
Role of Hippocampal Neurogenesis in Alcohol Withdrawal Seizures
Abstract
Chronic alcohol consumption results in alcohol use disorder (AUD). Interestingly, however, sudden alcohol withdrawal (AW) after chronic alcohol exposure also leads to a devastating series of symptoms, referred to as alcohol withdrawal syndromes. One key feature of AW syndromes is to produce phenotypes that are opposite to AUD. For example, while the brain is characterized by a hypoactive state in the presence of alcohol, AW induces a hyperactive state, which is manifested as seizure expression. In this review, we discuss the idea that hippocampal neurogenesis and neural circuits play a key role in neuroadaptation and establishment of allostatic states in response to alcohol exposure and AW. The intrinsic properties of dentate granule cells (DGCs), and their contribution to the formation of a potent feedback inhibitory loop, endow the dentate gyrus with a “gate” function, which can limit the entry of excessive excitatory signals from the cortex into the hippocampus. We discuss the possibility that alcohol exposure and withdrawal disrupts structural development and circuitry integration of hippocampal newborn neurons, and that this altered neurogenesis impairs the gate function of the hippocampus. Failure of this gate function is expected to alter the ratio of excitatory to inhibitory (E/I) signals in the hippocampus and to induce seizure expression during AW. Recent functional studies have shown that specific activation and inhibition of hippocampal newborn DGCs are both necessary and sufficient for the expression of AW-associated seizures, further supporting the concept that neurogenesis-induced neuroadaptation is a critical target to understand and treat AUD and AW-associated seizures.
INTRODUCTION
Alcohol use disorder (AUD) is a chronic relapsing disease characterized by alcohol abuse and dependence [1]. Excessive amounts of alcohol are consu-med in an uncontrolled manner during the alcohol abuse stage, and continued exposure to alcohol is required for the maintenance of physical and mental health during the alcohol dependence stage. According to the 2018 National Survey on Drug Use and Health, 14.4 million adults over the age of 18 suffer from AUD. This includes 9.2 million men and 5.3 million women [2]. More than 88,000 annual deaths have been attributed to AUD [3]. In addition to the severe health implications, continued alcohol abuse can also have immense negative consequences on personal, social, and economic well-being [4].
Alcoholism initially begins with casual alcohol use; however, the frequency and quantity of alcohol consumption gradually increases over time (Fig. 1). Initial alcohol use is primarily driven by the positive rein-forcement of alcohol [5]. The term reinforcement refers to the process by which a behavior or response is strengthened by previous experiences acquired from exposure to a given substance. For example, al-cohol’s positive reinforcement includes hedonic and rewarding effects, whereas sedation, motor impairments, and hangover represent the negative effects of alcohol [6]. The motivation to drink at the beginning of alcohol use is mainly driven by the net positive reinforcement of alcohol because its positive effects outweigh its adverse effects. However, as continued consumption of alcohol induces brain adaptations in structure and function, the brain becomes tolerant to alcohol, and its initial positive effects diminish, even in the presence of alcohol. Increased amounts of alcohol are required to maintain the same level of positive effects, and eventually, the continued presence of alcohol becomes necessary for mental and physical function in this alcohol-dependent state. These cycles of alcohol abuse and dependence are thought to be a mechanism underlying alcoholism.
Many attempts to stop alcohol abuse have often failed because a sudden attempt to cease alcohol consumption after chronic alcohol exposure results in a whole series of distinct and adverse symptoms (Fig. 1). Alcohol withdrawal (AW) syndromes differ from those associated with alcohol abuse and dependence, and the severity of these syndromes widely varies. Among AW syndromes, epileptic seizures and delirium tremens represent the most severe conditions [7]. AW syndromes have unique features, which are important to understand their impact on physical and mental health. First, the majority of alcoholics who visit clinics seek medical care to treat and manage AW syndromes [8, 9]. In fact, according to a surveillance report published by The National Institute on Alcohol Abuse and Alcoholism (NIAAA) in 2017, almost 50%of hospital admissions related to alcoholism resulted from AW symptoms, indicating that medical interventions mainly focus on treating AW syndromes. Therefore, understanding the mechanisms underlying symptoms induced by AW will provide a foundation to better understand how to treat AW syndromes. Second, a significant portion of alcoholics who attempt to stop consuming alcohol eventually relapse. This is because re-exposure to alcohol dramatically reduces the negative effects of AW, such as seizures and pain [10]. In addition, patients reuse alcohol in order to mitigate the anticipated psychological outcomes during abstinence, such as anxiety and depression. Therefore, repeated cycles of AW and relapse represent a downward spiral that leads to alcohol dependence [6, 11]. Lastly, AW syndromes display completely opposite effects to those of dependence, and the consequences of alcohol dependence are thought to emerge and express upon AW. Therefore, AW provides a unique and critical window to understand neuroadaptive changes that underlie alcohol dependence. Figure 1 summarizes the relationship between AUD and AW.
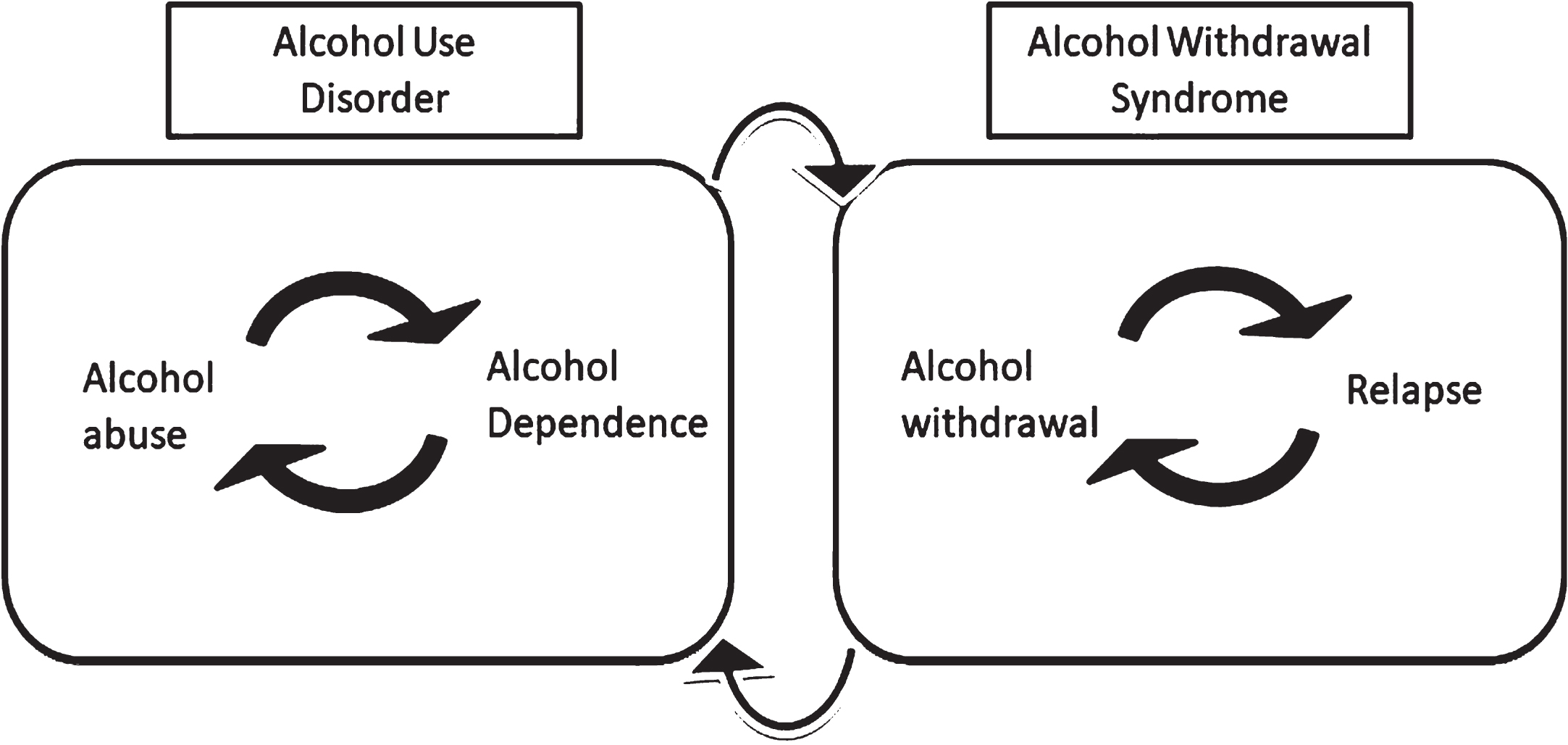
Fig. 1
Two major aspects of alcohol addiction: abuse and withdrawal. Alcohol use disorder or AUD is mainly comprised of alcohol exposure and dependence. Allostatic changes in the brain lead to alcohol tolerance, which bridges repeated cycles of alcohol abuse and dependence. On the other hand, stopping alcohol intake can cause alcohol withdrawal syndrome. Alcohol’s effect to alleviate alcohol withdrawal syndrome leads to a relapse. Repeated cycles of alcohol withdrawal and relapse can repeatedly induce a state of alcohol dependence.
