
Recovery of Hippocampal-Dependent Learning Despite Blunting Reactive Adult Neurogenesis After Alcohol Dependence
Abstract
Background:
The excessive alcohol drinking that occurs in alcohol use disorder (AUD) causes neurodegeneration in regions such as the hippocampus, though recovery may occur after a period of abstinence. Mechanisms of recovery are not clear, though reactive neurogenesis has been observed in the hippocampal dentate gyrus following alcohol dependence and correlates to recovery of granule cell number.
Objective:
We investigated the role of neurons born during reactive neurogenesis in the recovery of hippocampal learning behavior after 4-day binge alcohol exposure, a model of an AUD. We hypothesized that reducing reactive neurogenesis would impair functional recovery.
Methods:
Adult male rats were subjected to 4-day binge alcohol exposure and two approaches were tested to blunt reactive adult neurogenesis, acute doses of alcohol or the chemotherapy drug, temozolomide (TMZ).
Results:
Acute 5 g/kg doses of EtOH gavaged T6 and T7 days post binge did not inhibit significantly the number of Bromodeoxyuridine-positive (BrdU+) proliferating cells in EtOH animals receiving 5 g/kg EtOH versus controls. A single cycle of TMZ inhibited reactive proliferation (BrdU+ cells) and neurogenesis (NeuroD+ cells) to that of controls. However, despite this blunting of reactive neurogenesis to basal levels, EtOH-TMZ rats were not impaired in their recovery of acquisition of the Morris water maze (MWM), learning similarly to all other groups 35 days after 4-day binge exposure.
Conclusions:
These studies show that TMZ is effective in decreasing reactive proliferation/neurogenesis following 4-day binge EtOH exposure, and baseline levels of adult neurogenesis are sufficient to allow recovery of hippocampal function.
INTRODUCTION
Alcohol use disorder (AUD) is a chronic, relapsing disorder that impacts millions of Americans with nearly 14% of U.S. adults meeting diagnostic criteria in a given year [1]. Excessive alcohol use, the defining characteristic of an AUD, results in damage and neurodegeneration across many brain regions with binge patterns of alcohol drinking more likely to result in neurodegeneration [2–5]. One of the primary targets of alcohol neurotoxicity is the hippocampus [6, 7]. In humans, binge and heavy alcohol consumption have been associated with deficits in hippocampal-dependent tasks which correspond to cell and volume loss in the hippocampus [8–13]. Studies in animal models of AUDs strongly support causality between excessive alcohol exposure and loss of hippocampal integrity [14]. Numerous models demonstrate impairments in hippocampal-dependent learning and memory with corresponding deficits in hippocampal structure following alcohol dependence [7, 15–18]. However, recovery of brain structure and function may occur with abstinence from alcohol drinking [19–23]. The hippocampus is one such region where both structure and function have been reported to recover with abstinence, although results varied depending on the type of cognitive task or structural aspect measured as well as the timing of assessment relative to last drink [21, 24–26]. The mechanisms involved in recovery remain poorly understood, though data from human and animal models have long suggested various types of structural plasticity [17, 20, 27–29].
One means of hippocampal plasticity is the process of adult neurogenesis. The dentate gyrus of the hippocampus is one region of the brain where ongoing, adult neurogenesis is well accepted to contribute to hippocampal integrity in mammals, including humans [30–35]. Granule cells are born continuously from neural stem cells (NSC) in the neurogenic niche of the dentate gyrus subgranular zone, then migrate into the granule cell layer and integrate into hippocampal circuitry [27, 32, 33]. Newborn neurons play specific roles in learning and memory [33, 36–40]. For example, adult neurogenesis may be necessary for initial learning of a spatial task [33, 41–43], likely through their role in pattern separation, meaning the discrimination of contexts that are highly similar [37, 39, 44]. In support of this view, when adult neurogenesis is ablated or impaired, animals show increased interference when presented with variations to tasks or contexts that are similar to an initial one [39, 45–47]. Conversely, increasing adult neurogenesis is associated with improved learning and memory performance e.g. [35, 37] see also for review [32, 36]. In addition, many speculate on a role of adult neurogenesis in recovery from damage, especially in the hippocampus [48–50].
Alcohol exposure has dynamic effects on adult neurogenesis, contributing to alcohol-induced neurodegeneration, but also to recovery in abstinence [27, 50–52]. Intoxicating doses of alcohol inhibit proliferation and/or impact neuroblast survival to decrease adult neurogenesis, effects which have been observed extensively in animal models but also in humans [27, 50, 53–60]. However, while in withdrawal and abstinence after alcohol dependence, reactive increases in adult neurogenesis are associated with repair and recovery of hippocampal structure and function [51, 52, 61–63]. This reactive adult neurogenesis is initiated through the activation of Type 1 NSC proliferation in the dentate gyrus subgranular zone [61, 62]. However, the functional role of reactive neurogenesis in models of AUD is not known. Reactive neurogenesis may act as a compensatory mechanism in which rebound proliferation counteracts cells not generated during intoxication or helps to replace alcohol-killed granule cells [4, 29, 50, 51, 64–66]. Indeed, the concept of reactive adult neurogenesis is not exclusive to alcohol-induced neurotoxicity, similar increases have been found in other models of insult such as ischemia [67], traumatic brain injury [68], and seizure [69].
Though the observation of reactive neurogenesis after alcohol dependence correlates to the recovery of hippocampal structure in models of AUD, the relationship between the two has not been examined mechanistically [51, 61]. If reactive neurogenesis is responsible for recovery of hippocampal structure and function, then inhibiting reactive neurogenesis should block recovery in alcohol-exposed animals. Hippocampus-dependent learning tasks have been utilized to explore the relationship between alcohol and hippocampal integrity [15, 16, 70]. The Morris water maze (MWM) is a well-established hippocampal-dependent task, where rats exposed to a 4-day binge alcohol paradigm are impaired in learning the task [16, 18, 71; Chen et al., in prep]. Therefore, in order to test the hypothesis that recovery of hippocampal function is related to reactive, adult neurogenesis, two approaches were taken to blunt NSC proliferation and reactive adult neurogenesis, acute doses of alcohol or the chemotherapy drug temozolomide (TMZ), then performance on the hippocampal-dependent MWM was assessed.
METHODS
Subjects
142 male Sprague-Dawley rats weighing 275–300g upon arrival were used across four experiments (Charles River laboratory; PND 70). Eight rats were excluded due to failure to incorporate BrdU (seven rats from Experiment 1: three control+control (C + C), one EtOH+control (E + C), and three EtOH+EtOH (E + E), and one rat was removed from the T14 EtOH+saline group in Experiment 3). One rat was removed due to excess weight loss from the T35 TMZ group in Experiment 4. Animals were housed in a temperature- and humidity-controlled vivarium on a 12-hour light cycle with food and water available ad libitum unless otherwise noted. On arrival, rats were pair housed and acclimated to the vivarium for five days. During the acclimation period, rats were handled for three-minutes on three days. Experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky in accordance with The Guide for the Care and Use of Laboratory Animals [72].
Rat model of an alcohol use disorder
The Majchrowicz 4-day binge model of an AUD achieves high blood alcohol (EtOH) concentrations (BECs) characteristic of binge pattern drinking in addition to producing tolerance, physical dependence, neurodegeneration and reactive, adult neurogenesis [51, 61, 73, 74]. In all experiments the binge model was performed as previously described [51, 61, 74]: rats were gavaged initially with either a 5 g/kg dose of EtOH in diet (25% w/v EtOH in Vanilla Ensure Plus,® Abbott Laboratories, Columbus, OH) or an isocaloric control diet (dextrose in Vanilla Ensure Plus®). Subsequent doses were titrated based on intoxication behavior which was scored according to a 0 (normal) to 5 (highly intoxicated) scale modified from Majchrowicz [73, 74]. Each rat then receives the corresponding dose of EtOH via gavage (5 g/kg –0 g/kg respectively). Controls are administered the average volume given to EtOH animals. Unhandled, ungavaged control rats were not included as no differences in neurogenesis endpoints have been observed between rats gavaged with control diet compared to unhandled controls given ad libitum access to food and water in this model [54]. After four days of EtOH exposure, rats were observed for withdrawal behavior for 30 minutes of each hour and withdrawal behavior was scored for 17 hours beginning 10 hours after the final EtOH dose as described previously [74]. For each 30-minute period, the highest scored behavior observed was averaged across the 17 hours (mean withdrawal severity) as well as the overall highest scored behavior (peak withdrawal severity).
Blood ethanol concentration
90 minutes following the seventh EtOH dose, tail blood was collected from each EtOH animal for BEC determination [74]. Blood samples were centrifuged at 1800x g for five minutes, then stored at –20°C until analysis on an AM1 Alcohol Analyser (Analox Instruments, Lunenburg MA). Samples were run in triplicate and frequent calibration of the Analyser was performed using a 300 mg/dl EtOH standard. BECs are reported as mg/dl± standard error of the mean (SEM).
2.4Experiment 1: Ethanol-induced inhibition of reactive proliferation
The first experiment attempted to use EtOH to decrease reactive, adult neurogenesis in the 4-day binge model. Previous work has shown that a 5 g/kg acute dose of EtOH decreases NSC proliferation and adult neurogenesis [54, 75]. Therefore, animals were given two 5g/kg doses of EtOH because rats were not naïve. Hence, after four days of binge exposure, rats received either two 5 g/kg doses of EtOH or control diet, the first dose was administered on day six of abstinence (T6) and the second eight hours later at the beginning of T7 (Fig. 1). The three experimental conditions were as follows (see also Table 1): 4-day binge with control diet plus two doses of control diet (Control+control, C + C; n = 8), 4-day binge with EtOH diet+two doses of control diet (EtOH+control, E + C; n = 6), and 4-day binge with EtOH diet plus two doses of 5 g/kg EtOH (EtOH+EtOH, E + E; n = 8). Rats were then injected intraperitoneally (i.p.) with a 300 mg/kg (mean volume 5.1±0.1 ml) saturating dose of bromodeoxyuridine (BrdU; Roche, Mannheim, Germany) in 0.9% saline (20 mg/ml) to label dividing cells [76]. Two hours later, rats were then given a lethal dose of sodium pentobarbital (i.p.; Fatal Plus®, Vortech Pharmaceuticals, Dearborn, MI), transcardial perfusions were performed, and tissue harvested as described below.
Fig. 1
(A) The experimental timeline for Experiment 1: additional doses of 5g/kg EtOH or isocaloric control diet were given at T6- and T7-days post 4-day binge exposure, near the peak of reactive cell proliferation (grey arrows). On T7, BrdU was administered and rats were sacrificed 2 hours later (black arrow). (B-D) Representative images of BrdU immunoreactivity at T7 are shown for (B) E + C (n = 6), (C) C + C (n = 8), and (D) E + E (n = 8) groups. (E) Data are the mean number of BrdU+ cells per section in the subgranular zone with error bars reflecting SEM. *p < 0.05 versus control. GCL = granule cell layer. Scale bar = 100μm; 20μm in inset.
