
High Intensity Acute Aerobic Exercise Elicits Alterations in Circulating and Skeletal Muscle Tissue Expression of Neuroprotective Exerkines
Abstract
Background:
Cathepsin B (CTSB) and brain derived neurotrophic factor (BDNF) are increased with aerobic exercise (AE) and skeletal muscle has been identified as a potential source of secretion. However, the intensity of AE and the potential for skeletal muscle contributions to circulating CTSB and BDNF have not been fully studied in humans.
Objective:
Determine the effects of AE intensity on circulating and skeletal muscle CTSB and BDNF expression profiles.
Methods:
Young healthy subjects (n = 16) completed treadmill-based AE consisting of VO2max and calorie-matched acute AE sessions at 40%, 65% and 80% VO2max. Fasting serum was obtained before and 30-minutes after each bout of exercise. Skeletal muscle biopsies (vastus lateralis) were taken before, 30-minutes and 3-hours after the 80% bout. Circulating CTSB and BDNF were assayed in serum. CTSB protein, BDNF protein and mRNA expression were measured in skeletal muscle tissue.
Results:
Serum CTSB increased by 20±7% (p = 0.02) and 30±18% (p = 0.04) after 80% and VO2max AE bouts, respectively. Serum BDNF showed a small non-significant increase (6±3%; p = 0.09) after VO2max. In skeletal muscle tissue, proCTSB increased 3 h-post AE (87±26%; p < 0.01) with no change in CTSB gene expression. Mature BDNF protein decreased (31±35%; p = 0.03) while mRNA expression increased (131±41%; p < 0.01) 3 h-post AE. Skeletal muscle fiber typing revealed that type IIa and IIx fibers display greater BDNF expression compared to type I (p = 0.02 and p < 0.01, respectively).
Conclusions:
High intensity AE elicits greater increases in circulating CTSB compared with lower intensities. Skeletal muscle protein and gene expression corroborate the potential role of skeletal muscle in generating and releasing neuroprotective exerkines into the circulation.
NEW AND NOTEWORTHY: 1) CTSB is enriched in the circulation in an aerobic exercise intensity dependent manner. 2) Skeletal muscle tissue expresses both message and protein of CTSB and BDNF. 3) BDNF is highly expressed in glycolytic skeletal muscle fibers.
INTRODUCTION
Neurodegenerative diseases such as Alzheimer’s disease (AD) are the sixth leading cause of death in the United States among adults aged 65 or older [1]. Currently, AD is associated with significant financial burden, costing nearly $300 billion in 2020 within the United States [2]. Key pathological hallmarks of AD include increased amyloid beta plaque, neurofibrillary tangles, which lead to impairments in memory function and decreased quality of life [3– 6]. The risk of developing AD drastically increases with age, effecting nearly 35% of individuals above 85 years of age [7, 8]. Furthermore, both the population and proportion of adults above the age of 65 are projected to increase from 50 million to 80 million by 2050 [9]. Therefore, developing strategies to prevent onset and progression of cognitive impairments such as AD are of the utmost importance.
Aerobic exercise (AE) has been shown to have robust protective effects on cognitive function. Individuals who have been physically active throughout the lifespan have nearly 40% lower risk of cognitive decline compared to sedentary counterparts [10]. Cardiovascular fitness is associated with enhanced cognitive performance, reduced brain atrophy, and decreased risk of dementia [11– 16]. Additionally, prolonged AE training interventions have been shown to increase hippocampal volume and functional connectivity in cortical-hippocampal brain networks and improve memory performance in individuals with cognitive impairments [17– 19]. Although AE is a promising non-pharmaceutical modality to prevent the onset of cognitive decline, the mechanisms by which AE improves cognitive function remain to be fully elucidated. One hypothesis by which AE mediates cognitive benefits is via the release of bioactive factors, or exerkines, into the circulation. Exerkines are defined as metabolites, peptides or RNA released from peripheral tissue into the circulation and exert endocrine or paracrine effects on other cells, tissues, or organs in response to AE [20– 22]. Recently, skeletal muscle has drawn significant attention given its prominent metabolic role during AE. Importantly, a subset of exerkines derived from skeletal muscle possess robust neuroprotective effects [20, 23].
Cathepsin B (CTSB) is a lysosomal cysteine protease that has been shown to enhance memory function [24]. CTSB becomes enzymatically active (mature CTSB) after the proteolytic processing of its propeptide domain from its precursor form (proCTSB) [25]. CTSB can cross the blood brain barrier and increase expression of markers indicative of neurogenesis in tissue culture models [24, 26], although the current literature is contradictory on the precise biological functions of CTSB [27]. Some studies identify CTSB to have anti-amyloidogenic properties, while others show inhibition of CTSB decreases amyloid load and improves memory function [28, 29]. Importantly, CTSB is released from skeletal muscle cells in response to AMP kinase agonist [26]. In fact, AE-trained CTSB knockout mice failed to demonstrate the same beneficial cognitive adaptations from AE as wild type controls [24]. Further, lifelong physically active individuals have lower levels of circulating CTSB compared to sedentary counterparts [30]. Importantly, previous work demonstrates the ability of AE training interventions to increase circulating CTSB in humans [24, 31]. The effects of acute AE on circulating CTSB, however, remain inconclusive [32, 33]. Although CTSB plays a controversial role across the healthspan, AE induced CTSB release appears imperative in facilitating the cognitive adaptations associated with AE.
Like CTSB, brain derived neurotrophic factor (BDNF) is an exerkine and a critical mediator in neuronal maturation, survival and capable of crossing the blood brain barrier [34]. BDNF is derived from multiple tissues (i.e., platelets, neurons) and promotes synaptic plasticity, neuronal maturation and is potentially protective of hippocampal function [35– 37]. BDNF exists mainly in two isoforms, pro and mature BDNF, that elicit divergent signaling cascades [38]. BDNF is synthesized as ProBDNF, which ligates p75 neurotophin receptor and promotes cell apoptosis [39– 41]. ProBDNF is further processed to mature BDNF, which activates tyrosine receptor kinase B and induces hippocampal neurogenesis [42, 43]. Within the skeletal muscle BDNF appears to play a rather diverse role. Some reports suggest roles in regulating glycolytic fiber type identity [44, 45], fatty acid oxidation [46], satellite cell differentiation [47] and neuromuscular junction maintenance [48]. Regarding AE, it has been well established that circulating BDNF enrichment is intensity dependent [49– 51]. The potential contribution of skeletal muscle to circulating BDNF in response to AE remains to be elucidated.
Despite the promising role of these exerkines in facilitating cognitive benefits associated with AE, few studies to date have comprehensively investigated the effects of differential acute AE intensity on circulating CTSB and BDNF profiles [49– 51]. Additionally, the skeletal muscle regulation of BDNF and CTSB has not yet been fully investigated in humans. Therefore, the purpose of the current study was to determine the effects of AE intensity on circulating and skeletal muscle CTSB and BDNF expression profiles. We hypothesized that higher intensity (80% of VO2max and VO2max) acute AE would display the greatest magnitude of increase in circulating CTSB and BDNF profiles compared to lower intensity and that the protein expression of these neuromodulating exerkines within the skeletal muscle tissue would be inversely related to circulating changes.
METHODS
Study design
The current investigation is a retrospective analysis of a larger study that consisted of four visits [52]. All subjects were recruited from the greater Chicago Metropolitan area. Baseline measure such as height, weight, body composition via dual x-ray absorptiometry and maximal aerobic capacity (VO2max) were collected on visit 1. After visit 1, subjects completed sequential calorie matched AE sessions at 40%, 65% and 80% of their previously determined VO2max. Each AE bout was separated by 5– 7 days. Three days before each visit, participants were asked to complete diet and physical activity logs. Participants were also asked to refrain from vigorous exercise and alcohol consumptions 48 h prior to each visit, as well as caffeine consumption 24 h prior to each visit. All participants provided written, informed consent before participation. All procedures were approved by the Institutional Review Board at the University of Illinois, Chicago in accordance with the Declaration of Helsinki (IRB Approval #: 2015-0127).
Participants
Sixteen lean (10 male, 6 female, BMI < 25 kg/m2), healthy adults (age: 26±1 years) volunteered to participate in the study. Participants were excluded based on the following criteria: (1) current or past smoker within the last year or (2) the presence of cardiovascular, metabolic, or neurological disease. Fasting plasma glucose was measured via glucose oxidase reaction using an YSI STAT 2300 (YSI Life Sciences, Yellow Springs, OH). Fasting plasma insulin was determined via ELISA (9095, Crystal Chem, Elk Grove Village, IL).
Acute AE sessions
For the first AE session, VO2max was determined using treadmill ramp protocol during which the participants ran at a self-selected speed while the treadmill speed increased 2% every 2 minutes until maximum oxygen uptake was reached. All AE visits occurred between 0700–0900 hours to control for circadian alterations and lessen the participants’ burden of being fasted for the study. The order of the visits are as follows: VO2max on visit 1, 40% VO2max on visit 2, 65% VO2max on visit 3 and 80% VO2max on visit 4. All participants were instructed to maintain normal activities for the days between AE sessions. VO2max was achieved in all participants, which met 3 out of 4 following criteria: a plateau in VO2 despite an increase in workload; RPE > 17; RER > 1.1; HR > 85% of age-predicting maximal heart rate or volitional fatigue was reached. Expired air was collected and analyzed during the duration of the test via the PARVO Medics metabolic cart (Salt Lake City, UT). Heart rate was monitored with Polar heart rate fitness monitors. For the subsequent three submaximal AE exercise sessions, ACSM metabolic equations were used [53] to derive treadmill intensity for the calorie matched exercise bouts at 40%, 65% and 80% of VO2max. Participants’ expired air was collected throughout submaximal acute exercise sessions as described above. Adjustments to speed and incline were made as necessary to achieve desired VO2.
