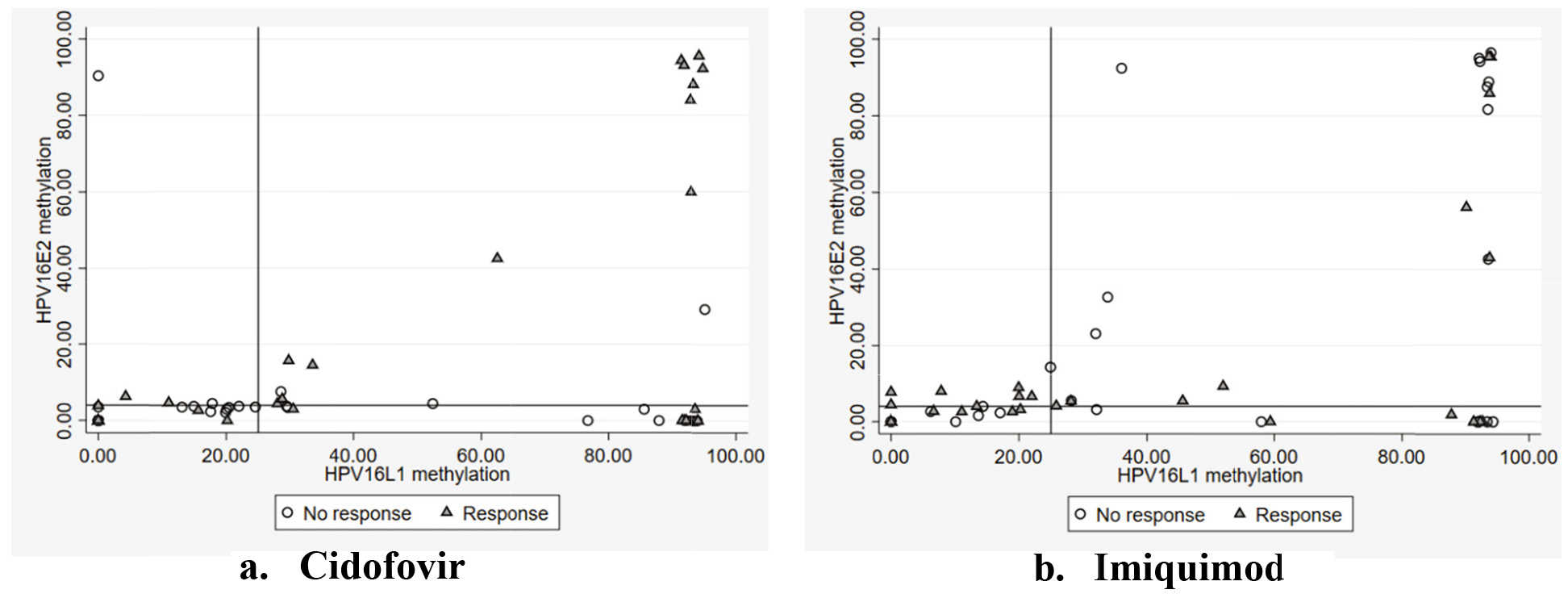
Figure 3.
Scatter plot of HPV 16 L1 and HPV 16 E2 methylation by treatment group.
3.2HPV 16 E2 methylation by assay
HPV 16
3.3Biomarkers of response
Using the data from the QMUL laboratory, Table 2 and Fig. 2 show the distribution of HPV factors (after imputing missing values as 0) by response in each treatment group. There was some evidence (
Although HPV 16
3.4HPV 16 L1 methylation as a biomarker for response with imiquimod
A ROC curve of HPV 16
3.5Combining HPV 16 L1 and HPV 16 E2 methylation
An HPV 16
3.6Sensitivity analyses exploring the validity of imputing missing values with zero
As described in the methods, in all the above analyses we imputed missing methylation values as 0 and included them in the HPV 16
4.Discussion
Cidofovir and imiquimod are topical treatments for vulval intraepithelial neoplasia; when used individually both treatments give complete response in approximately 50% of patients. HPV E2 methylation has been identified as a potential biomarker predictive of response to treatment, which could enable a more personalised approach with increased likelihood of efficacy [8]. A current limitation of the HPV E2 methylation assay is a high failure rate (approximately 50%). In this work, we demonstrated the problem of high failure rate could be overcome by combining HPV16
The assessment of HPV 16
It is noted that the overall HPV prevalence in our cohort (86.4%) is somewhat higher than many studies quoted in the literature, including a systematic review of 2,764 cases of VIN (76.3%) published in 2017 [1]. However, this systematic review also describes a wide range of prevalence (0%–100%) and a more recent trial in the same population found a prevalence of 89.7% [18]. The high prevalence rate seen in our study may be due to the application of two HPV detection assays that target different regions of the HPV genome (E6 PCR targets E6 and PapilloCheck targets E1) which is not a common approach in many of the published studies.
