
Lidocaine inhibited migration of NSCLCA549 cells via the CXCR4 regulation
Abstract
BACKGROUND:
Lidocaine is a local anesthetic that wildly used in surgical treatment and postoperative medical care for lung cancers. We hypothesized that lidocaine at clinical plasma concentration can inhibit CXCL12/CXCR4 axis-regulated cytoskeletal remodeling thereby reduce the migration of Non-small-cell lung cancers (NSCLC) cells.
METHODS:
We determined the effect of lidocaine at clinical plasma concentration on CXCL12-induced cell viability, apoptosis, cell death, monolayer cell wound healing rate, individual cell migration indicators, expression of CXCR4, CD44, and ICAM-1, intracellular Ca
RESULTS:
Lidocaine did not affect cell viability, apoptosis, and cell death but inhibited CXCL12-induced migration, intracellular Ca
CONCLUSION:
Lidocaine at clinical plasma concentrations inhibited CXCL12-induced CXCR4 activation, thereby reduced the intracellular Ca
List of abbreviations
| NSCLC: | Non-small cell lung cancers |
| CXCL12: | C-X-C Motif Chemokine Ligand 12 |
| CXCR4: | C-X-C chemokine receptor type 4 |
1.Introduction
Lung cancer is one of the most fatal cancer types with the most global death number among males and the second most global death number among females [1]. As the medical care for lung cancer develops, the mortality of lung cancer decreased in the USA and UK has decreased in recent years. However, in many industrialized nations, the emerging social smoking culture has resulted in higher lung cancer rates in these areas [2]. Among all lung cancer cases, more than four-fifths of clinical lung cancer diagnosed are non-small cell lung cancer (NSCLC) [3], a lung cancer type that has diverse pathological features and with an undesirable prognosis.
Clinical medical advancements have improved cancer therapy extensively in recent years [4]. As clinical diagnosis is critical for disease treatment [5], in-depth analyses of lung cancer subtypes have been developed for potential targeting therapy and customized treatment according to their genetic and cellular heterogeneity [6]. Surgical treatment is the most applicable intervention for early-stage lung cancer diagnoses and is critical for the further prescription of proper therapeutic options [7]. One of the clinical factors that might affect the outcome of surgical treatments is the anesthesia during the surgery [8, 9]. Many local anesthetics have been found to affect multiple cancer cells [10]. A local anesthetic, lidocaine, has been wildly used in surgical treatment [11] and postoperative medical care [12, 13]. Preclinical studies have revealed that lidocaine can inhibit the proliferation and migration of many cancer types, including lung cancer [14, 15], breast cancer [16, 17], gastric cancer [18], colon cancer [19, 20, 21], etc.
However, the doses of lidocaine used in most previous studies are much higher than the concentration in plasma during clinical use of lidocaine (we mentioned it as “plasma concentration”). In this study, we focused on a lower concentration range of lidocaine that is correspondent to clinical plasma concentration. Lidocaine at this concentration range has been reported to inhibit cytoskeletal remodeling and migration of breast cancer cell MDA-MB-231 [22]. We proposed that a similar effect of lidocaine on cell migration is also present in NSCLC cells.
The metastatic potential of NSCLC has been found to be strongly associated with the chemokine CXCL12 (C-X-C Motif Chemokine Ligand 12) and the activity of the CXCL12 receptor CXCR4 (C-X-C chemokine receptor type 4) [23]. CXCR4/CXCL12 axis controls the immunity of the body and the survival, invasion, and metastasis of cancer cells [24, 25]. CXCR4 has been found to be differently expressed in cancer cells and might contribute to the motility of cancer cells [26]. The CXCR4/CXCL12 axis has been proposed as a potential drug target for NSCLC [27]. In this study, we hypothesized that lidocaine at clinical plasma concentration can inhibit CXCL12/CXCR4 axis-regulated cytoskeletal remodeling, thereby decrease migration of NSCLC cells. This investigation can provide a better understanding of the potential pharmacological effects of lidocaine on the clinical treatment of NSCLC.
2.Methods and materials
2.1Cell line and cells culture
A549 cells were purchased from Biofeng (China). Cells were cultured using Ham’s F12K
2.2Knockdown and overexpression
CXCR4 knockdown was achieved by transfecting the CXCR4 shRNA (TRCN0000256866) plasmid into A549 cells to silence the expression of CXCR4. The plasmid was purchased from Sigma-Aldrich (USA). The Sequences of shRNA oligonucleotides are as follows: 5’-TCCTGTCCTGCTATTGCATTA-3’. CXCR4 overexpression was achieved by transfecting the CXCR4 expressing (hCXCR4-mTFP1) plasmid into A549 cells to overexpress CXCR4. The plasmid was purchased from Addgene (USA). The transfection method was described previously [28] The knowdown and overexpression of CXCR4 were validated by western blotting experiments.
2.3Testing reagents
Lidocaine HCl pre-made parenteral solution was purchased from Hospira Inc. (USA). Human CXCL12 and pertussis toxin were purchased from Sigma-Aldrich (USA). Fura-2 (Fura-2-acetoxymethyl ester) was purchased from Abcam (UK). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc (Kumamoto, Japan).
2.4CCK-8 assay
The CCK-8 assay is used to determine cell overall viability and cytotoxicity. The CCK-8 assay was conducted as previously described [29]. Briefly, cells were seeded in 96 well plates. After the incubation with testing drugs, 10
2.5Apoptosis and cell death detection
Apoptosis and cell death detection were determined using Bcl-2 ELISA kit (Abcam, UK) and Cell Death Detection ELISA plus (Roche, USA) respectively. The method of ELISA was described previously [30, 31]. The positive control was induced by high-temperature culture (55
2.6Western blotting
Membrane protein expression was determined using the western blotting assay Membrane proteins of A549 were extracted using Mem-PER™Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific, USA). The extraction was conducted as previously described [33, 34]. Subsequently, the western blotting assay was conducted as in previous studies [35, 36]. Briefly, SDS-PAGE gel electrophoresis was used to separate proteins and semi-dry protein transfer was conducted as described [37]. The membrane was then incubated with primary and secondary antibodies according to the recommended experimental condition. Na+/K+ ATPase protein was used as an internal reference gene to normalized the data. Antibodies used including CXCR4 Antibody (PA3-305), Anti-Alpha 1 Na+/K+ ATPase Antibody (#ANP-001), Recombinant Anti-CD44 antibody [EPR1013Y] (ab51037), Anti-ICAM1 antibody [EP1442Y] (ab53013), Goat Anti-Mouse IgG H&L (HRP) (ab6789), and Goat Anti-Rabbit IgG H&L (HRP) (ab6721). ECL reagent (Thermo Scientific, USA) was used to visualize the target protein.
2.7QPCR
The CXCR4 mRNA level was determined using a QPCR assay. The extraction was conducted as previously described [38, 39]. Briefly, Trizol buffer (Sigma-Aldrich, USA) was used to isolate cellular RNA. An Agilent Bioanalyzer (Agilent 2100; Agilent Technologies, Inc, USA) was used to assess the concentration and integrity of RNA extracted. QuantiTect Reverse Transcription Kit (QIAGEN, USA) and miRCURY LNA SYBR Green PCR Kit (QIAGEN, USA) were used to conduct RT-PCR. The protocol of PCR reaction was described previously [40]. The GAPDH gene was used as an internal reference gene to normalize the data. CXCR4 Human qPCR Primer Pair (NM_003467) and GAPDH Human qPCR Primer Pair (NM_002046) were purchased from OriGene Technologies (USA).
2.8Wound healing assay
Cells migration was determined using the wound healing assay. The method was described previously [41, 42]. Briefly, cells were grown in six-well plates at over 90% confluency. A scratch wound was created in the cell monolayer using a 200-
2.9Chemotaxis assay
Real-time cell migration of individual A549 cells was recorded using the m-Slide chemotaxis system (ibidi, Germany). The method was described previously [43, 44]. Briefly, A549 cells were cultured on the central channel of the chemotaxis slide at 10% confluency for 8 h to allow adherence. Testing drugs were applied when the assay started and the images of cells were recorded for 15 hours with a time-lapse Micro-Imager. Single-cell tracking was analyzed using the ImageJ software. Spider plots representing the aggregated trajectories of cells. Forward migration indexes and cell velocity were analyzed using the Ibidi software.