Blood sample collection
Participants were seated upright and rested quietly for 30 minutes prior to all blood draws to account for changes in postural-mediated changes in plasma volume. Fasting blood samples were collected before and 30-minutes after the completion of each AE bout. Samples for serum analyses were collected into a 10 mL SST vacutainer, clotted for 30-minutes at room temperature, then centrifuged at 3500 rpm for 10 minutes at 4°C. Serum was aliquoted and stored at – 80°C for future analyses.
Skeletal muscle biopsy
Skeletal muscle biopsies (∼200 mg) of the vastus lateralis were obtained under local anesthesia (2% lidocaine HCl) using a 5 mm Bergstrom needle with suction [52, 54– 56]. Biopsies were taken before, 30-minutes post and 3-hours after the completion of AE at 80% VO2max, as previously described [52]. Muscle tissue was cleared of all visible connective tissue and fat, blotted with gauze to remove blood, and immediately flash frozen in liquid nitrogen or preserved in RNA later and stored at – 80°C until future analysis.
Tissue homogenization and protein concentration determination
Skeletal muscle homogenization protocols were followed as previously described [52, 55]. Briefly, 20– 30 mg of muscle tissue was weighed and homogenized in ceramic beads (Lysing Matrix D; Fastprep®-24, Santa Ana, CA) in 1:20 ratio of ice cold 1x Cell Lysis Buffer (#9803, Cell Signaling Technology, Danvers, MA) supplemented with 1x Protease/Phosphatase inhibitor cocktail (#5872, Cell Signaling Technology, Danvers, MA). Protein concentration for each sample was determined by bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL).
BDNF and CTSB Enzyme Linked Immunosorbent Assay (ELISA)
BDNF and CTSB quantification were determined from serum using mature BDNF ELISA (BEK-2211, Biosensis, Australia) and total CTSB ELISA (ab119584, Abcam, Cambridge, UK) following manufacturer’s instructions. Frozen serum samples were thawed on ice and samples were diluted in assay buffer 1:20 and 1:200 for CTSB and BDNF ELISAs, respectively. Individual participant serum samples were loaded in duplicate for each timepoint and incubated following manufacturers guidelines. Each assay included recombinant protein as a positive control. Plates were read using SpectraMax ID5 Microplate Reader (Molecular Devices, Sunnyvale, CA). Average inter-assay variability was 13.4% and 3.2% for CTSB and BDNF, respectively.
Western blotting
Aliquots of muscle homogenates containing 15μg of total protein diluted in equal volumes of 2x Laemmli Buffer (#1610737, Bio-Rad, Hercules, CA) with 5% β-mercaptoethanol (βME) prior to heating at 90°C for 10 minutes. Denatured samples were brought to room temperature and loaded onto a 10% or 4– 20% polyacrylamide gel. After gel run, samples were transferred onto nitrocellulose membranes via Trans-blot Turbo transfer system (Bio-Rad). Membranes were blocked with Odyssey Blocking Buffer (OBB) (92750000, Li-Cor, Lincoln, NE) for 1 h at room temperature. Membranes were then incubated with primary antibodies CTSB (1:300, ab58802, Abcam, Cambridge, UK) or BDNF (1:1000, ab108319, Abcam, Cambridge, UK) overnight at 4°C in OBB with 0.1% Tween-20. BDNF and CTSB antibodies allowed for detection of both pro and mature isoforms. BDNF and CTSB banding patterns were confirmed using human recombinant CTSB (ab283434, Abcam, Cambridge, UK) or BDNF (ab9794 Abcam, Cambridge, UK) protein as shown in Figs. 3A and 4A, respectively. For CTSB, bands were identical with a more prominent signal in recombinant proCTSB as this recombinant protein contained proCTSB exclusively. In regard to BDNF, a slight mobility shift was seen when comparing skeletal muscle with recombinant protein. A band at the molecular weight observed in our skeletal muscle samples is consistent with previously published literature [45]. This observed mobility shift may be a result of unique post-translational modifications to mature BDNF within skeletal muscle tissue. Additionally, UniProt identifies 3 isoforms of human proBDNF between 28.9 and 31.1 kDa, which we quantified on our blots. Following primary antibody incubation, membranes were washed with TBST and incubated with anti-rabbit fluorophore-conjugated secondary antibody (1:20,000, 926-32213, Li-Cor, Lincoln, NE) in OBB with 0.1% Tween-20 for 1 h at room temperature. Membranes were then washed with TBST followed by TBS, then scanned on Odyssey CLx Imaging System (Li-Cor, Lincoln, NE) and quantified on Image Studio software (V4.0.21, Li-Cor, Lincoln, NE). Mature, pro, total and pro:mature forms of BDNF and CTSB were quantified. Total CTSB and BDNF was calculated by addition of pro and mature signal. The ratio of pro to mature ratio of CTSB and BDNF is represented as pro:mature. All targets were normalized to Revert Total Protein Stain (926-11016, Li-Cor, Lincoln, NE) and imaged via Odyssey Imaging System and quantified with Image Studio Software.
RNA extraction and reverse transcription
At the time of collection, a portion of the muscle biopsy sample (∼30 mg) was fixed in RNAlater (AM7021, ThermoFisher, Waltham, MA), immediately frozen in liquid nitrogen and stored at – 80°C until RNA extraction. RNA extraction was performed using RNeasy isolation kit (Qaigen) as per kit protocol with modifications. Briefly, RNAlater fixed samples were homogenized in Qaizol via bead homogenization as described above, added to QiaShredder spin column (Qaigen), and centrifuged at 8,000×G for 1 minute. Samples were transferred to a new tube and 100μL of gDNA eliminator (Qaigen) was added. Samples were then vortexed and 180μL chloroform was added. The homogenate then sat at room temperature for 3 minutes and centrifuged at 12,000×G for 15-minutes at 4°C. Supernatants were transferred to a sterile microcentrifuge tube and 450μL ethanol was then added to each sample which then was transferred onto the RNeasy spin column. Ethanol extracts were collected via centrifugation at 9,000×G for 30 seconds at room temp, and the protocol provided by Qiagen were followed thereafter. 2μL of each extraction was analyzed for RNA concentration via Nanodrop (ThermoFisher). Average RNA concentration was 128±9.9 ng/μL. Reverse transcription was performed with iScript Advanced reverse transcriptase kit (Bio-Rad) via manufacturer protocols to generate 150 ng of cDNA which was then diluted to 1:4 with nuclease free water.
Droplet digital PCR (ddPCR)
ddPCR (Bio-Rad) was used to quantify transcripts of CTSB and BDNF. Primers were designed for each target using Roche’s universal probe library (UPL) assay design center and SnapGene software. ddPCR assays were performed by combining 2μl cDNA with ddPCR mix for probes (Bio-Rad), along with appropriate probes, primers and nuclease free water resulting in a 20μl reaction. A duplicate of no-template negative controls in which cDNA was substituted with nuclease free water was performed alongside each of the assays. The 20μl reaction was combined with 70μl of droplet oil for probes (Bio-Rad) and droplets were generated using a droplet generator (Bio-Rad). The resultant droplet suspension (40μl) was then loaded onto a 96 well plate which was then sealed and placed on a thermocycler where 40 cycles of PCR were performed. The droplets were analyzed by QX200 ddPCR system (Bio-Rad) by counting droplets positive for FAM fluorescent probes. All transcript data are presented as copy number per ng cDNA input. The following primer and probe sequences were used: BDNF forward: AAAAGGCATTGGAACTCCCA; BDNF reverse: TGCTATCCATGGTAAGGGCC; BDNF probe: 56-FAM-AACTACCCAGTCGTAC-3_BHQ_1; CTSB forward: CTAGGATCCGGCTTCCAAC; CTSB reverse: CGACAGGGGATGGAAAGAG; CTSB probe: 56-FAM-CTGCTGCCTGCTGGTGTTGG -3_BHQ_1.
Immunofluorescence
At the time of collection, ∼20 mg of each muscle sample was embedded in OCT (Thermofisher) media and frozen in liquid nitrogen cooled isopentane (2-methyl butane) for histological analysis. Multiple 6μm thick skeletal muscle sections were serially generated for all samples. All sections were fixed with 1:1 acetone/methanol for 10 minutes at room temperature, washed with PBST and blocked for 1 hour with 5% donkey serum in PBS. Sections were incubated with BDNF (1:200, Ab108319, Abcam, Cambridge, UK) or CTSB (1:100, ab58802, Abcam, Cambridge, UK; 1:100, 31718, Cell Signaling, Danvers, MA and 1:100, A0967, Abclonal, Woburn, MA) overnight at 4°C. Following primary antibody incubation, sections were washed and incubated with secondary antibody (1:500, A21245, Thermofisher). Sections were then washed and stained with DAPI (1:1000, 62248, Thermofisher) for 3 minutes at room temperature and washed. All sections were mounted, and cover slipped with ProLong Antifade (P36980, Thermofisher). For myosin heavy chain fiber type analysis, separate serial sections were fixed, blocked and incubated with myosin heavy chain (MHC) IIa antibody (1:80, A4.74, Developmental Studies Hybridoma Bank, Iowa City, IA) for 30 minutes at 37°C, washed, and incubated with secondary antibody (1:500, Abcam, ab150106, Cambridge, UK). Following MHC IIa antibody incubation, sections were blocked with 5% donkey serum in PBS and incubated with MHC I antibody (1:100, BA-D5-s, Developmental Studies Hybridoma Bank, Iowa City, IA) for 30 minutes at 37°C. Sections were then incubated with secondary antibody (1:500, ab150107, Abcam, Cambridge, UK), washed and stained with DAPI (1:1000, 62248, Thermofisher, Waltham, MA) washed again, then imaged.
