
Modulation of Neural Networks by Interleukin-1
Abstract
Interleukin-1 (IL-1) is an inflammatory cytokine that has been shown to modulate neuronal signaling in homeostasis and diseases. In homeostasis, IL-1 regulates sleep and memory formation, whereas in diseases, IL-1 impairs memory and alters affect. Interestingly, IL-1 can cause long-lasting changes in behavior, suggesting IL-1 can alter neuroplasticity. The neuroplastic effects of IL-1 are mediated via its cognate receptor, Interleukin-1 Type 1 Receptor (IL-1R1), and are dependent on the distribution and cell type(s) of IL-1R1 expression. Recent reports found that IL-1R1 expression is restricted to discrete subpopulations of neurons, astrocytes, and endothelial cells and suggest IL-1 can influence neural circuits directly through neuronal IL-1R1 or indirectly via non-neuronal IL-1R1. In this review, we analyzed multiple mechanisms by which IL-1/IL-1R1 signaling might impact neuroplasticity based upon the most up-to-date literature and provided potential explanations to clarify discrepant and confusing findings reported in the past.
INTRODUCTION
Interleukin-1 (IL-1) is a master inflammatory cytokine that regulates all aspects of immunological responses of the host against infection and injury [1]. Its neuromodulatory role was first discovered in studies investigating the mechanisms of mammalian sickness symptoms, such as fever, lethargy, social withdrawal, anhedonia, hypophagia, increased secretion of glucocorticoids, and increased slow wave sleep [2]. Sickness symptoms are a set of auxiliary adaptive anti-infection changes controlled by the central nervous system (CNS) to conserve energy, avoid further opportunities of infection, and shift host’s overall biology towards anti-infection metabolism and activity [2]. Even when sickness symptoms/behaviors are caused by immune activity confined to the periphery, the expression of IL-1 can be found in the brain, where it orchestrates many of these responses [3, 4]. For example, brain IL-1 can activate neurons of the preoptic area-raphe pallidus circuit to induce fever [5, 6], stimulate the paraventricular nucleus of the hypothalamus to activate the hypothalamus-pituitary-adrenal gland axis (HPA) to induce glucocorticoids secretion [7], and modulate the hypothalamic feeding control center to cause hypophagia [8, 9]. These neuromodulatory actions are also operative when there are inflammatory activities in the brain itself, such as those during brain injury [10] or after bacterial or viral infection of the CNS [4, 11, 12]. These evidences suggest neural functions of IL-1 are designed to coordinate immunological activities with physiological and behavioral outputs through its actions in the CNS. Interestingly, these adaptive changes may even occur under conditions that are not directly related to an immune activation. For example, acute [13] or chronic [14] psychological stress can also induce brain IL-1 expression, which causes some of the responses resembling sickness symptoms. Thus, neuromodulatory functions of IL-1 may be called upon under a broader set of circumstances that imply the presence of danger to produce a shared set of CNS-controlled host defense responses.
In general, the neural responses to acute sickness are brief and occur immediately when homeostasis is disturbed by infection or stress, suggesting IL-1 could serve as an additional input to neural circuits regulating the relevant physiological and behavioral systems to transiently alter their outputs, producing the protective sickness symptoms. Further investigation of the CNS functions of IL-1 revealed that IL-1 has neuroregulatory roles beyond transitory activities during isolated inflammatory or stressful events. For example, IL-1 levels were found to vacillate with circadian rhythm [15] and influence normal sleep [16, 17]. In addition, a sickness event can cause delayed and lasting changes in cognition and IL-1 was found to play a critical role in the altered processes of learning and memory [18], the consequences of which could affect the nervous system long after the initial triggering infectious or stressful events. For such temporally discontinuous neuromodulatory actions of IL-1 to occur, IL-1 cannot simply act as an additional input to be conducted through existing neuro-circuitries, it must be able to alter the structure of neurocircuitry itself. This possibility is highlighted by the fact that during development, a period when neurocircuits are built and extensively modified, induction of IL-1 causes profound changes in the trajectory of neurodevelopment, resulting in behavioral phenotypes resembling psychological disorders later in life [19–21]. Alteration of neural networks can also occur during adult life and this phenomenon has been termed neuroplasticity. How IL-1 might cause changes in neuroplasticity is the focus of this review.
BASICS OF NEUROPLASTICITY
Classical neuroplasticity was originally conceived by Hebb who suggested coordinated activity between the pre- and postsynaptic neurons strengthens their connection [22]. This “fire together, wire together” hypothesis suggests that certain types of neuronal activities can result in changes of neuronal networks such that dynamic responses of neurons are shaped by prior activation history. Since then, a large literature has been developed to investigate the molecular and structural underpinnings of neuroplasticity and to define the ways neuronal connectivity can be modified. Presently, neuroplasticity is examined from two perspectives: 1) functional plasticity—changes in neuronal excitability; 2) structural plasticity—anatomical changes to neuronal circuits, such as addition or removal of cells or synapses. Overall, neuroplasticity encodes pro-survival information, allowing organisms to learn, retain memories, and predict outcomes based upon prior experiences. It is critical for the maintenance of the learned behavior.
Birth and death of neurons
The founder of modern neuroscience, Ramon y Cajal, famously stated in 1928: “Once the development was ended, the founts of growth and regeneration of the axons and dendrites dried up irrevocably. In the adult centers, the nerve paths are something fixed, ended, and immutable. Everything may die, nothing may be regenerated. It is for the science of the future to change, if possible, this harsh decree [23]”. This sentiment was held firm by neuroscientists for decades. However, breaking the historical status quo, Altman and Das showed incorporation of centrally injected thymidine-H3 into the DNA of proliferating cells of adult rat dentate gyrus and suggested that new neurons can be generated in adult brain [24]. Later studies substantiated this notion of adult neurogenesis using other proliferation markers, such as Bromodeoxyuridine (BrdU) [25, 26]. These studies located adult neurogenesis in discrete areas of the CNS, neurogenic niches. The best-known neurogenic niches are located in the subventricular zone and the hippocampal subgranular zone. The newly generated neurons can migrate from the neurogenic niches to incorporate into existing neuronal networks [27], creating a form of structural neuroplasticity. This neuroplastic process may be crucial for cognitive and affective behaviors. Experimental studies have shown: 1) chemical or genetic ablation of neural progenitor cells (NPC) impairs learning of normal animals [28] and cognitive recovery in animals with brain injury [29]; 2) preservation or enhancement of neurogenesis ameliorates depressive-like behaviors in animal models of stress [30, 31] and enhanced learning in exercising animals [32]. Neurogenesis is now confirmed in many species including fish, reptiles, birds, rodents, non-human primates and humans [40]. Thus far, neurogenesis has been implicated in the consolidation of new memories [33, 34], modification of cognition [35, 36], generation of positive affect [37], and reorganization of neural circuits after injury [38, 39]. Typically, neurogenesis is visualized by immunohistochemical labeling of proliferation markers, proliferation related proteins, immature cell markers, or by retroviral fluorescent labeling of dividing cells [41]. Although neuroplasticity is typically associated with the birth of new neurons, it is significantly influenced by the death of neurons as well because both addition and subtraction of neurons to neural networks alters neural structures. The loss of neurons via necrosis and apoptosis occur following traumatic brain injuries [42], excitotoxic injuries [43], substance abuse [44], and ischemia [45]. In animal models of disease and injury, presence of dying cells correlates with the deficits in regulation of memory and affect, implicating neuronal death in functions dependent on neuroplasticity.
Synaptic structure
Synapses are physical spaces between neurons that allow them to transmit chemical signals from one cell to the next. Synaptic communication or neurotransmission consists of pre-synaptic neurotransmitter production and release followed by a neurotransmitter receptor mediated response in the postsynaptic neuron. Synaptic plasticity is a driving force that contributes to dynamic changes in neurocircuitry that underlie complex cognitive behaviors [46, 47]. Aberrant changes to synaptic structures can be the cause of behavioral abnormalities, such as depression and anxiety [48, 49]. Enduring alterations in synaptic structure can occur in one of three ways: 1) creation and elimination of synapses, 2) alteration of synaptic morphology, and 3) changes to synaptic adhesion. Synaptogenesis, the generation of new synapses, which often manifests as newly formed synaptic spines, occurs on the timescale of days to weeks, thus providing a delayed and lasting effect on neural networks. The importance of proper synaptic network formation is exemplified in neurodevelopmental disorders such as autism spectrum disorder and X-linked mental retardation, where affected individuals have either vastly more [50, 51] or less dendritic spines [52], respectively. In addition, appropriate synaptic elimination, synaptic pruning, is critical to sculpt efficient neural circuits. For example, Huttenlocher et al. found experience-dependent visual learning requires elimination of under-used synapses [53]. Interestingly, synaptic pruning is modulated by glial cells. Recent studies have shown microglia actively engulf synapses in both health [54] and disease [55, 56] and was found essential for memory creation [57] and forgetting [47].
The morphology of synapses, especially spine morphology, can be visualized and analyzed. Dendritic spines can be classified as stubby, mushroom, thin, and filipodia. Each type of spine is suggestive of the maturity of a dendritic spine. Typically, mushroom spines are stable excitatory synapses whereas thin spines or filipodia are immature and unstable [58, 59]. Abnormal amount of certain type(s) of spine morphology have been observed in humans with neurodevelopmental disease, schizophrenia, and Alzheimer’s disease [60, 61]. Another parameter of synaptic structure is synaptic adhesion. Synaptic adhesion proteins contribute to the formation and maintenance of synapses [62]. Perturbations of neuronal adhesion are associated with impaired [63, 64] or exaggerated synaptic growth [65] which have been found in brains of patients with mental health diseases like addiction [66] and schizophrenia [67]. Synapses can be visualized by immunolabeling of synaptic proteins, by viral or genetic neuronal expression of reporter fluorescent proteins, and by electron microscopy. Synaptic adhesion has been studied in cultured cells expressing synaptic adhesion molecules.
Neuronal excitability
From the functional perspective, neuroplasticity is measured by the altered relationship between presynaptic inputs and postsynaptic responses —amplified or diminished excitability. Classic examples of functional plasticity are long term potentiation (LTP) and long term depression (LTD). During LTP the postsynaptic response is enhanced by a prior tetanus stimulus, a high frequency stimulation. Following the tetanus stimulation, influx of high levels of calcium is induced in the postsynaptic neuron to activate downstream signaling and change the postsynaptic receptor composition, especially AMPA receptors (AMPAR) and NMDA receptors (NMDAR), resulting in increased excitability of the postsynaptic membrane. During LTD, on the other hand, the postsynaptic response is suppressed after a low frequency stimulus conditions the postsynaptic neuron to hypo-respond to input stimulation. Influx of low levels of calcium following the low frequency stimulation causes phosphatases in the postsynaptic neuron to inhibit pathways that recruit AMPAR, thus causing decreased postsynaptic responses [68]. LTP and LTD play crucial roles in the generation of lasting memory, in cognitive function, and in mood disorder [69]. Beyond LTP and LTD, changes in resting membrane potential, ion channels, surface receptors, and rate of neurotransmitter clearance can also influence neuronal excitability. These influencers of excitability are clinically significant. Treatment for bipolar disorder alters resting neuronal membrane potential [70]; mutations in ion channels are known to contribute to the etiology of epilepsy [71]; alterations of AMPA/NMDA receptor ratio influences memory [72]; and abnormal neurotransmitter clearance is implicated in both depression [73] and excitotoxic injury [74]. Traditionally, LTP and LTD are measured using electrophysiological recordings in a slice preparation or in vivo. Other electrophysiological methods, such as whole cell patch clamp, are used to measure neuronal excitability. Additionally, imaging techniques have been employed to visualize neuronal calcium influx in vitro and in vivo [75].