To understand the impact of AW on physiology and psychology and to develop better treatments for AW syndromes, we will discuss the theories and mechanisms underlying AW symptoms using alcohol withdrawal seizure (AWS) as an example. Tonic- clonic seizures represent the most severe and prevalent condition among AW syndromes, and 9–25%of cases ultimately develop to status epilepticus (SE). In this review, we will discuss how hippocampal neurogenesis has a direct effect on the activity of hippocampal as well as global neural circuits associated with AWS. Neurogenesis is a process that includes the production and integration of newborn neurons into pre-existing neural circuits, and this process provides a significant level of plasticity that has a direct impact on the anatomy and function of the hippocampus. In particular, it has been proposed that newborn neurons contribute to the “gate” function of the dentate gyrus of the hippocampus by limiting the entry of excessive excitatory signals into the hippocampus and thereby maintaining the excitatory-to-inhibitory (E/I) signal balance in the hippocampus [12, 13]. Our overarching hypothesis is that alcohol exposure and withdrawal alters the neuronal development and connectivity of hippocampal newborn neurons, and that this disrupts the “gate” function of the hippocampus, thereby increasing the E/I signal and synchronized hyperactivity of neural circuits underlying AWS.
Brain adaption associated with alcohol dependence and AWS
More than 80 years ago, Himmelsbach proposed the theory that the physiological ability to maintain homeostasis in response to addictive substances may underlie drug tolerance [14]. This theory predicts that the brain undergoes adaptation to maintain homeostasis in response to such substances. This neuroadaptation is not apparently visible because altered homeostasis represents a “new normal” or allostasis in the presence of the substance. Interestingly, allostatic changes are unveiled and emerge when the substance in question is absent, and this disinhibition effect in the absence of addictive substances results in withdrawal syndromes [14]. In line with this view, it has been proposed that alcohol-induced brain adaptation and allostasis underlie alcohol dependence and withdrawal [15, 16]. Allostasis is defined as the process of maintaining stability by changing the body physiology to a new state in response to environmental changes; a chronic deviation of the regulatory systems from the normal state of operation establishes a new set point, or allostatic state [17–20]. This model predicts the development and progression of AWS as follows. At the beginning of alcohol consumption, alcohol exerts stimulatory effects in the brain, and alcohol’s positive effects are a major driving force towards alcohol consumption. However, alcohol eventually functions as a depressant for the central nervous system (CNS). In order to counterbalance alcohol’s effect as a depressant, the brain undergoes neuroadaptive changes in a direction that makes the brain hyperactive; however, this new normal state (i.e. hyperactive) or allostatic state is masked due to the continued action of alcohol’s depressive functions. The consequence of such allostatic changes is that the brain becomes tolerant to the previous amount of alcohol, and thus increased amounts of alcohol are needed to produce the same level of positive reinforcement. Despite the increased alcohol consumption, the brain becomes tolerant again and reaches the next level of allostatic state, which will motivate more alcohol consumption to retain alcohol’s positive effects. This repeated cycle of alcohol exposure and tolerance is expected to ultimately drive towards an alcohol dependence state. In this alcohol-dependent state, the equilibrium has been shifted significantly to make the brain more excitable in order to counterbalance alcohol’s depressive functions, but these neuroadaptations are veiled by the continued presence and action of alcohol [15, 16, 21].
The consequences of allostatic state in alcohol dependence emerge and are expressed when alcohol consumption is suddenly terminated [22]. Upon AW, alcohol’s function as a CNS depressant is removed, and the compensatory changes that have made the brain more excitable during an alcohol dependence state are expressed. This resultant hyperexcitability of the brain is thought to be manifested in the form of AWS, raising the hypothesis that alterations to the E/I balance in the brain may result in AWS. In this context, increased excitatory and/or decreased inhibitory neurotransmission will be discussed as potential mechanisms that can increase the E/I and shift the brain equilibrium to a hyperexcitable state. In addition, we will review how hippocampal neurogenesis, a process that provides a significant level of plasticity to the brain, contributes to the maintenance of E/I balance, and how disrupted neurogenesis may underlie AWS.
Increased excitatory neurotransmission during AWS
Increased excitatory neurotransmitter, enhanced expression and function of glutamate receptors, and altered receptor subtypes and subunits can contri-bute to the hyper-glutamatergic state during AW [23–28]. Here we use selected examples to discuss how increased glutamatergic neurotransmission can be attributed to alterations to the E/I balance.
Extracellular glutamate concentrations in various brain regions have been measured by microdialysis after AW. In one study, the concentration of extracellular glutamate was significantly increased in the striatum within 12 hours of AW compared to that of sucrose-treated controls [29]. Glutamate levels remained elevated for the subsequent 12 hours and returned to control levels within 36 hours of AW. Other microdialysis studies found increased glutamate concentrations in the nucleus accumbens and the hippocampus at 12 hours of AW [30, 31]. Interestingly, repeated cycles of ethanol exposure and withdrawal significantly increased glutamate concentration, i.e., glutamate concentration is much higher in the hippocampus after a third cycle of ethanol exposure and withdrawal compared to that of the first cycle [31].
Glutamate receptors include both the ionotropic (iGluRs) and metabotropic (mGluRs) families [32]. iGluRs include three major receptor subtypes: N-methyl-d-aspartate (NMDA) receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (AMPARs), and kainic acid receptors. Eight members of the G-protein-coupled mGluRs have been identified (mGluR1–8) and they are categorized into group I (mGluR1 and 5), group II (mGluR2 and 3), and group III (mGluR4, 6, 7, and 8). NMDARs have a tetrameric structure that consists of NR1, NR2A–D or NR3A-B subunits. Typically, NMDARs are composed of two NR1 subunits and any combination of NR2 and NR3 subunits. The AMPAR family is heteromeric and consists of four major subunits (GluR1-4). In particular, GluR2 pl-ays an important role in the control of neuronal activity because AMPARs containing GluR2 reduce Ca2 + permeability [33, 34]. The number and subunit compositions of both NMDARs and AMPARs vary among different brain regions and/or neuronal cell types, and changes in composition are associated with development, synaptic plasticity, and various pathological states [33, 35, 36]. AW can alter glu-tamate receptor-mediated synaptic transmission. Acute exposure and withdrawal induced long-term potentiation (LTP) of NMDAR-mediated postsynaptic currents in the dorsal striatum (DS) [37]. Six-teen hours after a 7-day ethanol exposure to rats, NMDAR currents were significantly increased in dorsomedial striatal (DMS) neurons. Moreover, an extended ethanol administration period from 7 to 14 days upregulated NMDAR activity, which lasted for up to 40 hours after the last administration [38].
A chronic intermittent ethanol (CIE) exposure is characterized by repeated cycles of alcohol exposure and withdrawal that result in a more persistent and severe form of AW syndrome. This effect is often referred as to a kindling effect [39, 40]. Because each cycle of alcohol exposure and withdrawal aggravates withdrawal symptoms as the cycle proceeds, this model closely recapitulates key aspects of human alcoholism [41]. For example, 48 hours after CIE exposure, enhanced glutamatergic transmission was reported in layer II/III pyramidal neurons of the medial prefrontal cortex (mPFC) [42]. Withdrawal from CIE has been shown to increase the NMDA/AMPA current ratio of layer V pyramidal neurons in the mPFC [43]. The NMDA/AMPA current ratio is often used to measure differential expression or synaptic transmission of NMDAR or AMPAR dependent currents. Thus, the NMDA/AMPA current ratio has been linked to synaptic plasticity, as well as synapse formation, gr-owth, and stability [44]. This increase in NMDA/ AMPA current ratio was associated with increased NR1 and NR2B subunit expression [43]. Prolonged withdrawal after CIE also induced a redistribution of AMPAR subunit composition by increasing GluR2-lacking AMPARs in the postsynaptic site. GluR2-lacking AMPARs are calcium permeable, have a higher single-channel conductance, and is associated with increased glutamatergic synaptic strength [45]. Another study has shown that CIE caused an increase in cell surface expression of GluR2/3, whereas withdrawal resulted in an increase in both GluR1 and GluR2/3 in the basolateral amygdala(BLA) [46]. Together, these cases support the concept that AW induces differential changes in glutamate release, glutamate receptor subtypes, and glutamate receptor subunits in response to alcohol exposure or withdrawal.