2.10Fura-2based intracellular Ca2 +
Fura-2-based fluorescence was used to determine the intracellular Ca
2.11Actin polymerization detection
The cytoskeleton remodeling was indicated by the determination of actin polymerization using Fluorescein Phalloidin staining. The method was described previously [47]. Briefly, cells were fixed in 4% paraformaldehyde for 10 min at 4
2.12Plotting and statistical analysis
Means and standard deviations are displayed in the figures. A T-test or ANOVA was used to analyze the significance of the difference (
Figure 1.
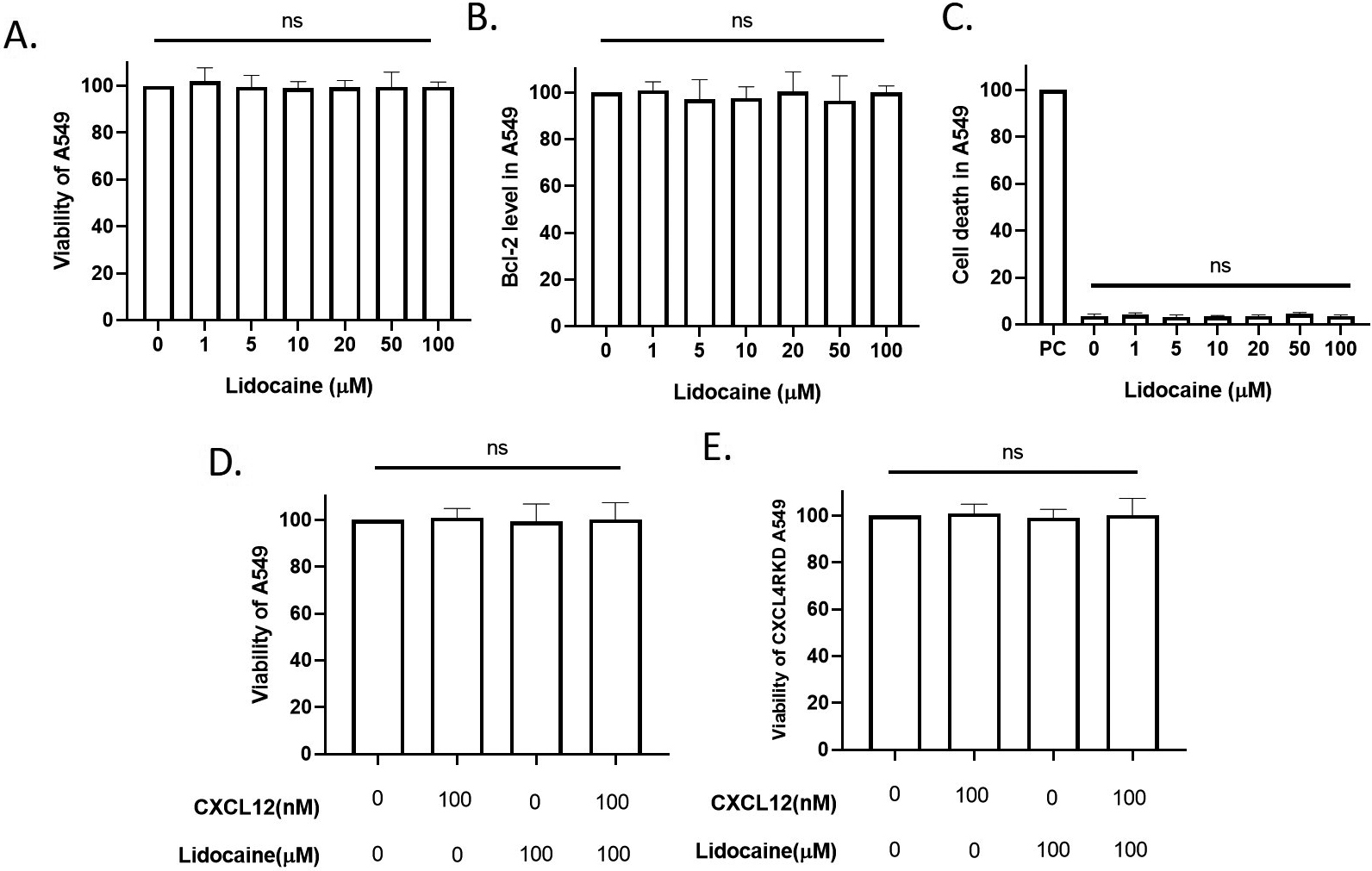
Effect of lidocaine at clinical plasma concentration on A549 viability. A549 were exposed to tested drugs for 24 hours. A. Effect of clinical-plasma-dose lidocaine on A549 viability. B. Effect of lidocaine on A549 apoptosis. C. Effect of lidocaine on A549 cell death. PC: positive control induced by high-temperature culture (55
3.Results
3.1Lidocaine did not affect A549 cell viability at the clinical plasma concentrations
The anti-arrhythmia plasma concentration and approximately equipotent nerve block concentrations of lidocaine are around 10
3.2lidocaine decreased CXCR4 expression
In this study, we focused on the effect of the CXCR4/CXCL12 signal on cancer cell migration. CXCR4/CXCL12 signal has been wildly accepted as a critical signal for the migration of lung cancer cells [49, 50, 51] and CXCR4 has been thought to be a drug target for the inhibition of lung cancer migration [27, 52]. We extracted the membrane expression of CXCR4 in A549 and observed the effect of lidocaine on the surface expression of CXCR4 in A549 cells. Results showed that lidocaine significantly decreased the surface expression of CXCR4 and the total expression of CXCR4 mRNA (Fig. 2). In the CXCR4 knockdown experiments, we knocked down over 80% CXCR4 mRNA (Fig. 2C) and the surface CXCR4 expression was very low that was almost not detectable in the western blotting (Fig. 2A and B). After CXCR4 knockdown, the effect of lidocaine on CXCR4 expression was eliminated (Fig. 2) In
Figure 2.
Effect of lidocaine on CXCR4 expression in A549. AB. membrane expression of CXCR4 protein in A549. C. CXCR4 mRNA expression in A549. (Significant differences compared to both control (CXCL12 and lidocaine are 0) are indicated by
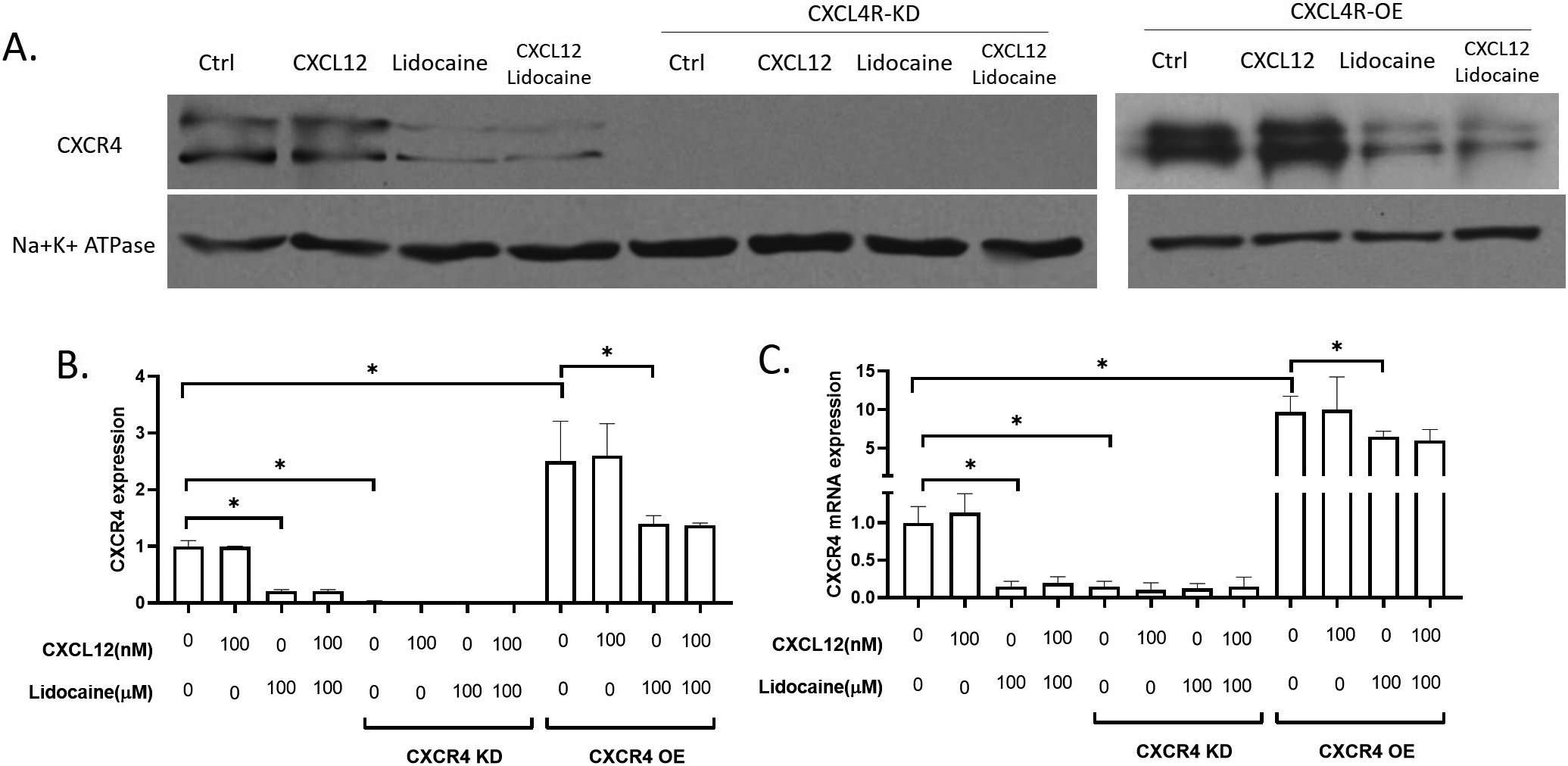
the CXCR4 overexpressing expression, we increased the mRNA of CXCR4 by about 8 times, and the CXCR4 protein expression was increased by about 2 times. At the presence of lidocaine, both the CXCR4 mRNA and protein levels were significantly decreased compared to the control (Fig. 2). Thus, we suggested that lidocaine down-regulated CXCR4 expression when it was highly expressed, but did not affect CXCR4 when it was at a low expression level.