INTERLEUKIN-1 AND NEUROPLASTICITY
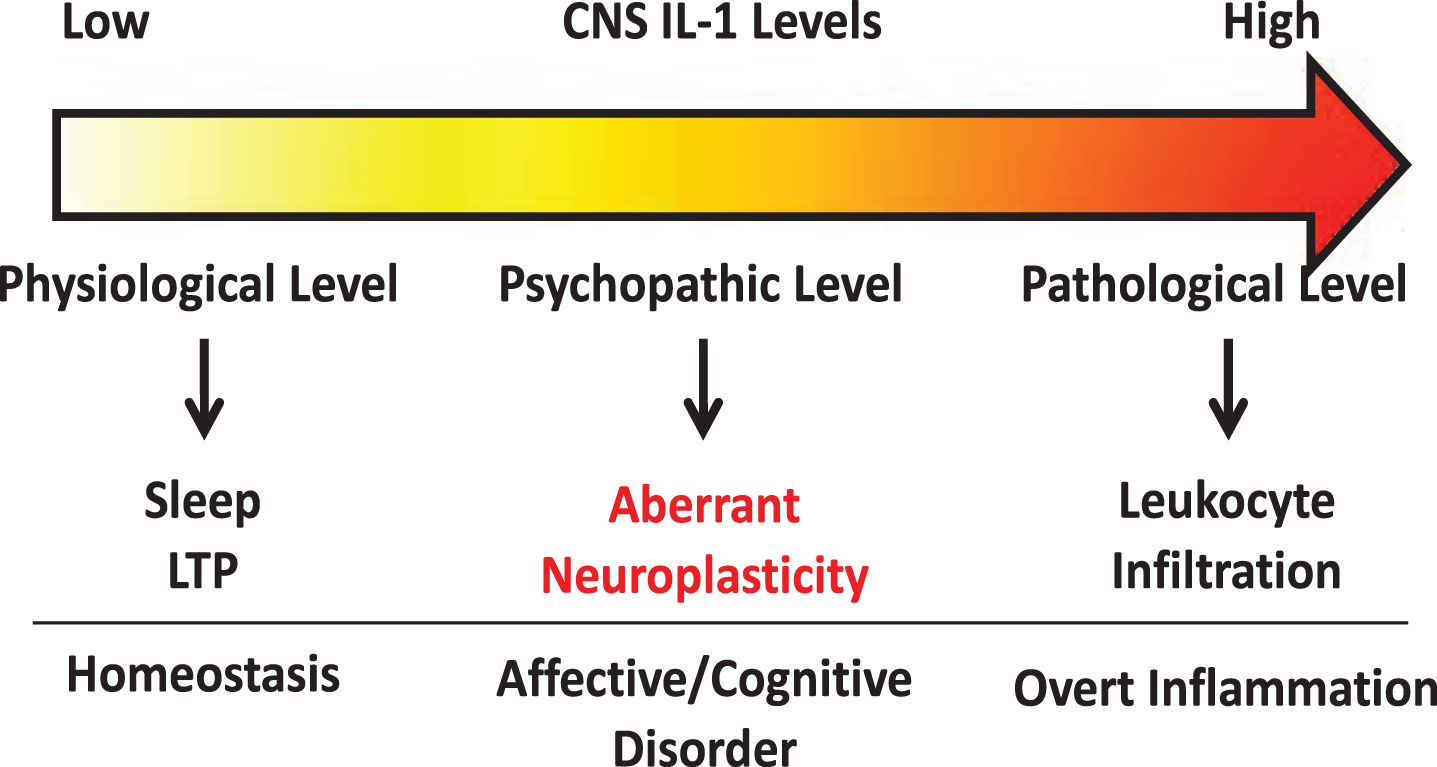
Several lines of research suggest IL-1 modulates neuroplasticity. Under physiological conditions, IL-1 levels are known to oscillate throughout the day [15]. Deletion of IL-1 receptor decreases sleep while central administration of IL-1, at low picogram levels, increases slow wave sleep in rodents [76, 77]. IL-1 has been found to be induced by learning processes [78] and is critical for spatial [79], but not associative learning [80], suggesting specific neural circuits are susceptible to modulation by IL-1. Furthermore, blockade of IL-1 signaling has been shown to impair memory consolidation [78, 81]. Therefore, these findings support a role for IL-1 in regulating physiological neuroplasticity in the generation of memory. In contrast, in models of severe stress [14, 82], brain injury [83, 84], or IL-1 overexpression [85], IL-1 was found to impair memory. These conditions are associated with significantly induced brain IL-1 expression, suggesting IL-1 at pathological levels might exert opposite effects on memory versus its physiological effects. At nanogram levels, central IL-1 elicits robust neuroinflammatory responses, such as leukocyte infiltration, microglial activation, and additional cytokine production [86]. Interestingly, mental health diseases like chronic fatigue [87, 88], depression [89, 90], anxiety [91, 92], and panic disorder [93] have all been linked with elevated brain IL-1. However, in these diseases overt neuroinflammatory responses are often not found, but above normal IL-1 levels are present [90]. Thus, a spectrum of IL-1 activities may exist depending on its concentration. We propose that very low IL-1 concentrations regulate normal physiological functions whereas very high IL-1 concentrations cause overt neuroinflammation. In between, chronically elevated IL-1 above physiological levels may alter neuronal plasticity and is important for the pathogenesis of psychopathology (See Fig. 1). In this review, we will dissect the diverse modalities by which IL-1 regulates neuroplasticity.
Fig. 1
Schematic of concentration dependent IL-1 signaling within the CNS.
Essential to understanding how IL-1 influences neuroplasticity, it is important to understand how IL-1 mediates intracellular signaling and how IL-1 receptors are distributed in the brain. IL-1 exists in two isoforms, IL-1α and IL-1β. They both bind to a single receptor, the type 1 Interleukin-1 Receptor (IL-1R1) to mediate signaling. IL-1/IL-1R1 complex recruits IL-1R1 accessory proteins (IL-1RAcP). IL-1RAcP also exists in two isoforms, IL-1RAcP and IL-1RAcPb. IL-1RAcP is known to recruit the adaptor protein, MyD88, which initiates activation of pathways such as NFκB and p38-MAPK. IL-1RAcPb prevents the recruitment of MyD88 and IL-1R1/IL-1AcPb complex signals through pathways such as AKT [94] or Src [95]. Interestingly, IL-1RAcPb is expressed specifically in neurons [96] and is up to 1000 times more sensitive to IL-1 than IL-1RAcP [95]. Therefore, IL-1 could stimulate neurons and non-neuronal cells very differently via different downstream signaling pathways and elicit dramatically different sensitivities on these cell types. Originally, IL-1R1 expression was thought to be ubiquitously expressed throughout the CNS [97]. However, using advanced genetic reporter we showed that IL-1R1 expression is restricted to specific endothelial cells, ventricular cells, astrocytes, and neurons in discrete subpopulations of the CNS [86, 98]. Restricted neuronal IL-1R1 expression suggests direct neuronal modulation by IL-1 is limited to select neuronal circuits. In this review, we will utilize this new information to analyze the direct versus indirect mechanisms by which IL-1 alters neuroplasticity.
IL-1 and neurogenesis
In non-neuronal cells, IL-1 can induce the proliferation of bone marrow derived mesenchymal stem cells [99, 100]. In neural tissue, on the contrary, in vitro data shows IL-1 is a potent inhibitor of NPC proliferation [101–104] suggesting the action of IL-1 is anti-neurogenic. Indeed, CNS IL-1 induction by bacterial [105, 106] and viral [11] infection, stress [107, 108], radiation [109, 110] and neurodegeneration [111] is correlated with decreases in brain NPCs. Furthermore, direct stimulation with IL-1 in the hippocampal neurogenic niche decreases neurogenesis [85, 86, 112]. The potent anti-neurogenic actions of IL-1 can be inhibited with IL-1R antagonist (IL-1Ra) [11, 111, 113], abrogated by IL-1R1 knockout [86, 114], or suppressed by voluntary exercise which reduced IL-1 expression [115, 116]. Surprisingly, neurogenesis is inhibited in transgenic mice that overexpresses IL-1Ra [117], but not in IL-1R1 knockout mice [86]. It is possible that the anti-neurogenic effect of transgenic overexpression IL-1Ra is due to mechanisms independent of IL-1R1.
Two mechanisms have been proposed to explain IL-1-mediated inhibition of neurogenesis: 1) inhibition of NPC proliferation, and 2) alteration of the cell fate of NPCs. The first mechanism is supported by the following evidences: 1) IL-1 was found to decrease incorporation of thymidine analogues in NPC [11, 86, 107]; 2) IL-1 activates signaling pathways associated with cell cycle arrest [118, 119]; and 3) IL-1 decreases the number of immature neurons [85, 86, 112]. The second mechanism is supported by: 1) In vitro stimulation of NPCs with IL-1α and IL-1β caused an increase in the amount of astroglia-like cells and a decrease of differentiated neurons [120, 121]; 2) In vivo, in a disease model of West Nile Virus, an increase in proliferating astrocytes and a decrease in neurogenesis were found in an IL-1R1 dependent manner [11].
Recently we have found IL-1R1 expression in dentate granule neurons near the hippocampal subgranular zone, but not in the adjacent NPCs, suggesting the anti-neurogenic effect of IL-1 must be indirect [86]. Indeed, conditional knockout of NPC MyD88 did not attenuate IL-1-induced suppression of neurogenesis [85]. Furthermore, Liu et al. showed that IL-1 induced anti-neurogenesis can be mediated indirectly via endothelial and myeloid IL-1R1 [86].
IL-1 and neuronal cell death
Historically, IL-1 was first found to alter neural networks by distorting neuronal connectivity via neuronal death, especially in brain injury and neurodegenerative diseases. Under these conditions, the expression of IL-1 coincides with loss of neurons. In in vitro studies, high levels of IL-1 can directly induce neuronal apoptosis [118, 122–124]. However, in these studies, neurons were cultured in the presence of glial cells and it is possible that IL-1 promoted neuronal death indirectly through glial cells. Indeed, Thornton et al. found the neurotoxic effect of IL-1 was abrogated when glial cells were removed from the culture [124, 125]. Consistently, IL-1 was found to stimulate glial production of both reactive oxygen and nitrogen species (ROS and NOS) which are harmful to neurons [124, 126]. In contrast, other studies found IL-1 can stimulate glial cells to produce nerve growth factor that is potentially neuroprotective [127, 128]. These in vitro results suggest IL-1 is capable to produce opposite effects on neuronal survival via glial cells.
Surprisingly, intracerebral administration of IL-1 in vivo does not affect neuronal survival [86, 129–131], suggesting IL-1 alone does not cause neuronal death even in the presence of glial cells in vivo. However, blockade of IL-1 signaling by genetic deletion of IL-1R1 or over-expression of IL-1Ra reduced neuronal damage in animal models of stroke [84] and epilepsy [132], suggesting that the neurotoxic effect of IL-1 will manifest if other injurious factors are present. Indeed, when IL-1 was administered during CNS injury [133], stroke [134], or co-administered with excitotoxins [130, 135], neuronal apoptosis was exacerbated.