Decreased inhibitory neurotransmission during AWS
Alcohol is a CNS depressant and modulates the function of gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the mammalian brain. Acute and chronic alcohol exposure modulates GABA receptors and GABA release in many different areas. The GABA receptor (GABAR) family consists of GABAAR, GABABR, and GABACR. [47]. GABAARs are ionotropic receptors that gate chloride ion channels, whereas GABABRs are met-abotropic G-protein-coupled receptors. Although different alcohol exposure paradigms differently modulate the activity of GABARs, it has been generally accepted that alcohol exposure upregulates GABAARs and downregulates GABABRs [48]. Different combinations of 19 GABAAR subunits (α1–6, β1–3, γ1–3, δ, ɛ, θ, π, and ρ1–3) can constitute different forms of GABAARs [48, 49]. GABAARs are comprised of two α and two β subunits, along with a single γ2 or δ subunit. The composition of GABAAR subunits determines their functional properties and localization at synaptic or extrasynaptic sites. For example, the γ subunit mediates phasic inhibition and is mostly localized within the synapse, whereas receptors containing α4, α5, α6, or δ subunits are largely located at extra- or non-synaptic sites [48, 50]. Tonic GABAAR-mediated currents are produced by extrasynaptic GABAARs containing the δ receptor subunit [51, 52]. GABAB1a, GABAB1b, and GABAB2 are the three main GABABR subunits, and a combination of GABAB2 subunit with a GABAB1a or GABAB1b subunit makes functional GABABRs [53]. In the following paragraph we will discuss some examples where AW affects GABAergic transmission.
In the hippocampus, kindling-like effect after CIE exposure was associated with hypofunction of GABAARs [54, 55]. At 2 days of AW after CIE ex-posure, mRNA expression of the α4 subunit of GABAARs was significantly increased in the DG, CA3, and CA1 of the hippocampus. As this upregulation of α4 subunit transcription has been observed in several animal models of temporal lobe epilepsy [56–58], it is plausible that elevated levels of GABAAR α4 subunits may be associated with AWS risk [59]. Similar alterations in GABA subunit composition were observed in the amygdala during the withdrawal phase of CIE [60]. While decreased surface expression of GABAAR subunits α1 and δ was observed in the basolateral amygdala (BLA) at 40 days after CIE treatment, surface expressions of α4 and γ2 subunits of GABAAR were increased. The majority of the GABAergic postsynaptic currents in BLA are mediated by α1- and α2-containing GABAARs. Thus, this decrease in α1-containing GABAARs may contribute to GABAergic transmission during AW [61].
Adult hippocampal neurogenesis
It was popularly thought that the adult brain is hard-wired and that no new neurons are regenerated. However, it has been established that new neurons are continuously generated and integrated into neural circuits in two structures of most mammalian brains, referred to as neurogenic niches [62–65]. One neurogenic niche is the subventricular zone (SVZ) of the lateral ventricle [66–68]. Neural stem cells (NSCs) that have the potential to self-renew and differentiate into multiple neural cells reside and produce immature neurons in the SVZ. Immature neurons migrate following the rostral migratory stream, mature and become interneurons, and integrate into the olfactory bulb. The second neurogenic niche is the subgranular zone (SGZ) of the dentate gyrus (DG) of the hippocampus [69–75]. NSCs are populated in the SGZ and differentiate into excitatory neurons or dentate granule cells (DGCs). Unlike the production of interneurons that are born in the SVZ and migrate to the olfactory bulb, adult-born DGCs are born in and are also integrated into the DG. Newborn neurons in both the olfactory bulb and DG continuously remodel local neural circuits. This life-long production and circuitry integration of newborn neurons provides a great deal of plasticity at the level of cell genesis, and this plasticity is required for cognition and emotion [76–80]. In physiology, integration of newborn DGCs plays a critical role in the maintenance of the E/I signal balance in the hippocampus. Because neurogenesis in the hippocampus plays a key role in cognitive and emotion as well as in maintaining the balance of the E/I signals, hippocampal neurogenesis has been implicated in characteristic features of AUD and AWS.
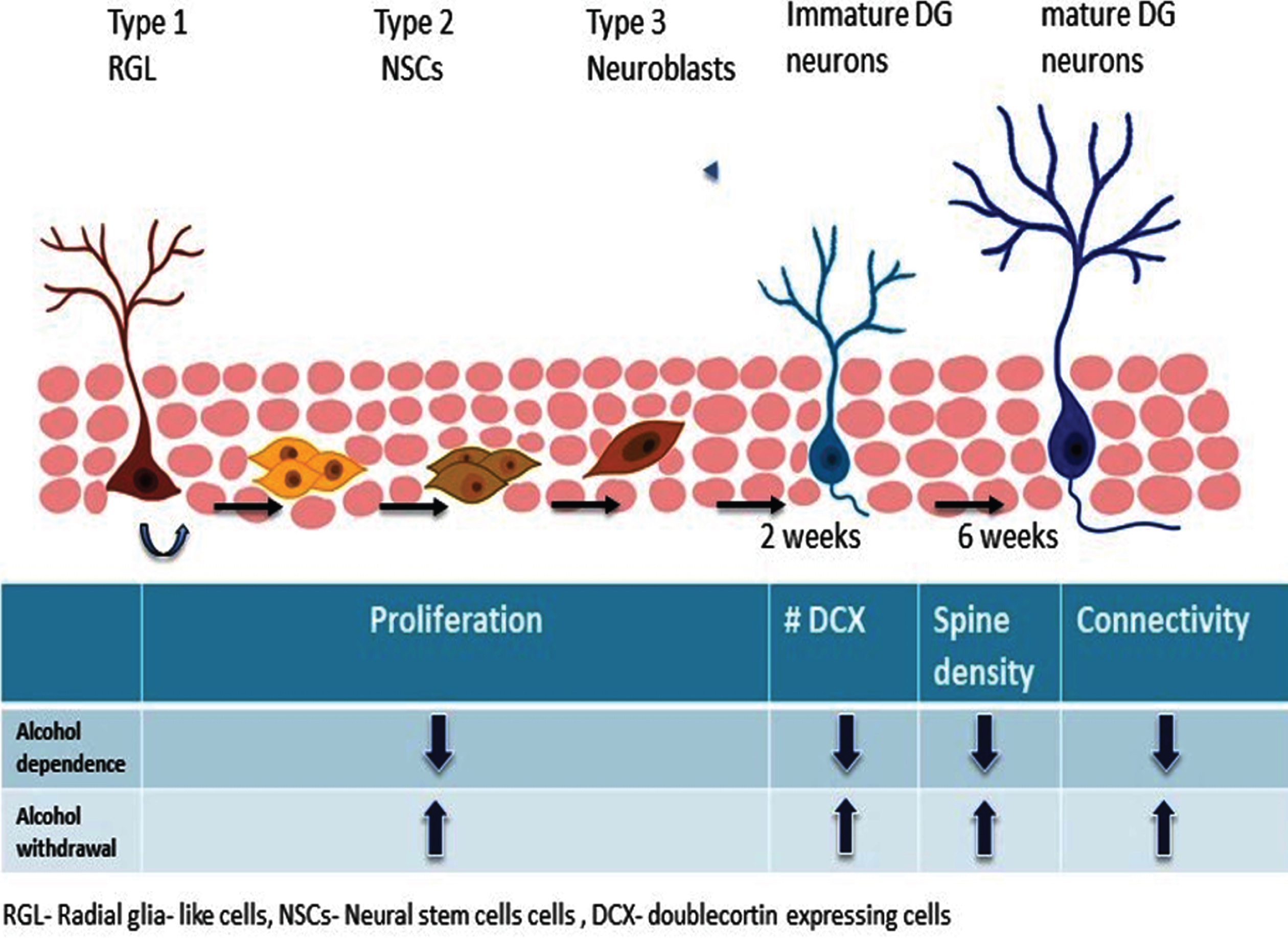
NSCs in the SGZ represent a heterogeneous cell population because they differ in morphology, marker expression, proliferation kinetics, and differentiation potential [74, 81, 82] (Fig. 2). Radial glial (RG) cells or Type-1 cells have radial morphology and represent slowly-dividing NSCs that can give rise to identical RG cells and Type-2 NSCs that subsequently produce neurons and astrocytes in the hippocampus [65, 81–83]. Type-2 NSCs do not have the distinct radial morphology, but they represent rapidly-dividing NSCs that have the potential to generate Type-3 cells and downstream neural cells. Type-3 cells represent neuroblast cells that transiently proliferate and ultimately differentiate into DGCs in the hippocampus. Different signaling mechanisms along with various neurotransmitters released at local synaptic circuits regulate the continued proliferation and activation into more differentiated cells [84]. The different stages of neurogenesis are shown in Fig. 2.