3.3Lidocaine inhibited CXCL12 induced migration
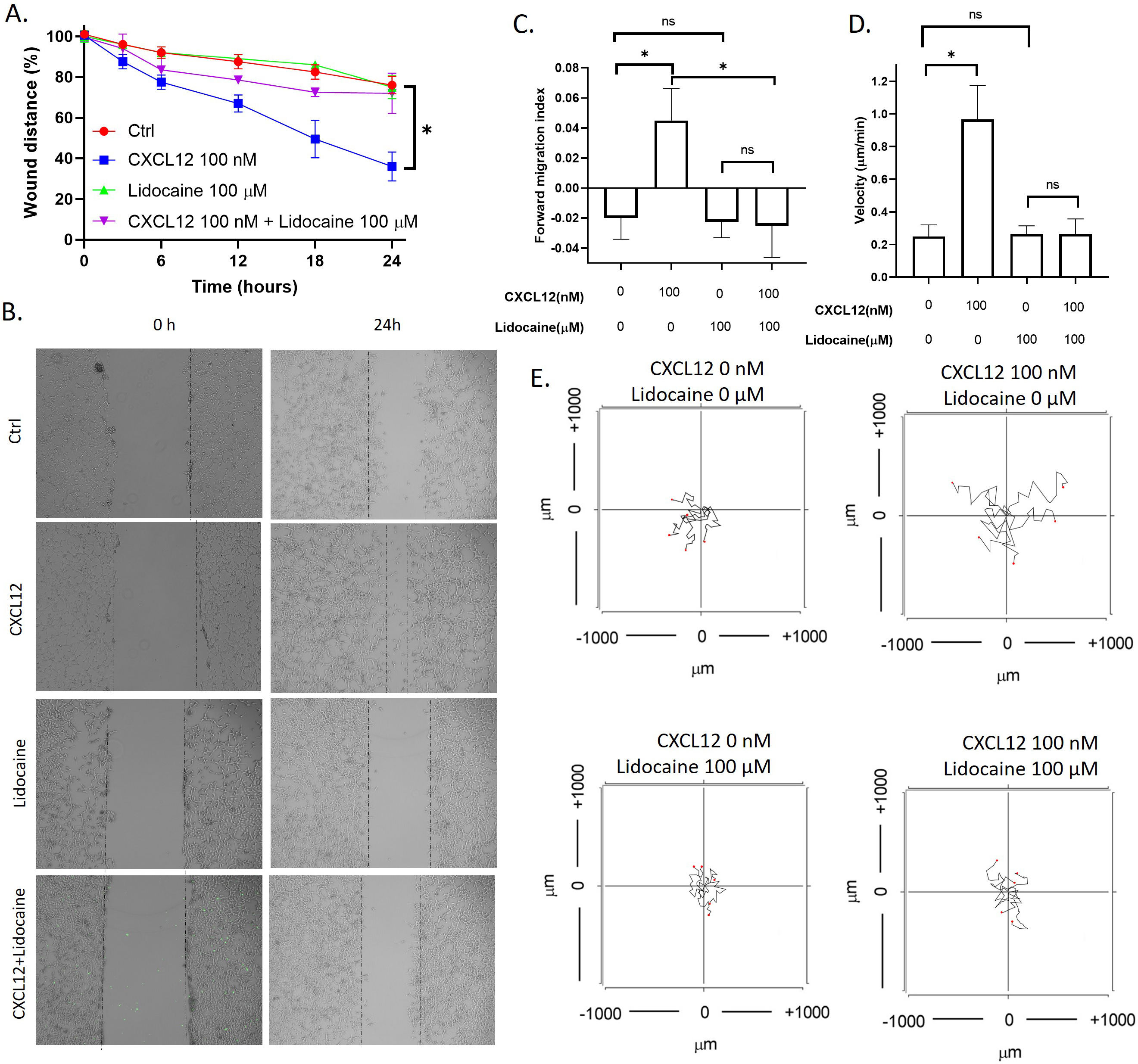
CXCL12 is the agonist of the CXCR4 and has been wildly used for studying chemokinesis or chemotaxis of cells [53]. To tested if lidocaine affected CXCL12 induced migration of A549 cells, we performed both the wound healing assay to observed the migration of monolayer A549 cells as a group and in vitro chemotaxis assay to observe the migration of individual A549 cells. Results showed that CXCL12 significantly increased the A549 wound healing rate. In addition, CXCL12 also increased the migration index and velocity of individual cells. As shown in the aggregated trajectories of individual A549 cells, A549 migrated a longer distance at the presence of CXCL12. At the presence of lidocaine, the A549 wound healing rate was the same as control, but the wound healing rate decreased compared with CXCL12-induced cells (Fig. 3). These results revealed that lidocaine did not affect A549 cell migration but blocked the stimulation of CXCL12.
Figure 3.