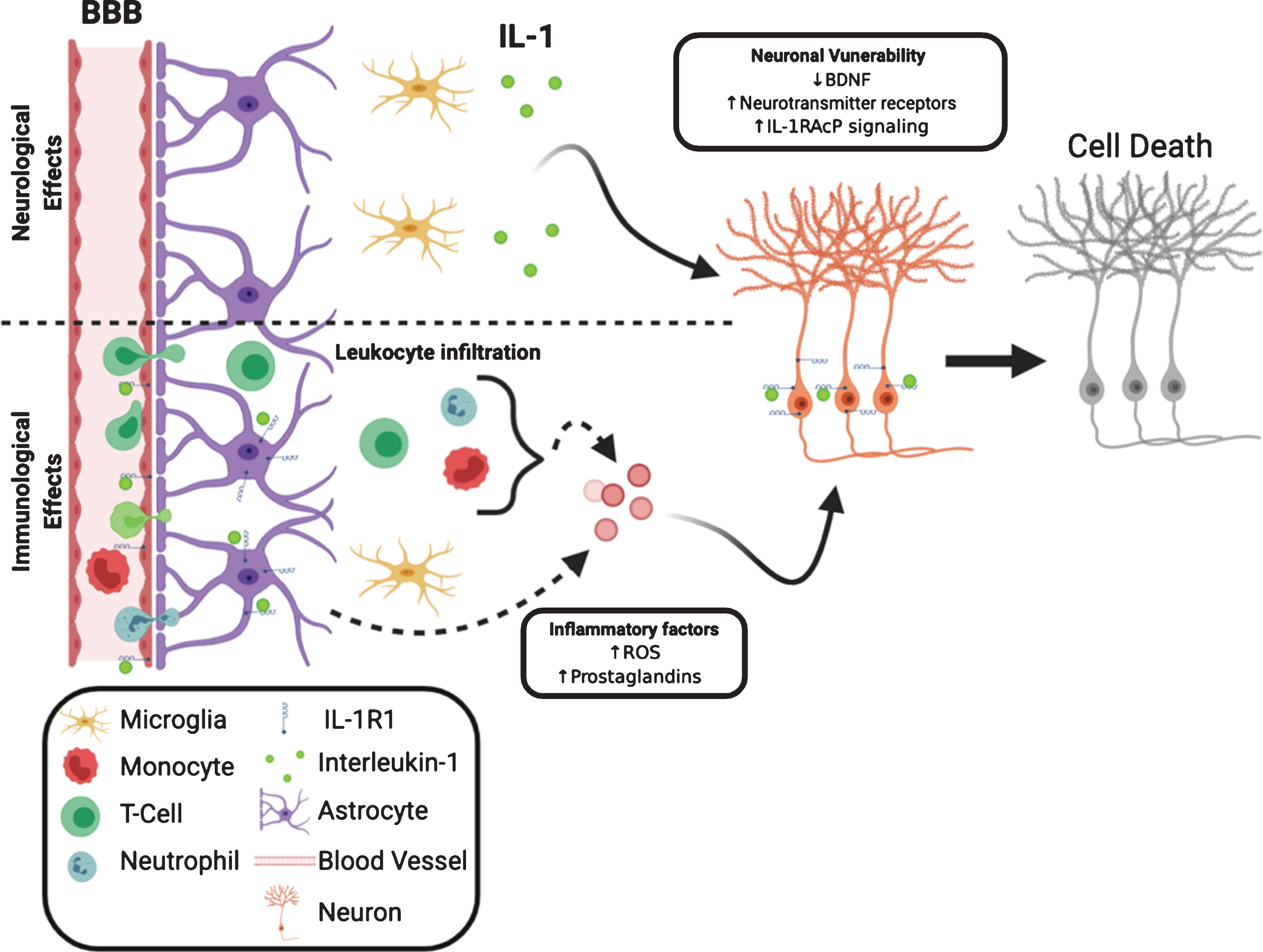
One mechanism for the exacerbation of neuronal death could be IL-1-mediated leukocyte infiltration into the CNS. In experimental models of neurodegeneration, such as epilepsy [136], ischemia [137], and experimental autoimmune encephalomyelitis (EAE) [138], IL-1 recruits leukocytes to the brain and is crucial for disease progression. Blockade of recruitment of leukocytes reduces neuronal death in these conditions. A second mechanism by which IL-1 exacerbates neuronal death is the induction of inflammatory factors such as ROS, NOS, and prostaglandins. Multiple studies found glial ROS and NOS induced by IL-1 cause neuronal death [124–126]. Interestingly, IL-1-induced endothelial prostaglandin E2 production is neurotoxic whereas IL-1 induced prostacyclin produced from neurons is neuroprotective [139]. The third mechanism is IL-1 could make neurons vulnerable by reducing neuronal viability. The maintenance of neuronal survival is associated with the presence of neurotrophic factors, such as brain derived neurotrophic factor (BDNF). BDNF is known to promote neuronal survival and a decrease of BDNF levels can make neurons more susceptible to injury [140]. IL-1 has been shown to impair the production of BDNF mRNA in vivo [141] and BDNF protein in vitro. IL-1 also interferes with BDNF-mediated signaling in cortical neurons, which results in decreased neuronal survival [123]. In addition, IL-1 can induce upregulation of neuronal excitatory neurotransmitter receptors thus conferring an increased sensitivity to excitotoxin [142]. Therefore, both the immunological (leukocyte recruitment and induction of inflammatory factors) and the neurological (enhancement of neuronal vulnerability) effects of IL-1 may converge to account for IL-1 mediated exacerbation of neuronal death during tissue injury. We have diagrammed these interactions in Fig. 2. The neurotoxic effects of IL-1 are most likely mediated by the canonical IL-1R1 signaling pathway. Blockade of NFκB and p38 abrogated neuronal death in models of injury [143, 144]. Recently a new IL-1R-mediated pathway has been discovered. In this pathway IL-1R1 is associated with IL-1RAcPb, not IL-1RAcP. Current studies indicate the IL-1R1/IL-1RAcPb signaling is neuroprotective because deletion of IL-1RAcPb enhanced neuronal death after excitotoxic [145] or immunological challenges [96]. IL-1RAcPb is neuronal specific; therefore, the IL-1-mediated neuroprotective effects would occur only in IL-1R1 and IL-1AcPb expressing neurons. We recently found IL-1R1 expression is limited to discrete subpopulations of neurons, suggesting the neuroprotective effects of IL-1 signaling in neurons is not universal. The balance between the canonical IL-1R1/IL-1RAcP and the alternative IL-1R1/IL-1RAcPb signaling could determine whether IL-1 exerts a neurotoxic or neuroprotective effect. Indeed during aging, where neurons are more susceptible to injury, increased IL-1RAcP/IL-1RAcPb ratio is correlated with increased inflammatory signaling and neuronal death [145].
Fig. 2
Neurological and immunological IL-1 effects converge to influence neuronal survival. IL-1R1 can directly influence neuronal viability via decreasing neurotrophic factors, increasing membrane-bound neurotransmitter receptor and increasing inflammatory signaling. The decreased neuronal viability is coupled with increased release of neurotoxic inflammatory factors from non-neuronal cells, such as leukocytes, astrocytes and endothelia.
Synaptic changes and IL-1
IL-1 has been shown to impact three aspects of synaptic structure: 1) neurotransmitter receptor composition, 2) spine density, and 3) synaptic adhesion. In vitro, IL-1 was shown to induce an increase expression of NMDARs [146] and reduced expression of AMPARs [147]. These IL-1 induced changes were confirmed in an in vivo model of hyperammonemia. Furthermore, these IL-1-induced changes in neurotransmitter receptor composition can be abrogated ex vivo in hippocampal slices by treatment with IL-1Ra [148]. Only three studies have investigated the effect of IL-1 on the density of synapses. In one study, IL-1 applied to cultured primary hippocampal neurons at a high concentration (3 ng/mL for 24 hrs) caused a decrease in postsynaptic densities measured by PSD95 immunolabeling [149]. In another, low levels (0.05 ng/mL for 30 mins) of IL-1β did not alter synaptic density [146]. In the third study, IL-1R1 deletion resulted in an increase of postsynaptic markers, suggesting endogenous IL-1 acts to reduce the amount of postsynaptic sites [150]. In terms of synaptic adhesion, Yoshida et al. found IL-1RAcP and IL-1RAcPb can couple, trans-synaptically, with protein tyrosine phosphatase receptor type delta (PTPRδ) to serve as synaptic adhesion molecules thus guiding synaptic formation [151]. In a cell culture system, expression of IL-1RAcP increased the protein expression of presynaptic markers, Bassoon and VGLUT1 and knockdown of IL-1RAcP reduced synapse formation. Interestingly, these two isoforms of IL-1RAcP appear to play distinct roles in synaptogenesis: whereas IL-1RAcP is important for presynaptic differentiation, IL-1RAcPb is important for postsynaptic differentiation [152]. Related to IL-1RAcP, Interleukin-1 Receptor Accessory Protein-Like 1 (IL-1RAPL1) is known to be critical in the formation of synapses. IL-1RAPL1 shares key features of the extracellular domains, transmembrane domain and signaling domain with IL-1RAcPb and is known to mediate synaptogenesis modulated by IL-1 [153]. Similar to IL-1RAcPb, IL-1RAPL1 also contains an extended intracellular C-terminus domain that prevents the activation of NFκB [154]. Mutations or genetic deletions in IL-1RAPL1 cause learning disabilities and autism-like syndromes in humans and mice in association with abnormal synaptic morphology [153, 155]. Neurons with IL-1RAPL1 knockdown or neurons from IL-1RAPL1 knockout mice have decreased dendritic arborization, decreased spine density, and decreased excitatory synapses [151, 156]. IL-1AcP knockout mice show decreased in synaptic spines within the cortex and hippocampus [152], however, it is unknown whether deletion of IL-1RAcPb could cause similar effects and whether IL-1 is involved in the regulation of synaptic adhesion.
LTP/LTD and IL-1
Influence of IL-1 on functional synaptic plasticity, such as LTP and LTD, has also been studied. Katsuki et al. were the first to show IL-1 can inhibit LTP in the CA3 neurons of the hippocampus [157]. Since this seminal finding, others studies have also shown IL-1 can inhibit LTP in the neurons of the CA1 [158, 159], CA3, and dentate gyrus of the hippocampus [160–163].
On the contrary, others suggest that IL-1 is critical for induction of LTP and its maintenance. When LTP was induced with electrical stimulation of hippocampal slices or in vivo, an increase in IL-1 was observed [164, 165]. Likewise, the act of learning was found to induce IL-1 production in the hippocampus suggesting learning associated neuronal activation can drive IL-1 expression. The level of IL-1 induction was correlated to the reduction of errors in a memory test suggesting IL-1 aids in the learning process [78, 164]. In addition, when IL-1R1 signaling was pharmacologically blocked by IL-1Ra, LTP maintenance was reduced and learning was impaired [165, 166]. In line with these findings, Avital et al. found LTP in dentate gyrus or CA1 neurons was not inducible in IL-1R1 KO mice [81]. IL-1 may also modulate LTD. In one study, application of IL-1 reduced basal neuronal responsiveness, preventing subsequent induction of LTD [167].
