
Preventing and Treating Neurological Disorders with the Flavonol Fisetin
Abstract
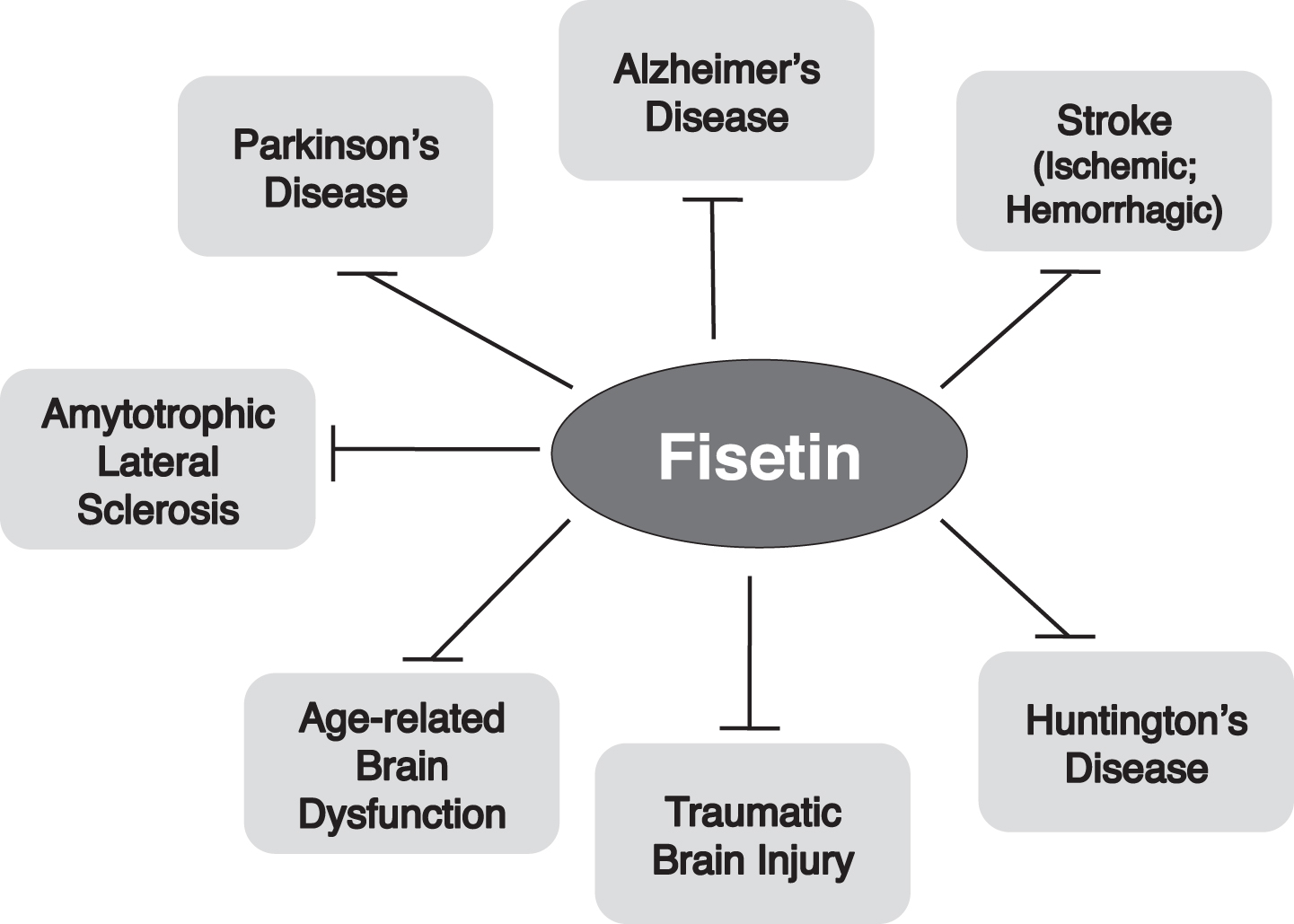
Neurological disorders, including neurodegenerative diseases, have a significant negative impact on both patients and society at large. Since the prevalence of most of these disorders increases with age, the consequences for our aging population are only going to grow. It is now acknowledged that neurological disorders are multi-factorial involving disruptions in multiple cellular systems. While each disorder has specific initiating mechanisms and pathologies, certain common pathways appear to be involved in most, if not all, neurological disorders. Thus, it is becoming increasingly important to identify compounds that can modulate the multiple pathways that contribute to disease development or progression. One of these compounds is the flavonol fisetin. Fisetin has now been shown in preclinical models to be effective at preventing the development and/or progression of multiple neurological disorders including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, stroke (both ischemic and hemorrhagic) and traumatic brain injury as well as to reduce age-associated changes in the brain. These beneficial effects stem from its actions on multiple pathways associated with the different neurological disorders. These actions include its well characterized anti-inflammatory and anti-oxidant effects as well as more recently described effects on the regulated cell death oxytosis/ferroptosis pathway, the gut microbiome and its senolytic activity. Therefore, the growing body of pre-clinical data, along with fisetin’s ability to modulate a large number of pathways associated with brain dysfunction, strongly suggest that it would be worthwhile to pursue its therapeutic effects in humans.