To make this biomarker clinically useful, an approach to those patients generating no methylation (missing methylation) result is required. In this analysis we treated missing methylation values as “zero” i.e. low. This may not be biologically accurate since they may represent cases with high levels of viral integration, a feature typically associated with higher levels of methylation. However, we found the same pattern of better response with imiquimod in patients who were HPV 16 positive with missing methylation and in those patients who were HPV 16 negative with missing methylation. Consequently, the data support giving imiquimod to all patients with low and missing methylation, and cidofovir to those with both HPV 16
S5 was not a significant marker of response in either imiquimod or cidofovir patients. The S5 classifier was initially designed as a triage tool for detection of CIN and cervical cancer and although it uses HPV and host methylation to do this, it was not designed as a biomarker to predict response to treatment. We noticed that the methylation levels of EPB41L3 were low (5–10%) in VIN patients as opposed to cervical precancer (10–20%) or cancer patients (above 20%) [19]. It is possible that any discriminatory power of the S5 classifier was diluted by inclusion of EPB41L3 which does not seem to differentiate responses in this population. This may reflect differences in the natural history and pathobiology of VIN and CIN.
HPV L1 methylation is a component of the S5 classifier and this study demonstrated that low HPV 16
Hypotheses to explain why high methylation is associated with response to cidofovir, and low methylation is associated with response to imiquimod, have previously been described [20]. These include that cidofovir may act as a demethylating agent. This is speculative but is consistent with cidofovir being a nucleoside analogue with similar structure to the established demethylating agent decitabine (used in the treatment of myelodysplastic bone conditions). This possibility is supported by a study of cases of failed cidofovir treatment in recurrent respiratory papillomatosis (caused by HPV 11), which correlated treatment failure with uniformly low levels of methylation [12]. Alternatively, E2 and L1 methylation may be surrogate markers of another relevant process, e.g. they may be associated with more advanced infections with lower levels of p53 protein. Although E6/E7 expression to some level is present at all times in an HPV infected cells independent of the presence of a ‘dormant’ infection or a disease causing infection, it is broadly accepted that it is deregulated E6/E6 expression that contributes to the development of a transforming (i.e. malignant potential) infection through the ubiquitination of p53/pRb rendering it dysfunctional. E2 DNA methylation is widely accepted as one means by which, E6/E7 gene expression becomes deregulated and consequently p53/pRb dysfunctional. This would be consistent with the suggestion that the selectivity of cidofovir for transformed cells is due to the absence, or perturbation, of normal DNA repair pathways associated with dysfunctional p53 mediated signalling [21]. Cidofovir has been shown to generate double-stranded breaks in cellular DNA, which can be repaired in normal cells, but not in tumour cells [22]. In HPV-infected cells, the level of p53 is reduced through ubiquitination and proteosomal degradation mediated by the HPV E6 oncoprotein, expression of which can become deregulated as a result of HPV integration and/or HPV DNA methylation [23]. HPV integration and increased methylation could therefore be more common in cells that have lower levels of p53/pRb and may be more likely to respond to cidofovir. The correlation between increased methylation and response to cidofovir could therefore be because methylation is a surrogate marker of absent/low-level p53/pRb.
Contrary to the case with cidofovir,
In conclusion, this study refines the use of assessment of HPV methylation as a potential predictive marker in treatment in VIN for all patients from whom a sample is taken. It demonstrates that reproducibility of this method of assessment of methylation, that addition of human genes adds little to the predictive power of the marker panel, and that high levels of HPV gene methylation are consistently associated with good response to treatment with cidofovir and that low or missing levels are associated with good response to treatment with imiquimod. These findings justify validation in a prospective trial.
Acknowledgments
This work was funded by the South Wales Gynaecology Cancer Fund and supported by a Cancer Research UK core funding at the Centre for Trials Research, Cardiff University.
Author contributions
Conception: SJ, CH, BN.
Interpretation or analysis of data: CH, BN, SJ.
Preparation of the manuscript: SJ, CH, BN.
Revision for important intellectual content: CH, BN, CA, NP, AT, SJ.