Several confounding factors need to be considered when assessing the role of IL-1 in LTP and LTD. Firstly, most electrophysiology studies are done in acute slices that are known to induce the expression of endogenous IL-1 in the injured slices. This injury associated increase in IL-1 may mask the effect of physiological levels of IL-1 in the slice. To reduce this potential artifact, two methods have been employed: 1) lower the incubation and recording temperatures and 2) increase the pre-test incubation duration. Ross et al. studied LTP in slice preparation at room temperature because injury related IL-1 release is higher at physiological temperature [166]. Schneider et al. incubated the slice for more than 5 hours before LTP recording to allow for injury-induced IL-1 to diminish [165]. Therefore, results from in vitro studies done at different temperatures or incubation preparations may be divergent. Secondly, the use of IL-1Ra to verify the effect of IL-1 could be confounded by the off-target effects. This unexpected possibility was suggested by Loscher et al. after showing application of IL-1Ra impaired LTP in synaptosomes from IL-1R1 deficient mice [168].
Most recent analysis suggests that IL-1 may modulate LTP in a synapse-specific manner. In hippocampal neurocircuitry, dentate gyrus granule neurons project to CA3 neurons via mossy fibers and CA3 neurons project to CA1 neurons via Schaffer collaterals. IL-1 has been shown to inhibit Schaffer/CA1 LTP, but not dentate/CA3 LTP [159]. Our studies have revealed that neuronal IL-1R1 expression is restricted to dentate granule cells in hippocampus. Therefore, the effects seen by Hoshino et al. in the CA1 cells are most likely indirect because CA3 and CA1 neurons do not express IL-1R1. The same conclusion may apply to most of the aforementioned studies because LTP is recorded in neurons in brain regions that do not express neuronal IL-1R1. Indirect mechanisms of modulation of LTP by IL-1 have been examined in previous literature. For example, IL-1-induced ROS has been found to mediate LTP impairment, which is rescued with antioxidant treatments [163].
Neuronal excitability and IL-1
The influence of IL-1 on neuronal excitability has been studied extensively; but the reported results are discrepant and confusing. Numerous studies have shown IL-1 suppresses neuronal excitability. IL-1 has been found to hyperpolarize neurons in the amygdala [169], anterior hypothalamus [170], dorsal motor nucleus [171], and hippocampus [81, 167], suggesting IL-1 can reduce the excitability of neurons. In support, IL-1 has been found to enhance the hyperpolarizing chloride [172, 173] and potassium currents [94] and inhibit the depolarizing sodium [174–176] and calcium currents [173, 176–179]. In addition, IL-1 has been shown to reduce the release of the excitatory neurotransmitter glutamate [163, 178, 180] and enhance the release and signaling of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) [172, 173, 181–183]. These findings are consistent with studies that show IL-1 can reduce excitatory postsynaptic potentials [169]. Finally, IL-1 suppressed spreading depolarization in a model of cortical ischemia-reperfusion [184]. Taken together this set of evidences suggests IL-1 is inhibitory for neuronal excitability.
Conversely, evidences exist to support that IL-1 can enhance neuronal excitability. IL-1 has been shown to induce depolarization in neurons in the hypothalamus [170, 185], subfornical organ [186], and trigeminal ganglion [187]. IL-1 has been shown to suppress the inhibitory chloride [188] and potassium currents [189, 190] and increase excitatory sodium currents [187]. In addition, IL-1 was found to increase neuronal intracellular calcium [142, 191, 192]. In models of three different diseases IL-1 was also found to increase neuronal excitability. In epilepsy, IL-1 suppressed GABA induced inhibitory currents [193]; in a prion disease model, IL-1 depolarized CA1 neurons [194]; and in multiple sclerosis, elevated IL-1 cerebrospinal fluid (CSF) level was correlated with an increased response to transcranial magnetic stimulation, suggesting the neurons are hyperexcitable [195]. The increased neuronal activity can be blocked by IL-1Ra suggesting IL-1R1 signaling mediates the neuronal excitability increase in these diseases [195–197]. Taken together this evidence suggest IL-1 is excitatory.
Beyond the apparent contradictory evidences reviewed above, Zhu et al. found that under basal conditions, IL-1 increased glutamate and GABA release, but suppressed neurotransmission once neurons were activated [198]. This suggests IL-1 modulates neuronal activity depending on state of activation.
Here we propose three possible explanations for the discrepancy in the literature. Firstly, IL-1 signaling may differentially influence neuronal excitability due to concentration-dependent activities of IL-1. Multiple studies found the kinetics of IL-1 signaling on neuronal excitability acts as a U-shaped curve, such that low and high concentrations of IL-1 exert opposite effects on neuronal excitability. This is demonstrated by two studies showing low concentrations of IL-1 and high concentrations of IL-1 causes opposite effects on neuronal excitability [177, 186]. In addition, neuronal IL-1R1 has been shown to have higher sensitivity to IL-1 compared to non-neuronal IL-1R1 [86, 199]. Therefore, different levels of IL-1 present during recording could engage different biochemical and cellular targets. Secondly, the presence or absence of non-neuronal IL-1R1 signaling could confound the interpretation of IL-1-mediated changes in neuronal excitability. For example, IL-1 induced hyperpolarization of dorsal motor neurons was found to be dependent on prostaglandin synthesis [171] and hyperpolarization of hypothalamic [170] or amygdala neurons [181] was shown to be dependent on MyD88 signaling. These signaling cascades are typically observed downstream of IL-1R1 activation in non-neuronal cells such as endothelia and astrocytes, suggesting non-neuronal cells near the recorded neurons could be the main driver of the observed IL-1 effects in these studies. Thirdly, previous studies were hindered by the lack of identification of IL-1R1 on the recorded neurons. Even assuming non-neuronal IL-1R1 does not participate in the IL-1 induced fast electrophysiological effects, the recorded effects on a neuron without IL-1R could still be resulted from changes occurred in a nearby neuron that does have IL-1R. This notion is supported a previous data that showed in certain brain regions, such as the striatum, neuronal activity was not altered by IL-1 [200]. We have observed in our IL-1R1 reporter mice, neuronal IL-1R1 is absent in the striatum (unpublished data). Future studies need to clarify the role of IL-1 on modulating neuronal excitability by identifying the precise cellular IL-1R involved in the studied effects.
Influence of IL-1 on neuronal excitability could impact neuroplasticity. Recent literature describes a new paradigm of non-Hebbian neuroplasticity where the alteration of neuronal excitability can influence how apt a circuit is to undergo plasticity, which is termed intrinsic plasticity. For example, quick calcium influx in hippocampal CA1 neurons can elicit place cell formation without repeated wiring of the circuit to produce rather instant spatial memory [201]. Whether IL-1 can alter intrinsic neuroplasticity has yet to be determined.
SUMMARY/CONCLUSION
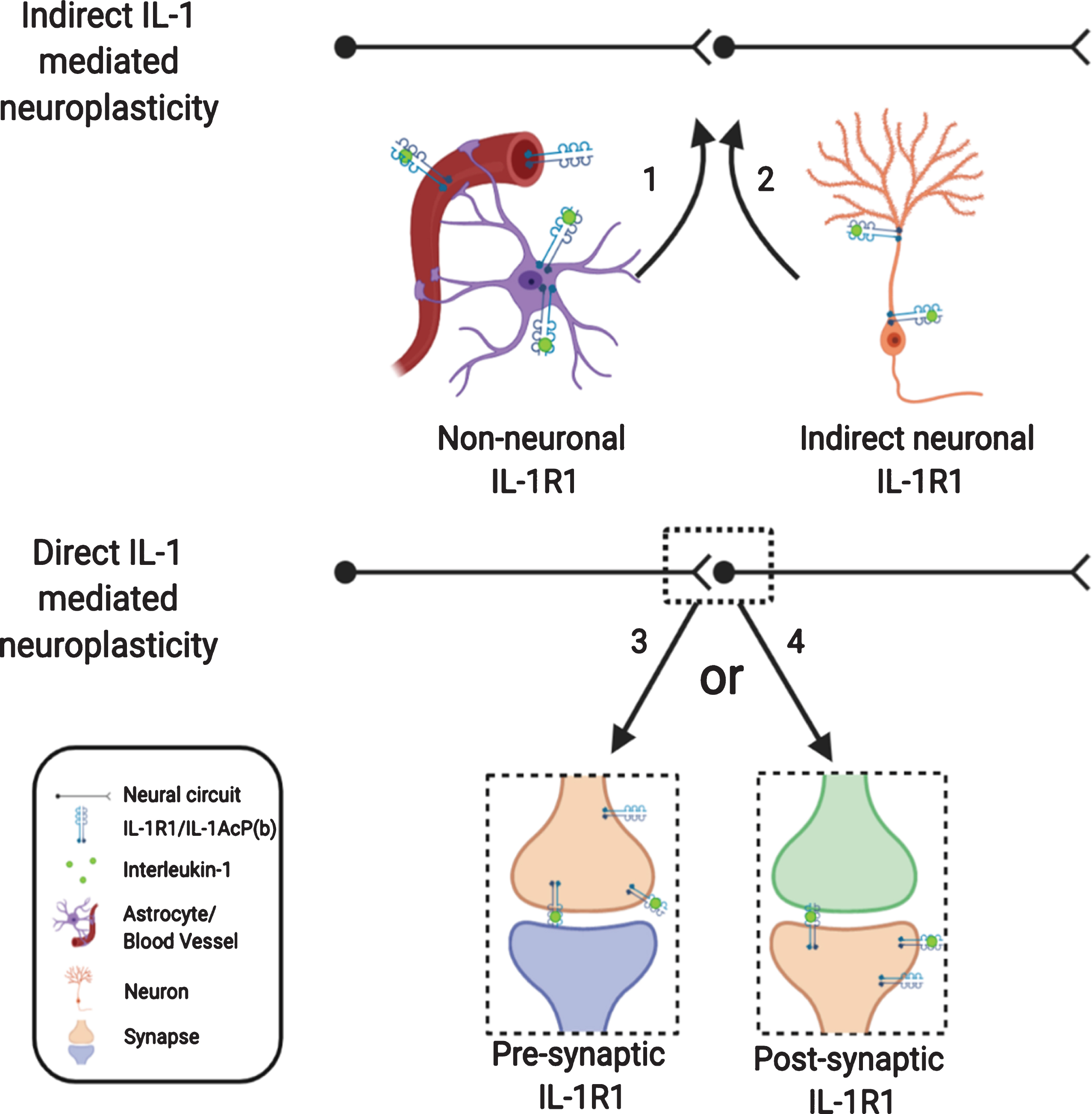
In this review, we analyzed the role of IL-1 in modulating neuroplasticity from the current literature. While the significance of IL-1-mediated changes in neuroplasticity is becoming increasingly clear in neurophysiology, neuropathology, and the pathogenesis of psychological and psychiatric disorders, how IL-1 causes these changes are complex and multi-faceted. Indirect pathways utilizing non-neuronal IL-1R1, which have been somewhat discounted in the past, could play predominant roles in shaping neuroplasticity. The direct influence of IL-1 on neuroplasticity through neuronal IL-1R1 is also more intricate than previously thought, due to restricted and discrete expression of neuronal IL-1R1 in subpopulations of neurons. How presynaptic IL-1R1 and postsynaptic IL-1R1 differentially modify neuroplasticity remains to be determined. In addition, different levels of IL-1 produced under physiological, psychopathological, and inflammatory conditions could modulate neuroplasticity through different modalities by engaging different IL-1R1 expressing cell types or different IL-1R1 expressing neurons. These conceptualizations are diagramed in Fig. 3.
Fig. 3
Nonconventional IL-1R1 mediated neuroplasticity. IL-1 may exert its effect indirectly via non-neuronal IL-1R1 or via IL-1R1 on outlying neurons. IL-1 may mediate its effect directly on neurons with IL-1R1, however, IL-1R1 signaling may initiate various responses depending on whether IL-1R1 is expressed pre- or postsynaptically.