All slides were imaged using BZ-X710 microscope (Keyence, Itasca, IL). Fluorescent signal was recorded using Cy5 (Excitation: 620/60 nm, Emission 700/75 mn) and Texas Red (Excitation: 560/40 nm, Emission: 630/75) filters for BDNF and MHC I, and for MHC IIA, respectively. Skeletal muscle cross section digital images were collected at 20x magnification. Signal intensity for the CTSB and BDNF was quantified in all discernable muscle fibers within the 20x field of view. An average of 43±7 fibers were quantified for per sample. BDNF and CTSB content within the skeletal muscle was expressed as relative intensity (total fiber intensity normalized to total fiber area).
Statistical analysis
Paired t-tests were used to examine differences in circulating CTSB and BDNF at each exercise intensity. Sex-specific differences in skeletal muscle fiber type distribution and changes (Δ) circulating BDNF and CTSB, were examined by independent samples t-test. When appropriate, either one-way repeated measures ANOVA or mixed effect model regression analyses were performed (when data set was missing time points due to limited sample availability) for skeletal muscle variables. Linear mixed-model regressions were performed using mixed effect terms for time and random effects terms controlling for participant variability and sexual dimorphism. Mixed effects model analyses were also performed on sex-stratified skeletal muscle fiber type distribution. Bonferroni correction was used for all post-hoc comparisons. Pearson’s correlation coefficient (r) was used to explore relationships between variables. Statistical analyses were performed in either Prism 4.0 software (GraphPad Software, Inc., La Jolla, CA) or RStudio package lme4 (Boston, MA) and effect sizes were calculated via G*Power 3.1 (Düsseldorf, Germany) and RStudio. Threshold for significance was set at p < 0.05 for all statistical tests. All data are presented as mean±standard error of the mean (SEM). T-test effect sizes are indicated by Cohen’s d, whereas ANOVA effect sizes are indicated by Cohen’s f2, and linear mixed effect model effect sizes are reported as R2 of fixed factors.
RESULTS
Clinical characteristics
Subject demographic and clinical characteristics are reported in Table 1. By design, all participants were young (age < 30 years) and had a lean body composition (BMI < 25 kg/m2) with normal insulin sensitivity. Additionally, all subjects displayed moderate levels of physical fitness and were characterized as recreationally active.
Table 1
Subject Characteristics
| Variable, Units | n = 16 |
| Age, years | 26±1 |
| Sex, M/F | 10/6 |
| Weight, kg | 63.3±2.4 |
| BMI, kg/m2 | 22.4±0.7 |
| Body Fat, % | 23.1±1.5 |
| Glucose, mg/dL | 91.5±2.1 |
| Insulin, mU/L | 4.9±0.5 |
| HOMA-IR | 1.1±0.1 |
| VO2MAX, mU/kg/min | 47.7±1.9 |
Data are represented as mean±SEM. HOMA-IR = fasting glucose(mg/dL) *fasting insulin (mU/L) / 405.
High intensity bouts of AE elicit increases in circulating CTSB
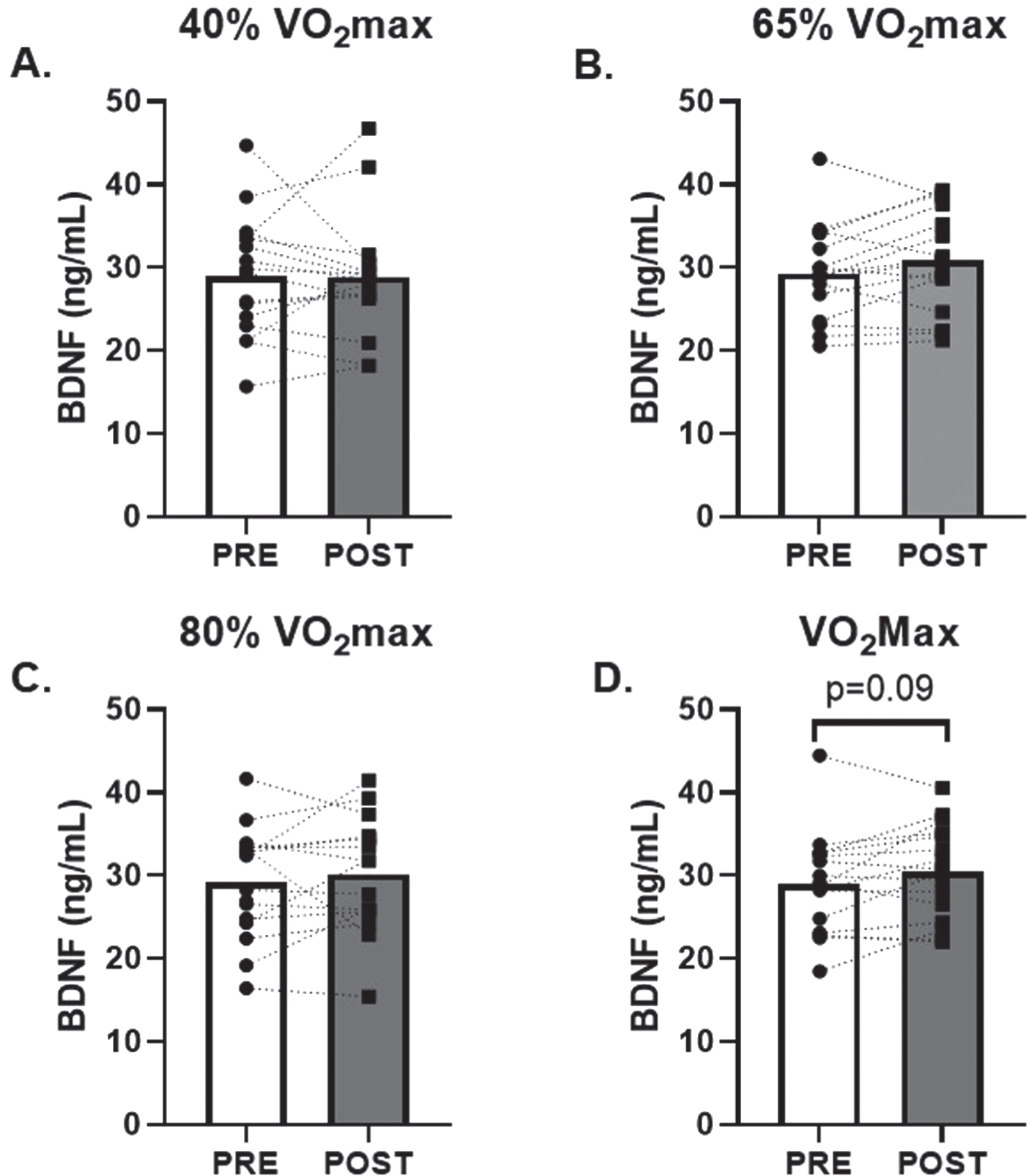
No change in serum CTSB was observed in response to low and moderate (40% and 65% of VO2max; d = 0.02 and 0.30, respectively) intensity AE (Fig. 1A and B). However, there was a 20±7% (p = 0.02; d = 0.65) and 30±18% (p = 0.04; d = 0.55) increase in circulating CTSB in response to 80% and VO2max AE, respectively (Fig. 1C and D). There were no changes in circulating BDNF observed after 40%, 65% or 80% (d = 0.01, 0.45, 0.22, respectively) VO2max AE bouts. A modest non-significant 6±3% increase in circulating BDNF was seen after VO2max (p = 0.09; d = 0.45, Fig. 2D).
Fig. 1
Effects of Acute Aerobic Exercise Intensity on Circulating CTSB. Circulating CTSB before and 30-minutes post AE A) 40% of VO2max, B) 65% of VO2max, C) 80% of VO2max and D) at VO2max. n = 15 (9M/6F) 80% VO2max, n = 16 (10M/6F) all other timepoints. All data are represented as mean±SEM. *p < 0.05 vs PRE.