References
[1] | M.T. Faber, F.L. Sand, V. Albieri, B. Norrild, S.K. Kjaer and F. Verdoodt, Prevalence and type distribution of human papillomavirus in squamous cell carcinoma and intraepithelial neoplasia of the vulva, Int J Cancer 141: ((2017) ), 1161–1169. |
[2] | T.S. Shylasree, V. Karanjgaokar, A. Tristram, A.R. Wilkes, A.B. MacLean and A.N. Flander, Contribution of demographic, psychological and disease-related factors to quality of life in women with high-grade vulval intraepithelial neoplasia, Gynecol Oncol 110: ((2008) ), 185–189. |
[3] | W.M. Likes, C. Stegbauer, T. Tillmanns and J. Pruett, Pilot study of sexual function and quality of life after excision for vulvar intraepithelial neoplasia, J Reprod Med 52: ((2007) ), 23–27. |
[4] | E.M. van Esch, M.C. Dam, M.E. Osse, H. Putter, B.J. Trimbos, G. Fleuren, S.H. van der Burg and M.I. van Poelgeest, Clinical characteristics associated with development of recurrence and progression in usual-type vulvar intraepithelial neoplasia, International Journal of Gynecological Cancer 23: ((2013) ), 1476–1483. |
[5] | M.K. Fehr, M. Baumann, M. Mueller, D. Fink, S. Heinzl, P. Imesch and K. Dedes, Disease progression and recurrence in women treated for vulvovaginal intraepithelial neoplasia, J Gynecol Oncol 24: ((2013) ), 236–241. |
[6] | A. Tristram, C.N. Hurt, T. Madden, N. Powell, S. Man, S. Hibbitts, P. Dutton, S. Jones, A.J. Nordin, R. Naik, A. Fiander and G. Griffiths, Activity, safety, and feasibility of cidofovir and imiquimod for treatment of vulval intraepithelial neoplasia (RT(3)VIN): A multicentre, open-label, randomised, phase 2 trial, Lancet Oncol 15: ((2014) ), 1361–1368. |
[7] | C.N. Hurt, S. Jones, T.A. Madden, A. Fiander, A.J. Nordin, R. Naik, N. Powell, M. Carucci and A. Tristram, Recurrence of vulval intraepithelial neoplasia following treatment with cidofovir or imiquimod: Results from a multicentre, randomised, phase II trial (RT3VIN), BJOG 125: ((2018) ), 1171–1177. |
[8] | S.E.F. Jones, S. Hibbitts, C.N. Hurt, D. Bryant, A.N. Fiander, N. Powell and A.J. Tristram, Human Papillomavirus DNA Methylation Predicts Response to Treatment Using Cidofovir and Imiquimod in Vulval Intraepithelial Neoplasia 3, Clinical Cancer Research, (2017) . |
[9] | S.I. Collins, C. Constandinou-Williams, K. Wen, L.S. Young, S. Roberts, P.G. Murray and C.B. Woodman, Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: A longitudinal cohort study, Cancer Res 69: ((2009) ), 3828–3832. |
[10] | D. Bryant, T. Onions, R. Raybould, S. Jones, A. Tristram, S. Hibbitts, A. Fiander and N. Powell, Increased methylation of Human Papillomavirus type 16 DNA correlates with viral integration in Vulval Intraepithelial Neoplasia, J Clin Virol 61: ((2014) ), 393–399. |
[11] | A.T. Ramirez, G.I. Sanchez, B. Nedjai, M.C. Agudelo, A.R. Brentnall, K. Cuschieri, K.M. Castaneda, J. Cuzick, A.T. Lorincz and A.-U.-C.T. Group, Effective methylation triage of HPV positive women with abnormal cytology in a middle-income country, Int J Cancer 148: ((2021) ), 1383–1393. |
[12] | K. Louvanto, K. Aro, B. Nedjai, R. Butzow, M. Jakobsson, I. Kalliala, J. Dillner, P. Nieminen and A. Lorincz, Methylation in predicting progression of untreated high-grade cervical intraepithelial neoplasia, Clinical Infectious Diseases 70: ((2020) ), 2582–2590. |
[13] | L. Mirabello, M. Schiffman, A. Ghosh, A.C. Rodriguez, N. Vasiljevic, N. Wentzensen, R. Herrero, A. Hildesheim, S. Wacholder, D. Scibior-Bentkowska, R.D. Burk and A.T. Lorincz, Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia, Int J Cancer 132: ((2013) ), 1412–1422. |