ACKNOWLEDGMENTS
Thanks to Sam McGovern for language and format edits. Figures within this review were created with BioRender.com.
FUNDING
This study was supported by National Institute of Mental Health (NIMH) R01-MH109165, Comprehensive Training in Oral and Craniofacial Sciences (CTOC) T32-DE014320, and National Institute of Neurological Disorders and Stroke (NINDS) R01-NS11694.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
REFERENCES
[1] | Garlanda C , Dinarello CA , Mantovani A . The Interleukin-1 Family: Back to the Future. Immunity. (2013) ;39: (6):1003–18. |
[2] | Konsman JP , Parnet P , Dantzer R . Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. (2002) ;25: (3):154–9. |
[3] | Quan N , Whiteside M , Herkenham M . Time course and localization patterns of interleukin-ib messenger rna expression in brain and pituitary after peripheral administration of lipopolysaccharide. 1998;13. |
[4] | Quan N , Sundar SK , Weiss JM . Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J Neuroimmunol. (1994) ;49: (1):125–34. |
[5] | Saper CB , Romanovsky AA , Scammell TE . Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat Neurosci. (2012) ;15: (8):1088–95. |
[6] | Xin L , Blatteis CM . Blockade by interleukin-1 receptor antagonist of IL-1β-induced neuronal activity in guinea pig preoptic area slices. Brain Res. (1992) ;569: (2):348–52. |
[7] | Goshen I , Yirmiya R . Interleukin-1 (IL-1): A central regulator of stress responses. Front Neuroendocrinol. (2009) ;30: (1):30–45. |
[8] | Dunn AJ , Swiergiel AH . The Role of Cyclooxygenases in Endotoxin- and Interleukin-1-Induced Hypophagia. Brain Behav Immun. (2000) ;14: (3):141–52. |
[9] | Dunn AJ , Swiergiel AH . The Reductions in Sweetened Milk Intake Induced by Interleukin-1 and Endotoxin Are Not Prevented by Chronic Antidepressant Treatment. Neuroimmunomodulation. (2001) ;9: (3):163–9. |
[10] | Murray KN , Parry-Jones AR , Allan SM . Interleukin-1 and acute brain injury. Front Cell Neurosci [Internet]. 2015 Feb 6 [cited 2020 Apr 11];9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4319479/ |
[11] | Garber C , Vasek MJ , Vollmer LL , Sun T , Jiang X , Klein RS . Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat Immunol. (2018) ;19: (2):151–61. |
[12] | Orr AG , Sharma A , Binder NB , Miller AH , Pearce BD . Interleukin-1 Mediates Long-Term Hippocampal Dentate Granule Cell Loss Following Postnatal Viral Infection. J Mol Neurosci. (2010) ;41: (1):89–96. |
[13] | Blandino P , Barnum CJ , Solomon LG , Larish Y , Lankow BS , Deak T . Gene expression changes in the hypothalamus provide evidence for regionally-selective changes in IL-1 and microglial markers after acute stress. Brain Behav Immun. (2009) ;23: (7):958–68. |
[14] | McKim DB , Niraula A , Tarr AJ , Wohleb ES , Sheridan JF , Godbout JP . Neuroinflammatory Dynamics Underlie Memory Impairments after Repeated Social Defeat. J Neurosci. (2016) ;36: (9):2590–604. |
[15] | Taishi P , Bredow S , Guha-Thakurta N , Obál F , Krueger JM . Diurnal variations of interleukin-1β mRNA and β-actin mRNA in rat brain. J Neuroimmunol. (1997) ;75: (1):69–74. |
[16] | Ingiosi AM , Raymond RM , Pavlova MN , Opp MR . Selective contributions of neuronal and astroglial interleukin-1 receptor 1 to the regulation of sleep. Brain Behav Immun. (2015) ;48: :244–57. |
[17] | Opp MR , Obal F , Krueger JM . Interleukin 1 alters rat sleep: temporal and dose-related effects. Am J Physiol-Regul Integr Comp Physiol. (1991) ;260: (1):R52–8. |
[18] | Moraes CA , Santos G , Spohr TCL de S e , D’Avila JC , Lima FRS , Benjamim CF , et al. Activated Microglia-Induced Deficits in Excitatory Synapses Through IL-1β: Implications for Cognitive Impairment in Sepsis. Mol Neurobiol. (2015) ;52: (1):653–63. |
[19] | Gilmore JH , Fredrik Jarskog L , Vadlamudi S , Lauder JM . Prenatal Infection and Risk for Schizophrenia: IL-1β, IL-6, and TNFα Inhibit Cortical Neuron Dendrite Development. Neuropsychopharmacology. (2004) ;29: (7):1221–9. |
[20] | Giovanoli S , Weber-Stadlbauer U , Schedlowski M , Meyer U , Engler H . Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav Immun. (2016) ;55: :25–38. |
[21] | Lin Y-L , Wang S . Prenatal lipopolysaccharide exposure increases depression-like behaviors and reduces hippocampal neurogenesis in adult rats. Behav Brain Res. (2014) ;259: :24–34. |
[22] | Donald O Hebb . The Organization of Behavior: A Neuropsychological Theory. John Wiley; (1949) . |
[23] | Cajal SR y . Degeneration and Regeneration of the Nervous System. 1928; pp. 396. |
[24] | Altman J . Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. (1965) ;124: (3):319–35. |
[25] | Eriksson PS , Perfilieva E , Björk-Eriksson T , Alborn A-M , Nordborg C , Peterson DA , et al. Neurogenesis in the adult human hippocampus. Nat Med. (1998) ;4: (11):1313–7. |
[26] | Kuhn HG , Dickinson-Anson H , Gage FH . Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci Off J Soc Neurosci. (1996) ;16: (6):2027–33. |
[27] | Boldrini M , Fulmore CA , Tartt AN , Simeon LR , Pavlova I , Poposka V , et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell. (2018) ;22: (4):589–599.e5. |
[28] | Saxe MD , Battaglia F , Wang J-W , Malleret G , David DJ , Monckton JE , et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci. (2006) ;103: (46):17501–6. |
[29] | Blaiss CA , Yu T-S , Zhang G , Chen J , Dimchev G , Parada LF , et al. Temporally Specified Genetic Ablation of Neurogenesis Impairs Cognitive Recovery after Traumatic Brain Injury. J Neurosci. (2011) ;31: (13):4906–16. |
[30] | Hill AS , Sahay A , Hen R . Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology. (2015) ;40: (10):2368–78. |
[31] | Yun S , Reynolds RP , Petrof I , White A , Rivera PD , Segev A , et al. Stimulation of entorhinal cortex-dentate gyrus circuitry is antidepressive. Nat Med. (2018) ;24: (5):658–66. |
[32] | van Praag H , Christie BR , Sejnowski TJ , Gage FH . Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci. (1999) ;96: (23):13427–31. |
[33] | Kitamura T , Inokuchi K . Role of adult neurogenesis in hippocampal-cortical memory consolidation. Mol Brain. (2014) ;7: (1):13. |
[34] | Terranova JI , Ogawa SK , Kitamura T . Adult hippocampal neurogenesis for systems consolidation of memory. Behav Brain Res. (2019) ;372: :112035. |
[35] | Bagnall-Moreau C , Chaudhry S , Salas-Ramirez K , Ahles T , Hubbard K . Chemotherapy-Induced Cognitive Impairment Is Associated with Increased Inflammation and Oxidative Damage in the Hippocampus. Mol Neurobiol. (2019) ;56: (10):7159–72. |
[36] | Zhang R-R , Cui Q-Y , Murai K , Lim YC , Smith ZD , Jin S , et al. Tet1 Regulates Adult Hippocampal Neurogenesis and Cognition. Cell Stem Cell. (2013) ;13: (2):237–45. |
[37] | Sahay A , Hen R . Adult hippocampal neurogenesis in depression. Nat Neurosci. (2007) ;10: (9):1110–5. |
[38] | Parent JM , Elliott RC , Pleasure SJ , Barbaro NM , Lowenstein DH . Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol. (2006) ;59: (1):81–91. |
[39] | Shetty AK . Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: Can early neural stem cell grafting intervention provide protection? Epilepsy Behav. (2014) ;38: :117–24. |
[40] | Augusto-Oliveira M , Arrifano G , Malva J , Crespo-Lopez M . Adult Hippocampal Neurogenesis in Different Taxonomic Groups: Possible Functional Similarities and Striking Controversies. Cells. (2019) ;8: (2):125. |
[41] | Deng W , Aimone JB , Gage FH . New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. (2010) ;11: (5):339–50. |
[42] | Truttmann AC , Ginet V , Puyal J . Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models. Front Cell Dev Biol [Internet]. 2020 [cited 2020 Apr 12];8. Available from: https://www.frontiersin.org/articles/10.3389/fcell.2020.00027/full |
[43] | Henshall DC , Murphy BM . Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol. (2008) ;8: (1):75–81. |
[44] | Zhu JPQ , Xu W , Angulo JA . Methamphetamine-induced cell death: Selective vulnerability in neuronal subpopulations of the striatum in mice. Neuroscience. (2006) ;140: (2):607–22. |
[45] | Wang J , Gan Y , Han P , Yin J , Liu Q , Ghanian S , et al. Ischemia-induced Neuronal Cell Death Is Mediated by Chemokine Receptor CX3CR1. Sci Rep. (2018) ;8: (1):1–11. |
[46] | Bliss TVP , Collingridge GL . A synaptic model of memory: long-term potentiation in the hippocampus. Nature. (1993) ;361: (6407):31–9. |
[47] | Wang C , Yue H , Hu Z , Shen Y , Ma J , Li J , et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science. (2020) ;367: (6478):688–94. |
[48] | Duman RS . Synaptic plasticity and mood disorders. Mol Psychiatry. (2002) ;7: (1):S29–34. |
[49] | Lee Y , Zhang Y , Kim S , Han K . Excitatory and inhibitory synaptic dysfunction in mania: an emerging hypothesis from animal model studies. Exp Mol Med. (2018) ;50: (4):1–11. |
[50] | Habela CW , Song H , Ming G . Modeling synaptogenesis in Schizophrenia and Autism using human iPSC derived neurons. Mol Cell Neurosci. (2016) ;73: :52–62. |
[51] | Koyama R , Ikegaya Y . Microglia in the pathogenesis of autism spectrum disorders. Neurosci Res. (2015) ;100: :1–5. |
[52] | Montani C , Gritti L , Beretta S , Verpelli C , Sala C . The Synaptic and Neuronal Functions of the X-Linked Intellectual Disability Protein Interleukin-1 Receptor Accessory Protein Like 1 (IL1RAPL1). Dev Neurobiol. (2019) ;79: (1):85–95. |
[53] | Huttenlocher PR , de Courten C , Garey LJ , Van der Loos H . Synaptogenesis in human visual cortex —evidence for synapse elimination during normal development. Neurosci Lett. (1982) ;33: (3):247–52. |
[54] | Paolicelli RC , Bolasco G , Pagani F , Maggi L , Scianni M , Panzanelli P , et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science. (2011) ;333: (6048):1456–8. |
[55] | Hong S , Dissing-Olesen L , Stevens B . New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. (2016) ;36: :128–34. |