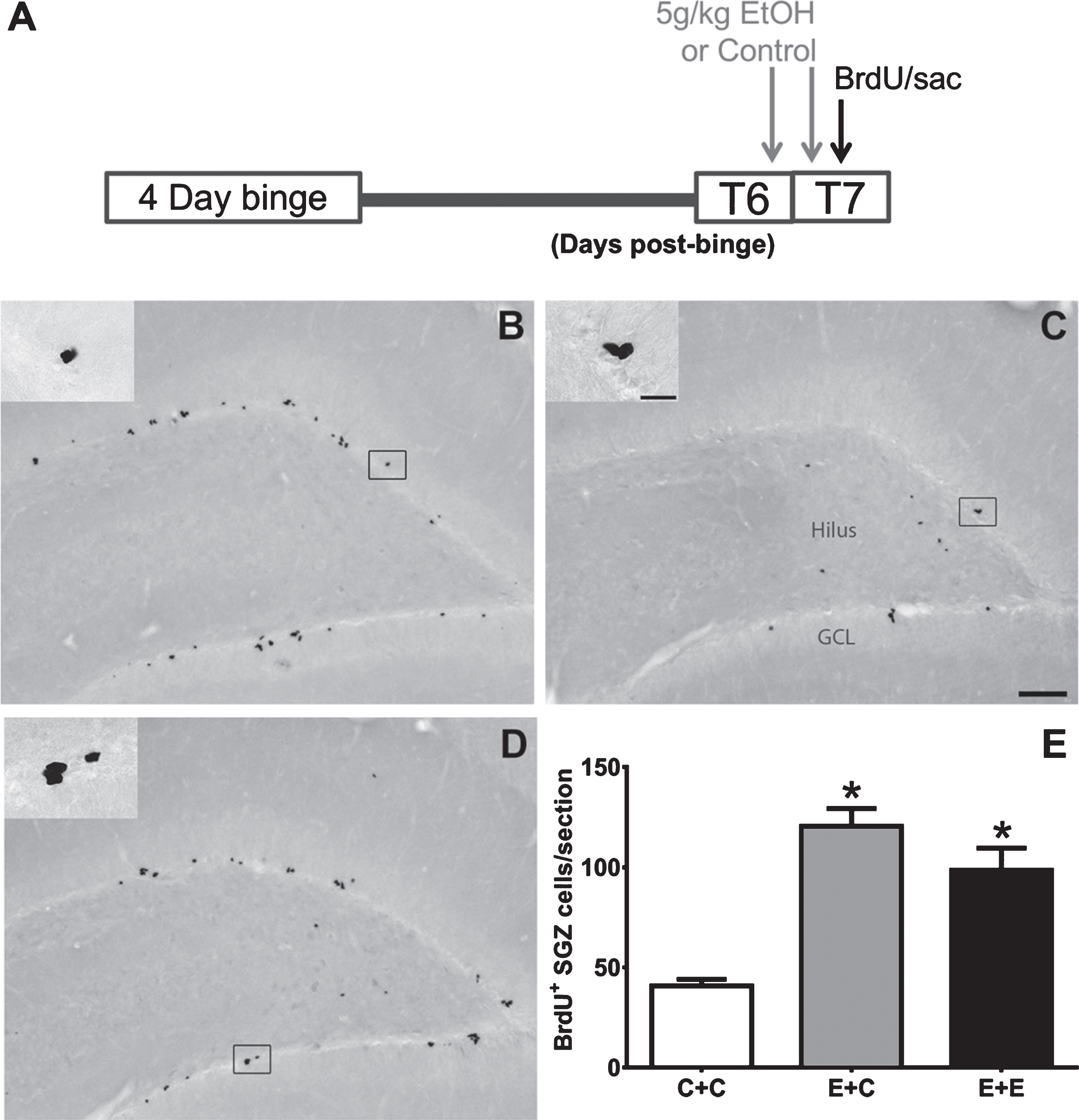
Table 1
Subject data for Experiment 1
| Binge Tx | Add’l Tx | Volume (ml/day) | Intox Score (1 –5) | Dose (g/kg/day) | BEC (mg/dl) | Mean WD | Peak WD | |
| E+E (n = 8) | EtOH | EtOH | 11.8±0.7 | 1.8±0.1 | 9.8±0.4 | 374.9±31.42 | 0.8±0.2 | 3.4±0.2 |
| E+C (n = 6) | EtOH | Control | 12.5±0.9 | 1.6±0.2 | 10.3±0.6 | 353.8±23.55 | 0.6±0.2 | 2.8±0.6 |
| C+C (n = 8) | Control | Control | 11.6±0.0 | n/a | n/a | n/a | n/a | n/a |
Experiment 2: Determine a dose of TMZ to inhibit reactive adult neurogenesis
TMZ (Cayman Chemicals, Ann Arbor, MI), a DNA alkylating agent typically utilized as an oral chemotherapy for glioblastoma, has been shown to decrease proliferation in adult neurogenesis studies [42, 45, 77]. Therefore, in experiment two we determined an effective dose of TMZ to inhibit reactive adult neurogenesis after alcohol dependence. Previous studies have used a 3 days on 4 days off cycle with doses ranging from 25–50 mg/kg [42, 45, 47]. As our lab has shown that following a 4-day binge, reactive cell proliferation occurs starting 5 days post binge and peaks at day 7 [61], we used a single 3-day cycle of TMZ starting day 3 through day 6. TMZ was made fresh per injection by dissolving 5 mg/ml TMZ in 0.9% normal saline, sonicating for 30 minutes in a 35°C water bath, then filter sterilizing. Control animals received an injection of a 0.9% saline vehicle solution. All animals received the same number of i.p. injections at approximately the same time. Volumes of TMZ administered averaged 1.6±0.02 ml and saline averaged 1.6±0.02 ml.
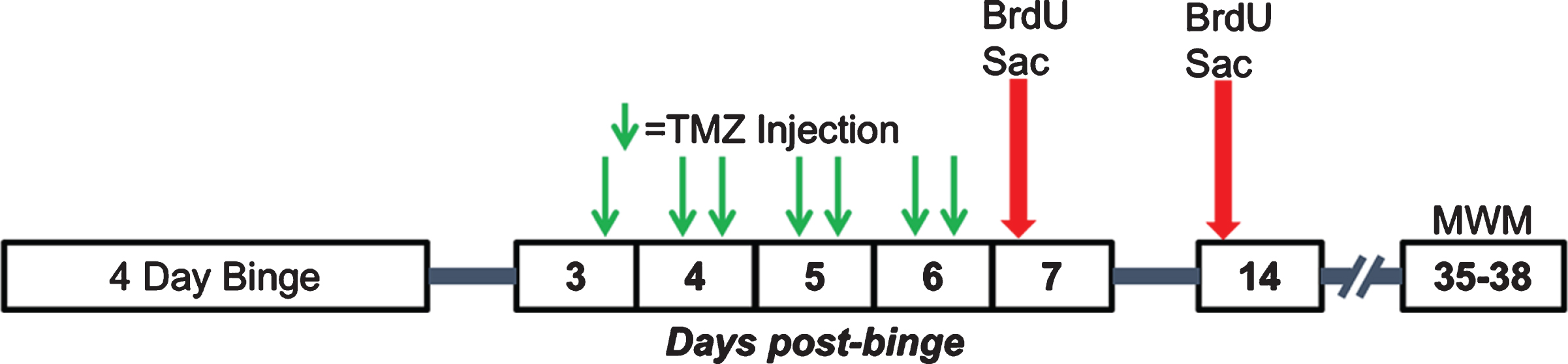
To establish an effective dose to decrease reactive proliferation, 43 rats (Table 2) received an injection of TMZ every 12 hours for 3.5 days starting 3.5 days after the last dose of EtOH as outlined in Fig. 2. Rats received either 50 mg/kg/day TMZ (25 mg/kg TMZ injection every 12 hours), 25 mg/kg/day TMZ (an alternating dose of 25 mg/kg TMZ then saline every 12 hours), or saline injection every 12 hours. Then, 12 hours after the last TMZ injection (T7), when reactive cell proliferation peaks [61], rats were injected with BrdU (300 mg/kg, i.p.) as above. Two hours after BrdU exposure, rats received a lethal dose of pentobarbital (Fatal Plus®) then were transcardially perfused and brains harvested as described below.
Table 2
Subject data for Experiments 2–4 (all TMZ studies)
| Group | Binge | TMZ | Volume | Intox | Dose | BEC | Mean | Peak | |
| Tx | (mg/kg) | (ml/day) | Score | (g/kg/day) | (mg/dl) | WD | WD | ||
| Exp. 2: T7 Proliferation | E+0 TMZ (n = 8) | EtOH | 0 | 11.5±0.4 | 1.9±0.1 | 9.4±0.3 | 353.2±16.0 | 2.1±0.3 | 3.5±0.1 |
| E+25 TMZ (n = 9) | EtOH | 25 | 11.8±0.7 | 1.7±0.1 | 9.8±0.4 | 341.7±15.3 | 1.6±0.3 | 3.5±0.1 | |
| E+50 TMZ (n = 8) | EtOH | 50 | 11.9±0.3 | 1.7±0.1 | 9.9±0.2 | 368.1±11.0 | 1.4±0.2 | 3.3±0.2 | |
| C+0 TMZ (n = 6) | Control | 0 | 11.7±0.0 | – | – | – | – | – | |
| C+25 TMZ (n = 6) | Control | 25 | 11.7±0.0 | – | – | – | – | – | |
| C+50 TMZ (n = 6) | Control | 50 | 11.7±0.0 | – | – | – | – | – | |
| Exp 3: T14 Neurogen. | E+0 TMZ (n = 7) | EtOH | 0 | 11.5±0.5 | 1.6±0.2 | 9.4±0.3 | 380.4±17.8 | 1.5±0.2 | 3.5±0.2 |
| E+50 TMZ (n = 8) | EtOH | 50 | 12.5±0.6 | 1.9±0.1 | 10.3±0.5 | 372.5±30.9 | 1.5±0.3 | 3.5±0.2 | |
| C+0 TMZ (n = 8) | Control | 0 | 11.8±0.0 | – | – | – | – | – | |
| C+50 TMZ (n = 8) | Control | 50 | 11.8±0.0 | – | – | – | – | – | |
| Exp 4: T35 MWM | E+0 TMZ (n = 9) | EtOH | 0 | 12.3±0.5 | 1.9±0.1 | 9.1±0.4 | 426.1±14.4 | 1.5±0.3 | 3.8±0.0 |
| E+50 TMZ (n = 10) | EtOH | 50 | 11.5±0.5 | 1.8±0.1 | 9.7±0.3 | 432.1±18.5 | 1.2±0.3 | 3.2±0.2 | |
| C+0 TMZ (n = 10) | Control | 0 | 11.5±0.0 | – | – | – | – | – | |
| C+50 TMZ (n = 10) | Control | 50 | 11.5±0.0 | – | – | – | – | – |
All binge parameters and subject data were statistically similar except peak withdrawal score. Peak withdrawal scores differed only between EtOH+saline and TMZ rats in the T35 group. As all groups learned at a similar rate regardless of treatment conditions, it is unlikely that peak withdrawal score had an effect.
Fig. 2
Experimental timeline for Experiments 2, 3, and 4. Following the 4-day binge, rats underwent TMZ or saline injections every 12 hours for 3.5 days between T3 and T6 days post-binge. Separate cohorts of rats were then injected with BrdU and sacrificed 2 hours later at either seven- or 14-days post-binge. A third cohort underwent MWM training 35–38 days post-binge.
Experiment 3: Verify that TMZ does not delay reactive neurogenesis
Based on the result of Experiment 2, 32 rats were given 50 mg/kg/day TMZ (i.e. 25 mg/kg of TMZ every 12 hours) for 3.5 days starting at 3.5 days post binge. Thus, for Experiment 3 (see timeline in Fig. 2), rats were injected with BrdU (300 mg/kg, i.p.) in 0.9% saline at T14, then two hours later sacrificed by pentobarbital overdose (Fatal Plus®), transcardially perfused, and brains extracted as described below. T14 was chosen because it has previously been shown that peak reactive proliferation occurs at T7 followed by a reactive increase in neurogenesis, as seen by early markers of neuronal fate (e.g. doublecortin; NeuroD), a week later at T14 [51, 61]. Furthermore, we wanted to rule out that TMZ administration did not merely delay reactive proliferation. Hence, we measured proliferation (BrdU) as well as neurogenesis markers (NeuroD) at T14.
Exp 4: Determine the effect of TMZ-inhibited reactive neurogenesis on Morris water maze behavior
In experiment 4, we examined whether inhibiting reactive, adult neurogenesis with TMZ prevented the recovery of 4-day binge alcohol impaired learning and memory behavior on the MWM. Following the experiment 3 above, we exposed 40 animals to 50 mg/kg/day TMZ (25 mg/kg i.p., every 12 hours) for 3.5 days starting 3.5 days post binge. As outlined in Fig. 2, starting at T35 hippocampus-dependent spatial learning and memory was assessed using the MWM as described previously [78]. Twenty-eight days between TMZ and testing were allotted because of numerous prior studies detailing that newborn granule cells require approximately 28 days to mature [79]. Furthermore, newborn neurons at around 28 days are recruited into activation during the MWM [80]. Briefly, rats were trained in the MWM for four days. Each day, a block of four 60 second trials were carried out in which rats searched for a 13 cm diameter platform that was submerged in a 180 cm diameter pool made opaque with black non-toxic paint. If the animal located the platform, he was allowed to rest on the platform for 15 seconds before being placed back in the home cage for four minutes between trials. The sequence of starting points for an individual day was consistent across animals, but the inter-day sequence was different and counterbalanced in a Latin square design. Animals were organized into blocks so that each trial contained a different treatment group to ensure that no group was run consecutively. On the final day, approximately one hour after the last training session, a probe trial was carried out in which the platform was removed, and rats were placed in the pool at a novel entry point. The probe trial was 60 seconds in length, unless the rat spent 5 seconds in the target zone (12.7 cm zone around the testing platform location) in which case the trial would end. A video camera, motion analyzer (EthoVision XT 10, version 10.1, Noldus, Wegeningen, The Netherlands), and computer (Dell Precision T3610) were used to measured swim path length (cm), latency to platform (seconds), and speed (cm/second). Daily measures were averaged for individual animal’s four trial blocks then averaged across treatment groups. Daily means are reported as length, latency, and speed±SEM.