Effect of lidocaine on CXCL12-induced A549 migration. A. Effect of lidocaine on CXCL12-induced A549 monolayer wound healing. B. Representative images of wound healing assay. C. Effect of lidocaine on CXCL12-induced A549 forward migration index. D. Effect of lidocaine on CXCL12-induced A549 migration velocity. E. Aggregated trajectories of individual A549 cells at different experimental conditions (10 hours). (Significant differences are indicated by
3.4CXCR4 knockdown block lidocaine effect in migration
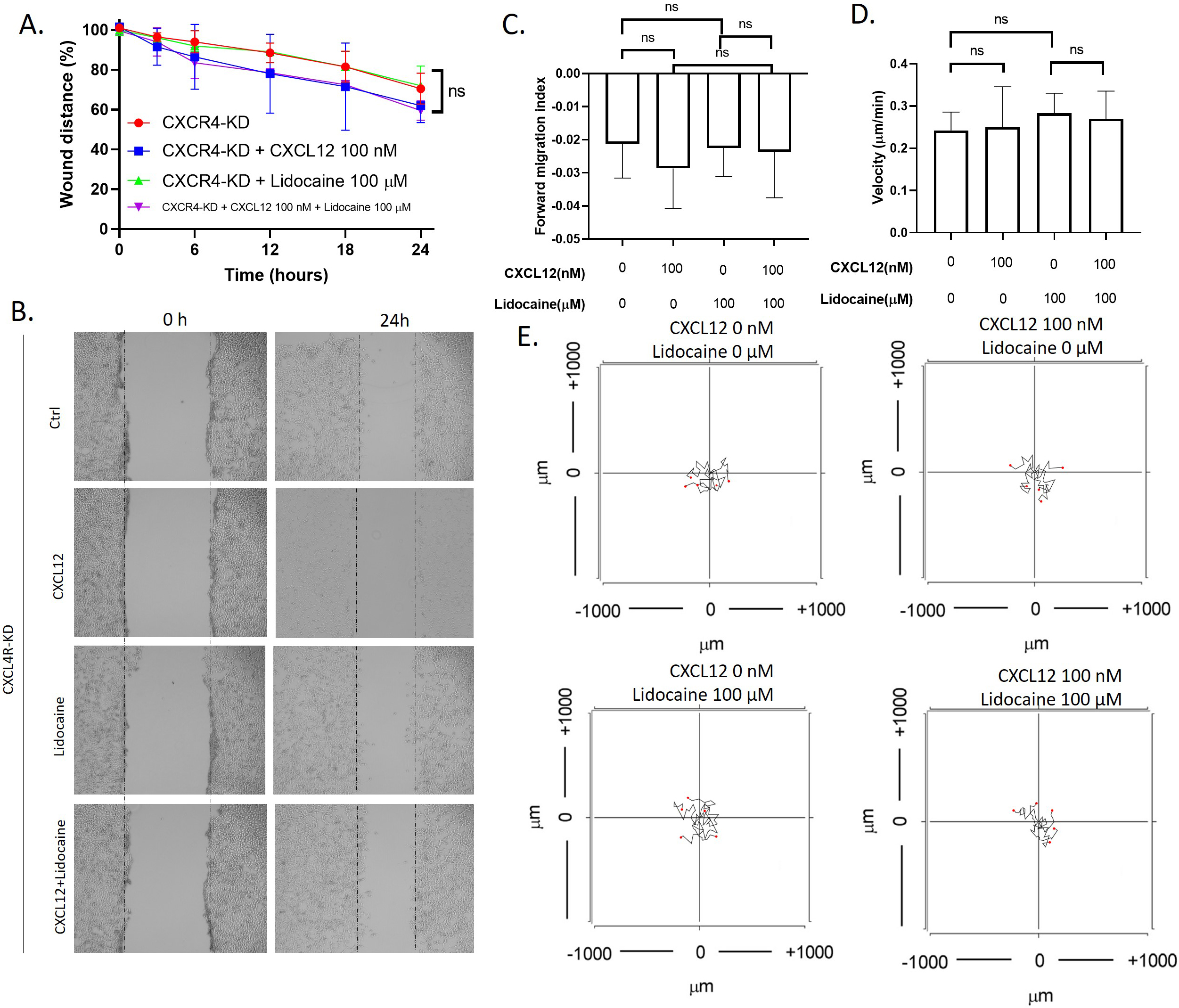
To investigate whether lidocaine affects migration through CXCR4, we also performed migration assays in CXCR4 knockdown A549 cells. Results showed that knockdown of CXCR4 eliminated the effects of CXCL12 or lidocaine on A549 migration. The exposure of CXCL12 or lidocaine had no significant effect on the wound healing speed and the migrate index and velocity of individual cells (Fig. 4). This suggested that CXCR4 was essential for the effects of CXCL12 and lidocaine.
Figure 4.
Effect of lidocaine on CXCL12-induced CXCR4 knocked down A549 cells migration. A. Effect of lidocaine on CXCL12-induced CXCR4- knocked down A549 monolayer wound healing. B. Representative images of wound healing assay. C. Effect of lidocaine on CXCL12-induced CXCR4-knocked down A549 forward migration index. D. Effect of lidocaine on CXCL12-induced CXCR4- knocked down A549 migration velocity. E. Aggregated trajectories of individual CXCR4- knocked down A549 cells at different experimental conditions (10 hours). (Significant differences are indicated by
3.5Lidocaine inhibited migration caused by CXCR4 overexpression
To further investigate the effect of lidocaine on CXCR4 mediated migration, we also performed migration assays in CXCR4 overexpressing A549 cells. Results showed that CXCL12 significantly increased CXCR4 overexpressing the A549 wound healing rate. In addition, CXCL12 also increased the migration index and velocity of individual CXCR4 overexpressing A549 cells. As shown in the aggregated trajectories of individual CXCR4 overexpressing A549 cells, these cells migrated a longer distance at the presence of CXCL12. At the presence of lidocaine, CXCR4 overexpressing the A549 wound healing rate was the same as control, but the wound healing rate decreased compared with CXCL12-induced cells (Fig. 5). These results revealed that lidocaine did not affect CXCR4 overexpressing A549 cell migration but blocked the stimulation of CXCL12 (Fig. 6). These results demonstrated that the effect of lidocaine was mediated by CXCR4/CXCL12 signal.
Figure 5.
Effect of lidocaine on CXCL12-induced CXCR4 overexpressing A549 cells migration. A. Effect of lidocaine on CXCL12-induced CXCR4 overexpressing A549 monolayer wound healing. B. Representative images of wound healing assay. C. Effect of lidocaine on CXCL12-induced CXCR4 overexpressing A549 forward migration index D. Effect of lidocaine on CXCL12-induced CXCR4 overexpressing A549 migration velocity. E. Aggregated trajectories of individual CXCR4 overexpressing A549 cells at different experimental conditions (10 hours). (Significant differences are indicated by
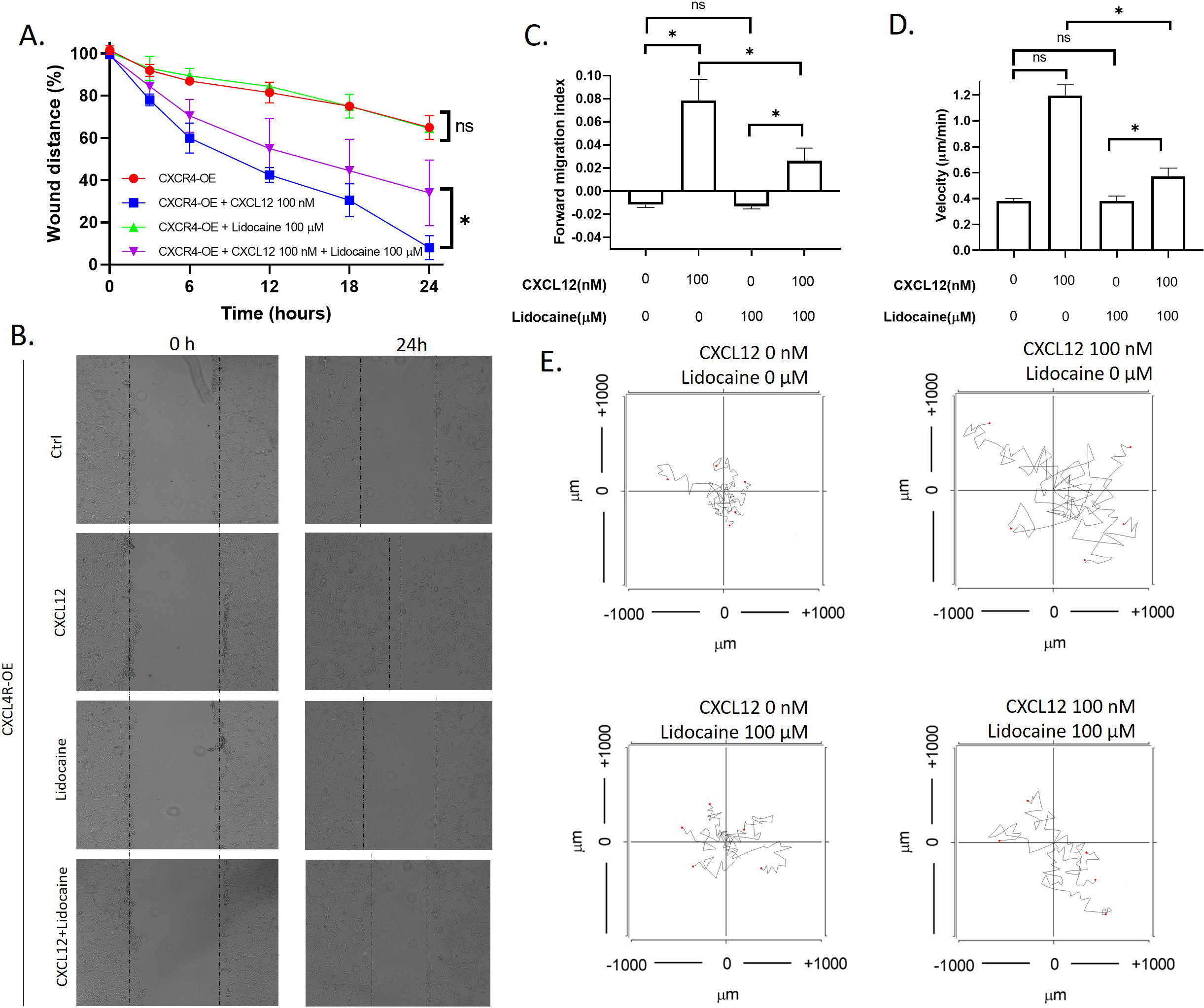
Figure 6.