[56] | Wake H , Moorhouse AJ , Jinno S , Kohsaka S , Nabekura J . Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J Neurosci. (2009) ;29: (13):3974–80. |
[57] | Parkhurst CN , Yang G , Ninan I , Savas JN , Yates JR , Lafaille JJ , et al. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell. (2013) ;155: (7):1596–609. |
[58] | Hering H , Sheng M . Dentritic spines: structure, dynamics and regulation. Nat Rev Neurosci. (2001) ;2: (12):880–8. |
[59] | Hlushchenko I , Koskinen M , Hotulainen P . Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton. (2016) ;73: (9):435–41. |
[60] | Xu X , Miller EC , Pozzo-Miller L . Dendritic spine dysgenesis in Rett syndrome. Front Neuroanat [Internet]. 2014 [cited 2020 Apr 21];8. Available from: https://www.frontiersin.org/articles/10.3389/fnana.2014.00097/full |
[61] | Penzes P , Cahill ME , Jones KA , VanLeeuwen J-E , Woolfrey KM . Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. (2011) ;14: (3):285–93. |
[62] | Yamagata M , Sanes JR , Weiner JA . Synaptic adhesion molecules. Curr Opin Cell Biol.. (2003) ;15: (5):621–32. |
[63] | Ranneva SV , Maksimov VF , Korostyshevskaja IM , Lipina TV . Lack of synaptic protein, calsyntenin-2, impairs morphology of synaptic complexes in mice. Synap N Y N. (2020) ;74: (2):e22132. |
[64] | Li M-Y , Miao W-Y , Wu Q-Z , He S-J , Yan G , Yang Y , et al. A Critical Role of Presynaptic Cadherin/Catenin/p140Cap Complexes in Stabilizing Spines and Functional Synapses in the Neocortex. Neuron. (2017) ;94: (6):1155–1172.e8. |
[65] | Dagar S , Gottmann K . Differential Properties of the Synaptogenic Activities of the Neurexin Ligands Neuroligin1 and LRRTM2. Front Mol Neurosci [Internet]. 2019 [cited 2020Apr 14];12.Available from: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00269/full |
[66] | Muskiewicz DE , Uhl GR , Hall FS . The Role of Cell Adhesion Molecule Genes Regulating Neuroplasticity in Addiction [Internet]. Vol. 2018, Neural Plasticity. Hindawi; 2018 [cited 2020 Apr 14]. pp. e9803764. Available from: http://www.hindawi.com/journals/np/2018/9803764/ |
[67] | Lips ES , Cornelisse LN , Toonen RF , Min JL , Hultman CM , International Schizophrenia Consortium, et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry. (2012) ;17: (10):996–1006. |
[68] | Lüscher C , Malenka RC . NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb Perspect Biol [Internet]. 2012 Jun [cited 2020Apr 12];4(6).Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3367554/ |
[69] | Malenka RC , Bear MF . LTP and LTD: An Embarrassment of Riches. Neuron. (2004) ;44: (1):5–21. |
[70] | Thiruvengadam A . Effect of lithium and sodium valproate ions on resting membrane potentials in neurons: an hypothesis. J Affect Disord. (2001) ;65: (1):95–9. |
[71] | Weinreich F , Jentsch TJ . Neurological diseases caused by ion-channel mutations. Curr Opin Neurobiol. (2000) ;10: (3):409–15. |
[72] | Li F , Tsien JZ . Memory and the NMDA Receptors. N Engl J Med. (2009) ;361: (3):302–3. |
[73] | Baganz NL , Blakely RD . A Dialogue between the Immune System and Brain, Spoken in the Language of Serotonin. ACS Chem Neurosci. (2013) ;4: (1):48–63. |
[74] | Bridges RJ , Natale NR , Patel SA . System xc- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. (2012) ;165: (1):20–34. |
[75] | Grienberger C , Konnerth A . Imaging Calcium in Neurons. Neuron. (2012) ;73: (5):862–85. |
[76] | Fang J , Wang Y , Krueger JM . Effects of interleukin-1β on sleep are mediated by the type I receptor. Am J Physiol-Regul Integr Comp Physiol. (1998) ;274: (3):R655–60. |
[77] | Krueger JM , Walter J , Dinarello CA , Wolff SM , Chedid L . Sleep-promoting effects of endogenous pyrogen (interleukin-1). Am J Physiol-Regul Integr Comp Physiol. (1984) ;246: (6):R994–9. |
[78] | Depino AM , Alonso M , Ferrari C , Rey A del , Anthony D , Besedovsky H , et al. Learning modulation by endogenous hippocampal IL- Blockade of endogenous IL-1 facilitates memory formation. Hippocampus. (2004) ;14: (4):526–35. |
[79] | Yirmiya R . Brain Interleukin-1 Is Involved in Spatial Memory and Passive Avoidance Conditioning. Neurobiol Learn Mem. (2002) ;78: (2):379–89. |
[80] | Pacheco-López G , Niemi M-B , Kou W , Baum S , Hoffman M , Altenburger P , et al. Central Blockade of IL-1 Does Not Impair Taste-LPS Associative Learning. Neuroimmunomodulation. (2007) ;14: (3-4):150–6. |
[81] | Avital A , Goshen I , Kamsler A , Segal M , Iverfeldt K , Richter-Levin G , et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. (2003) ;13: (7):826–34. |
[82] | Jones ME , Lebonville CL , Paniccia JE , Balentine ME , Reissner KJ , Lysle DT . Hippocampal interleukin-1 mediates stress-enhanced fear learning: A potential role for astrocyte-derived interleukin-1β . Brain Behav Immun. (2018) ;67: :355–63. |
[83] | Muccigrosso MM , Ford J , Benner B , Moussa D , Burnsides C , Fenn AM , et al. Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun. (2016) ;54: :95–109. |
[84] | Rothwell N , Allan S , Toulmond S . The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. (1997) ;100: (11):2648–52. |
[85] | Wu MD , Montgomery SL , Rivera-Escalera F , Olschowka JA , O’Banion MK . Sustained IL-1β expression impairs adult hippocampal neurogenesis independent of IL-1 signaling in nestin+neural precursor cells. Brain Behav Immun. (2013) ;32: :9–18. |
[86] | Liu X , Nemeth DP , McKim DB , Zhu L , DiSabato DJ , Berdysz O , et al. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity. (2019) ;50: (2):317–333.e6. |
[87] | Norheim KB , Harboe E , Gøransson LG , Omdal R . Interleukin-1 Inhibition and Fatigue in Primary Sjögren’s Syndrome - A Double Blind, Randomised Clinical Trial. Schillaci G, editor. PLoS ONE. (2012) ;7: (1):e30123. |
[88] | Cannon JG , Angel JB , Abad LW , Vannier E , Mileno MD , Fagioli L , et al. Interleukin-1β, Interleukin-1 Receptor Antagonist, and Soluble Interleukin-1 Receptor Type II Secretion in Chronic Fatigue Syndrome. J Clin Immunol. (1997) ;17: (3):253–61. |
[89] | Ellul P , Boyer L , Groc L , Leboyer M , Fond G . Interleukin-1 β-targeted treatment strategies in inflammatory depression: toward personalized care. Acta Psychiatr Scand. (2016) ;134: (6):469–84. |
[90] | Levine J , Barak Y , Chengappa KNR , Rapoport A , Rebey M , Barak V . Cerebrospinal Cytokine Levels in Patients with Acute Depression. Neuropsychobiology. (1999) ;40: (4):171–6. |
[91] | Koo JW , Duman RS . Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett. (2009) ;456: (1):39–43. |
[92] | Murray CL , Obiang P , Bannerman D , Cunningham C . Endogenous IL-1 in Cognitive Function and Anxiety: A Study in IL-1RI-/- Mice. PLOS ONE. (2013) ;8: (10):e78385. |
[93] | Quagliato LA , Nardi AE . Cytokine alterations in panic disorder: A systematic review. J Affect Disord. (2018) ;228: :91–6. |
[94] | Qian J , Zhu L , Li Q , Belevych N , Chen Q , Zhao F , et al. Interleukin-1R3 mediates interleukin-1–induced potassium current increase through fast activation of Akt kinase. Proc Natl Acad Sci. (2012) ;109: (30):12189–94. |
[95] | Huang Y , Smith DE , Ibáñez-Sandoval O , Sims JE , Friedman WJ . Neuron-Specific Effects of Interleukin-1β Are Mediated by a Novel Isoform of the IL-1 Receptor Accessory Protein. J Neurosci. (2011) ;31: (49):18048–59. |
[96] | Smith DE , Lipsky BP , Russell C , Ketchem RR , Kirchner J , Hensley K , et al. A Novel CNS-Restricted Isoform of the IL-1R Accessory Protein Modulates Neuronal Responses to IL-1. Immunity. (2009) ;30: (6):817–31. |
[97] | Farrar WL , Kilian PL , Ruff MR , Hill JM , Pert CB . Visualization and characterization of interleukin 1 receptors in brain. J Immunol Baltim Md 1950. (1987) ;139: (2):459–63. |
[98] | Liu X , Yamashita T , Chen Q , Belevych N , Mckim DB , Tarr AJ , et al. Interleukin 1 Type 1 Receptor Restore: A Genetic Mouse Model for Studying Interleukin 1 Receptor-Mediated Effects in Specific Cell Types. J Neurosci. (2015) ;35: (7):2860–70. |
[99] | Greco SJ , Rameshwar P . Enhancing Effect of IL-1α on Neurogenesis from Adult Human Mesenchymal Stem Cells: Implication for Inflammatory Mediators in Regenerative Medicine. J Immunol. (2007) ;179: (5):3342–50. |
[100] | Redondo-Castro E , Cunningham C , Miller J , Martuscelli L , Aoulad-Ali S , Rothwell NJ , et al. Interleukin-1 primes human mesenchymal stem cells towards an anti-inflammatory and pro-trophic phenotype in vitro . Stem Cell Res Ther. (2017) ;8: (1):79. |
[101] | Green HF , Nolan YM . Unlocking mechanisms in interleukin-1β-induced changes in hippocampal neurogenesis—a role for GSK-3β and TLX. Transl Psychiatry. (2012) ;2: (11):e194–e194. |
[102] | Green HF , Treacy E , Keohane AK , Sullivan AM , O’Keeffe GW , Nolan YM . A role for interleukin-1β in determining the lineage fate of embryonic rat hippocampal neural precursor cells. Mol Cell Neurosci. (2012) ;49: (3):311–21. |
[103] | Ryan SM , O’Keeffe GW , O’Connor C , Keeshan K , Nolan YM . Negative regulation of TLX by IL-1β correlates with an inhibition of adult hippocampal neural precursor cell proliferation. Brain Behav Immun.. (2013) ;33: :7–13. |
[104] | Zunszain PA , Anacker C , Cattaneo A , Choudhury S , Musaelyan K , Myint AM , et al. Interleukin-1 β: A New Regulator of the Kynurenine Pathway Affecting Human Hippocampal Neurogenesis. Neuropsychopharmacology. (2012) ;37: (4):939–49. |
[105] | Musaelyan K , Egeland M , Fernandes C , Pariante CM , Zunszain PA , Thuret S . Modulation of Adult Hippocampal Neurogenesis by Early-Life Environmental Challenges Triggering Immune Activation. Neural Plast. (2014) ;2014: :1–10. |
[106] | Valero J , Mastrella G , Neiva I , Sánchez S , Malva JO . Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front Neurosci [Internet]. 2014 Apr 21 [cited 2020 Feb 10];8. Available from: http://journal.frontiersin.org/article/10.3389/fnins.2014.00083/abstract |
[107] | Goshen I , Kreisel T , Ben-Menachem-Zidon O , Licht T , Weidenfeld J , Ben-Hur T , et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry.. (2008) ;13: (7):717–28. |