Histological methods
Tissue procurement
Rats were anesthetized by sodium pentobarbital overdose and transcardially perfused using 0.1M phosphate buffered saline followed by 4% paraformaldehyde pH 7.4 exactly as reported previously [81]. Brains were removed, post-fixed for 24 hours in 4% paraformaldehyde, stored in 0.1M phosphate buffered saline, and cut coronally into 40μm sections using a vibrating microtome (Leica VT1000S, Wetzlar, Germany). Sections were collected into cryoprotectant in an 1:12 series in 24-well plates which were stored at –20°C until processing.
Bromodeoxyuridine immunohistochemistry
Every 6th section was utilized for BrdU free-floating immunohistochemistry as described previously [54, 81]. Briefly, endogenous peroxidases were quenched by incubating in 0.6% hydrogen peroxide for 30 minutes. Antigen retrieval was then carried out with standard sodium citrate and 50% formamide for 2 hours at 65° C, followed by 2N HCl for 1 hour at 37° C. After neutralization and rinses in Tris-buffered saline (TBS), nonspecific binding was blocked (3% normal horse serum/0.1% Triton X-100/TBS; Vector Laboratories, Burlingame, CA) for 30 minutes. Sections were rinsed in a MgCl reaction buffer then DNase for 1 hour. Sections were incubated overnight at 4°C in primary antibody (1:5000 mouse anti-BrdU, MAB 3424 Millipore, Billerica, MA). The next day, sections were rinsed in blocking buffer 3x10 minutes, incubated in biotinylated horse anti-mouse secondary antibody for 1 hour (Vector Laboratories, Burlington, CA), then an avidin-biotin-complex (ABC Elite Kit, Vector Laboratories, Burlingame, CA) incubation for 1 hour at room temperature. Finally, tissue was processed with nickel-enhanced 3,3’-Diaminobenzidine (DAB, Polysciences, Warrington, PA) for visualization. Sections were mounted onto glass slides, allowed to dry overnight, counterstained lightly with cresyl violet (Acros Organics, Morris Plains, New Jersey) and sealed with glass coverslips and Cytoseal® (Stephens Scientific, Wayne, NJ).
Neurogenic differentiation factor 1 (NeuroD) immunohistochemistry
NeuroD1 reflects differentiation to neuronal fate [82]. Previous studies have shown increased NeuroD-positive (+) cells at T14 days post binge EtOH exposure [61]. Immunohistochemistry was performed on every 12th free floating section as described [61]. Briefly, tissue was incubated in 0.6% hydrogen peroxide for 30 minutes, Antigen Retrieval Citra Solution (BioGenex, Fremont, CA) for 1 hour at 65°C, blocking buffer (3% normal rabbit serum/0.1% triton-X/TBS; Vector Laboratories, Burlingame, CA) for 30 minutes, and finally primary antibody (1:1000 goat anti-NeuroD SC-1084, Santa Cruz Biotechnologies, Dallas, TX) in blocking buffer for two nights at 4°C. Following the ∼42 hour incubation, tissue was rinsed in blocking buffer, incubated in biotinylated rabbit anti-goat IgG secondary antibody (1:200) for 1 hour, avidin-biotin-complex (ABC Elite Kit) for 1 hour, and processed for nickel-enhanced DAB for visualization at room temperature. Sections were mounted onto glass slides, dried overnight and then sealed using glass coverslips and Cytoseal®.
Quantification
Slides were coded to blind experimenters of ex-perimental conditions. BrdU+ and NeuroD+ cells (240μm and 480μm between sections, respectively) were quantified on one hemisphere between –2.28 mm and –5.52 mm Bregma [83] along the subgranular zone, a band of cells around three cell bodies wide that runs between the granule cell layer and hilus of the dentate gyrus. For the dorsal hippocampus, this span results in 14–16 brain tissue sections for the 1:6 series (BrdU) or 6–8 sections in the 1:12 series (NeuroD). Due to issues in tissue integrity, sections were dropped rarely from quantification. Thus, on average 13.7±0.3 sections (Control) and 14.0±0.2 sections (EtOH) were profile counted across all BrdU+ experiments and 6.8±0.1 (Con) and 7±0.2 (EtOH) were counted for NeuroD. The number of sections did not differ between groups. Profile counts (immunopositive cells/section ± SEM) were obtained using a 100x objective lens on an Olympus BX41 microscope (Olympus, Center Valley, PA). Because the anterior portion of the dentate gyrus is a single blade compared to dual blades of the posterior portion, blades were quantified separately and then averaged across sections. A profile counting approach was chosen as it is more appropriate versus stereology due to immunopositive profiles being relatively few in number and non-homogenously distributed across the dentate gyrus [84, 85]. Our previous work has shown an identical percent change in profile counts compared to stereology for neurogenesis-related immunopositive profiles [84].
Statistics
Data were analyzed using GraphPad Prism® (version 7.0, GraphPad software, La Jolla, CA) or SPSS Statistics (version 22, IBM, Armonk, NY). Subject binge data were analyzed using the student’s t-test or one-way ANOVA for EtOH dose and BEC while the nonparametric Mann-Whitney test or Kruskal-Wallis test was used for intoxication and withdrawal scores as appropriate. Depending on the experiment and complexity of the design, histological data were analyzed by either ANOVA followed by Tukey’s multiple comparison tests or two-way ANOVA followed by planned comparisons (t-tests). In TMZ studies, planned comparisons via t-tests were chosen to test specific experimental questions important for the study because there was not an expectation of a significant interaction between diet and drug (TMZ would likely decrease proliferation in all conditions). Behavior outcomes on the MWM were analyzed by three-way repeated measures ANOVA and posthoc Bonferroni’s multiple comparisons test, where appropriate. The MWM probe trail was analyzed using a two-way ANOVA. A p-value of p < 0.05 was accepted as significant in all analyses. All data are reported as mean±SEM.
RESULTS
Experiment 1: Effect of additional doses of ethanol on reactive cell proliferation
Binge parameter data were collected during intoxication and withdrawal and compared between treatment groups as summarized in Table 1. Overall, 4-day binge EtOH-exposed rats had a mean intoxication score of 1.7±0.1 which corresponds to ataxia with an elevated abdomen. Rats therefore received an average of 10±0.3 g/kg/day EtOH which resulted in an average BEC of 365.9±20.0 mg/dl, values which are similar to those reported previously [74]. As shown in Table 1, no significant differences were found between E + E or E + C groups for 4-day binge subject parameters of average daily dose received or BECs (Student’s t-test) nor for intoxication scores (Mann-Whitney). Withdrawal behavior was scored across 17 hours. EtOH rats had a mean withdrawal severity score of 0.8±0.1 which corresponds with hyperactivity, while the mean peak withdrawal severity score achieved was 3.1±0.3 which corresponds with head tremor. No significant differences were found for mean or peak withdrawal scores between groups (Mann-Whitney test). Thus, on average, EtOH treated rats experienced a similar 4-day binge intoxication and withdrawal and were similar to our past reports.
BrdU immunohistochemistry was utilized to label and quantify proliferating cells in the subgranular zone of the hippocampal dentate gyrus. As shown in Fig. 1, BrdU+ cells can be found in clusters lining the subgranular zone in all groups as expected. One-way ANOVA on BrdU+ cells revealed a significant effect of 4-day binge treatment [F (2,19) =23.67; p < 0.0001]. Tukey’s multiple comparisons test showed that both E + C and E + E treatment conditions had increased BrdU+ cells (p < 0.05) compared to the C + C group. There was no significant difference found between E + C or E + E groups, however. These results indicate that two acute doses of 5 g/kg EtOH does not reduce alcohol-elevated BrdU+ cells seven days after 4-day binge EtOH exposure.
Experiment 2: TMZ normalizes alcohol-induced cell proliferation at T7
To determine an effective dose to blunt NSC proliferation after the 4-day binge, rats received 0, 25, or 50 mg/kg/day TMZ following either 4-day binge EtOH or isocaloric control diet (Table 2). Binge parameters were compared between treatment groups and no differences were found between EtOH groups for average daily dose or BEC (ANOVA), nor intoxication or withdrawal scores (Kruskal-Wallis). Overall, EtOH rats had an average intoxication score of 1.8±0.1, which corresponds to ataxia with an elevated abdomen. Rats received an average of 9.7±0.2 g/kg/day EtOH which resulted in an average BEC of 353.8±8.6 mg/d. These subject data values are similar to those reported previously for this model [74].
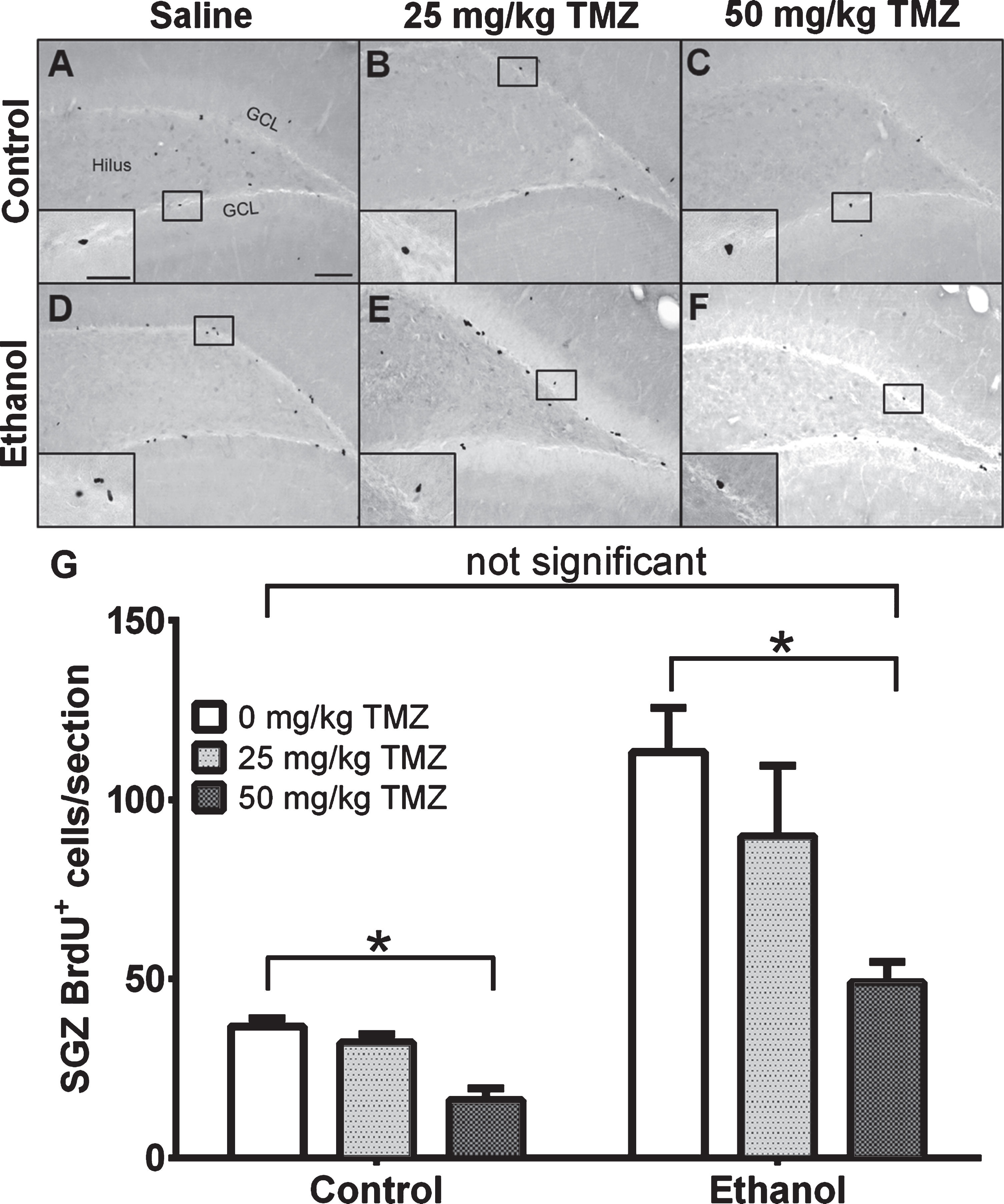
To measure the effect of TMZ on reactive cell proliferation in the subgranular zone at T7, 12 hours after the final dose of TMZ, BrdU was injected and rats were transcardially perfused 2 hours later. As expected, BrdU+ cells were clustered along the subgranular zone of all groups and quantified via profile counts (Fig. 3). Two-way ANOVA showed a main effect of both diet [F (1,37) =30.86; p < 0.0001] and drug [F (2,37) =6.098; p = 0.0051], but no significant interaction. Post-hoc planned comparisons via t-test, showed no significant differences between 25 mg/kg/day TMZ and saline for either EtOH or control groups. Importantly, in the EtOH group, there were significantly fewer BrdU+ cells/section found after 50 mg/kg/day TMZ compared to saline [t (14) =4.708; p = 0.0003], but this was also true for diet controls group [t (10) =5.316; p = 0.0003]. The 50 mg/kg/day cycle of TMZ decreased proliferation rates to that of control as planned t-test showed no significant difference between BrdU+ cell counts between 50 mg/kg/day EtOH+TMZ rats and control+saline rats (Fig. 3). Thus, four days of 50 mg/kg/day TMZ is an effective dose/pattern to reduce reactive proliferation to control levels in EtOH animals.