[14] | D. Bryant, A. Tristram, T. Liloglou, S. Hibbitts, A. Fiander and N. Powell, Quantitative measurement of Human Papillomavirus type 16 L1/L2 DNA methylation correlates with cervical disease grade, J Clin Virol 59: ((2014) ), 24–29. |
[15] | R. Hernandez-Lopez, A.T. Lorincz, L. Torres-Ibarra, C. Reuter, D. Scibior-Bentkowska, R. Warman, B. Nedjai, I. Mendiola-Pastrana, L. Leon-Maldonado, B. Rivera-Paredez, P. Ramirez-Palacios, E. Lazcano-Ponce, J. Cuzick, J. Salmeron and F.S. Group, Methylation estimates the risk of precancer in HPV-infected women with discrepant results between cytology and HPV16/18 genotyping, Clin Epigenetics 11: ((2019) ), 140. |
[16] | Y. Benjamini and Y. Hochberg, Controlling the false discovery rate: A practical and powerful approach to multiple testing, Journal of the Royal Statistical Society. Series B (Methodological) 57: ((1995) ), 289–300. |
[17] | J.M. Walboomers, M.V. Jacobs, M.M. Manos, F.X. Bosch, J.A. Kummer, K.V. Shah, P.J. Snijders, J. Peto, C.J. Meijer and N. Munoz, Human papillomavirus is a necessary cause of invasive cervical cancer worldwide, J Pathol 189: ((1999) ), 12–19. |
[18] | G. Trutnovsky, O. Reich, E.A. Joura, M. Holter, A. Ciresa-Konig, A. Widschwendter, C. Schauer, G. Bogner, Z. Jan, A. Boandl, M.S. Kalteis, S. Regauer and K. Tamussino, Topical imiquimod versus surgery for vulvar intraepithelial neoplasia: A multicentre, randomised, phase 3, non-inferiority trial, Lancet 399: ((2022) ), 1790–1798. |
[19] | B. Nedjai, C. Reuter, A. Ahmad, R. Banwait, R. Warman, J. Carton, S. Boer, J. Cuzick and A.T. Lorincz, Molecular progression to cervical precancer, epigenetic switch or sequential model, Int J Cancer 143: ((2018) ), 1720–1730. |
[20] | C. Gros, J. Fahy, L. Halby, I. Dufau, A. Erdmann, J.M. Gregoire, F. Ausseil, S. Vispe and P.B. Arimondo, DNA methylation inhibitors in cancer: Recent and future approaches, Biochimie 94: ((2012) ), 2280–2296. |
[21] | T. Gall, A. Kis, E. Feher, L. Gergely and K. Szarka, Virological failure of intralesional cidofovir therapy in recurrent respiratory papillomatosis is not associated with genetic or epigenetic changes of HPV11: Complete genome comparison of sequential isolates, Antiviral Res 92: ((2011) ), 356–358. |
[22] | G. Andrei, D. Topalis, T. De Schutter and R. Snoeck, Insights into the mechanism of action of cidofovir and other acyclic nucleoside phosphonates against polyoma- and papillomaviruses and non-viral induced neoplasia, Antiviral Res 114: ((2015) ), 21–46. |
[23] | T. De Schutter, G. Andrei, D. Topalis, S. Duraffour, T. Mitera, L. Naesens, van den Oord, P. Matthys and R. Snoeck, Cidofovir treatment improves the pathology caused by the growth of human papillomavirus-positive cervical carcinoma xenografts in athymic nude mice, Cancer Lett 329: ((2013) ), 137–145. |
[24] | J. Doorbar, Molecular biology of human papillomavirus infection and cervical cancer, Clin Sci (Lond) 110: ((2006) ), 525–541. |
[25] | M.A. Stanley, Imiquimod and the imidazoquinolones: Mechanism of action and therapeutic potential, Clin Exp Dermatol 27: ((2002) ), 571–577. |
[26] | C. Diaz-Arrastia, I. Arany, S.C. Robazetti, T.V. Dinh, Z. Gatalica, S.K. Tyring and E. Hannigan, Clinical and molecular responses in high-grade intraepithelial neoplasia treated with topical imiquimod 5%, Clinical Cancer Research 7: ((2001) ), 3031–3033. |
[27] | S.J. Bowden, I. Kalliala, A.A. Veroniki, M. Arbyn, A. Mitra, K. Lathouras, L. Mirabello, M. Chadeau-Hyam, E. Paraskevaidis, J.M. Flanagan and M. Kyrgiou, The use of human papillomavirus DNA methylation in cervical intraepithelial neoplasia: A systematic review and meta-analysis, EBioMedicine 50: ((2019) ), 246–259. |