INTRODUCTION
Neurological disorders are the leading cause of disability worldwide and the second leading cause of death [1]. Among the fifteen neurological disorders examined in a recent study, stroke and Alzheimer’s disease (AD) and other dementias were two of the top contributors to both disability adjusted life years (DALYs-the sum of years life lost and years lived with disability) and death. Importantly, the prevalence of non-communicable neurological disorders significantly increases with age. Thus, given the aging population worldwide, the burden of these neurological disorders will continue to increase and is likely to pose ever greater challenges to health care systems worldwide. This is especially true of neurodegenerative diseases which all lack effective treatments. Because these diseases are both complex and occur in the context of the aging brain, it is unlikely that targeting a single pathophysiological change will prove effective at preventing nerve cell damage and death. For example, there are numerous failed clinical trials testing single target drug candidates against the amyloid pathway for the treatment of AD [2, 3]. In addition, there is a strong possibility that the relative contributions of different pathophysiological changes will vary among individuals. Importantly, these changes interact with lifestyle, environmental and genetic risk factors with varying degrees of penetrance. Therefore, one approach is to use combinations of drugs directed against different disease targets. However, this approach is subject to a number of potential problems. These include pharmacokinetic and bioavailability challenges which in central nervous system (CNS) diseases are exacerbated by the difficulty of getting multiple compounds across the blood brain barrier and the potential for adverse drug-drug interactions. A better approach is to identify small molecules that have multiple biological activities that can impact the multiplicity of pathophysiological changes to the brain that contribute to neurological disorder development and progression [4].
One excellent source for multi-target compounds is the original pharmacopeia, plants. The earliest records describing the use of plants for medicinal purposes date back to 2600-2900 BC [5]. Still today, ∼25% of all prescribed drugs are derived from plants [6]. Plants synthesize a huge array of compounds called secondary metabolites that are not required for plant growth. These compounds are derived from a limited number of basic chemical scaffolds which are modified by specific types of substitutions. It has been suggested [6] that these compounds, as well as receptors, enzymes and regulatory proteins, originated from a relatively small number of parental molecules which may have co-evolved to interact with one another. Although their biological functions and structures have since diverged, structural features shared from their common past may be the reason that many interact with medically relevant targets.
Among the huge number of plant-derived secondary metabolites, several epidemiological studies have specifically highlighted the potential beneficial role of flavonoids for the prevention of neurodegenerative diseases. A retrospective study that looked at flavonoid intake versus DALYs due to dementia in 23 developed countries found that total combined flavonoid intake was significantly and negatively correlated with dementia [7]. Among the flavonoid groups, only flavonol consumption showed a significant, negative correlation with dementia. Consistent with these results, a prospective cohort study [8] found that the risk ratio for dementia between the highest and lowest tertiles of flavonoid intake was 0.49.
We originally identified the flavonol fisetin (Fig. 1) as a promising neuroprotective compound in a screen for compounds that could prevent a form of regulated cell death that we called oxytosis [9, 10] and has recently been renamed ferroptosis [11, 12]. Of the ∼30 flavonoids screened in this study, only two, fisetin and quercetin, were also able to maintain glutathione (GSH) levels in the presence of oxidative stress, indicating that this is not a common property of flavonoids. Further studies showed that fisetin also possessed neurotrophic activity, with low micromolar doses promoting the differentiation of PC12 cells, as defined by the production of neurites, via activation of the Ras-ERK cascade [13]. Again, this was a property that distinguished fisetin from almost all of the other ∼30 flavonoids tested. Only quercetin, isorhamnetin and luteolin showed some differentiation-inducing activity and they were all much less effective than fisetin. Together, these observations suggested that fisetin had multiple properties that might make it useful for the treatment of neurological diseases.