Effect of lidocaine on membrane expression of CD44 and ICAM-1 in A549. A. Representative images of western blotting. B. membrane expression of CD44 protein in A549. C. membrane expression of ICAM-1 protein in A549. (Significant differences are indicated by
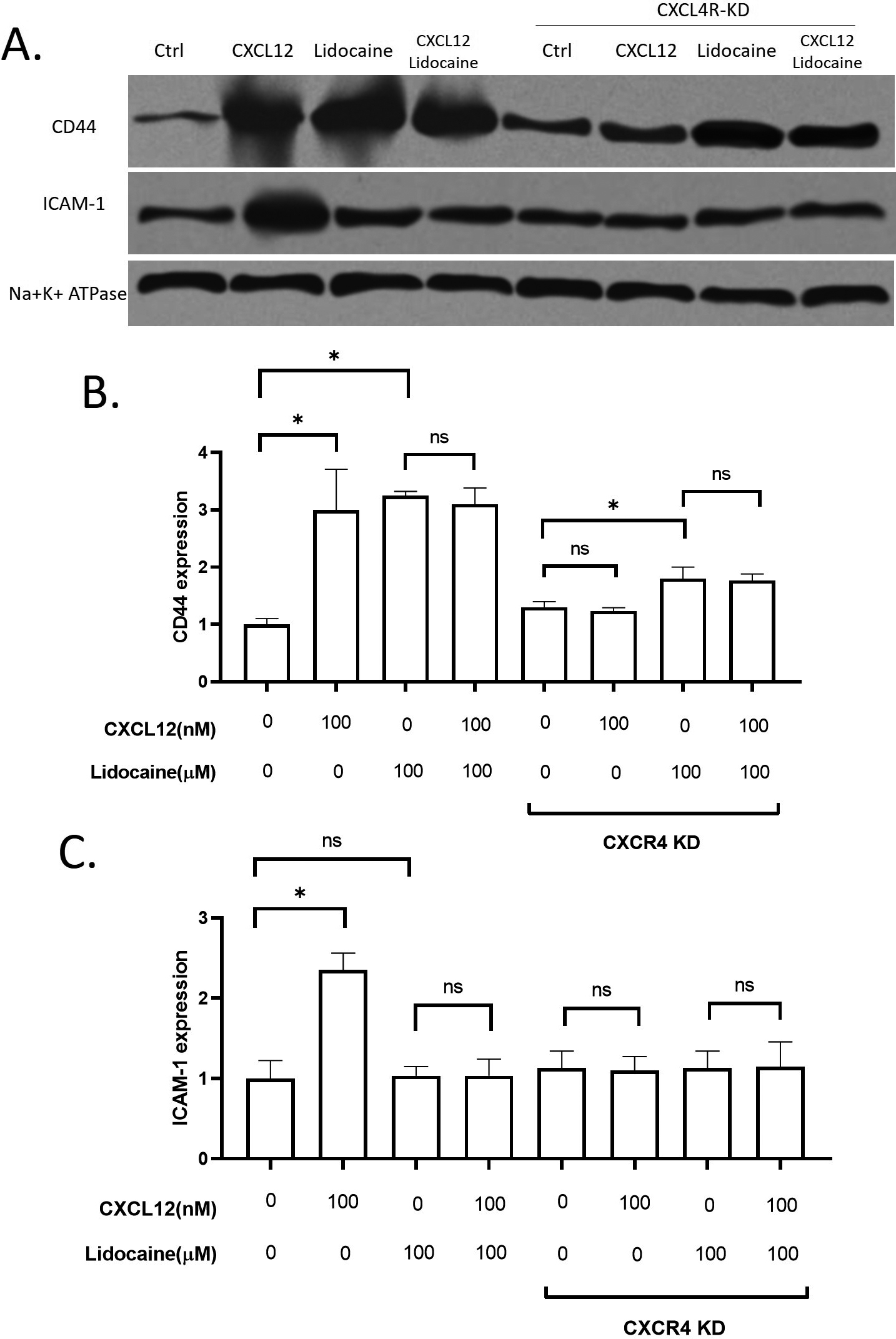
3.6Regulations of lidocaine on CD44 and ICAM-1
To further explore the potential mechanisms for lidocaine action on A549 migration, we investigated two critical adhesion molecules on the epithelial cell membrane, CD44 and ICAM-1. CD44 has been wildly used as a key migration-related biomarker for lung cancers [54, 55]. Another migration-related extracellular molecule, ICAM-1, is also thought to be critical in lung cancer cell migration [56, 57, 58]. Both CD44 and ICAM-1 levels were significantly increased by CXCL12. Lidocaine increased CD44 in both wild-type A549 and CXCR4 knockdown A549 cells in the absence or presence of CXCL12, but it did not affect ICAM-1 (Fig. 6). These results suggested that lidocaine can up-regulate CD44 but not ICAM-1. However, the regulation of lidocaine on CD44 was not essential for CXCL12 induced migration.
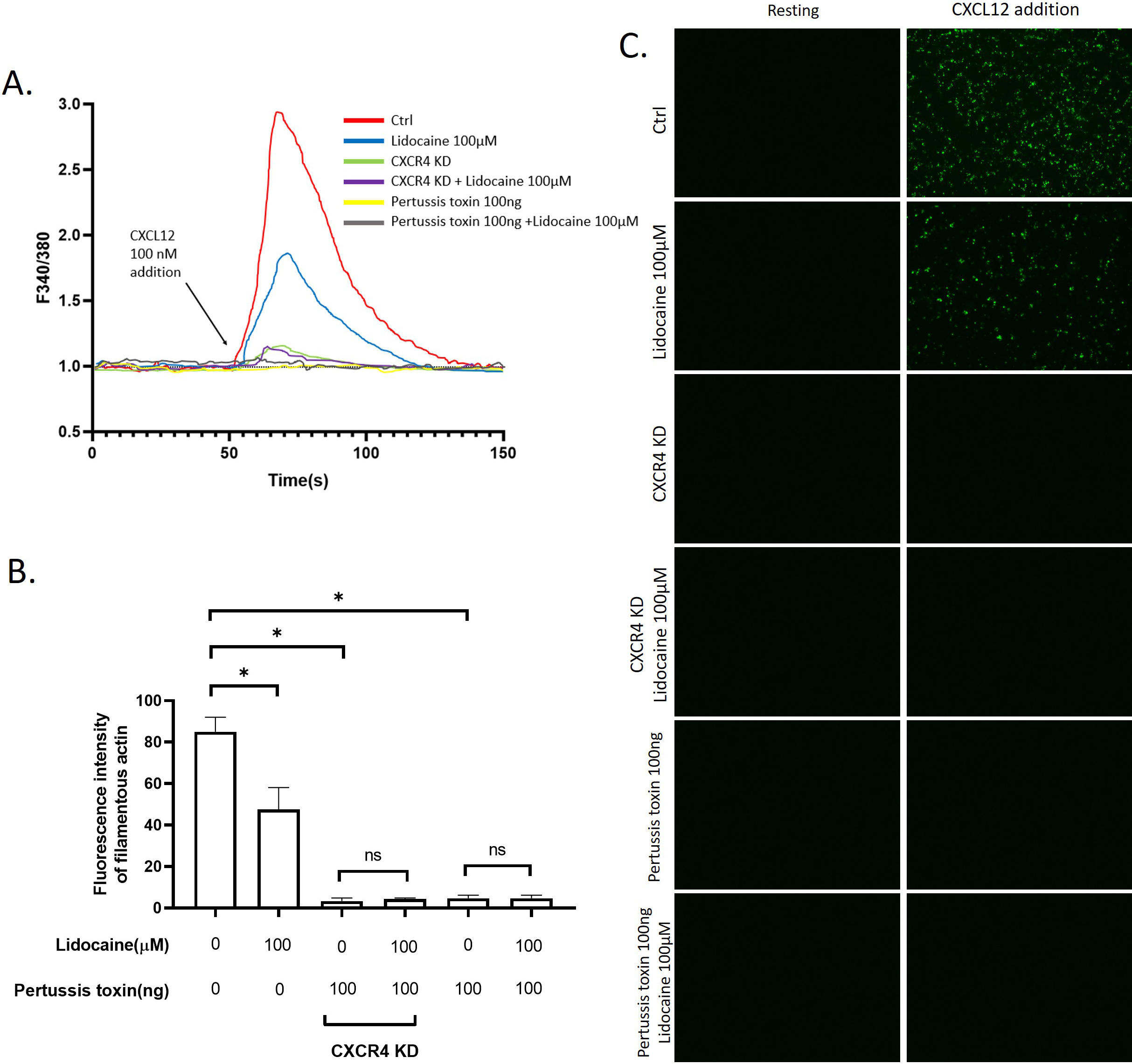
3.7Lidocaine inhibited CXCL12induced intracellular Ca2 +
Another potential mechanism we hypothesized underlying lidocaine’s effect on migration is the regulation of the cytoskeleton remodeling. To test this hypothesis, we monitored the effect of lidocaine on the intracellular Ca
Figure 7.