Fig. 3
TMZ reduces reactive cell proliferation indicated by BrdU+ cells. (A-E) Representative images of BrdU immunoreactivity at T7 (2 hours after BrdU injection) are shown for (A-C) control and (D-E) EtOH groups for 0 mg/kg (saline), 25 mg/kg, and 50 mg/kg TMZ. (G) Data are the mean number of BrdU+ cells in the subgranular zone with error bars reflecting SEM. *p < 0.05; n = 8-9 for EtOH groups and n = 6 for control groups. GCL = granule cell layer. Scale bar = 100μm; inset 40μm.
Experiment 3: TMZ normalizes alcohol-induced reactive neurogenesis at T14
After the four-day binge, rats were again given either saline or 50 mg/kg/day TMZ to decrease proliferation and neurogenesis. Intoxication behavior was scored and compared to ensure binge treatment was similar between groups (Table 2). No significant difference was found for average daily dose and BEC between EtOH groups using the Student’s t-test. In addition, no difference was found for intoxication or withdrawal scores using the non-parametric Mann-Whitney test. Mean intoxication score of all EtOH rats was found to be 1.7±0.1 which corresponds to ataxia and an elevated abdomen. Rats received an average daily dose of 9.8±0.3 g/kg/day EtOH which lead to a mean BEC of 376.4±17.2 mg/dl. Values are similar to those previously reported [74].
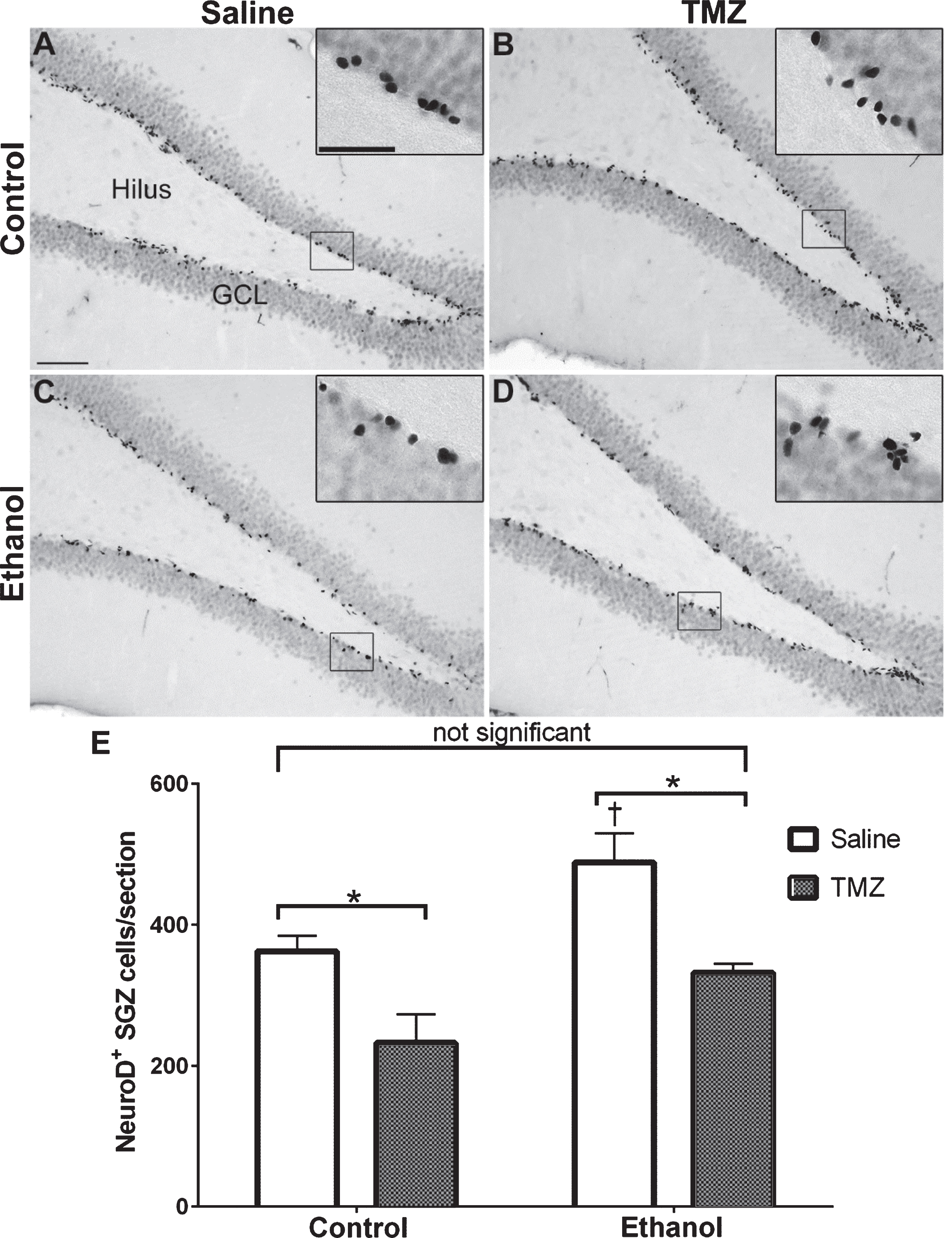
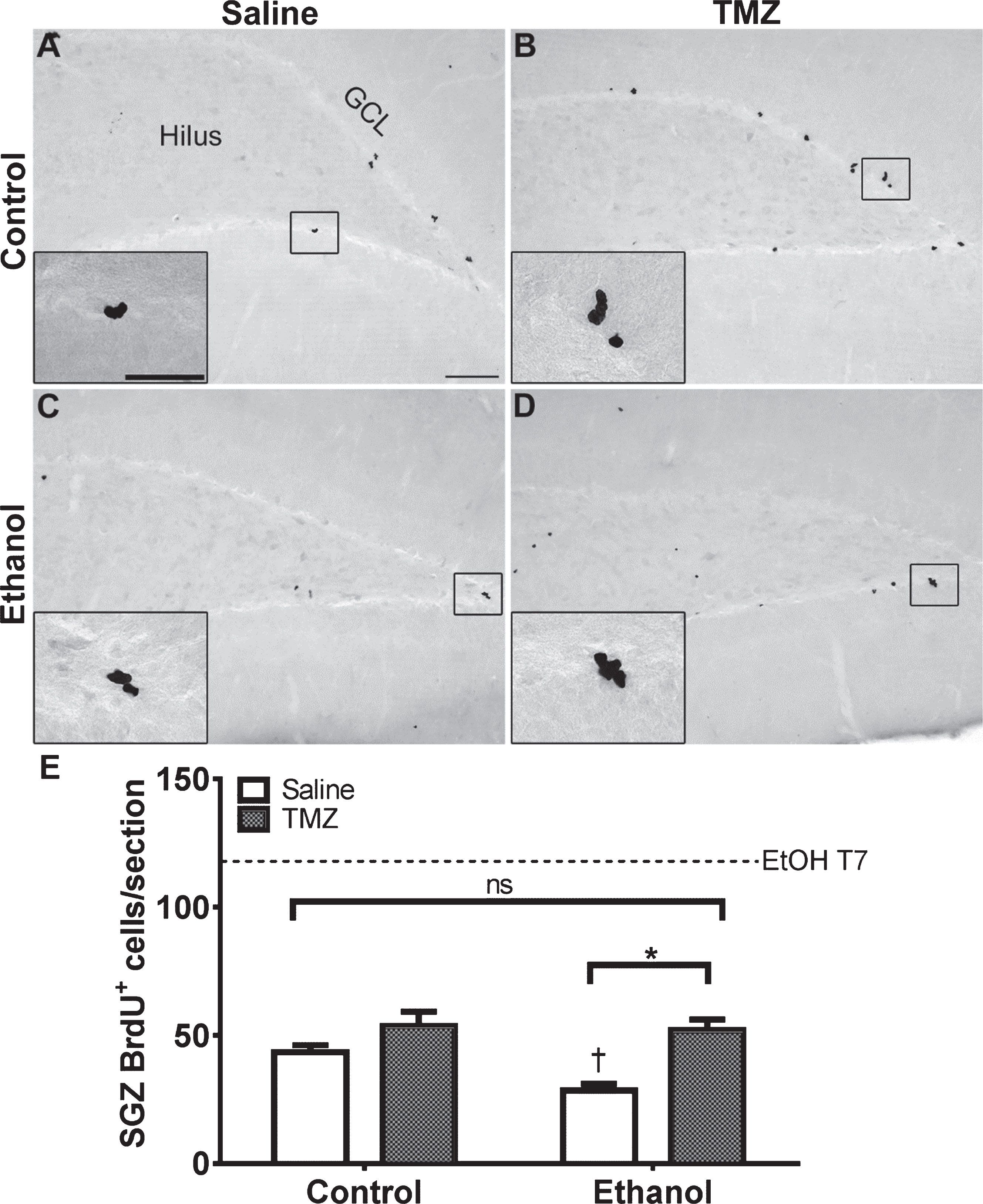
NeuroD is an early marker of progenitors com-mitted to a neuronal fate [82, 86], and immunohistochemistry was used to quantify NeuroD+ cells at T14 after EtOH exposure. NeuroD+ cells were seen in the subgranular zone and inner granule cell layer, consistent with markers of neuroblasts or immature neurons. Nuclear staining revealed decreases in 50 mg/kg/day TMZ compared to saline treated rats (Fig. 4). Two-way ANOVA revealed a main effect of both diet [F (1,28) =12.70; p = 0.0013] and drug [F (1,28) =20.41; p = 0.0001], but no significant interaction. Planned t-test comparisons showed that NeuroD+ cells were increased significantly at T14 in the EtOH+saline rats as opposed to control+saline [t (14) =2.694; p = 0.0175] consistent with our previous reports of reactive neurogenesis [51]. EtOH+TMZ rats had no significant difference in the number NeuroD+ cells compared to control+saline [t (14) =1.19; p = 0.25], indicating that TMZ blunts neurogenesis to levels similar to controls. Planned t-tests also showed TMZ administration decreased NeuroD+ cell counts of both control [t (14) =2.79; p = 0.014] and EtOH [t (14) =3.64; p = 0.0027] animals. Thus, regardless of diet, TMZ reduced neurogenesis compared to saline administered animals.
Fig. 4
TMZ reduces reactive neurogenesis indicated by NeuroD+ cells. (A-D) Representative images of NeuroD immunoreactivity at T14 are shown for (A, B) control and (C, D) EtOH following saline or TMZ (50 mg/kg). (E) Data are the number of NeuroD+ cells in the subgranular zone with error bars reflecting SEM. *p < 0.05; †p < 0.05 control-saline vs. ethanol-saline; n = 8/group. GCL = granule cell layer. Scale bar = 100μm; inset = 40μm.