[108] | Koo JW , Russo SJ , Ferguson D , Nestler EJ , Duman RS . Nuclear factor- B is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci. (2010) ;107: (6):2669–74. |
[109] | Raber J , Rola R , LeFevour A , Morhardt D , Curley J , Mizumatsu S , et al. Radiation-Induced Cognitive Impairments are Associated with Changes in Indicators of Hippocampal Neurogenesis. Radiat Res.. (2004) ;162: (1):39–47. |
[110] | Xue J , Dong J-H , Huang G-D , Qu X-F , Wu G , Dong X-R . NF-κB signaling modulates radiation-induced microglial activation. Oncol Rep. (2014) ;31: (6):2555–60. |
[111] | Ben-Menachem-Zidon O , Ben-Menahem Y , Ben-Hur T , Yirmiya R . Intra-Hippocampal Transplantation of Neural Precursor Cells with Transgenic Over-Expression of IL-1 Receptor Antagonist Rescues Memory and Neurogenesis Impairments in an Alzheimer’s Disease Model. Neuropsychopharmacology.. (2014) ;39: (2):401–14. |
[112] | Hueston CM , O’Leary JD , Hoban AE , Kozareva DA , Pawley LC , O’Leary OF , et al. Chronic interleukin-1β in the dorsal hippocampus impairs behavioural pattern separation. Brain Behav Immun. (2018) ;74: :252–64. |
[113] | Ben Menachem-Zidon O , Goshen I , Kreisel T , Ben Menahem Y , Reinhartz E , Ben Hur T , et al. Intrahippocampal Transplantation of Transgenic Neural Precursor Cells Overexpressing Interleukin-1 Receptor Antagonist Blocks Chronic Isolation-Induced Impairment in Memory and Neurogenesis. Neuropsychopharmacology. (2008) ;33: (9):2251–62. |
[114] | Wu MD , Hein AM , Moravan MJ , Shaftel SS , Olschowka JA , O’Banion MK . Adult murine hippocampal neurogenesis is inhibited by sustained IL-1β and not rescued by voluntary running. Brain Behav Immun. (2012) ;26: (2):292–300. |
[115] | Littlefield AM , Setti SE , Priester C , Kohman RA . Voluntary exercise attenuates LPS-induced reductions in neurogenesis and increases microglia expression of a proneurogenic phenotype in aged mice. J Neuroinflammation. (2015) ;12: (1):138. |
[116] | van Praag H . Exercise Enhances Learning and Hippocampal Neurogenesis in Aged Mice. J Neurosci. (2005) ;25: (38):8680–5. |
[117] | Spulber S , Oprica M , Bartfai T , Winblad B , Schultzberg M . Blunted neurogenesis and gliosis due to transgenic overexpression of human soluble IL-1ra in the mouse. Eur J Neurosci. (2008) ;27: (3):549–58. |
[118] | Guadagno J , Swan P , Shaikh R , Cregan SP . Microglia-derived IL-1 β triggers p53-mediated cell cycle arrest and apoptosis in neural precursor cells. Cell Death Dis. (2015) ;6: (6):e1779–e1779. |
[119] | Rossi S , Motta C , Studer V , Macchiarulo G , Volpe E , Barbieri F , et al. Interleukin-1β causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol Neurodegener. (2014) ;9: (1):56. |
[120] | Ajmone-Cat MA , Cacci E , Ragazzoni Y , Minghetti L , Biagioni S . Pro-gliogenic effect of IL-1α in the differentiation of embryonic neural precursor cells in vitro . J Neurochem. (2010) ;113: (4):1060–72. |
[121] | Zhang K , Xu H , Cao L , Li K , Huang Q . Interleukin-1β inhibits the differentiation of hippocampal neural precursor cells into serotonergic neurons. Brain Res. (2013) ;1490: :193–201. |
[122] | Friedlander RM , Gagliardini V , Rotello RJ , Yuan J . Functional role of interleukin 1 beta (IL-1 beta) in IL-1 beta-converting enzyme-mediated apoptosis. J Exp Med. (1996) ;184: (2):717–24. |
[123] | Tong L , Balazs R , Soiampornkul R , Thangnipon W , Cotman CW . Interleukin-1β impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. (2008) ;29: (9):1380–93. |
[124] | Hu S , Peterson PK , Chao CC . Cytokine-mediated neuronal apoptosis. Neurochem Int. (1997) ;30: (4):427–31. |
[125] | Thornton P , Pinteaux E , Gibson RM , Allan SM , Rothwell NJ . Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem. (2006) ;98: (1):258–66. |
[126] | Greenlund LJS , Deckwerth TL , Johnson EM . Superoxide dismutase delays neuronal apoptosis: A role for reactive oxygen species in programmed neuronal death. Neuron. (1995) ;14: (2):303–15. |
[127] | Lindholm D , Heumann R , Meyer M , Thoenen H . Interleukin-1 regulates synthesis of nerve growth factor in non-neuronal cells of rat sciatic nerve. Nature. (1987) ;330: (6149):658–9. |
[128] | Juric DM , Carman-Krzan M . Interleukin-1 beta, but not IL-1 alpha, mediates nerve growth factor secretion from rat astrocytes via type I IL-1 receptor. Int J Dev Neurosci Off J Int Soc Dev Neurosci. (2001) ;19: (7):675–83. |
[129] | Hein AM , Stasko MR , Matousek SB , Scott-McKean JJ , Maier SF , Olschowka JA , et al. Sustained hippocampal IL-1β overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun.. (2010) ;24: (2):243–53. |
[130] | Lawrence CB , Allan SM , Rothwell NJ . Interleukin-1β and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur J Neurosci. (1998) ;10: (3):1188–95. |
[131] | Proescholdt MG , Chakravarty S , Foster JA , Foti SB , Briley EM , Herkenham M . Intracerebroventricular but not intravenous interleukin-1β induces widespread vascular-mediated leukocyte infiltration and immune signal mRNA expression followed by brain-wide glial activation. Neuroscience. (2002) ;112: (3):731–49. |
[132] | Carlos AJ , Tong L , Prieto GA , Cotman CW . IL-1β impairs retrograde flow of BDNF signaling by attenuating endosome trafficking. J Neuroinflammation. (2017) ;14: (1):1–12. |
[133] | Wang X , Kong K , Qi W , Ye W , Song P . Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacol Sin. (2005) ;26: (8):934–42. |
[134] | Relton JK , Rothwell NJ . Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull. (1992) ;29: (2):243–6. |
[135] | Medel-Matus J-S , Álvarez-Croda D-M , Martínez-Quiroz J , Beltrán-Parrazal L , Morgado-Valle C , López-Meraz M-L . IL-1β increases necrotic neuronal cell death in the developing rat hippocampus after status epilepticus by activating type I IL-1 receptor (IL-1RI). Int J Dev Neurosci. (2014) ;38: :232–40. |
[136] | Varvel NH , Neher JJ , Bosch A , Wang W , Ransohoff RM , Miller RJ , et al. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A. (2016) ;113: (38):E5665–5674. |
[137] | Honjo M , Tanihara H , Nishijima K , Kiryu J , Honda Y , Yue BYJT , et al. Statin Inhibits Leukocyte-Endothelial Interaction and Prevents Neuronal Death Induced by Ischemia-Reperfusion Injury in the Rat Retina. Arch Ophthalmol. (2002) ;120: (12):1707–13. |
[138] | Lévesque SA , Paré A , Mailhot B , Bellver-Landete V , Kébir H , Lécuyer M-A , et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med. (2016) ;213: (6):929–49. |
[139] | An Y , Belevych N , Wang Y , Zhang H , Herschman H , Chen Q , et al. Neuronal and nonneuronal COX-2 expression confers neurotoxic and neuroprotective phenotypes in response to excitotoxin challenge. J Neurosci Res. (2014) ;92: (4):486–95. |
[140] | Jin Y , Sun LH , Yang W , Cui RJ , Xu SB . The Role of BDNF in the Neuroimmune Axis Regulation of Mood Disorders. Front Neurol [Internet]. 2019 [cited 2020 Apr 27];10. Available from: https://www.frontiersin.org/articles/10.3389/fneur.2019.00515/full |
[141] | Barrientos RM , Sprunger DB , Campeau S , Watkins LR , Rudy JW , Maier SF . BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1β administration. J Neuroimmunol. (2004) ;155: (1):119–26. |
[142] | Viviani B , Bartesaghi S , Gardoni F , Vezzani A , Behrens MM , Bartfai T , et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. J Neurosci. (2003) ;23: (25):8692–700. |
[143] | Kawasaki H , Morooka T , Shimohama S , Kimura J , Hirano T , Gotoh Y , et al. Activation and Involvement of p38 Mitogen-activated Protein Kinase in Glutamate-induced Apoptosis in Rat Cerebellar Granule Cells. J Biol Chem. (1997) ;272: (30):18518–21. |
[144] | Zhang W , Potrovita I , Tarabin V , Herrmann O , Beer V , Weih F , et al. Neuronal Activation of NF-κB Contributes to Cell Death in Cerebral Ischemia. J Cereb Blood Flow Metab. (2005) ;25: (1):30–40. |
[145] | Prieto GA , Snigdha S , Baglietto-Vargas D , Smith ED , Berchtold NC , Tong L , et al. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1β in the aged hippocampus. Proc Natl Acad Sci. (2015) ;112: (36):E5078–87. |
[146] | Gardoni F , Boraso M , Zianni E , Corsini E , Galli CL , Cattabeni F , et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1b and NMDA stimulation. 2011;6. |
[147] | Lai AY , Swayze RD , El-Husseini A , Song C . Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. (2006) ;175: (1):97–106. |
[148] | Taoro-Gonzalez L , Arenas YM , Cabrera-Pastor A , Felipo V . Hyperammonemia alters membrane expression of GluA1 and GluA2 subunits of AMPA receptors in hippocampus by enhancing activation of the IL-1 receptor: underlying mechanisms. J Neuroinflammation. (2018) ;15: (1):36. |
[149] | Mishra A , Kim HJ , Shin AH , Thayer SA . Synapse Loss Induced by Interleukin-1β Requires Pre- and Post-synaptic Mechanisms. J Neuroimmune Pharmacol. (2012) ;7: (3):571–8. |
[150] | Avdic U , Chugh D , Osman H , Chapman K , Jackson J , Ekdahl CT . Absence of interleukin-1 receptor 1 increases excitatory and inhibitory scaffolding protein expression and microglial activation in the adult mouse hippocampus. Cell Mol Immunol. (2015) ;12: (5):645–7. |
[151] | Yoshida T , Yasumura M , Uemura T , Lee S-J , Ra M , Taguchi R , et al. IL-1 Receptor Accessory Protein-Like 1 Associated with Mental Retardation and Autism Mediates Synapse Formation by Trans-Synaptic Interaction with Protein Tyrosine Phosphatase. J Neurosci. (2011) ;31: (38):13485–99. |
[152] | Yoshida T , Shiroshima T , Lee S-J , Yasumura M , Uemura T , Chen X , et al. Interleukin-1 Receptor Accessory Protein Organizes Neuronal Synaptogenesis as a Cell Adhesion Molecule. J Neurosci. (2012) ;32: (8):2588–600. |
[153] | Montani C , Ramos-Brossier M , Ponzoni L , Gritti L , Cwetsch AW , Braida D , et al. The X-Linked Intellectual Disability Protein IL1RAPL1 Regulates Dendrite Complexity. J Neurosci Off J Soc Neurosci. (2017) ;37: (28):6606–27. |