Fig. 2
Hippocampal neurogenesis, alcohol dependence and withdrawal. Alcohol exposure and withdrawal distinctly affect each step of hippocampal neurogenesis.
Hippocampal neurogenesis is tightly regulated by intrinsic as well as extrinsic factors. Among such regulators, brain activity that is translated by the expression of neurotransmitters plays a critical role in distinct stages of neurogenesis. In particular, GABA plays critical roles in the regulation of multiple steps of neurogenesis. First, it has been shown that GABA regulates proliferation of RG cells though GABAARs [85]. Specifically, GABA action on GABAARs containing γ2-subunit restricts proliferation of Type-1 RG cells, leading to decreased production of newborn DGCs. GABA also acts on GABAARs expressed in Type-2 cells and it is required to transform Type-2 cells into mature neurons [86, 87]. Interestingly, GABA depolarizes immature DGCs. This is due to higher intracellular chloride concentrations compared to the extracellular space. As a result, the opening of GABA receptors upon GABA binding onto its receptors allows chloride ions to move out, making immature DGCs depolarized [88–90]. This GABA-mediated depolarization induces the extension of apical dendrites of immature neurons into the molecular layer, where immature DGCs make synaptic connections with entorhinal cortical neurons and the axon projections to the CA3, forming the mossy fiber track [90]. Thus, GABA-mediated depolarization is essential for the integration of newborn neurons into hippocampal neural circuits.
Glutamate also plays a key role in the regulation of hippocampal neurogenesis. It has been shown that NMDA administration rapidly decreases cell proliferation, while NMDAR antagonists induce the opposite effect [91, 92]. Moreover, induction of LTP invoked by perforant path axon fibers of entorhinal neurons that project to DGCs promotes the proliferation of NSCs, further supporting the idea that glutamate has additional roles during neurogenesis [93, 94]. Specific deletion of an essential subunit of the NMDA glutamate receptor, NR1, in newborn neurons showed that glutamate-mediated excitation of newborn neurons is required for the survival of newborn DGCs that are integrated into neural circuits [95–97]. Synaptic connections from the perforant path can be detected within three to five weeks after their birth [98], whereas functional connections formed with efferent CA3 neurons can be observed four to six weeks after their birth [99]. Within a time window of four to six weeks, adult-born DGCs mature and form the characteristic tri-synaptic circuits connecting the entorhinal cortex to the DG, and the DG to the CA3. These four- to six-week-old neurons show reduced induction threshold and increased LTP amplitude in response to a physiological stimulation [100]. Thus, adult neurogenesis represents an ongoing developmental process during which the nervous system is continuously remodeled. This provides an expanded capacity of plasticity in response to experience [100].
“Gate” function of adult-born hippocampal neurons
Despite the fact that DGCs receive a significant amount of excitatory inputs, mainly from the entorhinal cortex, they are sparsely activated [101]. This unique function of hippocampal newborn DGCs stems from their distinct intrinsic properties and neuronal connectivity [100, 102–104]. The sparse activation of DGCs is achieved by feedback inhibition: DGCs project to different types of interneurons located mainly in the hilus, and these interneurons in turn project back to DGCs. This strong feedback inhibition endows hippocampal DGCs with a “gate” function that limits the excessive excitatory signals from entering the DG [12, 105, 106].
In addition to the contribution of DGCs to the for-mation of a feedback inhibitory loop, DGCs have unique intrinsic properties that facilitate sparse act-ivation. These include hyperpolarized resting membrane potential, low input resistance, and relatively high threshold for firing [107–109]. During DGC development, the action of GABA is critical to dec-rease the E/I ratio, suggesting that this inhibition is crucial for reduced spiking of maturing DGCs [103]. Immature DGCs are highly responsive due to low excitatory innervation, high intrinsic excitability, and high E/I ratio, whereas mature DGCs are minimally responsive to a broad array of cortical activity patterns due to feedback inhibition [13, 103]. Indeed, blocking neurogenesis in mice can elevate overall hippocampal activity, suggesting that the gate function of DGCs is critical for the functional physiology of the hippocampus [110].
Aberrant neurogenesis and seizures
Disrupted hippocampal neurogenesis and circuitry integration of hippocampal newborn DGCs alter the E/I ratio, which may underlie hippocampal hyperactivity manifested by the expression of seizures in some neurological conditions. Uncontrolled and synchronized bursts of neurons lead to electrical and behavioral seizures, and chronic conditions of seizure occurrence are key features of epilepsy. Dramatic changes in hippocampal neurogenesis have been reported in epilepsy. In rodent epilepsy models, seizure activity dramatically increased cell proliferation in the DG, leading to increased neurogenesis [111–113]. A number of mechanisms have been proposed to explain increased proliferation. It is possible that activation of GABA receptors expressed by NSCs [84] or differential epigenetic modification induced by seizures on the NSCs can also play important roles in their proliferation [114, 115]. Moreover, seizure-induced expression of trophic factors, such as brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF), and others can induce NSC proliferation [116–118). Of note, other studies have suggested that newborn neurons induced by seizures are prone to degenerate eventually 119, 120].
In addition to increased neurogenesis in rodent models of temporal lobe epilepsy (TLE), seizures also induced abnormal development of hippocampal newborn DGCs. While mature neurons are resistant to seizure-induced morphological changes, DGCs that are born immediately before and after SE develop abnormally [111, 121–124]. They display mossy fiber sprouting, abnormal migration and dispersion into the hilus and molecular layer, and ectopic basal dendrite formation. All of these structural abnormalities have been implicated in aberrant circuitry integration that leads to hyperactivity of the hippocampus [105, 123, 125, 126]. In particular, aberrant circuitry integration of hippocampal newborn DGCs is thought to disrupt the gate function of the DG. Zhou et al. showed that hippocampal newborn DGCs received increased excitatory inputs from the entorhinal cortex, as well as ectopic innervations from the CA3 and neighboring DGCs, while the connectivity ratio of newborn DGCs with hilar interneurons did not change [127]. This altered neuronal connectivity is expected to increase the flow of excessive excitatory signals from the entorhinal cortex to the DG and to amplify excitatory signals via de novo formation of recurrent excitatory loops between the DG and CA3 in epileptic brains [127, 128]. In addition, both normotopic and ectopic DGCs showed higher excitability and action potentials compared to control rats after SE [129]. These observations clearly suggest that abnormal development, integration, and neurophysiology of hippocampal newborn DGCs anatomically facilitate the formation of pro-epileptic neural circuits and physiologically disrupt the gate function of the DG, all of which contribute to hyperactivity of the hippocampus.
DIRECT ROLE OF HIPPOCAMPAL NEWBORN DGCS IN AWS
Alcohol exposure and withdrawal distinctly affect hippocampal neurogenesis. In general, a negative correlation has been found between alcohol consumption and neurogenesis. Acute binge consumption of alcohol reduced proliferation of NSCs in the DG [130], while alcohol withdrawal after a 4-day-binge alcohol exposure resulted in a burst of cell proliferation in rats [131]. One interesting aspect of changes in cell proliferation after binge alcohol exposure was the observation of sequential proliferation of different cell types: increased proliferation of microglia, astrocytes, and NSCs was observed at 2, 4, and 7 days of AW, respectively. These studies provide a critical time window to study the cascade of alcohol’s effect on different cell types [132–135]
Chronic alcohol consumption produced different results possibly due to the dosage, intake pattern, and/ or the duration of alcohol exposure as well as the species used. A majority of studies in this field con-fer that chronic alcohol exposure causes a reduction in NSC proliferation. In one study, chronic alcohol exposure resulted in decreased survival of newborn DGCs in mice while having no effect on proliferation of neuronal progenitors [136]. Another study showed that mice undergoing long-term alcohol self-administration for 10 weeks showed enhanced proliferation in the DG, with no change in cell sur-vival or differentiation [137]. Contrary to this experiment, chronic alcohol consumption for over 11 months in adolescent macaque monkeys resulted in a long-lasting reduction in the number of proliferating NSCs in the DG [138]. Repeated alcohol exposure in rats resulted in loss of neurons, displaying thinning of the granular cell layer [139–141]. In line with this study, chronic alcohol exposure via pair-fed alcohol exposure in mice reduced both proliferation of NSCs and survival of newborn neurons, which contributed to reduced neurogenesis [142]. In the same study, chronic alcohol exposure also impaired the structural development of newborn DGCs and cognitive function, suggesting that alcohol-induced deficits in synaptic connectivity may underlie cognitive impairments [142]. In human subjects, alcoholics have a reduced number of hippocampal neurons [143]. A recent study showed that the expression of Ki67, SOX2, and DCX, which represent molecular markers of proliferating cells, NSCs/neural progenitor cells, and immature neurons, respectively, was reduced in autopsied hippocampi from alcoholics [144]. These results suggest that chronic alcohol exposure is likely to reduce hippocampal neurogenesis by impairing cell proliferation of NSCs and survival of newborn neurons in both rodents and humans. Figure 2 shows how alcohol dependence and withdrawal differently af-fect cell proliferation, spine density and connectivity.