BrdU+ cells were also quantified at T14 post-binge exposure along the subgranular zone (Fig. 5). Two-way ANOVA showed a main effect for both diet [F (1,27) = 4.28; p = 0.048] and drug [F (1,27) = 17.69; p = 0.0003], but no significant interaction which reflects a significant difference between EtOH+TMZ and EtOH+saline rats according to planned posthoc t-tests [t (13) = 4.642; p = 0.0005] and control+saline versus EtOH+saline [t (13) = 3.919; p = 0.0018; Fig. 5]. Importantly, no significant difference was found between EtOH+TMZ and control+saline animals by planned t-test [t (14) = 1.759; p = 0.1], indicating TMZ blunts proliferation to that seen in controls. A dashed line is included in Fig. 5 to represent the extent of reactive proliferation seen in EtOH-exposed rats at T7. If TMZ delayed reactive proliferation by one week, the line indicates the number of BrdU+ cells that would be expected at T14.
Fig. 5
TMZ does not merely delay reactive NSC proliferation. (A-D) Representative images of BrdU immunoreactivity at T14 (2 hours after BrdU injection) are shown for (A, B) control and (C, D) EtOH following saline or TMZ (50 mg/kg). (E) Data are the number of BrdU+ cells in the subgranular zone with error bars reflecting SEM. *p < 0.05; †p < 0.05 saline control vs. saline ethanol. n = 7-8/group. GCL = granule cell layer. Scale bar = 100μm; inset = 40μm.
Experiment 4: TMZ blunting of reactive neurogenesis does not affect hippocampal-dependent learning
A hippocampus-dependent version of the MWM was utilized to determine how inhibiting neurogenesis would impact functional recovery of learning and memory task. As shown in Fig. 2, rats were exposed to 4-day binge alcohol or control diet and scored for withdrawal behavior. After the TMZ injection cycle, rats remained in their home cages for four weeks to allow newborn neurons to mature. Overall, the mean intoxication score was 1.9±0.1 which corresponds to ataxia with an elevated abdomen and did not differ between EtOH groups (Mann-Whitney test). This behavior resulted in rats receiving an average daily dose of 9.4±0.3 g/kg/day EtOH which produced a mean BEC of 429.0±11.3 mg/dl, a value that was slightly higher than previous studies [74]. No significant differences were found for average daily dose of EtOH or BEC between EtOH groups (Student’s t-test; Table 2). The grand mean withdrawal score was 1.3±0.2 and did not differ between EtOH groups (Mann-Whitney). However, peak withdrawal scores differed (U = 19.5; p = 0.028): EtOH+TMZ was 3.2±0.2 versus 3.8±0.03 in the EtOH+saline group. With the exception of a slightly higher BEC, all other subject binge data were similar to what has been found previously [74]. As TMZ was administered after alcohol exposure and withdrawal, the slight differences in the exposure model were not due to TMZ but merely chance in the random assignment of animals to groups.
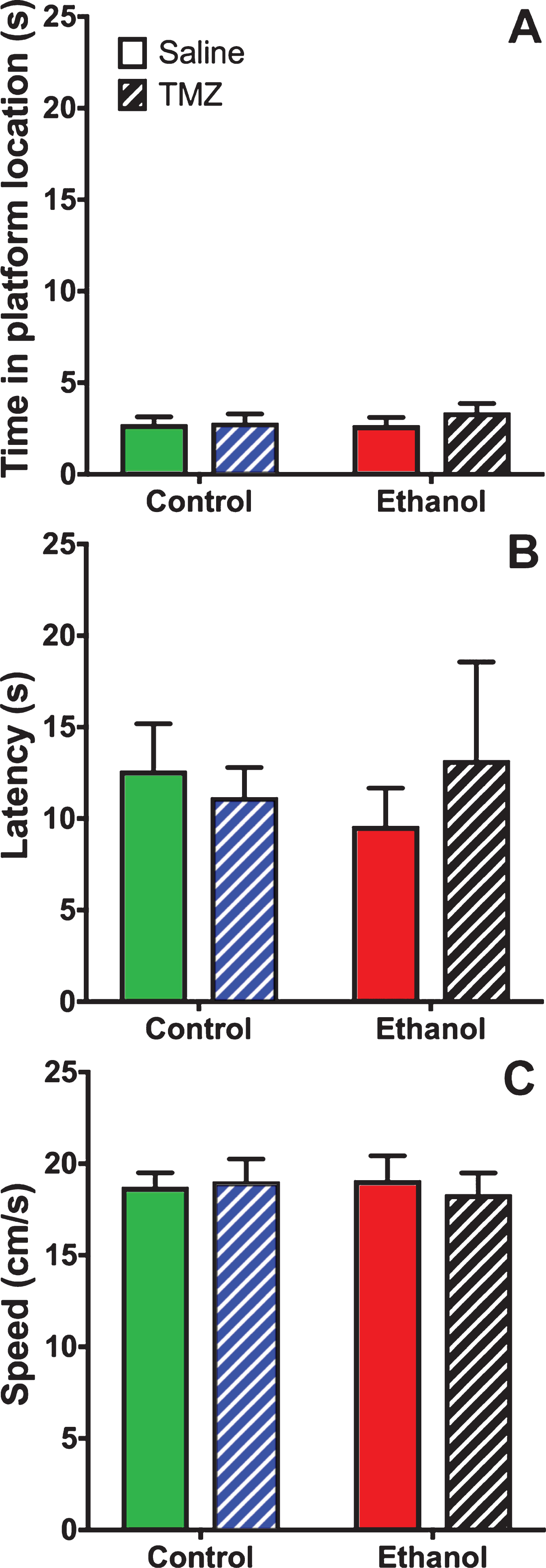
TMZ was used to blunt reactive neurogenesis following 4-day binge alcohol exposure. At T35 post binge, rats began MWM training (see Fig. 2 for timeline). This time point was used because it has been shown that newborn neurons mature at 28 days [79], and that 4-week-old neurons are activated during MWM [80]. As shown in Fig. 6, three-way repeated measures ANOVA (day x diet x drug) showed a main effect for day with distance to goal [F (3,108) =81.3; p < 0.001], time to goal [F (3,108) =93.8; p < 0.001] and speed [F (3,108) =48.7; p < 0.001]. No effect was found for diet or drug for any measure examined. To summarize, at T35 animals improved performance across training days, but no difference was found due to binge or TMZ exposure. A probe trial was also run even though we had not previous observed an effect of prior binge EtOH exposure on probe trial performance. As shown in Fig. 7, two-way ANOVA (diet x drug) showed no differences for the probe trial for time spent in the 12.7 cm goal zone, latency to approach the goal zone, or speed. Hence, treatment groups did not differ from one another and though TMZ normalized levels of neurogenesis, recovery of MWM performance was not blunted.
Fig. 6
TMZ did not affect MWM performance 5-week post-binge. Three aspects of MWM performance are shown: (A) distance to goal, (B) time to goal, and (C) speed. n = 9–11/group.
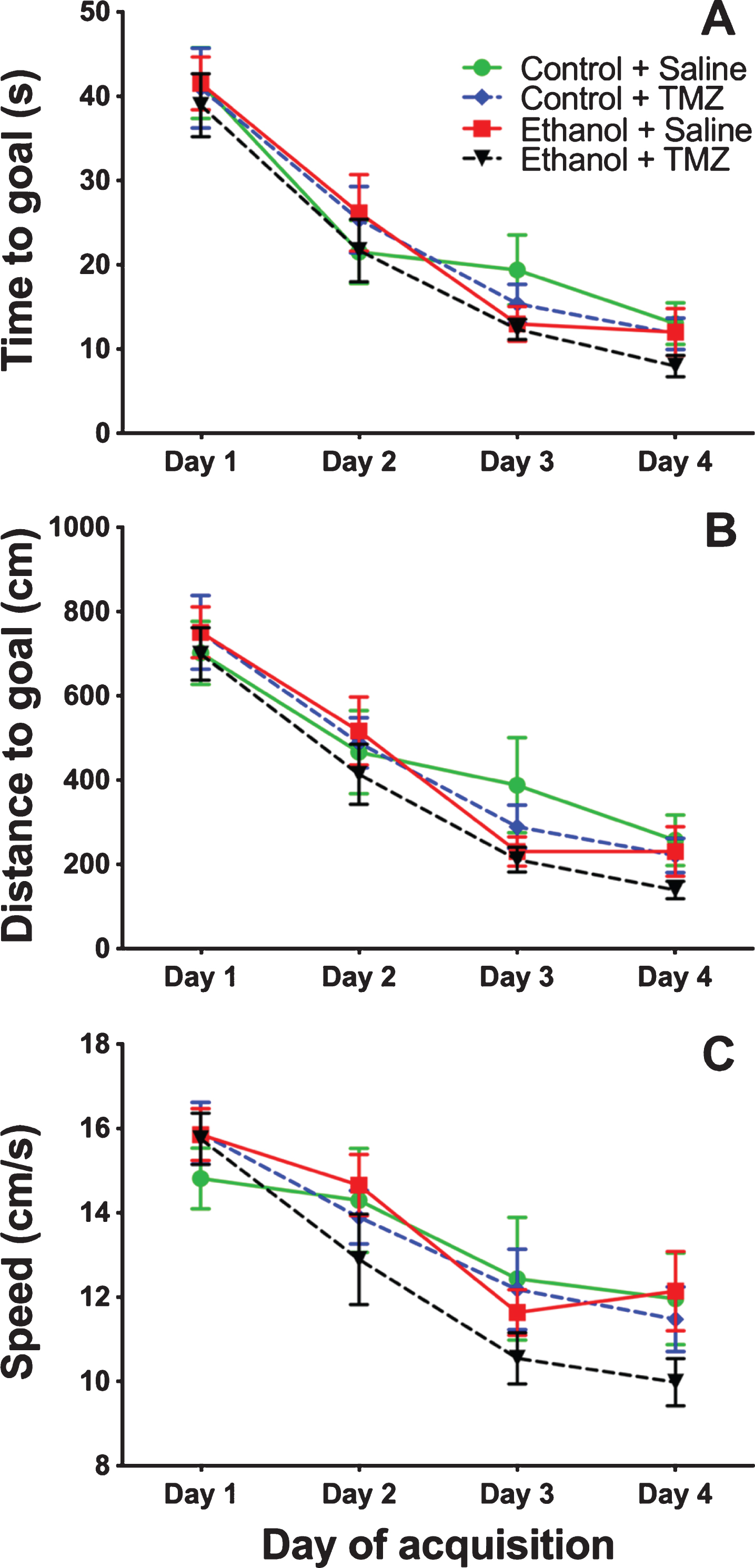
Fig. 7
TMZ did not affect MWM performance on the probe trail. Three measures of MWM probe trial performance are shown: (A) time spent in goal zone, (B) latency to goal zone, and (C) speed. n = 9–11/group.
DISCUSSION
Excessive alcohol consumption, one of the defining characteristics of an AUD, damages the brain and results in impairments across a variety of cognitive dimensions, though some deficits may recover with abstinence from alcohol drinking. The hippocampus is one such target of alcohol neurotoxicity [6, 8–13] that recovers with abstinence [24–26]. A variety of structural plasticity mechanisms have long been hypothesized to contribute to recovery in abstinence [20], but only within the last 20 years have hippocampal NSCs or adult neurogenesis been considered [50, 51]. The discovery of hippocampal NSCs and their role in hippocampal structure and function has made it enticing to speculate that adult neurogenesis could contribute to repair or repopulation of the dentate gyrus after damage by alcohol [4, 27, 29, 50, 66]. Our past reports that NSC activation drives a dramatic increase in adult neurogenesis after alcohol dependence further supports this speculation [51, 61, 62]. As such, we attempted to examine the role of these reactive, adult-born neurons in the recovery of hippocampal learning and memory in abstinence after alcohol dependence in a rat model. Two methods were examined for blunting reactive NSC proliferation and neurogenesis, (1) acute doses of alcohol and (2) TMZ, a brain penetrant DNA alkylating agent used in glioma [42]. TMZ administration, but not alcohol, successfully inhibited reactive neurogenesis to control levels. However, blunting of reactive neurogenesis to normal levels did not impact the recovery of hippocampal-dependent learning. Thus, despite reducing this potential mechanism of hippocampal recovery, learning and memory behavior still recovered and was identical to controls.