[154] | Bahi N , Friocourt G , Carrié A , Graham ME , Weiss JL , Chafey P , et al. IL1 receptor accessory protein like, a protein involved in X-linked mental retardation, interacts with Neuronal Calcium Sensor-1 and regulates exocytosis. Hum Mol Genet. (2003) ;12: (12):1415–25. |
[155] | Ramos-Brossier M , Montani C , Lebrun N , Gritti L , Martin C , Seminatore-Nole C , et al. Novel IL1RAPL1 mutations associated with intellectual disability impair synaptogenesis. Hum Mol Genet. (2015) ;24: (4):1106–18. |
[156] | Yasumura M , Yoshida T , Yamazaki M , Abe M , Natsume R , Kanno K , et al. IL1RAPL1 knockout mice show spine density decrease, learning deficiency, hyperactivity and reduced anxiety-like behaviours. Sci Rep. (2014) ;4: :6613. |
[157] | Katsuki H , Nakai S , Hirai Y , Akaji K , Kiso Y , Satoh M . Interleukin-1β inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol. (1990) ;181: (3):323–6. |
[158] | Bellinger FP , Madamba S , Siggins GR . Interleukin 1β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. (1993) ;628: (1):227–34. |
[159] | Hoshino K , Hasegawa K , Kamiya H , Morimoto Y . Synapse-specific effects of IL-1β on long-term potentiation in the mouse hippocampus. Biomed Res. (2017) ;38: (3):183–8. |
[160] | Coogan A , O’Connor JJ . Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1β . NeuroReport. (1997) ;8: (9):2107–10. |
[161] | Cunningham AJ , Murray CA , O’Neill LA , Lynch MA , O’Connor JJ . Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. (1996) ;203: (1):17–20. |
[162] | Murray CA , Lynch MA . Evidence That Increased Hippocampal Expression of the Cytokine Interleukin-1β Is a Common Trigger for Age- and Stress-Induced Impairments in Long-Term Potentiation. J Neurosci. (1998) ;18: (8):2974–81. |
[163] | Vereker E , O’Donnell E , Lynch MA . The Inhibitory Effect of Interleukin-1β on Long-Term Potentiation Is Coupled with Increased Activity of Stress-Activated Protein Kinases. J Neurosci. (2000) ;20: (18):6811–9. |
[164] | del Rey A , Balschun D , Wetzel W , Randolf A , Besedovsky HO . A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during long-term potentiation and learning. Brain Behav Immun. (2013) ;33: :15–23. |
[165] | Schneider H , Pitossi F , Balschun D , Wagner A , del Rey A , Besedovsky HO . A neuromodulatory role of interleukin-1 in the hippocampus. Proc Natl Acad Sci. (1998) ;95: (13):7778–83. |
[166] | Ross FM , Allan SM , Rothwell NJ , Verkhratsky A . A dual role for interleukin-1 in LTP in mouse hippocampal slices. J Neuroimmunol. (2003) ;144: (1):61–7. |
[167] | Ikegaya Y , Delcroix I , Iwakura Y , Matsuki N , Nishiyama N . Interleukin-1b abrogates long-term depression of hippocampal CA1 synaptic transmission. Synapse. (2003) ;47: (1):54–7. |
[168] | Loscher CE , Mills KHG , Lynch MA . Interleukin-1 receptor antagonist exerts agonist activity in the hippocampus independent of the interleukin-1 type I receptor. J Neuroimmunol. (2003) ;137: (1):117–24. |
[169] | Yu B , Shinnick-Gallagher P . Interleukin-1 beta inhibits synaptic transmission and induces membrane hyperpolarization in amygdala neurons. JPET.. (1994) ;271: (2):590–600. |
[170] | Tabarean IV , Korn H , Bartfai T . Interleukin-1β induces hyperpolarization and modulates synaptic inhibition in preoptic and anterior hypothalamic neurons. Neuroscience. (2006) ;141: (4):1685–95. |
[171] | Mo Z-L , Katafuchi T , Hori T . Effects of IL-1β on neuronal activities in the dorsal motor nucleus of the vagus in rat brain slices. Brain Res Bull. (1996) ;41: (4):249–55. |
[172] | Miller LG , Galpern WR , Dunlap K , Dinarello CA , Turner TJ . Interleukin-1 augments gamma-aminobutyric acidA receptor function in brain. Mol Pharmacol. (1991) ;39: (2):105–8. |
[173] | Miller LG , Fahey JM . Interleukin-1 Modulates GABAergic and Glutamatergic Function in Braina. Ann N Y Acad Sci. (1994) ;739: (1):292–8. |
[174] | Diem R , Hobom M , Grötsch P , Kramer B , Bähr M . Interleukin-1β protects neurons via the interleukin-1 (IL-1) receptor-mediated Akt pathway and by IL-1 receptor-independent decrease of transmembrane currents in vivo . Mol Cell Neurosci. (2003) ;22: (4):487–500. |
[175] | Qi C , Zhang W-W , Li X-N , Zhou C . [Interleukin-1β inhibits the amplitudes of voltage-gated Na(+) currents and action potential in cultured cortical neurons of rat]. Sheng Li Xue Bao. (2011) ;63: (2):131–7. |
[176] | Zhou C , Qi C , Zhao J , Wang F , Zhang W , Li C , et al. Interleukin-1β Inhibits Voltage-Gated Sodium Currents in a Time- and Dose-Dependent Manner in Cortical Neurons. Neurochem Res. (2011) ;36: (6):1116. |
[177] | Campbell V , Lynch MA . Biphasic modulation of intracellular Ca2+concentration by interleukin-1β in cortical synaptosomes: involvement of a pertussis toxin-sensitive G-protein and mitogen-activated protein kinase. NeuroReport. (1998) ;9: (9):1923–7. |
[178] | Murray CA , McGahon B , McBennett S , Lynch MA . Interleukin-1β Inhibits Glutamate Release in Hippocampus of Young, But Not Aged, Rats. Neurobiol Aging. (1997) ;18: (3):343–8. |
[179] | Plata-Salaman CR , Ffrench-Mullen MH . Interleukin- 1 Depresses Calcium Currents in CA1 Hippocampal Neurons at Pathophysiological Concentrations. 1992;3. |
[180] | Yang S , Liu Z-W , Wen L , Qiao H-F , Zhou W-X , Zhang Y-X . Interleukin-1β enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. (2005) ;1034: (1-2):172–9. |
[181] | Bajo M , Patel RR , Hedges DM , Varodayan FP , Vlkolinsky R , Davis TD , et al. Role of MyD88 in IL-1β and Ethanol Modulation of GABAergic Transmission in the Central Amygdala. Brain Sci. (2019) ;9: (12):361. |
[182] | Hellstrom IC , Danik M , Luheshi GN , Williams S . Chronic LPS exposure produces changes in intrinsic membrane properties and a sustained IL-β-dependent increase in GABAergic inhibition in hippocampal CA1 pyramidal neurons. Hippocampus. (2005) ;15: (5):656–64. |
[183] | Patel RR , Khom S , Steinman MQ , Varodayan FP , Kiosses WB , Hedges DM , et al. IL-1β expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala. Brain Behav Immun. (2019) ;75: :208–19. |
[184] | Richter F , Eitner A , Leuchtweis J , Bauer R , Lehmenkühler A , Schaible H-G . Effects of interleukin-1ß on cortical spreading depolarization and cerebral vasculature. J Cereb Blood Flow Metab. (2017) ;37: (5):1791–802. |
[185] | Ferri CC , Ferguson AV . Interleukin-1β Depolarizes Paraventricular Nucleus Parvocellular Neurones. J Neuroendocrinol. (2003) ;15: (2):126–33. |
[186] | Desson SE , Ferguson AV . Interleukin 1β modulates rat subfornical organ neurons as a result of activation of a non-selective cationic conductance. J Physiol. (2003) ;550: (Pt 1):113–22. |
[187] | Liu L , Yang TM , Liedtke W , Simon SA . Chronic IL-1β Signaling Potentiates Voltage-Dependent Sodium Currents in Trigeminal Nociceptive Neurons. J Neurophysiol. (2006) ;95: (3):1478–90. |
[188] | Wang S , Cheng Q , Malik S , Yang J . Interleukin-1β Inhibits γ-Aminobutyric Acid Type A (GABAA) Receptor Current in Cultured Hippocampal Neurons. J Pharmacol Exp Ther. (2000) ;292: (2):497–504. |
[189] | Takeda M , Kitagawa J , Takahashi M , Matsumoto S . Activation of interleukin-1beta receptor suppresses the voltage-gated potassium currents in the small-diameter trigeminal ganglion neurons following peripheral inflammation. Pain. (2008) ;139: (3):594–602. |
[190] | Zhang R , Yamada J , Hayashi Y , Wu Z , Koyama S , Nakanishi H . Inhibition of NMDA-induced outward currents by interleukin-1β in hippocampal neurons. Biochem Biophys Res Commun. (2008) ;372: (4):816–20. |
[191] | Ghosh B , Green MV , Krogh KA , Thayer SA . Interleukin-1β activates an Src family kinase to stimulate the plasma membrane Ca2+ pump in hippocampal neurons. J Neurophysiol. (2016) ;115: (4):1875–85. |
[192] | Yu W , Xuzhong R , Xuqun S . Effect of Interleukin-l on the Elevation of Cytoplasmic Free Calcium of the Cultured Hippocampal Neurons Induced by L- Glutamate. 1999;4. |
[193] | Roseti C , van Vliet EA , Cifelli P , Ruffolo G , Baayen JC , Di Castro MA , et al. GABAA currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis. (2015) ;82: :311–20. |
[194] | Skelly DT , Griffin ÉW , Murray CL , Harney S , O’Boyle C , Hennessy E , et al. Acute transient cognitive dysfunction and acute brain injury induced by systemic inflammation occur by dissociable IL-1-dependent mechanisms. Mol Psychiatry. (2019) ;24: (10):1533–48. |
[195] | Rossi S , Furlan R , De Chiara V , Motta C , Studer V , Mori F , et al. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol. (2012) ;71: (1):76–83. |
[196] | Bertani I , Iori V , Trusel M , Maroso M , Foray C , Mantovani S , et al. Inhibition of IL-1β Signaling Normalizes NMDA-Dependent Neurotransmission and Reduces Seizure Susceptibility in a Mouse Model of Creutzfeldt-Jakob Disease. J Neurosci. (2017) ;37: (43):10278–89. |
[197] | Maroso M , Balosso S , Ravizza T , Liu J , Bianchi ME , Vezzani A . Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J Intern Med. (2011) ;270: (4):319–26. |
[198] | Zhu G , Okada M , Yoshida S , Mori F , Ueno S , Wakabayashi K , et al. Effects of interleukin-1β on hippocampal glutamate and GABA releases associated with Ca2+-induced Ca2+releasing systems. Epilepsy Res. (2006) ;71: (2):107–16. |
[199] | Gosselin D , Bellavance M-A , Rivest S . IL-1RAcPb signaling regulates adaptive mechanisms in neurons that promote their long-term survival following excitotoxic insults. Front Cell Neurosci [Internet]. 2013 [cited 2020 Feb 9];7. Available from: http://journal.frontiersin.org/article/10.3389/fncel.2013.00009/abstract |
[200] | Allan SM , Lawrence CB , Rothwell NJ . Interleukin-1 β and interleukin-1 receptor antagonist do not affect glutamate release or calcium entry in rat striatal synaptosomes. Mol Psychiatry. (1998) ;3: (2):178–82. |
[201] | Bittner KC , Milstein AD , Grienberger C , Romani S , Magee JC . Behavioral time scale synaptic plasticity underlies CA1 place fields. Science. (2017) ;357: (6355):1033–6. |