Neurogenesis as a neural substrate for AWS
Generalized tonic–clonic seizures are one of the most prominent features of AW syndromes. Previous studies strongly suggested that neuroadaptations during alcohol exposure play a critical role in the expression of seizures when alcohol is not present. In addition to mechanisms discussed previously, abnormal hippocampal neurogenesis has been proposed as a neural substrate that transmits the effects of alcohol exposure and withdrawal into brain activity [142, 145].
In the study by Lee et al., animals had voluntary access to a nutritionally-adequate liquid diet containing ethanol for 4 weeks [145]. Upon sudden AW, mice displayed two distinct waves of seizures. The first wave consisted of a surge of multiple seizures and epileptic spikes that occurred immediately after AW and lasted for only a few days during abstinence. In the second wave, the frequency of the epileptic spikes was reduced, but they occurred for a protracted period of abstinence in a time-dependent manner. Starting from week 1 of abstinence, the frequency of spikes progressively increased, reaching a peak at week 4 and then gradually decreased and was abolished by week 8 of abstinence. AW also increased the spine density of hippocampal newborn DGCs, which correlated with an increase in epileptic spikes (i.e. 2 and 4 weeks of abstinence); however, dendritic spine density returned to the level of control mice when AWS disappeared. Changes in the density of mushroom spines were responsible for overall changes in spine densities during AW. Most importantly, DREADD (designer receptor activated by designer drug)-mediated functional studies revealed that hippocampal newborn neurons are necessary and sufficient for the expression of AWS [145]. DREADDs are genetically-engineered receptors that can be activated by administration of the exogenous ligand CNO (Clozapine N-oxide) [94, 146, 147]. The interaction of CNO with hM3Dq or hM4Di DREADDs results in the activation or inhibition of neuronal activity, respectively. This DREADD-mediated specific activation and inhibition of hippocampal newborn DGCs increased and decreased the expression of seizures during the second wave of AWS, respectively [145]. This functional study showed that hippocampal neural circuits, which are continuously remodeled by the integration of newborn DGCs, underlie the expression of AWS, suggesting that hippocampal neurogenesis and hippocampal newborn DGCs are involved in neuroadaptation during alcohol exposure and withdrawal.
CONCLUSIONS AND FUTURE DIRECTIONS
In this review, we discussed that hippocampal neurogenesis is one of the major neural substrates that undergo neuroadaptation in response to alcohol exposure and withdrawal. In particular, we propose that the ability of hippocampal newborn neurons to maintain the E/I ratio is a key to link the effects of alcohol to seizure expression during withdrawal, raising the possibility that dysfunction of DGCs as a ‘gatekeeper’ may account for the hyperexcitable state of the brain induced by AW. This result provides insight into new therapeutic strategies to treat epilepsy or AWS by specifically targeting hippocampal neurogenesis and neural circuits. Development or screening of new medicines or molecules to restore the E/I ratio in the hippocampus can be an immediate consideration. Alternatively, direct manipulation of the activity of hippocampal neural circuits can be an excellent approach. For example, transplantation of interneurons into the hippocampus showed dramatic effects in reducing epilepsy phenotype [148, 149]. DREADD has been successfully delivered to the hippocampus via the guidance of ultrasound in a non-invasive manner [150], and this method can be applied for targeted chemogenetics to normalize the activity of DGCs. A better understanding of the neural cell types and neural circuits affected by alcohol exposure and withdrawal will provide a strong foundation to understand and treat AUD and AWS.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Alcohol Abuse and Alcoholism (R01AA022377 and R01AA027766 to H.S.). We thank Dr. Christopher L. Nelson for editorial assistance.
REFERENCES
[1] | Koob GF . Neurocircuitry of alcohol addiction: synthesis from animal models. Handbook of Clinical Neurology. 125: Elsevier; (2014) . pp. 33–54. |
[2] | McCance-Katz EFJSA , May MHSAhwsgdsdfn-p–pA. The National Survey on Drug Use and Health: 2017. 2019;7. |
[3] | Jones C , Paulozzi L , Mack KJMMMWR . Centers for Disease Control and Prevention (CDC) Alcohol involvement in opioid pain reliever and benzodiazepine drug abuse-related emergency department visits and drug-related deaths-United States, 2010. (2014) ;63: (40):881–5. |
[4] | Kranzler HR , Soyka MJJ . Diagnosis and pharmacotherapy of alcohol use disorder: a review. (2018) ;320: (8):815–24. |
[5] | Koob GF . Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Behavioral neurobiology of alcohol addiction: Springer; (2011) . pp. 3–30. |
[6] | Becker HCJAR , Health. Health. Alcohol dependence, withdrawal, and relapse. 2008. |
[7] | Jesse S , Bråthen G , Ferrara M , Keindl M , Ben-Men-achem E , Tanasescu R , et al. Alcohol withdrawal syndrome: mechanisms, manifestations, and management. Acta neurologica Scandinavica. (2017) ;135: (1):4–16. |
[8] | Foy A , Kay J , Taylor A . The course of alcohol withdrawal in a general hospital. QJM : monthly journal of the Association of Physicians. (1997) ;90: (4):253–61. |
[9] | Kattimani S , Bharadwaj B . Clinical management of alcohol withdrawal: A systematic review. Industrial psychiatry journal. (2013) ;22: (2):100. |
[10] | Bottlender M , Soyka M . Impact of craving on alcohol relapse during, and 12 months following, outpatient treatment. Alcohol and alcoholism (Oxford, Oxfordshire). (2004) ;39: (4):357–61. |
[11] | Cui C , Noronha A , Warren KR , Koob GF , Sinha R , Thakkar M , et al. Brain pathways to recovery from alcohol dependence. Alcohol (Fayetteville, NY). (2015) ;49: (5):435–52. |
[12] | Hsu D . The dentate gyrus as a filter or gate: a look back and a look ahead. Progress in Brain Research. (2007) ;163: :601–13. |
[13] | Dieni CV , Panichi R , Aimone JB , Kuo CT , Wadiche JI , Overstreet-Wadiche L . Low Excitatory Innervation Balances High Intrinsic Excitability of Immature Dentate Neurons. Nature Communications. Nature Communications. (2016) ;7: (1):1–13. |
[14] | Himmelsbach C . The morphine abstinence syndrome, its nature and treatment. Annals of Internal Medicine. (1941) ;15: (5):829–39. |
[15] | Littleton J . Neurochemical mechanisms underlying alcohol withdrawal. Alcohol Health and Research World. (1998) ;22: (1):13. |
[16] | Koob GF . Neuroadaptive mechanisms of addiction: studies on the extended amygdala. European Neuropsychopharmacology: The journal of the European College of Neuropsychopharmacology. (2003) ;13: (6):442–52. |
[17] | Koob GF . Alcoholism: allostasis and beyond. Alcoholism, Clinical and Experimental Research. (2003) ;27: (2):232–43. |
[18] | Wise RA , Koob GF . The development and maintenance of drug addiction. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology. (2014) ;39: (2):254–62. |
[19] | Becker HC , Mulholland PJ . Neurochemical mechanisms of alcohol withdrawal. Handbook of clinical neurology. (2014) ;125: :133–56. |
[20] | Heilig M , Egli M , Crabbe JC , Becker HC . Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addiction Biology. (2010) ;15: (2):169–84. |
[21] | Koob GF , Le Moal M . Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology. (2001) ;24: (2):97–129. |
[22] | Saitz R . Introduction to alcohol withdrawal. Alcohol Health and Research World. (1998) ;22: (1):5. |
[23] | Carpenter-Hyland EP , Chandler LJ . Homeostatic plasticity during alcohol exposure promotes enlargement of dendritic spines. The European Journal of Neuroscience. (2006) ;24: (12):3496–506. |