Effect of lidocaine on CXCL12-induced intracellular Ca
4.Discussion
The therapeutic effect of surgery in metastatic NSCLC has been controversial [59, 60]. During the surgery, many clinical factors might contribute to the metastasis and recurrence of lung cancer surgery The application of lidocaine during the surgery or preoperational treatment results in a plasma lidocaine micro-environment for lung cancer cell migration and survival. Hence, the potential impact of lidocaine on metastasis and recurrence of lung cancer surgery should be further studied. However, so far, the effect of lidocaine at plasma concentration on cancers was less studied. The doses of lidocaine used in most previous studies are much higher than the clinical plasma concentration. Therefore, although many previous studies demonstrated the effect of lidocaine on lung cancer cells, they fail to convince clinical surgeons that lidocaine exerts a considerable impact on the surgery outcome. Lidocaine is known as a sodium channel blocker. Although the major target of lidocaine, the voltage-gated sodium channels, has been found to play a role in cancer developments [61], lidocaine might also affect cancer independent of sodium channel blockade
Although a previous study suggested that lidocaine at the “mM” concentration range inhibited proliferation [15] and induced apoptosis of A549 [62], our result showed that, at plasma concentration, lidocaine had almost no effect on cell proliferation, apoptosis, and cell death. The previous study also suggested that the migration of A549 was inhibited by lidocaine at 8 mM, but as shown by our data, the effect of lidocaine on migration was not significant when the doses of lidocaine decreased to 100
Different membrane surface proteins expressed on lung cancer cells as adhesion molecules can be critical in the migration of cells [63] CD44 has been reported to play roles in the metastasis of NSCLC cells [54]. In this study, the expression of CD44 was promoted by CXCL12 stimulation. Our results also showed that the surface expression of CD44 on A549 was up-regulated by lidocaine. However, the lidocaine did not further increase the CD44 expression at the presence of CXCL12 and the increase of CD44 in the lidocaine alone group did not affect cell migration. Thus, we suggested that CD44 was directly up-regulated by lidocaine bypassing the CXCL12/CXCR4 axis but the increase of CD44 was not essential for the migration of A549. In addition, we also determined another critical adhesion molecule for migration, the ICAM-1. The expression of ICAM-1 has been associated with lung cancer progression and prognosis [64]. A previous study reported that lidocaine affects the migration of a lung cancer cell line H838 by reducing ICAM-1 [65]. However, in this study, the lidocaine at plasma concentration did not affect ICAM-1 expression. The ICAM-1 expression was up-regulated by CXCR4 activation by CXCL12, hence, we suggested ICAM-1 might be a potential downstream target of lidocaine/CXCR regulation (Fig. 8). Further validation is required in the future.
Figure 8.
Effect of lidocaine on A549 migration.
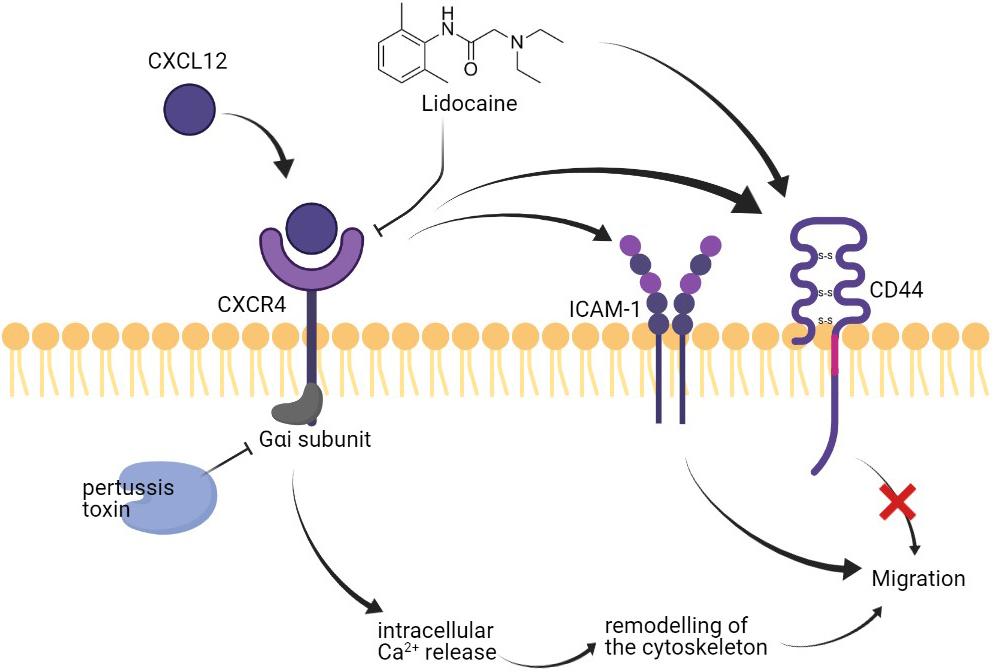
Another activity that might impact the migration of A549 was the remodeling of the cytoskeleton. The activation of CXCR4 by CXCL12 can trigger intracellular Ca
5.Conclusion
This study demonstrated that lidocaine at clinical plasma concentrations showed a significant inhibition effect on CXCL12-induced CXCR4 activation, thereby reduced the intracellular Ca
Ethics approval and consent to participate
This work was approved and consented by the Ethical Committee of Changzhi Medical College Affiliated Heping hospital.
Funding
This study received funding from the Changzhi Medical College Affiliated Heping hospital (No. is not available).
Availability of data and materials
The raw data of this study are provided from the corresponding author with a reasonable request.
Authors’ contributions
Interpretation or analysis of data: Baichun Xing.
Preparation of the manuscript: Baichun Xing and Linlin Yang.
Revision for important intellectual content: Yanan Cui.
Supervision: Yanan Cui.
Consent for publication
All the authors consent for this publication.
Conflict of interest
The authors claimed that there is no conflict of interest.