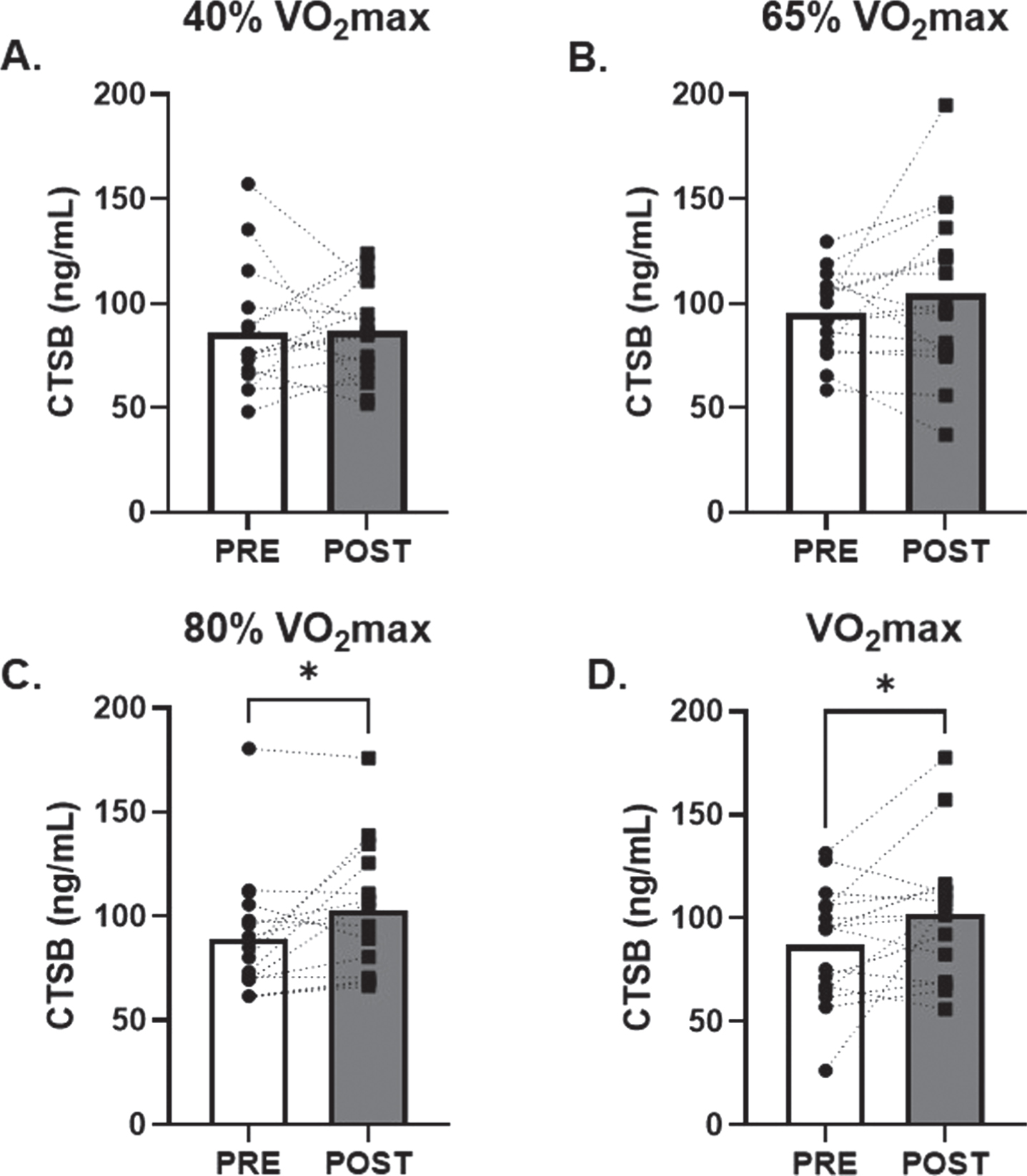
Fig. 2
Effects of Acute Aerobic Exercise Intensity on Circulating BDNF. Circulating BDNF before and 30-minutes post AE A) 40% of VO2max, B) 65% of VO2max, C) 80% of VO2max and D) at VO2max. n = 15 (9M/6F) 80% VO2max, n = 16 (10M/6F) all other timepoints. All data are represented as mean±SEM.
Skeletal muscle CTSB response to acute AE
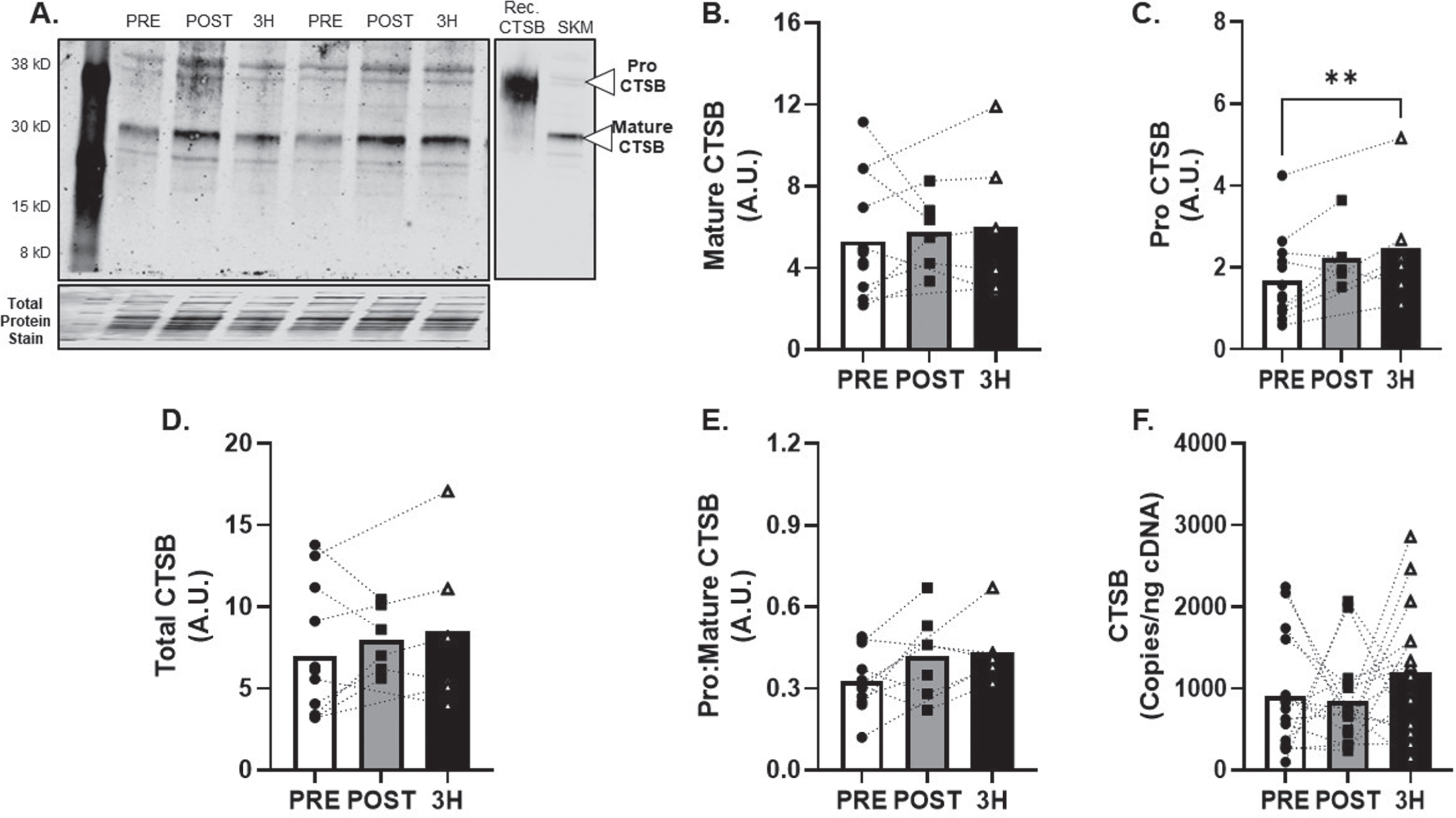
CTSB protein expression (Fig. 3A) was detected in all skeletal muscle tissue samples. Mature CTSB protein expression (Fig. 3B) was unchanged (R2= 0.09) but proCTSB expression increased by 87±26% at 3 h after AE at 80% VO2max (p < 0.01; R2= 0.03, Fig. 3C) compared to baseline. Additional metrics of skeletal muscle CTSB protein expression (total and pro:mature) were unchanged (R2= 0.06 and 0.20; Fig. 3D-E). CTSB mRNA expression was unchanged (f = 0.28) after AE (Fig. 3F).
Fig. 3
CTSB Skeletal Muscle Protein and Transcript Response to 80% Acute Aerobic Exercise. A) Representative western blot probing for CTSB before, 30-minutes post and 3-hours post 80% acute AE. All samples were normalized to total protein stain. Protein and transcript content of B) Mature CTSB protein, C) ProCTSB protein, D) Total CTSB protein and E) Pro:Mature CTSB protein ratio and F) CTSB transcript n = 15 (8M/7F). Mature CTSB is expected at 30 kD, ProCTSB is expected at 41 KD. All data are represented as mean±SEM. n = 13 PRE (7M/6F), n = 6 POST (5M/1F), and n = 6 3 h (4M/2F). *p < 0.05 vs PRE, **p < 0.01 vs PRE, #p < 0.05 main effect of time.