[24] | Crews FT , Morrow AL , Criswell H , Breese G . Effects of ethanol on ion channels. International review of neurobiology. International Review of Neurobiology. (1996) ;39: :283–367. |
[25] | De Witte P , Pinto E , Ansseau M , Verbanck P . Alcohol and withdrawal: from animal research to clinical issues. Neuroscience and Biobehavioral Reviews. (2003) ;27: (3):189–97. |
[26] | Grant K , Lovinger DM . Cellular and behavioral neurobiology of alcohol: receptor-mediated neuronal processes. Clinical Neuroscience (New York, NY). (1995) ;3: (3):155–64. |
[27] | Grant KA , Valverius P , Hudspith M , Tabakoff B . Ethanol withdrawal seizures and the NMDA receptor complex. European Journal of Pharmacology. (1990) ;176: (3):289–96. |
[28] | Kotlinska JH , Bochenski M , Danysz W . The role of group I mGlu receptors in the expression of ethanol-induced conditioned place preference and ethanol withdrawal seizures in rats. European Journal of Pharmacology. (2011) ;670: (1):154–61. |
[29] | Rossetti ZL , Carboni S . Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. European Journal of Pharmacology. (1995) ;283: (1-3):177–83. |
[30] | Dahchour A , Quertemont E , De Witte P . Taurine increases in the nucleus accumbens microdialysate after acute ethanol administration to naive and chronically alcoholised rats. Brain Research. (1996) ;735: (1):9–19. |
[31] | Dahchour A , De Witte P . Excitatory and inhibitory amino acid changes during repeated episodes of ethanol withdrawal: an in vivo microdialysis study. European Journal of Pharmacology. (2003) ;459: (2-3):171–8. |
[32] | Haugbøl SR , Ebert B , Ulrichsen J . Upregulation of glutamate receptor subtypes during alcohol withdrawal in rats. Alcohol and Alcoholism (Oxford, Oxfordshire). (2005) ;40: (2):89–95. |
[33] | Paoletti P , Bellone C , Zhou Q . NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience. (2013) ;14: (6):383–400. |
[34] | Kew JN , Kemp JA . Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology. (2005) ;179: (1):4–29. |
[35] | Groc L , Choquet DJS . Linking glutamate receptor movements and synapse function. Science (New York, NY). 2020;368(6496). |
[36] | Gan Q , Salussolia CL , Wollmuth LP . Assembly of AMPA receptors: mechanisms and regulation. The Journal of Physiology. (2015) ;593: (1):39–48. |
[37] | Wang J , Carnicella S , Phamluong K , Jeanblanc J , Ronesi JA , Chaudhri N , et al. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2007) ;27: (13):3593–602. |
[38] | Wang J , Lanfranco MF , Gibb SL , Yowell QV , Carnicella S , Ron DJJoN . Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2010) ;30: (30):10187–98. |
[39] | Olsen RW , Spigelman I . GABAA receptor plasticity in alcohol withdrawal. Jasper’s Basic Mechanisms of the Epilepsies [Internet] 4th edition: National Center for Biotechnology Information (US); 2012. |
[40] | Goddard GV . Development of epileptic seizures through brain stimulation at low intensity. Nature. (1967) ;214(5092):1020–1. |
[41] | Pinel JP . Alcohol withdrawal seizures: implications of kindling. Pharmacology, Biochemistry, and Behavior. (1980) ;13: (Suppl 1):225–31. |
[42] | Pleil KE , Lowery-Gionta EG , Crowley NA , Li C , Marcinkiewcz CA , Rose JH , et al. Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology. (2015) ;99: :735–49. |
[43] | Kroener S , Mulholland PJ , New NN , Gass JT , Becker HC , Chandler LJ . Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PloS one. (2012) ;7(5). |
[44] | Myme CI , Sugino K , Turrigiano GG , Nelson SBJ . The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. Journal of Neurophysiology.. (2003) ;90: (2):771–9. |
[45] | Spigelman I , Marty VN . Long-lasting alterations in membrane properties, K+currents, and glutamatergic synaptic currents of nucleus accumbens medium spiny neurons in a rat model of alcohol dependence. Frontiers in Neuroscience. (2012) ;6: :86. |
[46] | Christian DT , Alexander NJ , Diaz MR , Robinson S , McCool BA . Chronic intermittent ethanol and withdrawal differentially modulate basolateral amygdala AMPA-type glutamate receptor function and trafficking. Neuropharmacology. (2012) ;62: (7):2430–9. |
[47] | Olsen R , Sieghart W . Subtypes of gamma-amin-obutyric acid (A) receptors: classification on the basis of subunit composition, pharmacology, and function. Pharmacological Reviews. (2008) ;60: (3):243–60. |
[48] | Roberto M , Varodayan FP . Synaptic targets: chronic alcohol actions. Neuropharmacology. (2017) ;122: :85–99. |
[49] | Förstera B , Castro PA , Moraga-Cid G , Aguayo LG . Potentiation of gamma aminobutyric acid receptors (GABAAR) by ethanol: how are inhibitory receptors affected? Frontiers in Cellular Neuroscience. (2016) ;10: :114. |
[50] | McKernan RM , Whiting PJ . Which GABAA-receptor subtypes really occur in the brain? Trends in Neurosciences. (1996) ;19: (4):139–43. |
[51] | Hanchar HJ , Dodson PD , Olsen RW , Otis TS , Wallner M . Alcohol-induced motor impairment caused by increased extrasynaptic GABA A receptor activity. Nature Neuroscience. (2005) ;8: (3):339–45. |
[52] | Sieghart W , Sperk G . Subunit composition, distribution and function of GABA-A receptor subtypes. Current Topics in Medicinal Chemistry. (2002) ;2: (8):795–816. |
[53] | Gassmann M , Bettler B . Regulation of neuronal GABA B receptor functions by subunit composition. Nature Reviews Neuroscience. (2012) ;13: (6):380–94. |
[54] | Kang M , Spigelman I , Sapp DW , Olsen RW . Persistent reduction of GABAA receptor-mediated inhibition in rat hippocampus after chronic intermittent ethanol treatment. Brain Research. (1996) ;709: (2):221–8. |
[55] | Kang MH , Spigelman I , Olsen RW . Alteration in the sensitivity of GABAA receptors to allosteric modulatory drugs in rat hippocampus after chronic intermittent ethanol treatment. Alcoholism, Clinical and Experimental Research. (1998) ;22: (9):2165–73. |
[56] | Olsen RW , Spigelman I . GABA(A) Receptor Plasticity in Alcohol Withdrawal. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda (MD): National Center for Biotechnology Information (US)Copyright © 2012. Michael A Rogawski, Antonio V Delgado-Escueta, Jeffrey L Noebels, Massimo Avoli and Richard W Olsen.: (2012) . |
[57] | Olsen RW , Liang J . Role of GABA(A) receptors in alcohol use disorders suggested by chronic intermittent ethanol (CIE) rodent model. Molecular Brain. (2017) ;10: (1):45. |
[58] | Olsen RW , DeLorey TM , Gordey M , Kang MH . GABA receptor function and epilepsy. Advances in Neurology. (1999) ;79: :499–510. |
[59] | Devaud LL , Alele P . Differential effects of chronic ethanol administration and withdrawal on gamma-aminobutyric acid type A and NMDA receptor subunit proteins in male and female rat brain. Alcoholism, Clinical and Experimental Research. (2004) ;28: (6):957–65. |
[60] | Diaz MR , Christian DT , Anderson NJ , McCool BA . Chronic ethanol and withdrawal differentially modulate lateral/basolateral amygdala paracapsular and local GABAergic synapses. The Journal of Pharmacology and Experimental Therapeutics. (2011) ;337: (1):162–70. |
[61] | Lindemeyer AK , Liang J , Marty VN , Meyer EM , Suryanarayanan A , Olsen RW , et al. Ethanol-induced plasticity of GABA A receptors in the basolateral amygdala. Neurochemical Research. (2014) ;39: (6):1162–70. |
[62] | Gage FH . Mammalian neural stem cells. Science (New York, NY). (2000) ;287(5457):1433–8. |
[63] | Gross CG . Neurogenesis in the adult brain: death of a dogma. Nature Reviews Neuroscience. (2000) ;1: (1):67–73 . |
[64] | Alvarez-Buylla A , Lim DA . For the long run: maintaining germinal niches in the adult brain. Neuron. (2004) ;41: (5):683–6. |
[65] | Alvarez-Buylla A , Garcıa-Verdugo JM . Neurogenesis in adult subventricular zone. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. (2002) ;22: (3):629–34. |