In experiment one, it was hypothesized that two acute doses of EtOH at six and seven days after 4-day binge exposure –across the peak of reactive NSC activation - would blunt reactive neurogenesis, as alcohol dose-dependently decreases NSC proliferation [54, 56, 87]. Surprisingly, reactive proliferation was not suppressed by acute doses of EtOH. BrdU+ cell counts were reduced by only 18% (p = n.s.; Fig. 1) at T7 post-binge EtOH exposure. While one might consider that a lengthier EtOH exposure is required, reactive neurogenesis in binge exposed rats was not inhibited by a second 4-day binge, at least in female rats [63]. Alcohol may not directly target cell proliferation mechanisms, as with even the highest relevant doses, cell proliferation is typically only reduced ∼50–60% [50, 54, 56].
TMZ, however, decreased alcohol-induced reactive proliferation and neurogenesis. TMZ normalized proliferation as indicated by a reduction of BrdU+cells in the TMZ group to levels similar to control+saline at T7 (Fig. 3). Importantly, we confirmed that TMZ inhibited reactive proliferation without merely delaying the timeline of proliferation (Fig. 5; [61]). In this model, reactive proliferation is detectable 5 days after 4-day binge exposure (e.g. T5), peaks around T7 and returns to normal by T10 [51, 61, 88, 89]. Although the number of BrdU+ cells at T14 in TMZ treated animals was increased compared to EtOH+saline, the effect size may be due more to the atypically lower BrdU+ cells in the EtOH+saline group. Indeed, EtOH+saline have fewer BrdU+ cells than control+saline rats, an effect not previously observed at this time point. Importantly, it is a much smaller increase than found at T7 (dashed line, Fig. 5) and the TMZ group does not differ from the control+saline group. In addition, previous reports have not shown a difference between EtOH and control animals at T14 in this model [51, 61, 88, 89] while others have seen persistent decreases in other alcohol models [90].
Reactive neurogenesis was observed but also reduced to control levels by TMZ, according to the number of NeuroD+ cells, an endogenous immature neuron marker. First, the reactive increase in neurogenesis was confirmed as shown in Fig. 4 where the number of NeuroD+ cells is increased in EtOH+saline as opposed to control+saline. TMZ-reduction of reactive neurogenesis to control levels is demonstrated by similar NeuroD+ profiles in both EtOH+TMZ and control+saline groups (Fig. 4) at T14. These data are consistent with literature showing that TMZ can be utilized to reduce neurogenesis [42, 45, 47, 91] as well as reactive neurogenesis [92]. As such, TMZ also decreased neurogenesis in control+TMZ rats at T14, though this was expected [42, 45, 47, 91]. While any reduction in neurogenesis may result in learning and memory deficits, no differences were observed in MWM performance among any group (Fig. 6). Another important caveat is that TMZ may have reduced basal neurogenesis in addition to reactive neurogenesis in the EtOH rats. Currently, there is no way to identify and selectively inhibit populations of cells which reactively proliferate, especially in a rat model. Another caveat is that we only examined the short term impact of TMZ on cell proliferation and neurogenesis. Although we ensured that reactive proliferation did not merely shift forward a week (Fig. 5), we did not examine the state of proliferation and neurogenesis at the time of behavior. Based on the similar performance on the MWM, NSC proliferation and neurogenesis were likely similar between groups, but this could be examined in future studies.
The MWM results were especially surprising for TMZ animals whose neurogenesis levels were blunted to that of saline controls yet performed identical to all other groups (Fig. 6). While some studies show that a single cycle of TMZ administration does not affect learning or memory in a hippocampus-dependent MWM task, such as trace and long-delay eyeblink classical conditioning [42], the goal was not to impair learning and memory per se, but to prevent the reactive neurogenesis response beyond basal levels in order to explore its role in recovery. Unfortunately, the acute nature and timing of the NSC activation/reactive neurogenesis phenomena is not conducive to multiple cycles of TMZ. Plus, the effect of TMZ on proliferation/neurogenesis is transient. A more specific and longer-acting knock out approach such as the TK-GFAP “neurogenesis knock down” rats may be appropriate [93] and help to elucidate the role of these newborn cells in recovery. Alternatively, these data may suggest that basal levels of adult neurogenesis are sufficient to support the recovery of hippocampal function [91]. TMZ rats were similar to all other groups in acquisition of MWM performance at T35-38 as opposed to impairments in acquisition observed at T8-11 (Chen et al., in prep). Thus, either the basal level of neurogenesis was sufficient to support recovery of hippocampal function or existing neurons were able to compensate [91].
Newborn neurons born post insult may play a vastly different role in other pathological states. Reactive increases in adult neurogenesis have been shown to occur after insult in many conditions [67–69, 94] besides AUD [51, 52, 61, 95, 96]. In some traumatic brain injury and stroke models, reactive neurogenesis appears to contribute to recovery of hippocampus-dependent learning [97–99], but after seizure in models of epileptogenesis, increased neurogenesis may be detrimental [69]. Perhaps in more severe conditions where neurons appear aberrant, specifically located ectopically or with altered morphology, that these new cells contribute to deficits in hippocampal functions [69, 89, 92, 100, 101]. Further, adding a substantial number of immature neurons to hippocampal circuitry may be as detrimental as the loss of newborn cells due, in part, to the distinct role these new cells play in learning and memory [102, 103]. This aberrant neurogenesis is most evident in cases of seizure e.g. [69, 101], which is of particular note because in the AUD model utilized, withdrawal sequelae may include seizures, the severity of which correlates to the extent of reactive neurogenesis [51, 74].
These opposing effects of reactive neurogenesis associated with recovery versus aberrant neurogenesis in models of epilepsy support that different mechanisms of NSC activation, neurogenesis and/or neuron integration are likely involved. However, little is known about stem cell activation in either condition. Plus, increased neurogenesis in seizure models is accompanied by other pathologies not observed in AUD models such as mossy fiber sprouting/reorganization, ectopic granule cells and aberrant morphology of newborn cells [101, 104, 105], though see [89]. Additionally, following induction of seizure, long lasting decreases in neurogenesis have been shown after initial increase of neurogenesis [106] and those cells that do persist demonstrate both morphological and functional abnormalities [107]. Abnormal dendritic arborization, in particular, may be a hallmark of these aberrant neurons, hypothesized to prevent newborn neurons from responding to the excess electrical activity of a seizure [108]. Alcohol withdrawal seizures differ from those that occur in epilepsy, though some responses to overexcitation are common to both conditions [109]. However, essentially nothing is known about the morphology or function of cells born during reactive neurogenesis in alcohol models. Only effects of alcohol intoxication prior to withdrawal have been reported for newborn cell dendritic morphology: blunted dendritic trees of doublecortin-labeled immature neurons in a chronic drinking model of an AUD [110] and reduced spine density [59]. Blunted dendritic arborizations of granule cell neurons have long been known to occur in models of AUD [111, 112]. Thus, examining cell morphology and function of those cells born during reactive neurogenesis remains an important future direction to understand the role of these cells in recovery of hippocampal integrity after alcohol dependence.
In conclusion, a single cycle of TMZ inhibited reactive proliferation and neurogenesis following four-day binge EtOH exposure. TMZ treatment and reduction of reactive neurogenesis to control levels did not impair the recovery of performance in the MWM one month after TMZ treatment and 35 days after EtOH exposure as originally predicted. Rats still recovered learning and memory performance despite only baseline neurogenesis occurring. While initially unexpected there are other neuropathological events that occur with excess alcohol exposure that may recover and help explain these results. Gliosis, for example, is especially severe in the hippocampus initially in the model utilized [113]. Altogether, the effect of alcohol on adult neurogenesis in animal models explains a novel aspect of hippocampal pathology and recovery that corresponds to an emerging similar story in the human condition [6, 50, 51, 53, 54, 58, 60]. Hence, further work is needed to better characterize and differentiate how adult born neurons contribute to both hippocampal impairments in alcohol use and abuse but also recovery in abstinence.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors thank Deann Hopkins (U.K.) for technical assistance and Natalie Nawarawong, Ph.D. (U.T.) for critical reading of the manuscript. This work was supported by NIAAA and NIDA grants R01AA016959 (KN), F31AA023459 (CGN), T32DA016176 (Dwoskin) and the University of Kentucky Department of Pharmaceutical Sciences.
REFERENCES
[1] | Grant BF , Goldstein RB , Saha TD , Chou SP , Jung J , Zhang H , et al. Epidemiology of DSM-5 Alcohol Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. (2015) ;72: (8):757–66. doi:10.1001/jamapsychiatry.2015.0584 |
[2] | Zahr NM , Kaufman KL , Harper CG . Clinical and pathological features of alcohol-related brain damage. Nat Rev Neurol. (2011) ;7: (5):284–94. doi:10.1038/nrneurol.2011.42 |
[3] | Eckardt MJ , Martin PR . Clinical assessment of cognition in alcoholism. Alcohol Clin Exp Res. (1986) ;10: (2):123–7. doi:10.1111/j.1530-0277.1986.tb05058.x |
[4] | Crews FT , Nixon K . Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. (2009) ;44: (2):115–27. doi:10.1093/alcalc/agn079 |
[5] | Hunt WA . Are binge drinkers more at risk of developing brain damage? Alcohol. (1993) ;10: (6):559–61. doi:10.1016/0741-8329(93)90083-z |
[6] | Wilson S , Bair JL , Thomas KM , Iacono WG . Problematic alcohol use and reduced hippocampal volume: a meta-analytic review. Psych Med. (2017) ;47: (13):2288–301. doi:10.1017/S0033291717000721 |
[7] | Staples MC , Mandyam CD . Thinking after Drinking: Impaired Hippocampal-Dependent Cognition in Human Alcoholics and Animal Models of Alcohol Dependence. Front Psychiat. (2016) ;7: :162. doi:10.3389/fpsyt.2016.00162 |
[8] | Beresford TP , Arciniegas DB , Alfers J , Clapp L , Martin B , Du Y , et al. Hippocampus volume loss due to chronic heavy drinking. Alcohol Clin Exp Res. (2006) ;30: (11):1866–70. doi:10.1111/j.1530-0277.2006.00223.x |
[9] | Laakso MP , Vaurio O , Savolainen L , Repo E , Soininen H , Aronen HJ , et al. A volumetric MRI study of the hippocampus in type 1 and 2 alcoholism. Behav Brain Res. (2000) ;109: (2):177–86. doi:10.1016/s0166-4328(99)00172-2 |