Skeletal muscle BDNF response to acute AE
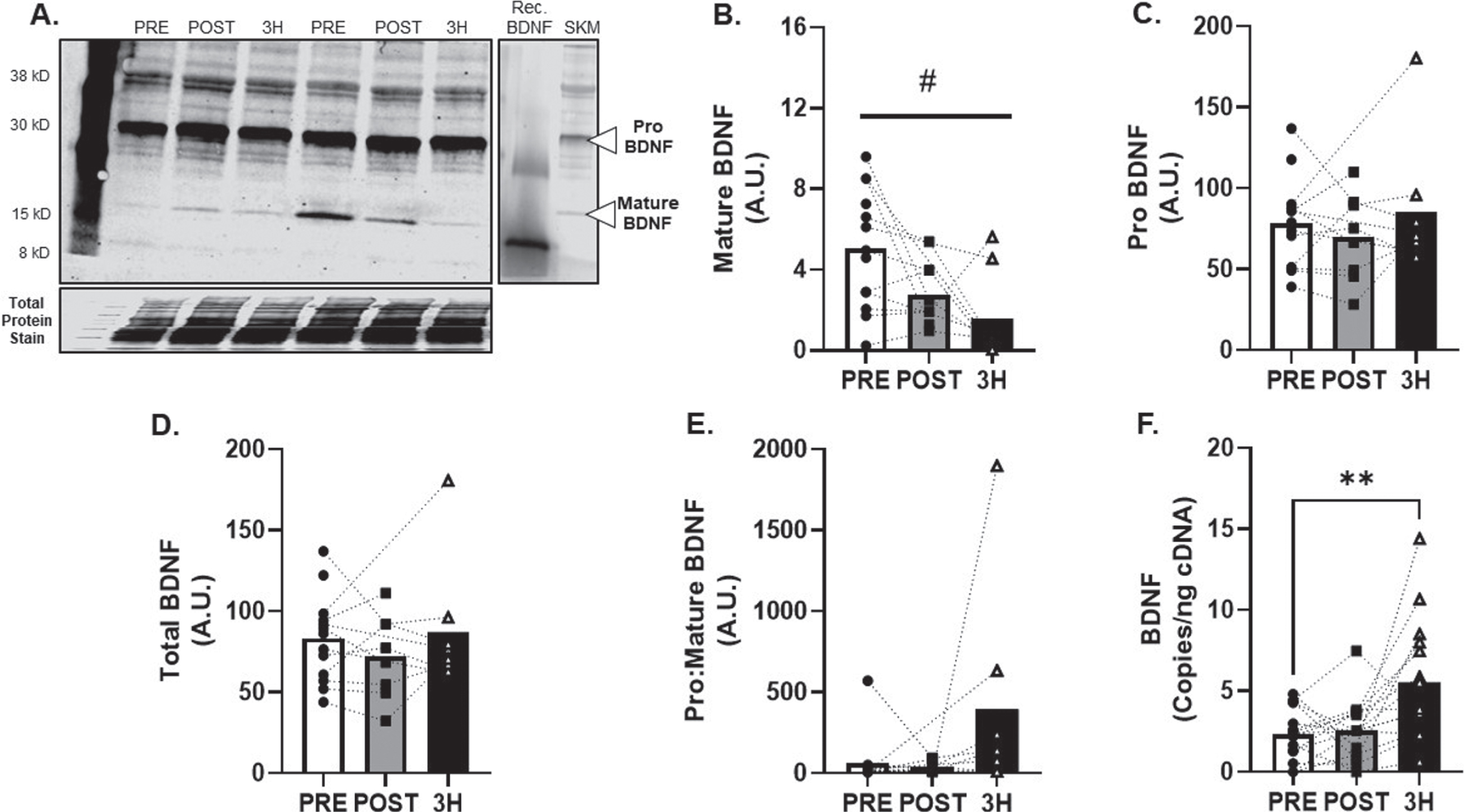
BDNF protein expression (Fig. 4A) was detected in all skeletal muscle tissue samples and mature BDNF was attenuated in the hours following AE at 80% VO2max (main effect of time; p = 0.03; R2= 0.38, Fig. 4B). Additional metrics of skeletal muscle BDNF protein expression (pro, total and pro:mature) were unchanged (R2= 0.21, 027. and 0.38; Fig. 3C-E). Conversely, BDNF mRNA increased by 131±41% at 3-hours after exercise (p < 0.01; f2= 0.82, Fig. 4F).
Fig. 4
BDNF Skeletal Muscle Protein and Transcript Response to 80% Acute Aerobic Exercise. A) Representative western blot probing for BDNF before, 30-minutes post and 3-hours post 80% acute AE. All samples were normalized to total protein stain. Protein and transcript content of B) Mature BDNF protein, C) ProBDNF protein, D) Total BDNF protein and E) Pro:Mature BDNF protein ratio and F) BDNF transcript n = 15 (8M/7F). Mature BDNF is expected at 15 kD, Pro BDNF is expected at 30 kD. All data are represented as mean±SEM. n = 13 PRE (7M/6F), n = 8 POST (5M/3F), and n = 8 3 h (4M/4F). *p < 0.05 vs PRE, **p < 0.01 vs PRE, #p < 0.05 main effect of time.
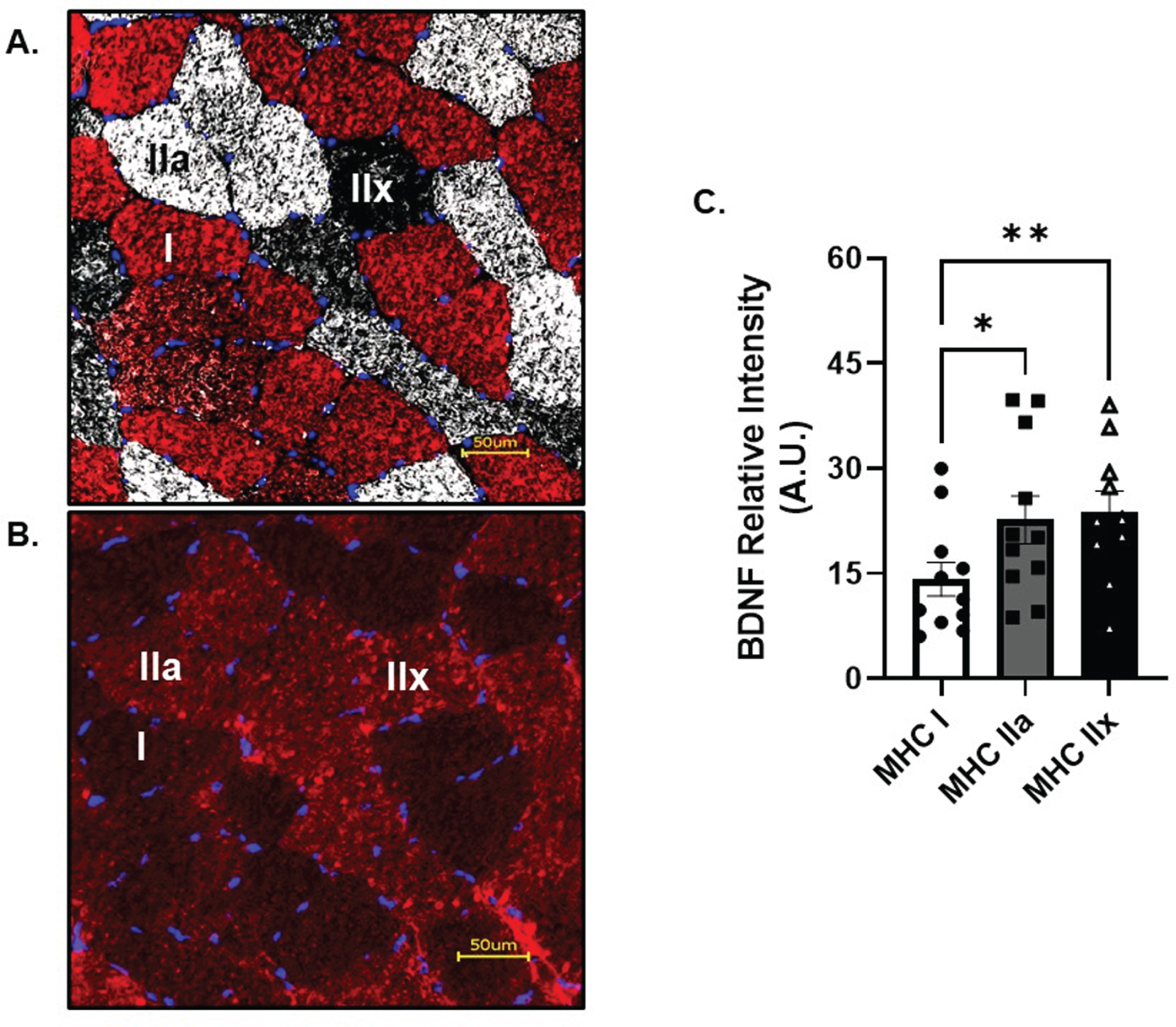
Skeletal muscle fiber type profile of BDNF
Skeletal muscle fiber type characteristics are reported in Table 2. A typical mixed fiber distribution was found between MHC I and MHC IIa, reflective of a young, healthy and recreationally active cohort. BDNF protein localization was most prominent in glycolytic, fast twitch MHC IIa (p = 0.02, R2= 0.19) and IIx fibers (p < 0.01, R2= 0.18) when compared to MHC I (Fig. 5A&B) at baseline. However, no change in BDNF protein localization was observed in the hours following AE (data not shown). CTSB was not able to be detected in skeletal muscle cross sections above background fluorescence (data not shown).
Table 2
Skeletal muscle distribution PRE acute exercise
| Fiber | Number of | Percent | Fiber | Total |
| Isoform | Fibers | (%) | Area | Area |
| per Section | (μm2) | (μm2) | ||
| MHC I | 18±4 | 41±4 | 4890±348 | 97923±18934 |
| MHC lIa | 19±3 | 46±4 | 6317±736 | 105966±14680 |
| MHC lIx | 6±2 | 13±4 | 1110±266 | 19442±5691 |
Data are represented as mean±SEM.
Fig. 5
Skeletal Muscle Fiber Type Analysis of BDNF. A) Representative image of skeletal muscle fiber type and B) BDNF. C) BDNF relative intensity per fiber type. Red fibers = MHC I, white fibers = MHC IIa and black fibers = MHC IIx All data are represented as mean±SEM. n = 11 (7M/4F) for MHC I and IIa, n = 10 (6M/4F) for MHC IIx. *p < 0.05, **p < 0.01.