[66] | Gil-Perotín S , García-Verdugo JM , Álvarez-Buylla A . Neurogenesis in the Intact Adult Mammalian Central Nervous System. Identification and Characterization of Neural Progenitor Cells in the Adult Mammalian Brain. Advances in Anatomy, Embryology, and Cell Biology. (2009) ;203: :1-101, ix. |
[67] | Quiñones-Hinojosa A , Sanai N , Soriano-Navarro M , Gonzalez-Perez O , Mirzadeh Z , Gil-Perotin S , et al. Cellular composition and cytoarchitecture of the adult human subventricular zone: a niche of neural stem cells. The Journal of Comparative Neurology. (2006) ;494: (3):415–34. |
[68] | Seki T , Sawamoto K , Parent JM , Alvarez-Buylla A . Neurogenesis in the adult brain I: Neurobiology: Springer Science & Business Media; 2011. |
[69] | Kempermann G . Adult neurogenesis: stem cells and neuronal development in the adult brain: Oxford University Press, USA; 2006. |
[70] | Kempermann G , Gage FH , editors. Neurogenesis in the adult hippocampus. Neural Transplantation in Neurodegenerative Disease: Current Status and New Directions: Novartis Foundation Symposium 231; 2000:Wiley Online Library. |
[71] | Kempermann G , Wiskott L , Gage FH . Functional significance of adult neurogenesis. Current Opinion in Neurobiology. (2004) ;14: (2):186–91. |
[72] | Ming G-l , Song H . Adult neurogenesis in the mammalian central nervous system. Annual Review of Neuroscience. (2005) ;28: :223–50. |
[73] | Ming G-l , Song H . Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. (2011) ;70: (4):687–702 . |
[74] | Suh H , Deng W , Gage FH . Signaling in adult neurogenesis. Annual Review of Cell and Developmental Biology. (2009) ;25: :253–75. |
[75] | Vivar C , Potter MC , Choi J , Lee J-y , Stringer TP , Callaway EM , et al. Monosynaptic inputs to new neurons in the dentate gyrus. Nature Communications. (2012) ;3: :1107;. |
[76] | Oppenheim RW . Adult hippocampal neurogenesis in mammals (and humans): the death of a central dogma in neuroscience and its replacement by a new dogma. Developmental Neurobiology. (2019) ;79: (3):268–80. |
[77] | Abbott LC , Nigussie F . Adult neurogenesis in the mammalian dentate gyrus. Anatomia, Histologia, Embryologia. (2020) ;49: (1):3–16. |
[78] | Toda T , Parylak SL , Linker SB , Gage FH . The role of adult hippocampal neurogenesis in brain health and disease. Molecular Psychiatry. (2019) ;24: (1):67–87. |
[79] | Gonçalves JT , Schafer ST , Gage FHJC . Adult neurogenesis in the hippocampus: from stem cells to behavior. (2016) ;167: (4):897–914. |
[80] | Anacker C , Hen R . Adult hippocampal neurogenesis and cognitive flexibility— linking memory and mood. Nature Reviews Neuroscience. (2017) ;18: (6):335. |
[81] | Gebara E , Bonaguidi MA , Beckervordersandforth R , Sultan S , Udry F , Gijs PJ , et al. Heterogeneity of radial glia-like cells in the adult hippocampus. Stem Cells (Dayton, Ohio). (2016) ;34: (4):997–1010. |
[82] | Kempermann G , Song H , Gage FH . Neurogenesis in the adult hippocampus. Cold Spring Harbor Perspectives in Biology. (2015) ;7: (9):a018812. |
[83] | Kriegstein A , Alvarez-Buylla A . The glial nature of embryonic and adult neural stem cells. Annual Review of Neuroscience. (2009) ;32: :149–84. |
[84] | Song JM , ChristianK, MingGl, SongH. Modification of hippocampal circuitry by adult neurogenesis. Developmental Neurobiology. (2012) ;72: (7):1032–43. |
[85] | Song J , Olsen RH , Sun J , Ming G-l , Song H . Neuronal circuitry mechanisms regulating adult mammalian neurogenesis. Cold Spring Harbor Perspectives in Biology. (2016) ;8: (8):a018937. |
[86] | Dieni CV , Chancey JH , Overstreet-Wadiche LS . Dynamic functions of GABA signaling during granule cell maturation. Frontiers in Neural Circuits. (2013) ;6: :113. |
[87] | Tozuka Y , Fukuda S , Namba T , Seki T , Hisatsune T . GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. (2005) ;47: (6):803–15. |
[88] | Ge S , Goh EL , Sailor KA , Kitabatake Y , Ming G-l , Song H . GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. (2006) ;439(7076):589–93. |
[89] | Zhao C , Teng EM , Summers RG , Ming G-l , Gage FH . Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2006) ;26: (1):3–11. |
[90] | Jin X . The role of neurogenesis during development and in the adult brain. The European Journal of Neuroscience. (2016) ;44: (6):2291–9. |
[91] | Cameron HA , McEwen BS , Gould E . Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (1995) ;15: (6):4687–92. |
[92] | Nacher J , Rosell DR , Alonso-Llosa G , McEwen BS . NMDA receptor antagonist treatment induces a long-lasting increase in the number of proliferating cells, PSA-NCAM-immunoreactive granule neurons and radial glia in the adult rat dentate gyrus. The European Journal of Neuroscience. (2001) ;13: (3):512–20. |
[93] | Cho T , Ryu JK , Taghibiglou C , Ge Y , Chan AW , Liu L , et al. Long-term potentiation promotes proliferation/survival and neuronal differentiation of neural stem/progenitor cells. PloS One. (2013) ;8: (10):e76860. |
[94] | Ray RS , Corcoran AE , Brust RD , Kim JC , Richerson GB , Nattie E , et al. Impaired respiratory and body temperature control upon acute serotonergic neuron inhibition. Science (New York, NY). (2011) ;333(6042):637–42. |
[95] | Bruel-Jungerman E , Davis S , Rampon C , Laroche S . Long-term potentiation enhances neurogenesis in the adult dentate gyrus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2006) ;26: (22):5888–93. |
[96] | Chun SK , Sun W , Park J-J , Jung MW . Enhanced proliferation of progenitor cells following long-term potentiation induction in the rat dentate gyrus. Neurobiology of Learning and Memory. (2006) ;86: (3):322–9. |
[97] | Tashiro A , Sandler VM , Toni N , Zhao C , Gage FH . NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature. (2006) ;442(7105):929–33. |
[98] | Deshpande A , Bergami M , Ghanem A , Conzelmann K-K , Lepier A , Götz M , et al. Retrograde monosynaptic tracing reveals the temporal evolution of inputs onto new neurons in the adult dentate gyrus and olfactory bulb. Proceedings of the National Academy of Sciences of the United States of America. (2013) ;110: (12):E1152–E61. |
[99] | Restivo L , Niibori Y , Mercaldo V , Josselyn SA , Frankland PW . Development of adult-generated cell connectivity with excitatory and inhibitory cell populations in the hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2015) ;35: (29):10600–12. |
[100] | Ge S , Yang C-h , Hsu K-s , Ming G-l , Song H . A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. (2007) ;54: (4):559–66. |
[101] | Esther PY , Dengler CG , Frausto SF , Putt ME , Yue C , Takano H , et al. Protracted postnatal development of sparse, specific dentate granule cell activation in the mouse hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2013) ;33: (7):2947–60. |
[102] | Marín-Burgin A , Schinder AF . Requirement of adult-born neurons for hippocampus-dependent learning. Behavioural Brain Research. (2012) ;227: (2):391–9. |
[103] | Dieni CV , Nietz AK , Panichi R , Wadiche JI , Overstreet-Wadiche L . Distinct determinants of sparse activation during granule cell maturation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2013) ;33: (49):19131–42. |
[104] | Brunner J , Neubrandt M , Van-Weert S , Andrási T , Borgmann FBK , Jessberger S , et al. Adult-born granule cells mature through two functionally distinct states. eLife. (2014) ;3: :e03104. |
[105] | Danzer SC . Depression, stress, epilepsy and adult neurogenesis. Experimental Neurology. (2012) ;233: (1):22–32. |
[106] | Krook-Magnuson E , Armstrong C , Bui A , Lew S , Oijala M , Soltesz I . In vivo evaluation of the dentate gate theory in epilepsy. The Journal of Physiology. (2015) ;593: (10):2379–88. |
[107] | Lothman E , Stringer J , Bertram E . The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Research Supplement. (1992) ;7: :301–13. |
[108] | Coulter DA . Chronic epileptogenic cellular alterations in the limbic system after status epilepticus. Epilepsia. (1999) ;40: :s23–s33. |