[10] | Parada M , Corral M , Caamano-Isorna F , Mota N , Crego A , Holguin SR , et al. Binge drinking and declarative memory in university students. Alcohol Clin Exp Res. (2011) ;35: (8):1475–84. doi:10.1111/j.1530-0277.2011.01484.x |
[11] | Agartz I , Momenan R , Rawlings RR , Kerich MJ , Hommer DW . Hippocampal volume in patients with alcohol dependence. Arch Gen Psychiat. (1999) ;56: (4):356–63. doi:10.1001/archpsyc.56.4.356 |
[12] | Dhanabalan G , Le Maitre TW , Bogdanovic N , Alkass K , Druid H . Hippocampal granule cell loss in human chronic alcohol abusers. Neurobiol Dis. (2018) ;120: :63–75. doi:10.1016/j.nbd.2018.08.011 |
[13] | Sullivan EV , Marsh L , Mathalon DH , Lim KO , Pfefferbaum A . Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol Clin Exp Res. (1995) ;19: (1):110–22. doi:10.1111/j.1530-0277.1995.tb01478.x |
[14] | Walker DW , Barnes DE , Zornetzer SF , Hunter BE , Kubanis P . Neuronal loss in hippocampus induced by prolonged ethanol consumption in rats. Science. (1980) ;209: (4457):711–3. doi:10.1126/science.7394532 |
[15] | Cippitelli A , Damadzic R , Frankola K , Goldstein A , Thorsell A , Singley E , et al. Alcohol-induced neurodegeneration, suppression of transforming growth factor-beta, and cognitive impairment in rats: prevention by group II metabotropic glutamate receptor activation. Biol Psych. (2010) ;67: (9):823–30. doi:10.1016/j.biopsych.2009.12.018 |
[16] | Obernier JA , White AM , Swartzwelder HS , Crews FT . Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharm Biochem Behav. (2002) ;72: (3):521–32. doi:10.1016/s0091-3057(02)00715-3 |
[17] | Cadete-Leite A , Tavares MA , Uylings HB , Paula-Barbosa M . Granule cell loss and dendritic regrowth in the hippocampal dentate gyrus of the rat after chronic alcohol consumption. Brain Res. (1988) ;473: (1):1–14. doi:10.1016/0006-8993(88)90309-5 |
[18] | Maynard ME , Barton EA , Robinson CR , Wooden JI , Leasure JL . Sex differences in hippocampal damage, cognitive impairment, and trophic factor expression in an animal model of an alcohol use disorder. Brain Struct Function. (2018) ;223: (1):195–210. doi:10.1007/s00429-017-1482-3 |
[19] | Pfefferbaum A , Sullivan EV , Mathalon DH , Shear PK , Rosenbloom MJ , Lim KO . Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol Clin Exp Res. (1995) ;19: (5):1177–91. doi:10.1111/j.1530-0277.1995.tb01598.x |
[20] | Carlen PL , Wortzman G , Holgate RC , Wilkinson DA , Rankin JC . Reversible cerebral atrophy in recently abstinent chronic alcoholics measured by computed tomography scans. Science. (1978) ;200: (4345):1076–8. doi:10.1126/science.653357 |
[21] | Brandt J , Butters N , Ryan C , Bayog R . Cognitive loss and recovery in long-term alcohol abusers. Arch Gen Psychiat. (1983) ;40: (4):435–42. doi:10.1001/archpsyc.1983.01790040089012 |
[22] | Gazdzinski S , Durazzo TC , Meyerhoff DJ . Temporal dynamics and determinants of whole brain tissue volume changes during recovery from alcohol dependence. Drug Alcohol Depend. (2005) ;78: (3):263–73. doi:10.1016/j.drugalcdep.2004.11.004 |
[23] | Mann K , Gunther A , Stetter F , Ackermann K . Rapid recovery from cognitive deficits in abstinent alcoholics: a controlled test-retest study. Alcohol Alcohol. (1999) ;34: (4):567–74. doi:10.1093/alcalc/34.4.567 |
[24] | Zou X , Durazzo TC , Meyerhoff DJ . Regional Brain Volume Changes in Alcohol-Dependent Individuals During Short-Term and Long-Term Abstinence. Alcohol Clin Exp Res. (2018) ;42: (6):1062–72. doi:10.1111/acer.13757 |
[25] | Gazdzinski S , Durazzo TC , Yeh PH , Hardin D , Banys P , Meyerhoff DJ . Chronic cigarette smoking modulates injury and short-term recovery of the medial temporal lobe in alcoholics. Psychiat Res. (2008) ;162: (2):133–45. doi:10.1016/j.pscychresns.2007.04.003 |
[26] | Bartels C , Kunert HJ , Stawicki S , Kroner-Herwig B , Ehrenreich H , Krampe H . Recovery of hippocampus-related functions in chronic alcoholics during monitored long-term abstinence. Alcohol Alcohol. (2007) ;42: (2):92–102. doi:10.1093/alcalc/agl104 |
[27] | Olsufka R , Peng H , Newton J , Nixon K . Alcohol effects on adult neural stem cells – a novel mechanism of neuro- toxicity in alcohol use disorders. In: Rasmussen T, editor. Stem Cells in Toxicology and Teratology. New York: John Wiley and Sons; (2018) . |
[28] | Mon A , Durazzo TC , Gazdzinski S , Hutchison KE , Pennington D , Meyerhoff DJ . Brain-derived neurotrophic factor genotype is associated with brain gray and white matter tissue volumes recovery in abstinent alcohol-dependent individuals. Genes Brain Behav. (2013) ;12: (1):98–107. doi:10.1111/j.1601-183X.2012.00854.x |
[29] | Kuhn S , Charlet K , Schubert F , Kiefer F , Zimmermann P , Heinz A , et al. Plasticity of hippocampal subfield volume cornu ammonis 2+3 over the course of withdrawal in patients with alcohol dependence. JAMA Psychiat. (2014) ;71: (7):806–11. doi:10.1001/jamapsychiatry.2014.352 |
[30] | Boldrini M , Fulmore CA , Tartt AN , Simeon LR , Pavlova I , Poposka V , et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell. (2018) ;22: (4):589–99 e5. doi:10.1016/j.stem.2018.03.015 |
[31] | Eriksson PS , Perfilieva E , Bjork-Eriksson T , Alborn AM , Nordborg C , Peterson DA , et al. Neurogenesis in the adult human hippocampus. Nat Med. (1998) ;4: (11):1313–7. doi:10.1038/3305 |
[32] | Goncalves JT , Schafer ST , Gage FH . Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell. (2016) ;167: (4):897–914. doi:10.1016/j.cell.2016.10.021 |
[33] | Imayoshi I , Sakamoto M , Ohtsuka T , Takao K , Miyakawa T , Yamaguchi M , et al. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. (2008) ;11: (10):1153–61. doi:10.1038/nn.2185 |
[34] | van Praag H , Schinder AF , Christie BR , Toni N , Palmer TD , Gage FH . Functional neurogenesis in the adult hippocampus. Nature. (2002) ;415: (6875):1030–4. doi:10.1038/4151030a |
[35] | van Praag H , Christie BR , Sejnowski TJ , Gage FH . Running enhances neurogenesis, learning, and long-term potentiation in mice. PNAS. (1999) ;96: (23):13427–31. doi:10.1073/pnas.96.23.13427 |
[36] | Snyder JS , Drew MR . Functional neurogenesis over the years. Behav Brain Res. (2020) ;382: :112470. doi:10.1016/j.bbr.2020.112470 |
[37] | Sahay A , Scobie KN , Hill AS , O’Carroll CM , Kheirbek MA , Burghardt NS , et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. (2011) ;472: (7344):466–70. doi:10.1038/nature09817 |
[38] | Akers KG , Martinez-Canabal A , Restivo L , Yiu AP , De Cristofaro A , Hsiang HL , et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science. (2014) ;344: (6184):598–602. doi:10.1126/science.1248903 |
[39] | Clelland CD , Choi M , Romberg C , Clemenson GD Jr , Fragniere A , Tyers P , et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. (2009) ;325: (5937):210–3. doi:10.1126/science.1173215 |
[40] | Shors TJ , Miesegaes G , Beylin A , Zhao M , Rydel T , Gould E . Neurogenesis in the adult is involved in the formation of trace memories. Nature. (2001) ;410: (6826):372–6. doi:10.1038/35066584 |
[41] | Gan H , Zhang Q , Zhu B , Wu S , Chai D . Fluoxetine reverses brain radiation and temozolomide-induced anxiety and spatial learning and memory defect in mice. J Neurophysiol. (2019) ;121: (1):298–305. doi:10.1152/jn.00581.2018 |
[42] | Nokia MS , Anderson ML , Shors TJ . Chemotherapy disrupts learning, neurogenesis and theta activity in the adult brain. Eur J Neurosci. (2012) ;36: (11):3521–30. doi:10.1111/ejn.12007 |
[43] | Saxe MD , Battaglia F , Wang JW , Malleret G , David DJ , Monckton JE , et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. PNAS. (2006) ;103: (46):17501–6. doi:10.1073/pnas.0607207103 |
[44] | Danielson NB , Kaifosh P , Zaremba JD , Lovett-Barron M , Tsai J , Denny CA , et al. Distinct Contribution of Adult-Born Hippocampal Granule Cells to Context Encoding. Neuron. (2016) ;90: (1):101–12. doi:10.1016/j.neuron.2016.02.019 |
[45] | Garthe A , Behr J , Kempermann G . Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PloS one. (2009) ;4: (5):e5464. doi:10.1371/journal.pone.0005464 |
[46] | Denny CA , Kheirbek MA , Alba EL , Tanaka KF , Brachman RA , Laughman KB , et al. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron. (2014) ;83: (1):189–201. doi:10.1016/j.neuron.2014.05.018 |
[47] | Niibori Y , Yu TS , Epp JR , Akers KG , Josselyn SA , Frankland PW . Suppression of adult neurogenesis impairs population coding of similar contexts in hippocampal CA3 region. Nat Comm. (2012) ;3: :1253. doi:10.1038/ncomms2261 |
[48] | Kozorovitskiy Y , Gould E . Adult neurogenesis: a mechanism for brain repair? J Clin Exp Neuropsych. (2003) ;25: (5):721–32. doi:10.1076/jcen.25.5.721.14580 |
[49] | Quadrato G , Elnaggar MY , Di Giovanni S . Adult neurogenesis in brain repair: cellular plasticity vs. cellular replacement. Front Neurosci. (2014) ;8: :17. doi:10.3389/fnins.2014.00017 |
[50] | Nixon K . Alcohol and adult neurogenesis: roles in neuro-degeneration and recovery in chronic alcoholism. Hippo-campus. (2006) ;16: (3):287–95. doi:10.1002/hipo.20162 |
[51] | Nixon K , Crews FT . Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J Neurosci. (2004) ;24: (43):9714–22. doi:10.1523/JNEUROSCI.3063-04.2004 |
[52] | Fannon MJ , Mysore KK , Williams J , Quach LW , Purohit DC , Sibley BD , et al. Hippocampal neural progenitor cells play a distinct role in fear memory retrieval in male and female CIE rats. Neuropharm. (2018) ;143: :239–49. doi:10.1016/j.neuropharm.2018.09.045 |
[53] | Le Maitre TW , Dhanabalan G , Bogdanovic N , Alkass K , Druid H . Effects of Alcohol Abuse on Proliferating Cells, Stem/Progenitor Cells, and Immature Neurons in the Adult Human Hippocampus. Neuropsychopharm. (2018) ;43: (4):690–9. doi:10.1038/npp.2017.251 |
[54] | Nixon K , Crews FT . Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J Neurochem. (2002) ;83: (5):1087–93. doi:10.1046/j.1471-4159.2002.01214.x |
[55] | Richardson HN , Chan SH , Crawford EF , Lee YK , Funk CK , Koob GF , et al. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol Dis. (2009) ;36: (1):1–10. doi:10.1016/j.nbd.2009.05.021 |
[56] | Crews FT , Mdzinarishvili A , Kim D , He J , Nixon K . Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience. (2006) ;137: (2):437–45. doi:10.1016/j.neuroscience.2005.08.090 |
[57] | Herrera DG , Yague AG , Johnsen-Soriano S , Bosch-Morell F , Collado-Morente L , Muriach M , et al. Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. PNAS. (2003) ;100: (13):7919–24. doi:10.1073/pnas.1230907100 |
[58] | Taffe MA , Kotzebue RW , Crean RD , Crawford EF , Edwards S , Mandyam CD . Long-lasting reduction in hippocampal neurogenesis by alcohol consumption in adolescent nonhuman primates. PNAS. (2010) ;107: (24):11104–9. doi:10.1073/pnas.0912810107 |
[59] | Golub HM , Zhou QG , Zucker H , McMullen MR , Kokiko-Cochran ON , Ro EJ , et al. Chronic Alcohol Exposure is Associated with Decreased Neurogenesis, Aberrant Integration of Newborn Neurons, and Cognitive Dysfunction in Female Mice. Alcohol Clin Exp Res. (2010) ;39: (10):1967–77. doi:10.1111/acer.12843 |
[60] | Geil CR , Hayes DM , McClain JA , Liput DJ , Marshall SA , Chen KY , et al. Alcohol and adult hippocampal neurogenesis: promiscuous drug, wanton effects. Prog Neuropsychopharm Biol Psych. (2014) ;54: :103–13. doi:10.1016/j.pnpbp.2014.05.003 |