Analyses of sexual dimorphism
When data were stratified by sex, a dimorphic response was identified in the Δ BDNF at 80% VO2max where females displayed a – 7±5% reduction compared to a 12±7% increase (p = 0.027, d = 1.2) in males. This response was confirmed using regression analyses when investigating the interaction of sex and time (p = 0.012). No additional sex-specific differences were identified at the remaining exercise intensities for CTSB or BDNF. Myosin heavy chain isoforms were not found to be different by sex. No changes were observed with fiber-type specific BDNF expression when stratified by sex.
CTSB and BDNF correlational analysis
CTSB and BDNF profiles in circulation were not found to be correlated with one another, or with metabolic and anthropometric measures at any exercise intensity. Table 3 displays the correlations performed between circulating factors (Baseline and Δ CTSB and BDNF) and skeletal muscle expression metrics (mature, pro, pro:mature, total and transcript) for each target (Baseline and Δ CTSB and BDNF). No correlations were identified between CTSB and BDNF expression metrics in tissue.
Table 3
Correlation analyses
| Baseline Serum | Baseline Serum | ||||||
| CTSB | BDNF | ||||||
| Baseline CTSB Muscle | r | p-value | n | Baseline BDNF Muscle Expression | r | p-value | n |
| Mature | 0.253 | 0.427 | 12 | mature | – 0.19 | 0.545 | 12 |
| pro | 0.015 | 0.963 | 12 | pro | 0.255 | 0.424 | 12 |
| pro:mature | – 0.56 | 0.058 | 12 | pro:mature | 0.425 | 0.168 | 12 |
| total | 0.199 | 0.536 | 12 | total | 0.241 | 0.451 | 12 |
| transcript | 0.495 | 0.072 | 14 | transcript | 0.203 | 0.487 | 14 |
| Δ Serum CTSB | Δ Serum BDNF | ||||||
| Δ CTSB Muscle | r | p-value | n | Δ BDNF Muscle | r | p-value | n |
| mature | – 0.28 | 0.596 | 6 | mature | – 0.46 | 0.301 | 7 |
| pro | – 0.25 | 0.630 | 6 | pro | 0.452 | 0.309 | 7 |
| pro:mature | –0.03 | 0.951 | 6 | pro:mature | 0.08 | 0.864 | 7 |
| Total | –0.33 | 0.530 | 6 | Total | 0.437 | 0.327 | 7 |
| transcript | 0.168 | 0.567 | 14 | transcript | 0.34 | 0.234 | 14 |
r = Pearson’s correlation coefficient, n = sample size.
DISCUSSION
It is well known that AE facilitates positive cognitive adaptations. Yet, the mechanisms by which these adaptations occur are not completely understood. The current findings show that CTSB is enriched in the circulation in an AE intensity dependent manner. Further, the data indicates that skeletal muscle tissue expresses both message and protein of CTSB and BDNF, and BDNF is highly expressed in glycolytic skeletal muscle fibers.
The current findings are one of the first to report the effect of high intensity (80% VO2max and VO2max) acute AE on circulating CTSB. Comparatively, individuals undergoing acute high intensity circuit training demonstrated increases in circulating CTSB immediately post exercise [57]. However, authors employed both aerobic and resistance exercise performed during the same training session, which has differential effects on skeletal muscle signaling, and potentially, CTSB release [58, 59]. To the contrary, several additional reports did not observe changes in circulating CTSB sampled from a sedentary population undergoing both acute and chronic high intensity interval exercise modalities [32, 33]. Importantly, AE training programs of moderate-to-vigorous intensities for at least 16-weeks demonstrate increases in circulating CTSB in both healthy and cognitively impaired populations [24, 31]. A unique study by Gökçe et al. [60] examined CTSB responses in a mixed sport, highly trained endurance athlete cohort (average VO2max of ∼55 ml/kg/min), and a control cohort (average VO2max of ∼45 ml/kg/min) where circulating CTSB was increased in response to moderate intensity exercise by 27– 55% in the athlete groups but only 17% in the control cohort. In parallel to the data presented herein, the non-athlete, recreationally active cohort did not achieve an elevated CTSB response to moderate intensity aerobic exercise. Given the chronic aerobic trained state of the mixed sport athlete group, an extended training timeline or higher cardiorespiratory fitness may be necessary to achieve increases in circulating CTSB at lower exercise intensities. However, lifelong exercisers have lower levels of circulating CTSB compared to their sedentary, age-matched peers [30], bringing into question the precise role of CTSB throughout the lifespan.
We next sought to determine the effects of an acute, high-intensity AE stimulus on skeletal muscle CTSB expression profiles. Although we found no changes in mature CTSB protein, we did find an increase in proCTSB protein 3-hours following AE at 80% VO2max. As cleavage of CTSB propeptide is necessary for enzymatic activation of CTSB [25], increases in mature CTSB protein may happen > 3-hours after AE. Despite the increase in proCTSB post-AE, transcript levels were unchanged. The lack of change in CTSB transcript may be explained by transcription factors that regulate CTSB gene expression. Activation of both AMPK and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) led to significant increases in CTSB transcript in skeletal muscle cells [24, 61]. Therefore, acute AE may facilitate skeletal muscle CTSB expression via AMPK-PGC1α signaling. However, in a previous report we failed to find changes in AMPK activation in our cohort, thus potentially explaining the lack of increased CTSB transcript in the current study. Alternatively, PGC1α convergence with transcription factors that influence lysosomal gene expression, such as transcription factor EB [62], may regulate CTSB transcript in response to AE. Together, these findings suggest that high acute AE promotes CTSB production within the skeletal muscle.
In addition to CTSB, we also investigated the effects of acute AE on circulating BDNF. Previous research has highlighted BDNF to be released in an AE intensity dependent manner [49– 51], critical for mediating neuronal maturation and improving cognitive function [35– 37]. In contrast, we found no significant increases in circulating BDNF after acute AE at any intensity. Although the current study did not observe a significant increase in circulating BDNF, our findings align with previous work suggesting circulating BDNF returns to basal levels 30 minutes post AE [63]. It is important to note that transient increases in circulating BDNF after acute AE were not detected in older trained populations [45]. Albeit other reports detect acute AE mediated increases in circulating BDNF in older, cognitively impaired persons [64, 65]. Given the young healthy cohort in the current study, future studies are warranted to determine the effect of biological age on acute AE mediated increases in circulating BDNF.
Although a significant amount of BDNF is stored and released from multiple tissues [66– 68], the ability of skeletal muscle to release BDNF into the circulation in response to AE remains controversial. We observed a decrease in skeletal muscle mature BDNF in the hours following exercise. Mature BDNF contributes to a relatively small amount of the total skeletal muscle BDNF pool, possibly explaining why no changes in total BDNF were observed in the current study. However, previous results have shown increases in total BDNF protein occur around 24 hours following acute AE [46]. This is consistent with increases in BDNF transcript in response to AE found in our study and could be explained by a possible repletion of an intracellular mature BDNF protein.
Further to the BDNF regulation in skeletal muscle, we found fast-twitch glycolytic fibers (MHC IIa and IIx) expressed higher levels of BDNF when compared to oxidative fibers (MHC I). Although we did not find significant changes in circulating BDNF after AE, glycolytic fibers rather than oxidative may be partially responsible for contributing to the AE-induced circulating BDNF pool immediately after AE. This is further supported by high intensity acute AE inducing larger increases in circulating BDNF when compared to low intensity [49– 51]. It is important to note that previous studies employing anaerobic exercise (Wingate test) found conflicting results pertaining to circulating BDNF post anaerobic exercise [69, 70]. During high intensity aerobic exercise however, a larger proportion of glycolytic fibers are recruited according to Henneman’s size principle [71, 72]. Based upon this principle, aerobic exercise at a higher intensity would elicit contraction from a higher proportion of glycolytic fibers when compared to low intensity. As a result, this may be why previous investigations found more robust increases in circulating BDNF after high intensity aerobic exercise. Alternatively, previous work indicates skeletal muscle BDNF in knockout mice exhibited delayed differentiation and regeneration in response to injury [73]. One interpretation for this phenotype could be the ability of skeletal muscle to regenerate and proliferate after muscular injury such as intense exercise in our young healthy cohort. However, we did not find changes in BDNF protein after AE.
No correlations were found between circulating or skeletal muscle CTSB or BDNF and clinical, anthropometric, or metabolic measures. One explanation for this could be our study population. By design we recruited young, recreationally active healthy individuals, which is concomitant with optimal cognitive health. However, cross sectional studies and studies of individuals with cognitive impairments undergoing AE training interventions report positive correlations between hippocampal volume, aerobic fitness and BDNF, as well as aerobic fitness and CTSB [18, 31, 74]. These correlations may be not significant in healthy individuals or take extended periods of AE training to develop.
Our study was not without limitations and our data should be interpreted with caution. Certainly, our small sample size may have prevented us from realizing statistical differences being a-priori power analyses were not performed in this retrospective analysis of previously collected human samples. However, we have reported effect sizes for all analyses and future adequately powered prospective studies can be informed from our work. Unfortunately, due to depleted skeletal muscle tissue, we reported a reduced number of subjects, with some subjects being unmatched across all time points. The tissue yields of human skeletal muscle biopsies are often a limitation for comprehensive molecular studies; notwithstanding the subject burden of these invasive tests. Future, prospective studies of human skeletal muscle or animal studies will illuminate the precise molecular underpinnings of CTSB and BDNF regulation.