References
[1] | L.A. Torre, R.L. Siegel and A. Jemal, Lung Cancer Statistics, Adv Exp Med Biol 893: ((2016) ), 1–19. |
[2] | J.A. Barta, C.A. Powell and J.P. Wisnivesky, Global Epidemiology of Lung Cancer, Ann Glob Health 85: ((2019) ). |
[3] | J. Skřučková, B. Kadlec, O. Venclíček and Z. Merta, Lung cancer, Cas Lek Cesk 157: ((2018) ), 226–236. |
[4] | H. Liu, Effect of Traditional Medicine on Clinical Cancer, Biomedical Journal of Scientific & Technical Research 30: ((2020) ), 23548–23551. |
[5] | W. Haixia, M. Shu, Y. Li, W. Panpan, S. Kehuan, X. Yingquan, L. Hengrui, L. Xiaoguang, W. Zhidi and O. Ling, Effectiveness associated with different therapies for senile osteopo-rosis: a network Meta-analysis, J Tradit Chin Med 40: ((2020) ), 17–27. |
[6] | Z. Chen, C.M. Fillmore, P.S. Hammerman, C.F. Kim and K.K. Wong, Non-small-cell lung cancers: a heterogeneous set of diseases, Nat Rev Cancer 14: ((2014) ), 535–46. |
[7] | H. Hoy, T. Lynch and M. Beck, Surgical treatment of lung cancer, Crit Care Nurs Clin North Am 31: ((2019) ), 303–313. |
[8] | R. Li, Y. Huang, H. Liu, J.P. Dilger and J. Lin, Comparing volatile and intravenous anesthetics in a mouse model of breast cancer metastasis, American Association for Cancer Research, (2018) , p. 2162. |
[9] | R. Li, H. Liu, J.P. Dilger and J. Lin, Effect of Propofol on breast Cancer cell, the immune system, and patient outcome, BMC Anesthesiol 18: ((2018) ), 77. |
[10] | H. Liu, J.P. Dilger and J. Lin, Effects of local anesthetics on cancer cells, Pharmacology & Therapeutics 212: ((2020) ), 107558. |
[11] | H. Liu, A clinical mini-review: Clinical use of Local anesthetics in cancer surgeries, The Gazette of Medical Sciences 1: ((2020) ), 030–034. |
[12] | M. Akkuş and E. Öner, Can local infiltration of lidocaine reduce the postoperative atrial fibrillation rate in patients undergoing lobectomy for lung cancer? Acta Chir Belg 120: ((2020) ), 265–270. |
[13] | S.N. Watanabe, K. Imai, T. Kimura, Y. Saito, S. Takashima, I. Matsuzaki, N. Kurihara, M. Atari, T. Matsuo, H. Iwai, Y. Sato, S. Motoyama, K. Nomura, T. Nishikawa and Y. Minamiya, Effect of lidocaine cream analgesia for chest drain tube removal after video-assisted thoracoscopic surgery for lung cancer: a randomized clinical trial, Reg Anesth Pain Med ((2019) ). |
[14] | L. Zhang, R. Hu, Y. Cheng, X. Wu, S. Xi, Y. Sun and H. Jiang, Lidocaine inhibits the proliferation of lung cancer by regulating the expression of GOLT1A, Cell Prolif 50: ((2017) ). |
[15] | H. Sun and Y. Sun, Lidocaine inhibits proliferation and metastasis of lung cancer cell via regulation of miR-539/EGFR axis, Artif Cells Nanomed Biotechnol 47: ((2019) ), 2866–2874. |
[16] | T.P. Wall, P.D. Crowley, A. Sherwin, A.G. Foley and D.J. Buggy, Effects of Lidocaine and Src Inhibition on Metastasis in a Murine Model of Breast Cancer Surgery, Cancers (Basel) 11: ((2019) ). |
[17] | H. Liu, J.P. Dilger and J. Lin, Lidocaine suppresses viability and migration of human breast cancer cells: TRPM7 as a target for some breast cancer cell lines, Cancers (Basel) 13: ((2021) ), 234. |
[18] | L. Ye, Y. Zhang, Y.J. Chen and Q. Liu, Anti-tumor effects of lidocaine on human gastric cancer cells in vitro, Bratisl Lek Listy 120: ((2019) ), 212–217. |
[19] | H. Liu, A Prospective for the Potential Effect of Local Anesthetics on Stem-Like Cells in Colon Cancer, Biomedical Journal of Scientific & Technical Research 25: ((2020) ), 18927–18930. |
[20] | W. Siekmann, E. Tina, A.K. Von Sydow and A. Gupta, Effect of lidocaine and ropivacaine on primary (SW480) and metastatic (SW620) colon cancer cell lines, Oncol Lett 18: ((2019) ), 395–401. |
[21] | A.C. Bundscherer, M. Malsy, D.I. Bitzinger, C.H. Wiese, M.A. Gruber and B.M. Graf, Effects of lidocaine on HT-29 and SW480 colon cancer cells in vitro, Anticancer Res 37: ((2017) ), 1941–1945. |
[22] | G. D’Agostino, A. Saporito, V. Cecchinato, Y. Silvestri, A. Borgeat, L. Anselmi and M. Uguccioni, Lidocaine inhibits cytoskeletal remodelling and human breast cancer cell migration, Br J Anaesth 121: ((2018) ), 962–968. |
[23] | S. Cavallaro, CXCR4/CXCL12 in non-small-cell lung cancer metastasis to the brain, Int J Mol Sci 14: ((2013) ), 1713–27. |
[24] | M.C. Smith, K.E. Luker, J.R. Garbow, J.L. Prior, E. Jackson, D. Piwnica-Worms and G.D. Luker, CXCR4 regulates growth of both primary and metastatic breast cancer, Cancer Res 64: ((2004) ), 8604–12. |
[25] | F. Bachelerie, A. Ben-Baruch, A.M. Burkhardt, C. Combadiere, J.M. Farber, G.J. Graham, R. Horuk, A.H. Sparre-Ulrich, M. Locati, A.D. Luster, A. Mantovani, K. Matsushima, P.M. Murphy, R. Nibbs, H. Nomiyama, C.A. Power, A.E. Proudfoot, M.M. Rosenkilde, A. Rot, S. Sozzani, M. Thelen, O. Yoshie and A. Zlotnik, International Union of Basic and Clinical Pharmacology [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors, Pharmacol Rev 66: ((2014) ), 1–79. |
[26] | B. Furusato, A. Mohamed, M. Uhlén and J.S. Rhim, CXCR4 and cancer, Pathol Int 60: ((2010) ), 497–505. |
[27] | Z. Wang, J. Sun, Y. Feng, X. Tian, B. Wang and Y. Zhou, Oncogenic roles and drug target of CXCR4/CXCL12 axis in lung cancer and cancer stem cell, Tumour Biol 37: ((2016) ), 8515–28. |
[28] | X. Li, B. Peng, X. Zhu, P. Wang, Y. Xiong, H. Liu, K. Sun, H. Wang, L. Ou, Z. Wu, X. Liu, H. He, S. Mo, X. Peng, Y. Tian, R. Zhang and L. Yang, Changes in related circular RNAs following ERbeta knockdown and the relationship to rBMSC osteogenesis, Biochem Biophys Res Commun 493: ((2017) ), 100–107. |
[29] | Z. Wu, L. Ou, C. Wang, L. Yang, P. Wang, H. Liu, Y. Xiong, K. Sun, R. Zhang and X. Zhu, Icaritin induces MC3T3-E1 subclone14 cell differentiation through estrogen receptor-mediated ERK1/2 and p38 signaling activation, Biomed Pharmacother 94: ((2017) ), 1–9. |
[30] | X. Liu, H. Liu, Y. Xiong, L. Yang, C. Wang, R. Zhang and X. Zhu, Postmenopausal osteoporosis is associated with the regulation of SP, CGRP, VIP, and NPY, Biomed Pharmacother 104: ((2018) ), 742–750. |
[31] | J.S. Plested, P.A. Coull and M.A. Gidney, ELISA, Methods Mol Med 71: ((2003) ), 243–61. |
[32] | Y. Zhang, X. Zhan, J. Xiong, S. Peng, W. Huang, R. Joshi, Y. Cai, Y. Liu, R. Li, K. Yuan, N. Zhou and W. Min, Temperature-dependent cell death patterns induced by functionalized gold nanoparticle photothermal therapy in melanoma cells, Sci Rep 8: ((2018) ), 8720. |
[33] | J. Phuchareon, F. McCormick, D.W. Eisele and O. Tetsu, EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function, Proc Natl Acad Sci U S A 112: ((2015) ), E3855–63. |
[34] | L. Farilla, A. Bulotta, B. Hirshberg, S. Li Calzi, N. Khoury, H. Noushmehr, C. Bertolotto, U. Di Mario, D.M. Harlan and R. Perfetti, Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets, Endocrinology 144: ((2003) ), 5149–58. |
[35] | H. Liu, Y. Xiong, H. Wang, L. Yang, C. Wang, X. Liu, Z. Wu, X. Li, L. Ou, R. Zhang and X. Zhu, Effects of water extract from epimedium on neuropeptide signaling in an ovariectomized osteoporosis rat model, J Ethnopharmacol 221: ((2018) ), 126–136. |
[36] | L. Li, L. He, Y. Wu and Y. Zhang, Carvacrol affects breast cancer cells through TRPM7 mediated cell cycle regulation, Life Sciences 266: ((2021) ), 118894. |
[37] | P. Mehta, Semi-dry protein transfer and immunodetection of P-selectin using an antibody to its C-terminal tag, Methods Mol Biol 536: ((2009) ), 229–35. |