[61] | Hayes DM , Nickell CG , Chen KY , McClain JA , Heath MM , Deeny MA , et al. Activation of neural stem cells from quiescence drives reactive hippocampal neurogenesis after alcohol dependence. Neuropharm. (2018) ;133: :276–88. doi:10.1016/j.neuropharm.2018.01.032 |
[62] | Nickell CRG , Peng H , Hayes DM , Chen KY , McClain JA , Nixon K . Type 2 Neural Progenitor Cell Activation Drives Reactive Neurogenesis after Binge-Like Alcohol Exposure in Adolescent Male Rats. Front Psychiat. (2017) ;8: :283. doi:10.3389/fpsyt.2017.00283 |
[63] | Maynard ME , Leasure JL . Exercise enhances hippocampal recovery following binge ethanol exposure. PloS one. (2013) ;8: (9):e76644. doi:10.1371/journal.pone.0076644 |
[64] | Obernier JA , Bouldin TW , Crews FT . Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res. (2002) ;26: (4):547–57. |
[65] | Leasure JL , Nixon K . Exercise neuroprotection in a rat model of binge alcohol consumption. Alcohol Clin Exp Res. (2010) ;34: (3):404–14. doi:10.1111/j.1530-0277.2009.01105.x |
[66] | Mandyam CD , Koob GF . The addicted brain craves new neurons: putative role for adult-born progenitors in promoting recovery. Trends Neurosci. (2012) ;35: (4):250–60. doi:10.1016/j.tins.2011.12.005 |
[67] | Liu J , Solway K , Messing RO , Sharp FR . Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. (1998) ;18: (19):7768–78. |
[68] | Dash PK , Mach SA , Moore AN . Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. (2001) ;63: (4):313–9. doi:10.1002/1097-4547(20010215)63:4<313::AID-JNR1025>3.0.CO;2-4 |
[69] | Parent JM , Yu TW , Leibowitz RT , Geschwind DH , Sloviter RS , Lowenstein DH . Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. (1997) ;17: (10):3727–38. |
[70] | Kuzmin A , Liljequist S , Meis J , Chefer V , Shippenberg T , Bakalkin G . Repeated moderate-dose ethanol bouts impair cognitive function in Wistar rats. Addict Biol. (2012) ;17: (1):132–40. doi:10.1111/j.1369-1600.2010.00224.x |
[71] | West RK , Wooden JI , Barton EA , Leasure JL . Recurrent binge ethanol is associated with significant loss of dentate gyrus granule neurons in female rats despite concomitant increase in neurogenesis. Neuropharm. (2019) ;148: :272–83. doi:10.1016/j.neuropharm.2019.01.016 |
[72] | NRC. Guide for the Care and Use of Laboratory Animals. Washington, D.C.: The National Academies Press; (1996) . |
[73] | Majchrowicz E . Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia. (1975) ;43: (3):245–54. doi:10.1007/BF00429258 |
[74] | Morris SA , Kelso ML , Liput DJ , Marshall SA , Nixon K . Similar withdrawal severity in adolescents and adults in a rat model of alcohol dependence. Alcohol. (2010) ;44: (1):89–98. doi:10.1016/j.alcohol.2009.10.017 |
[75] | Khatri D , Laroche G , Grant ML , Jones VM , Vetreno RP , Crews FT , et al. Acute Ethanol Inhibition of Adult Hippocampal Neurogenesis Involves CB1 Cannabinoid Receptor Signaling. Alcohol Clin Exp Res. (2018) ;42: (4):718–26. doi:10.1111/acer.13608 |
[76] | Cameron HA , McKay RD . Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. Journal Comp Neurol. (2001) ;435: (4):406–17. doi:10.1002/cne.1040 |
[77] | Garthe A , Roeder I , Kempermann G . Mice in an enriched environment learn more flexibly because of adult hippocampal neurogenesis. Hippocampus. (2016) ;26: (2):261–71. doi:10.1002/hipo.22520 |
[78] | Verbois SL , Hopkins DM , Scheff SW , Pauly JR . Chronic intermittent nicotine administration attenuates traumatic brain injury-induced cognitive dysfunction. Neuroscience. (2003) ;119: (4):1199–208. doi:10.1016/s0306-4522(03)00206-9 |
[79] | Jessberger S , Kempermann G . Adult-born hippocampal neurons mature into activity-dependent responsiveness. Eur J Neurosci. (2003) ;18: (10):2707–12. doi:10.1111/j.1460-9568.2003.02986.x |
[80] | Kee N , Teixeira CM , Wang AH , Frankland PW . Imaging activation of adult-generated granule cells in spatial memory. Nat Protoc. (2007) ;2: (12):3033–44. doi:10.1038/nprot.2007.415 |
[81] | McClain JA , Hayes DM , Morris SA , Nixon K . Adolescent binge alcohol exposure alters hippocampal progenitor cell proliferation in rats: effects on cell cycle kinetics. J Comp Neurol. (2011) ;519: (13):2697–710. doi:10.1002/cne.22647 |
[82] | Gao Z , Ure K , Ables JL , Lagace DC , Nave KA , Goebbels S , et al. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat Neurosci. (2009) ;12: (9):1090–2. doi:10.1038/nn.2385 |
[83] | Paxinos G , Watson C . The rat brain in stereotaxic coordinates / George Paxinos, CharlesWatson. Compact 6th ed.5 London: Elsevier Academic; (2009) . |
[84] | Crews FT , Nixon K , Wilkie ME . Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcoho. (2004) ;33: (1):63–71. doi:10.1016/j.alcohol.2004.04.005 |
[85] | Noori HR , Fornal CA . The appropriateness of unbiased optical fractionators to assess cell proliferation in the adult hippocampus. Front Neurosci. (2011) ;5: :140. doi:10.3389/fnins.2011.00140 |
[86] | Pataskar A , Jung J , Smialowski P , Noack F , Calegari F , Straub T , et al. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. (2016) ;35: (1):24–45. doi:10.15252/embj.201591206 |
[87] | Jang MH , Shin MC , Kim EH , Kim CJ . Acute alcohol intoxication decreases cell proliferation and nitric oxide synthase expression in dentate gyrus of rats. Toxicol Lett. (2002) ;133: (2-3):255–62. doi:10.1016/s0378-4274(02)00129-7 |
[88] | Nixon K , Kim DH , Potts EN , He J , Crews FT . Distinct cell proliferation events during abstinence after alcohol dependence: microglia proliferation precedes neurogenesis. Neurobiol Dis. (2008) ;31: (2):218–29. doi:10.1016/j.nbd.2008.04.009 |
[89] | McClain JA , Morris SA , Marshall SA , Nixon K . Ectopic hippocampal neurogenesis in adolescent male rats following alcohol dependence. Addict Biol. (2014) ;19: (4):687–99. doi:10.1111/adb.12075 |
[90] | Broadwater MA , Liu W , Crews FT , Spear LP . Persistent loss of hippocampal neurogenesis and increased cell death following adolescent, but not adult, chronic ethanol exposure. Dev Neurosci. (2014) ;36: (3-4):297–305. doi:10.1159/000362874 |
[91] | Martinez-Canabal A , Akers KG , Josselyn SA , Frankland PW . Age-dependent effects of hippocampal neurogenesis suppression on spatial learning. Hippocampus. (2013) ;23: (1):66–74. doi:10.1002/hipo.22054 |
[92] | Cuartero MI , de la Parra J , Perez-Ruiz A , Bravo-Ferrer I , Duran-Laforet V , Garcia-Culebras A , et al. Abolition of aberrant neurogenesis ameliorates cognitive impairment after stroke in mice. J Clin Invest. (2013) ;129: (4):1536–50. doi:10.1172/JCI120412 |
[93] | Snyder JS , Grigereit L , Russo A , Seib DR , Brewer M , Pickel J , et al. A Transgenic Rat for Specifically Inhibiting Adult Neurogenesis. eNeuro. (2016) ;3: (3). doi:10.1523/ENEURO.0064-16.2016 |
[94] | Zhang K , Wang F , Zhao Y , He M , Luo Y , Cheng Y , et al. The regulative effects of levetiracetam on adult hippocampal neurogenesis in mice via Wnt/beta-catenin signaling. Neurochem Int. (2020) ;133: :104643. doi:10.1016/j.neuint.2019.104643 |
[95] | Somkuwar SS , Fannon MJ , Staples MC , Zamora-Martinez ER , Navarro AI , Kim A , et al. Alcohol dependence-induced regulation of the proliferation and survival of adult brain progenitors is associated with altered BDNF-TrkB signaling. Brain Struct Function. (2016) ;221: (9):4319–35. doi:10.1007/s00429-015-1163-z |
[96] | Hansson AC , Nixon K , Rimondini R , Damadzic R , Sommer WH , Eskay R , et al. Long-term suppression of forebrain neurogenesis and loss of neuronal progenitor cells following prolonged alcohol dependence in rats. Intl J Neuropsychopharm. (2010) ;13: (5):583–93. doi:10.1017/S1461145710000246 |
[97] | Sun C , Sun H , Wu S , Lee CC , Akamatsu Y , Wang RK , et al. Conditional ablation of neuroprogenitor cells in adult mice impedes recovery of poststroke cognitive function and reduces synaptic connectivity in the perforant pathway. J Neurosci. (2013) ;33: (44):17314–25. doi:10.1523/JNEUROSCI.2129-13.2013 |
[98] | Sun D , Daniels TE , Rolfe A , Waters M , Hamm R . Inhibition of injury-induced cell proliferation in the dentate gyrus of the hippocampus impairs spontaneous cognitive recovery after traumatic brain injury. J Neurotrauma. (2015) ;32: (7):495–505. doi:10.1089/neu.2014.3545 |
[99] | Blaiss CA , Yu TS , Zhang G , Chen J , Dimchev G , Parada LF , et al. Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J Neurosci. (2011) ;31: (13):4906–16. doi:10.1523/JNEUROSCI.5265-10.2011 |
[100] | Ibrahim S , Hu W , Wang X , Gao X , He C , Chen J . Traumatic Brain Injury Causes Aberrant Migration of Adult-Born Neurons in the Hippocampus. Sci Rep. (2016) ;6: :21793. doi:10.1038/srep21793 |
[101] | Cho KO , Lybrand ZR , Ito N , Brulet R , Tafacory F , Zhang L , et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Comm. (2015) ;6: :6606. doi:10.1038/ncomms7606 |
[102] | Aimone JB , Wiles J , Gage FH . Computational influence of adult neurogenesis on memory encoding. Neuron. (2009) ;61: (2):187–202. doi:10.1016/j.neuron.2008.11.026 |
[103] | Bielefeld P , Dura I , Danielewicz J , Lucassen PJ , Baekelandt V , Abrous DN , et al. Insult-induced aberrant hippocampal neurogenesis: Functional consequences and possible therapeutic strategies. Behav Brain Res. (2019) ;372: :112032. doi:10.1016/j.bbr.2019.112032 |
[104] | Zhu K , Yuan B , Hu M , Feng GF , Liu Y , Liu JX . Reduced abnormal integration of adult-generated granule cells does not attenuate spontaneous recurrent seizures in mice. Epilepsy Res. (2017) ;133: :58–66. doi:10.1016/j.eplepsyres.2017.04.004 |
[105] | Varma P , Brulet R , Zhang L , Hsieh J . Targeting Seizure-Induced Neurogenesis in a Clinically Relevant Time Period Leads to Transient But Not Persistent Seizure Reduction. J Neurosci. (2019) ;39: (35):7019–28. doi:10.1523/JNEUROSCI.0920-19.2019 |
[106] | Sierra A , Martin-Suarez S , Valcarcel-Martin R , Pascual-Brazo J , Aelvoet SA , Abiega O , et al. Neuronal hyper-activity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell. (2015) ;16: (5):488–503. doi:10.1016/j.stem.2015.04.003 |
[107] | Scharfman HE . Epilepsy as an example of neural plasticity. Neuroscientist. (2002) ;8: (2):154–73. doi:10.1177/107385840200800211 |
[108] | Dieni CV , Chancey JH , Overstreet-Wadiche LS . Dynamic functions of GABA signaling during granule cell maturation. Front Neural Cir. (2012) ;6: :113. doi:10.3389/fncir.2012.00113 |
[109] | Hall W , Zador D . The alcohol withdrawal syndrome. Lancet. (1997) ;349: (9069):1897–900. doi:10.1016/S0140-6736(97)04572-8 |
[110] | He J , Nixon K , Shetty AK , Crews FT . Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Euro J Neurosci. (2005) ;21: (10):2711–20. doi:10.1111/j.1460-9568.2005.04120.x |
[111] | Riley JN , Walker DW . Morphological alterations in hippocampus after long-term alcohol consumption in mice. Science. (1978) ;201: (4356):646–8. doi:10.1126/science.566953 |
[112] | Staples MC , Kim A , Mandyam CD . Dendritic remodeling of hippocampal neurons is associated with altered NMDA receptor expression in alcohol dependent rats. Mol Cell Neurosci. (2015) ;65: :153–62. doi:10.1016/j.mcn.2015.03.008 |
[113] | Kelso ML , Liput DJ , Eaves DW , Nixon K . Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience. (2011) ;197: :381–93. doi:10.1016/j.neuroscience.2011.09.019 |