Our study population included healthy, active, young individuals representing optimal health. Unfortunately, sex distribution was not matched, nor were sexually dimorphic changes part of our original study design and menstrual cycle was not controlled for in female participants. Future studies are warranted to determine if sexual dimorphic changes exist and the potential influence of reproductive hormones on CTSB and BDNF responses to exercise. Future studies should also interrogate responses in sedentary, older, and cognitively impaired cohorts. It should be noted that acute AE sessions were separated by 5– 7 days. This time frame was chosen to washout any lasting effects from the previous exercise bout. However, the fluctuations in washout period may have effects on peripheral CTSB and BDNF levels. Pertaining to fiber type-specific protein expression, we were not able to detectable CTSB in skeletal muscle cross sections. CTSB is a cysteine protease, primarily localized to lysosomal compartments, where it is involved in turnover of intracellular and extracellular proteins [25]. Therefore, because CTSB is encapsulated within the lysosome, it may have been masked from epitope recognition via our histological methodology. However, our CTSB skeletal muscle homogenate detection is supported by previous reports that were able to detect intracellular and secreted CTSB in murine skeletal muscle culture models [24]. This was further confirmed in our laboratory, as we were able to detect intracellular CTSB via immunoblot and CTSB within media of fully differentiated human skeletal muscle myotubes (data not shown). Although we utilized multiple antibodies compatible with immunofluorescence without success, we did not perform an exhaustive approach. Therefore, more work is needed to determine the skeletal muscle fiber specific profile of CTSB. Finally, many other exerkines may facilitate cognitive benefits associated with exercise aside from CTSB and BDNF (i.e., GPLD1, IL-6, Klotho). While these additional exerkines may also be regulated in an exercise intensity-specific manner, these targets were outside the scope of our current work and we refer the readers to more comprehensive reviews on this subject [20, 22, 23, 75, 76].
CONCLUSIONS
This is the first study to identify the effects of differential AE intensity on circulating CTSB and BDNF as well as and skeletal muscle protein and gene expression patterns of CTSB and BDNF at rest and following moderate-vigorous aerobic exercise. Importantly the findings from our current study will allow for a better understanding of the molecular underpinnings regulating CTSB and BDNF by establishing the profiles of a healthy young adult. Our findings suggest that high intensity AE enriches circulating CTSB more so than low intensity. CTSB and BDNF are promising therapeutic targets to delay the onset and progression of cognitive impairments and future studies are needed to elucidate the mechanisms regulating the release, processing, and fiber type specific role of CTSB and BDNF.
ACKNOWLEDGMENTS
None.
FUNDING
Supported in part by National Institutes of Health Grant R01DK109948 and a charitable gift from the Sarns’ Family.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. (2021) ;17: (3):327–406. |
[2] | Wong W . Economic burden of Alzheimer disease and managed care considerations. Am J Manag Care. (2020) ;26: (8 Suppl):S177–S83. |
[3] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT . Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. (2011) ;1: (1):a006189. |
[4] | Parnetti L , Chiasserini D , Eusebi P , Giannandrea D , Bellomo G , De Carlo C , et al. Performance of aβ1-40, aβ1-42, total tau, and phosphorylated tau as predictors of dementia in a cohort of patients with mild cognitive impairment. J Alzheimers Dis. (2012) ;29: (1):229–38. |
[5] | Chen TB , Lee YJ , Lin SY , Chen JP , Hu CJ , Wang PN , et al. Plasma Aβ42 and total tau predict cognitive decline in amnestic mild cognitive impairment. Sci Rep. (2019) ;9: (1):13984. |
[6] | Bloom GS . Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. (2014) ;71: (4):505–8. |
[7] | Rajan KB , Weuve J , Barnes LL , McAninch EA , Wilson RS , Evans DA . Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020-2060). Alzheimers Dement. (2021) . |
[8] | Kahle-Wrobleski K , Ye W , Henley D , Hake AM , Siemers E , Chen YF , et al. Assessing quality of life in Alzheimer’s disease: Implications for clinical trials. Alzheimers Dement (Amst). (2017) ;6: :82–90. |
[9] | Sanda L , Colby JMO . Projections of the Size and Composition of the US Population: 2014 to 2060. Population Estimates and Projections. Current Population Reports: US Census Bureau; (2015) . p. 25–1143. |
[10] | Sofi F , Valecchi D , Bacci D , Abbate R , Gensini GF , Casini A , et al. Physical activity and risk of cognitive decline: A meta-analysis of prospective studies. J Intern Med. (2011) ;269: (1):107–17. |
[11] | Aberg MA , Pedersen NL , Torén K , Svartengren M , Bäckstrand B , Johnsson T , et al. Cardiovascular fitness is associated with cognition in young adulthood. Proc Natl Acad Sci U S A. (2009) ;106: (49):20906–11. |
[12] | Burns JM , Cronk BB , Anderson HS , Donnelly JE , Thomas GP , Harsha A , et al. Cardiorespiratory fitness and brain atrophy in early Alzheimer disease. Neurology. (2008) ;71: (3):210–6. |
[13] | Stillman CM , Uyar F , Huang H , Grove GA , Watt JC , Wollam ME , et al. Cardiorespiratory fitness is associated with enhanced hippocampal functional connectivity in healthy young adults. Hippocampus. (2018) ;28: (3):239–47. |
[14] | Alfini AJ , Weiss LR , Nielson KA , Verber MD , Smith JC . Resting cerebral blood flow after exercise training in mild cognitive impairment. J Alzheimers Dis. (2019) ;67: (2):671–84. |
[15] | Bossers WJ , van der Woude LH , Boersma F , Hortobágyi T , Scherder EJ , van Heuvelen MJ . A 9-week aerobic and strength training program improves cognitive and motor function in patients with dementia: A randomized, controlled trial. Am J Geriatr Psychiatry. (2015) ;23: (11):1106–16. |
[16] | Suzuki T , Shimada H , Makizako H , Doi T , Yoshida D , Ito K , et al. A randomized controlled trial of multicomponent exercise in older adults with mild cognitive impairment. PLoS One. (2013) ;8: (4):e61483. |
[17] | ten Brinke LF , Bolandzadeh N , Nagamatsu LS , Hsu CL , Davis JC , Miran-Khan K , et al. Aerobic exercise increases hippocampal volume in older women with probable mild cognitive impairment: A 6-month randomised controlled trial. Br J Sports Med. (2015) ;49: (4):248–54. |
[18] | Erickson KI , Voss MW , Prakash RS , Basak C , Szabo A , Chaddock L , et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. (2011) ;108: (7):3017–22. |
[19] | Voss MW , Weng TB , Narayana-Kumanan K , Cole RC , Wharff C , Reist L , et al. Acute exercise effects predict training change in cognition and connectivity. Med Sci Sports Exerc. (2020) ;52: (1):131–40. |
[20] | Chow LS , Gerszten RE , Taylor JM , Pedersen BK , van Praag H , Trappe S , et al. Exerkines in health, resilience and disease. Nat Rev Endocrinol. (2022) ;18: (5):273–89. |
[21] | Thyfault JP , Bergouignan A . Exercise and metabolic health: Beyond skeletal muscle. Diabetologia. (2020) ;63: (8):1464–74. |
[22] | Townsend LK , MacPherson REK , Wright DC . New horizon: Exercise and a focus on tissue-brain crosstalk. J Clin Endocrinol Metab. (2021) ;106: (8):2147–63. |
[23] | Pedersen BK , Febbraio MA . Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat Rev Endocrinol. (2012) ;8: (8):457–65. |
[24] | Moon HY , Becke A , Berron D , Becker B , Sah N , Benoni G , et al. Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab. (2016) ;24: (2):332–40. |
[25] | Mort JS , Buttle DJ , Cathepsin B . Int J Biochem Cell Biol. (1997) ;29: (5):715–20. |
[26] | Moon HY , Javadi S , Stremlau M , Yoon KJ , Becker B , Kang SU , et al. Conditioned media from AICAR-treated skeletal muscle cells increases neuronal differentiation of adult neural progenitor cells. Neuropharmacology. (2019) ;145: (Pt A):123–30. |
[27] | Embury CM , Dyavarshetty B , Lu Y , Wiederin JL , Ciborowski P , Gendelman HE , et al. Cathepsin B improves ß-amyloidosis and learning and memory in models of Alzheimer’s disease. J neuroimmune pharmacol. (2017) ;12: (2):340–52. |
[28] | Hook G , Hook V , Kindy M . The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J Alzheimers Dis. (2011) ;26: (2):387–408. |
[29] | Hook VY , Kindy M , Hook G . Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J Biol Chem. (2008) ;283: (12):7745–53. |
[30] | De la Rosa A , Solana E , Corpas R , Bartrés-Faz D , Pallàs M , Vina J , et al. Long-term exercise training improves memory in middle-aged men and modulates peripheral levels of BDNF and Cathepsin B. Sci Rep. (2019) ;9: (1):3337. |
[31] | Gaitán JM , Moon HY , Stremlau M , Dubal DB , Cook DB , Okonkwo OC , et al. Effects of aerobic exercise training on systemic biomarkers and cognition in late middle-aged adults at risk for Alzheimer’s disease. Front Endocrinol (Lausanne). (2021) ;12: :660181. |