[109] | Heinemann U , Beck H , Dreier J , Ficker E , Stabel J , Zhang C . The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Research Supplement. (1992) ;7: :273–80. |
[110] | Tuncdemir SN , Lacefield CO , Hen R . Contributions of adult neurogenesis to dentate gyrus network activity and computations. Behavioural Brain Research. (2019) :112112. |
[111] | Parent JM , Timothy WY , Leibowitz RT , Geschwind DH , Sloviter RS , Lowenstein DH . Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (1997) ;17: (10):3727–38. |
[112] | Gray WP , Sundstrom LE . Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Research. (1998) ;790: (1-2):52–9. |
[113] | Jessberger S , Römer B , Babu H , Kempermann G . Seizures induce proliferation and dispersion of doubl-ecortin-positive hippocampal progenitor cells. Experimental Neurology. (2005) ;196: (2):342–51. |
[114] | Huang Y , Doherty JJ , Dingledine R . Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2002) ;22: (19):8422–8. |
[115] | Jessberger S , Nakashima K , Clemenson GD , Mejia E , Mathews E , Ure K , et al. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2007) ;27: (22):5967–75. |
[116] | Isackson PJ , Huntsman MM , Murray KD , Gall CM . BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. (1991) ;6: (6):937–48. |
[117] | Gall CM . Seizure-induced changes in neurotrophin expression: implications for epilepsy. Experimental Neurology. (1993) ;124: (1):150–66. |
[118] | Newton SS , Collier EF , Hunsberger J , Adams D , Terwilliger R , Selvanayagam E , et al. Gene profile of elect-roconvulsive seizures: induction of neurotrophic and angiogenic factors. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2003) ;23: (34):10841–51. |
[119] | Hattiangady B , Shetty AK . Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia. (2008) ;49: :26–41. |
[120] | Kuruba R , Hattiangady B , Shetty AK . Hippocampal neurogenesis and neural stem cells in temporal lobe epilepsy. Epilepsy & Behavior: E&B. (2009) ;14: (Suppl 1):65–73. |
[121] | Parent JM , Elliott RC , Pleasure SJ , Barbaro NM , Lowenstein DH . Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Annals of Neurology. (2006) ;59: (1):81–91. |
[122] | Jessberger S , Zhao C , Toni N , Clemenson GD , Li Y , Gage FH . Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2007) ;27: (35):9400–7. |
[123] | Jessberger S , Parent JM . Epilepsy and adult neurogenesis. Cold Spring Harbor Perspectives in Biology. (2015) ;7(12). |
[124] | Danzer SC . Adult Neurogenesis in the Development of Epilepsy. Epilepsy Currents. (2019) ;19: (5):316–20. |
[125] | Hester MS , Danzer SC . Hippocampal granule cell pathology in epilepsy— a possible structural basis for comorbidities of epilepsy? Epilepsy & Behavior: E&B. (2014) ;38: :105–16. |
[126] | Parent JM , Lowenstein DH . Seizure-induced neurogenesis: are more new neurons good for an adult brain? Progress in brain research. (2002) ;135: :121–31. |
[127] | Zhou Q-G , Nemes AD , Lee D , Ro EJ , Zhang J , Nowacki AS , et al. Chemogenetic silencing of hippocampal neurons suppresses epileptic neural circuits. The Journal of Clinical Investigation. (2019) ;129: (1):310–23. |
[128] | Scharfman HE . The CA3 “backprojection” to the dentate gyrus. Progress in Brain Research. (2007) ;163: :627–37. |
[129] | Althaus A , Sagher O , Parent JM , Murphy GG . Intrinsic neurophysiological properties of hilar ectopic and normotopic dentate granule cells in human temporal lobe epilepsy and a rat model. Journal of Neurophysiology. (2015) ;113: (4):1184–94. |
[130] | Nixon K , Crews FT . Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. Journal of Neurochemistry. (2002) ;83: (5):1087–93. |
[131] | Nixon K , Crews FT . Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2004) ;24: (43):9714–22. |
[132] | Nixon K , Kim D , Potts E , He J , Crews F . Distinct cell proliferation events during abstinence after alcohol dependence: microglia proliferation precedes neurogenesis. Neurobiology of Disease. (2008) ;31: (2):218–29. |
[133] | McClain JA , Hayes DM , Morris SA , Nixon K . Adolescent binge alcohol exposure alters hippocampal progenitor cell proliferation in rats: effects on cell cycle kinetics. The Journal of Comparative Neurology. (2011) ;519: (13):2697–710. |
[134] | Kelso M , Liput D , Eaves D , Nixon K . Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience. (2011) ;197: :381–93. |
[135] | McClain JA , Morris SA , Deeny MA , Marshall SA , Hayes DM , Kiser ZM , et al. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain, Behavior, and Immunity. (2011) ;25: (Suppl 1):S120–8. |
[136] | Herrera DG , Yagüe AG , Johnsen-Soriano S , Bosch-Morell F , Collado-Morente L , Muriach M , et al. Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. Proceedings of the National Academy of Sciences of the United States of America. (2003) ;100: (13):7919–24. |
[137] | Åberg E , Hofstetter CP , Olson L , Brené S . Moderate ethanol consumption increases hippocampal cell proliferation and neurogenesis in the adult mouse. The International Journal of Neuropsychopharmacology. (2005) ;8: (4):557–67. |
[138] | Taffe MA , Kotzebue RW , Crean RD , Crawford EF , Edwards S , Mandyam CD . Long-lasting reduction in hippocampal neurogenesis by alcohol consumption in adolescent nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America. (2010) ;107: (24):11104–9. |
[139] | Richardson HN , Chan SH , Crawford EF , Lee YK , Funk CK , Koob GF , et al. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiology of Disease. (2009) ;36: (1):1–10. |
[140] | Satriotomo I , Miki T , Itoh M , Ameno K , Ijiri I , Takeuchi YJ . Short-term ethanol exposure alters calbindin D28k and glial fibrillary acidic protein immunoreactivity in hippocampus of mice. Brain research. (2000) ;879: (1-2):55–64. |
[141] | Lukoyanov NV , Brandão F , Cadete-Leite A , Madeira MD , Paula-Barbosa MM . Synaptic reorganization in the hippocampal formation of alcohol-fed rats may compensate for functional deficits related to neuronal loss. Alcohol (Fayetteville, NY). (2000) ;20: (2):139–48. |
[142] | Golub HM , Zhou QG , Zucker H , McMullen MR , Kokiko-Cochran ON , Ro EJ , et al. Chronic alcohol exposure is associated with decreased neurogenesis, aberrant integration of newborn neurons, and cognitive dysfunction in female mice. Alcoholism, Clinical and Experimental Research. (2015) ;39: (10):1967–77. |
[143] | Bengochea O , Gonzalo L . Effect of chronic alcoholism on the human hippocampus. Histology and Histopathology. (1990) ;5: (3):349–57. |
[144] | Le Maître TW , Dhanabalan G , Bogdanovic N , Alkass K , Druid H . Effects of alcohol abuse on proliferating cells, stem/progenitor cells, and immature neurons in the adult human hippocampus. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology. (2018) ;43: (4):690–9. |
[145] | Lee D , Krishnan B , Zhang H , Park HR , Ro EJ , Jung Y-N , et al. Activity of hippocampal adult-born neurons regulates alcohol withdrawal seizures. JCI Insight. (2019) ;4(19). |
[146] | Urban DJ , Roth BL . DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annual Review of Pharmacology and Toxicology. (2015) ;55: :399–417. |
[147] | Armbruster BN , Li X , Pausch MH , Herlitze S , Roth BL . Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proceedings of the National Academy of Sciences of the United States of America. (2007) ;104: (12):5163–8. |
[148] | Cunningham M , Cho JH , Leung A , Savvidis G , Ahn S , Moon M , et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell stem cell. (2014) ;15: (5):559–73. |
[149] | Hunt RF , Baraban SC . Interneuron transplantation as a treatment for epilepsy. Cold Spring Harbor Perspectives in Medicine. (2015) ;5(12). |
[150] | Szablowski JO , Lee-Gosselin A , Lue B , Malounda D , Shapiro MG . Acoustically targeted chemogenetics for the non-invasive control of neural circuits. Nature Biomedical Engineering. (2018) ;2: (7):475–84. |