[38] | H. Liu, Y. Xiong, X. Zhu, H. Gao, S. Yin, J. Wang, G. Chen, C. Wang, L. Xiang, P. Wang, J. Fang, R. Zhang and L. Yang, Icariin improves osteoporosis, inhibits the expression of PPARgamma, C/EBPalpha, FABP4 mRNA, N1ICD and jagged1 proteins, and increases Notch2 mRNA in ovariectomized rats, Exp Ther Med 13: ((2017) ), 1360–1368. |
[39] | J. Song, W. Song and L. Zhang, LncRNA RP1-85F18. 6 affects osteoblast cells by regulating the cell cycle, Open Life Sciences 15: ((2020) ), 951–958. |
[40] | G. Chen, C. Wang, J. Wang, S. Yin, H. Gao, L.U. Xiang, H. Liu, Y. Xiong, P. Wang, X. Zhu, L.I. Yang and R. Zhang, Antiosteoporotic effect of icariin in ovariectomized rats is mediated via the Wnt/beta-catenin pathway, Exp Ther Med 12: ((2016) ), 279–287. |
[41] | F. Cappiello, B. Casciaro and M.L. Mangoni, A novel in vitro wound healing assay to evaluate cell migration, J Vis Exp ((2018) ). |
[42] | M. Ding, H. Zhan, X. Liao, A. Li, Y. Zhong, Q. Gao, Y. Liu, W. Huang and Z. Cai, Enhancer RNA - P2RY2e induced by estrogen promotes malignant behaviors of bladder cancer, Int J Biol Sci 14: ((2018) ), 1268–1276. |
[43] | E.E. Pepperell and S.M. Watt, A novel application for a 3-dimensional timelapse assay that distinguishes chemotactic from chemokinetic responses of hematopoietic CD133(+) stem/progenitor cells, Stem Cell Res 11: ((2013) ), 707–20. |
[44] | R. Zantl and E. Horn, Chemotaxis of slow migrating mammalian cells analysed by video microscopy, Methods Mol Biol 769: ((2011) ), 191–203. |
[45] | H. Liu, J.P. Dilger and J. Lin, The role of transient receptor potential Melastatin 7 (TRPM7) in cell viability: A potential target to suppress breast cancer cell cycle, Cancers (Basel) 12: ((2020) ). |
[46] | M.W. Roe, J.J. Lemasters and B. Herman, Assessment of Fura-2 for measurements of cytosolic free calcium, Cell Calcium 11: ((1990) ), 63–73. |
[47] | B. Chazotte, Labeling cytoskeletal F-actin with rhodamine phalloidin or fluorescein phalloidin for imaging, Cold Spring Harb Protoc 2010: ((2010) ), pdbprot4947.. |
[48] | R. Li, C. Xiao, H. Liu, Y. Huang, J.P. Dilger and J. Lin, Effects of local anesthetics on breast cancer cell viability and migration, BMC Cancer 18: ((2018) ), 666. |
[49] | T. Liang, B. Wang, J. Li and Y. Liu, LINC00922 accelerates the proliferation, migration and invasion of lung cancer via the miRNA-204/CXCR4 axis, Med Sci Monit 25: ((2019) ), 5075–5086. |
[50] | Y. Wu, Q.W. Shen, Y.X. Niu, X.Y. Chen, H.W. Liu and X.Y. Shen, LncNORAD interference inhibits tumor growth and lung cancer cell proliferation, invasion and migration by down-regulating CXCR4 to suppress RhoA/ROCK signaling pathway, Eur Rev Med Pharmacol Sci 24: ((2020) ), 5446–5455. |
[51] | J. Zuo, M. Wen, S. Li, X. Lv, L. Wang, X. Ai and M. Lei, Overexpression of CXCR4 promotes invasion and migration of non-small cell lung cancer via EGFR and MMP-9, Oncol Lett 14: ((2017) ), 7513–7521. |
[52] | J.A. Burger, D.J. Stewart, O. Wald and A. Peled, Potential of CXCR4 antagonists for the treatment of metastatic lung cancer, Expert Rev Anticancer Ther 11: ((2011) ), 621–30. |
[53] | W. Zhou, S. Guo, M. Liu, M.E. Burow and G. Wang, Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy, Curr Med Chem 26: ((2019) ), 3026–3041. |
[54] | J.Y. Guo, C.H. Chiu, M.J. Wang, F.A. Li and J.Y. Chen, Proteoglycan serglycin promotes non-small cell lung cancer cell migration through the interaction of its glycosaminoglycans with CD44, J Biomed Sci 27: ((2020) ), 2. |
[55] | C.Y. Wang, C.S. Huang, Y.P. Yang, C.Y. Liu, Y.Y. Liu, W.W. Wu, K.H. Lu, K.H. Chen, Y.L. Chang, S.D. Lee and H.C. Lin, The subpopulation of CD44-positive cells promoted tumorigenicity and metastatic ability in lung adenocarcinoma, J Chin Med Assoc 82: ((2019) ), 196–201. |
[56] | H.B. Zhang, B. Shen, Z.C. Ma, Y.Y. Xu, Y.L. Lou and M. Chen, MiR-593-5p inhibited proliferation and migration of lung adenocarcinoma by targeting ICAM-1, Eur Rev Med Pharmacol Sci 24: ((2020) ), 4298–4305. |
[57] | Z. He, L. Jia, Y. Lyu, G. Dong, Y. Jiang and J. Zheng, [Increased expression of transendothelial migration-related molecules CCL4, ICAM-1 and VCAM-1 in lung tissue after surgical removal of mouse tumor-bearing lymph node], Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 36: ((2020) ), 699–703. |
[58] | Q.M. Shen, H.Y. Wang and S. Xu, MARCH9 Suppresses Lung Adenocarcinoma Progression by Downregulating ICAM-1, Cell Physiol Biochem 50: ((2018) ), 92–107. |
[59] | F. Le Pimpec Barthes, P. Mordant, C. Pricopi, C. Foucault, A. Dujon and M. Riquet, [The place of surgery in metastatic non-small cell lung cancer], Rev Mal Respir 29: ((2012) ), 376–83. |
[60] | T. Liu, H. Liu, G. Wang, C. Zhang and B. Liu, Survival of M1a non-small cell lung cancer treated surgically: A retrospective single-center study, Thorac Cardiovasc Surg 63: ((2015) ), 577–82. |
[61] | H. Liu, Nav channels in cancers: Nonclassical roles, Global Journal of Cancer Therapy 6: ((2020) ), 5. |
[62] | H.W. Wang, L.Y. Wang, L. Jiang, S.M. Tian, T.D. Zhong and X.M. Fang, Amide-linked local anesthetics induce apoptosis in human non-small cell lung cancer, J Thorac Dis 8: ((2016) ), 2748–2757. |
[63] | M. Yousefi, T. Bahrami, A. Salmaninejad, R. Nosrati, P. Ghaffari and S.H. Ghaffari, Lung cancer-associated brain metastasis: Molecular mechanisms and therapeutic options, Cell Oncol (Dordr) 40: ((2017) ), 419–441. |
[64] | E.A. Kotteas, P. Boulas, I. Gkiozos, S. Tsagkouli, G. Tsoukalas and K.N. Syrigos, The intercellular cell adhesion molecule-1 (icam-1) in lung cancer: implications for disease progression and prognosis, Anticancer Res 34: ((2014) ), 4665–72. |
[65] | T. Piegeler, E.G. Votta-Velis, G. Liu, A.T. Place, D.E. Schwartz, B. Beck-Schimmer, R.D. Minshall and A. Borgeat, Antimetastatic potential of amide-linked local anesthetics: inhibition of lung adenocarcinoma cell migration and inflammatory Src signaling independent of sodium channel blockade, Anesthesiology 117: ((2012) ), 548–59. |
[66] | K. Princen, S. Hatse, K. Vermeire, E. De Clercq and D. Schols, Evaluation of SDF-1/CXCR4-induced Ca2+ signaling by fluorometric imaging plate reader (FLIPR) and flow cytometry, Cytometry A 51: ((2003) ), 35–45. |
[67] | N.W. Ikebuchi and D.M. Waisman, Calcium-dependent regulation of actin filament bundling by lipocortin-85, J Biol Chem 265: ((1990) ), 3392–400. |
[68] | A.R. van Vliet, F. Giordano, S. Gerlo, I. Segura, S. Van Eygen, G. Molenberghs, S. Rocha, A. Houcine, R. Derua, T. Verfaillie, J. Vangindertael, H. De Keersmaecker, E. Waelkens, J. Tavernier, J. Hofkens, W. Annaert, P. Carmeliet, A. Samali, H. Mizuno and P. Agostinis, The ER stress sensor perk coordinates ER-plasma membrane contact site formation through interaction with Filamin-A and F-Actin remodeling, Mol Cell 65: ((2017) ), 885–899e6. |
[69] | T. Oda, M. Iwasa, T. Aihara, Y. Maéda and A. Narita, The nature of the globular- to fibrous-actin transition, Nature 457: ((2009) ), 441–5. |
[70] | D. Yang and J. Kim, Emerging role of transient receptor potential (TRP) channels in cancer progression, BMB Rep 53: ((2020) ), 125–132. |
[71] | H. Liu, A prospective for the role of two-pore channels in breast cancer cells, Global Journal of Cancer Therapy 6: ((2020) ), 001–003. |