[32] | Nicolini C , Toepp S , Harasym D , Michalski B , Fahnestock M , Gibala MJ , et al. No changes in corticospinal excitability, biochemical markers, and working memory after six weeks of high-intensity interval training in sedentary males. Physiol Rep. (2019) ;7: (11):e14140. |
[33] | Nicolini C , Michalski B , Toepp SL , Turco CV , D’Hoine T , Harasym D , et al. A single bout of high-intensity interval exercise increases corticospinal excitability, brain-derived neurotrophic factor, and uncarboxylated osteolcalcin in sedentary, healthy males. Neuroscience. (2020) ;437: :242–55. |
[34] | Griffin É , Mullally S , Foley C , Warmington SA , O’Mara SM , Kelly AM . Aerobic exercise improves hippocampal function and increases BDNF in the serum of young adult males. Physiol Behav. (2011) ;104: (5):934–41. |
[35] | Lu B , Nagappan G , Lu Y . BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol. (2014) ;220: :223–50. |
[36] | Voss MW , Erickson KI , Prakash RS , Chaddock L , Kim JS , Alves H , et al. Neurobiological markers of exercise-related brain plasticity in older adults. Brain Behav Immun. (2013) ;28: :90–9. |
[37] | Nascimento CM , Pereira JR , Pires de Andrade L , Garuffi M , Ayan C , Kerr DS , et al. Physical exercise improves peripheral BDNF levels and cognitive functions in mild cognitive impairment elderly with different bdnf Val66Met genotypes. J Alzheimers Dis. (2015) ;43: (1):81–91. |
[38] | Lu B , Pang PT , Woo NH . The yin and yang of neurotrophin action. Nat Rev Neurosci. (2005) ;6: (8):603–14. |
[39] | Bamji SX , Majdan M , Pozniak CD , Belliveau DJ , Aloyz R , Kohn J , et al. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. (1998) ;140: (4):911–23. |
[40] | Woo NH , Teng HK , Siao CJ , Chiaruttini C , Pang PT , Milner TA , et al. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. (2005) ;8: (8):1069–77. |
[41] | Teng HK , Teng KK , Lee R , Wright S , Tevar S , Almeida RD , et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. (2005) ;25: (22):5455–63. |
[42] | Lee J , Duan W , Mattson MP . Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem. (2002) ;82: (6):1367–75. |
[43] | Chan JP , Cordeira J , Calderon GA , Iyer LK , Rios M . Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol Cell Neurosci. (2008) ;39: (3):372–83. |
[44] | Delezie J , Weihrauch M , Maier G , Tejero R , Ham DJ , Gill JF , et al. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc Natl Acad Sci U S A. (2019) ;116: (32):16111–20. |
[45] | Máderová D , Krumpolec P , Slobodová L , Schön M , Tirpáková V , Kovaničová Z , et al. Acute and regular exercise distinctly modulate serum, plasma and skeletal muscle BDNF in the elderly. Neuropeptides. (2019) ;78: :101961. |
[46] | Matthews VB , Aström MB , Chan MH , Bruce CR , Krabbe KS , Prelovsek O , et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia. (2009) ;52: (7):1409–18. |
[47] | Mousavi K , Jasmin BJ . BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J Neurosci. (2006) ;26: (21):5739–49. |
[48] | Hurtado E , Cilleros V , Nadal L , Simó A , Obis T , Garcia N , et al. Muscle contraction regulates BDNF/TrkB signaling to modulate synaptic function through presynaptic cPKCα and cPKCβI. Front Mol Neurosci. (2017) ;10: :147. |
[49] | Schmolesky MT , Webb DL , Hansen RA . The effects of aerobic exercise intensity and duration on levels of brain-derived neurotrophic factor in healthy men. J Sports Sci Med. (2013) ;12: (3):502–11. |
[50] | Saucedo Marquez CM , Vanaudenaerde B , Troosters T , Wenderoth N . High-intensity interval training evokes larger serum BDNF levels compared with intense continuous exercise. J Appl Physiol. (2015) ;119: (12):1363–73. |
[51] | Kovacevic A , Fenesi B , Paolucci E , Heisz JJ . The effects of aerobic exercise intensity on memory in older adults. Appl Physiol Nutr Metab. (2020) ;45: (6):591–600. |
[52] | Chaves AB , Miranda ER , Mey JT , Blackburn BK , Fuller KNZ , Stearns B , et al. Exercise reduces the protein abundance of TXNIP and its interacting partner REDD1 in skeletal muscle: Potential role for a PKA-mediated mechanism. J Appl Physiol. (1985) . 2021. |
[53] | Stephen Glass GBD . ACSM’S metabolic calculations handbook. Philadelphia, PA: Lippincott Williams & Wilkins; (2007) . |
[54] | Mahmoud AM , Szczurek MR , Blackburn BK , Mey JT , Chen Z , Robinson AT , et al. Hyperinsulinemia augments endothelin-1 protein expression and impairs vasodilation of human skeletal muscle arterioles. Physiol Rep. (2016) ;4: (16). |
[55] | Mey JT , Blackburn BK , Miranda ER , Chaves AB , Briller J , Bonini MG , et al. Dicarbonyl stress and glyoxalase enzyme system regulation in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. (2018) ;314: (2):R181–R90. |
[56] | Mey JT , Solomon TPJ , Kirwan JP , Haus JM . Skeletal muscle Nur77 and NOR1 insulin responsiveness is blunted in obesity and type 2 diabetes but improved after exercise training. Physiol Rep. (2019) ;7: (6):e14042. |
[57] | Micielska K , Kortas JA , Gmiat A , Jaworska J , Kozlowska M , Lysak-Radomska A , et al. Habitually inactive physically - a proposed procedure of counteracting cognitive decline in women with diminished insulin sensitivity through a high-intensity circuit training program. Physiol Behav. (2021) ;229: :113235. |
[58] | Coffey VG , Jemiolo B , Edge J , Garnham AP , Trappe SW , Hawley JA . Effect of consecutive repeated sprint and resistance exercise bouts on acute adaptive responses in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol.R. (2009) ;297: (5):1441–51. |
[59] | Hickso RC . Interference of strength development by simultaneously training for strength and endurance. Eur J Appl Physiol Occup Physiol. (1980) ;45: (2-3):255–63. |
[60] | Gökçe E , Güneş E , Arı F , Hayme S , Nalçacı E . Comparison of the effects of open- and closed-skill exercise on cognition and peripheral proteins: A cross-sectional study. PLoS One. (2021) ;16: (6):e0251907. |
[61] | Karlsson L , González-Alvarado MN , Motalleb R , Wang Y , Börjesson M , Zhu C , et al. Constitutive PGC-1α overexpression in skeletal muscle does not contribute to exercise-induced neurogenesis. Mol Neurobiol. (2021) ;58: (4):1465–81. |
[62] | Erlich AT , Brownlee DM , Beyfuss K , Hood DA . Exercise induces TFEB expression and activity in skeletal muscle in a PGC-1α-dependent manner. Am J Physiol Cell Physiol. (2018) ;314: (1):C62–C72. |
[63] | Rojas Vega S , Strüder HK , Vera Wahrmann B , Schmidt A , Bloch W , Hollmann W . Acute BDNF and cortisol response to low intensity exercise and following ramp incremental exercise to exhaustion in humans. Brain Res. (1121) ;1 59–65. |
[64] | Devenney KE , Guinan EM , Kelly Á , Mota BC , Walsh C , Olde Rikkert M , et al. Acute high-intensity aerobic exercise affects brain-derived neurotrophic factor in mild cognitive impairment: A randomised controlled study. BMJ Open Sport Exerc Med. (2019) ;5: (1):e000499. |
[65] | Nilsson J , Ekblom Ö , Ekblom M , Lebedev A , Tarassova O , Moberg M , et al. Acute increases in brain-derived neurotrophic factor in plasma following physical exercise relates to subsequent learning in older adults. Sci Rep. (2020) ;10: (1):4395. |
[66] | Fujimura H , Altar CA , Chen R , Nakamura T , Nakahashi T , Kambayashi J , et al. Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost. (2002) ;87: (4):728–34. |
[67] | Rasmussen P , Brassard P , Adser H , Pedersen MV , Leick L , Hart E , et al. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol. (2009) ;94: (10):1062–9. |
[68] | Antony R , Li Y . BDNF secretion from C2C12 cells is enhanced by methionine restriction. Biochem Biophys Res Commun. (2020) ;533: (4):1347–51. |
[69] | Murawska-Cialowicz E , Wojna J , Zuwala-Jagiello J . Crossfit training changes brain-derived neurotrophic factor and irisin levels at rest, after wingate and progressive tests, and improves aerobic capacity and body composition of young physically active men and women. J Physiol Pharmacol. (2015) ;66: (6):811–21. |
[70] | Murawska-Ciałowicz E , de Assis GG , Clemente FM , Feito Y , Stastny P , Zuwała-Jagiełło J , et al. Effect of four different forms of high intensity training on BDNF response to Wingate and Graded Exercise Test. Sci Rep. (2021) ;11: (1):8599. |
[71] | Henneman E . Relation between size of neurons and their susceptibility to discharge. Science. (1957) ;126: (3287):1345–7. |
[72] | Mcphedran AM , Wuerker RB , Henneman E . PRoperties of motor units in a homogeneous red muscle (soleus) of the cat. J Neurophysiol. (1965) ;28: :71–84. |
[73] | Clow C , Jasmin BJ . Brain-derived neurotrophic factor regulates satellite cell differentiation and skeltal muscle regeneration. Mol Biol Cell. (2010) ;21: (13):2182–90. |
[74] | Erickson KI , Prakash RS , Voss MW , Chaddock L , Hu L , Morris KS , et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus. (2009) ;19: (10):1030–9. |
[75] | Pedersen BK . Physical activity and muscle-brain crosstalk. Nat Rev Endocrinol. (2019) ;15: (7):383–92. |
[76] | Isaac AR , Lima-Filho RAS , Lourenco MV . How does the skeletal muscle communicate with the brain in health and disease? Neuropharmacology. (2021) ;197: :108744. |