Fig. 1
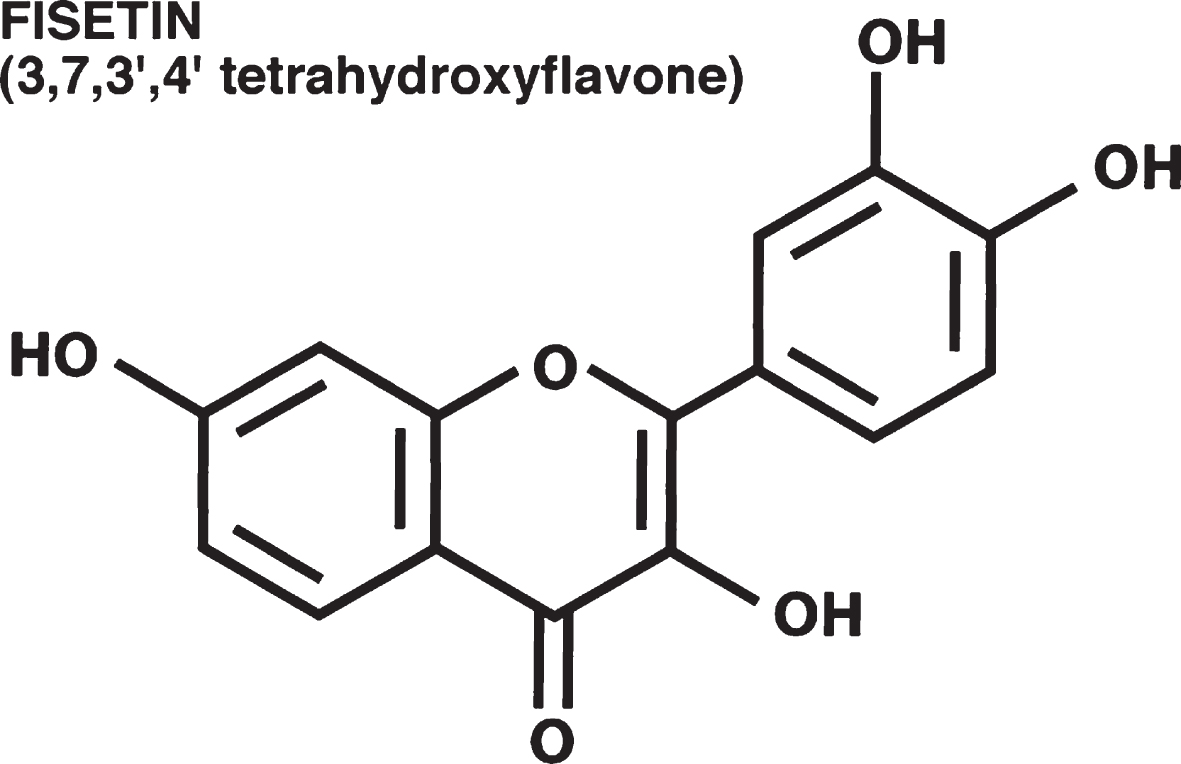
Unlike many of the better studied flavonoids such as quercetin, fisetin is not particularly abundant in fruits and vegetables. The highest levels (160μg/g) are found in strawberries [14] with 5–10 fold lower levels in apples and persimmons. Small amounts are also found in kiwi fruit, peaches, grapes, tomatoes, onions and cucumbers [15]. The bioavailability of fisetin from these sources has not yet been studied. Furthermore, the levels of fisetin in these dietary sources are too low to achieve the equivalent human levels of the concentrations that were effective in the various animal studies. However, fisetin is currently marketed in the US by several nutraceutical companies in 100 mg capsules. For these products, fisetin is isolated from Rhus succedanea or Cotinus coggygria which provide more abundant sources. Some of the companies provide information on the purity of the fisetin on their websites. However, a “generally recognized as safe” (GRAS) determination by the US Food and Drug Administration (FDA) has not been made for fisetin.
In the following paragraphs, I will describe the results of many pre-clinical studies and, in one case, a clinical study that looked at the beneficial effects of fisetin in models of neurological disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), ischemic stroke, hemorrhagic stroke and traumatic brain injury (TBI) and then discuss several novel mechanisms of action. For each disease/disorder, I will begin by providing a brief overview, describe the models used to study the effects of potential protective compounds on the disease/disorder and then describe the results. The effects of fisetin in neurological disorders were last reviewed by me in 2015 [16] so this review will mainly focus on those studies conducted since the prior review and on diseases/disorders that were not discussed in the earlier review.
NEUROLOGICAL DISEASES/DISORDERS AND FISETIN
Fisetin and Alzheimer’s disease and dementia
Alzheimer’s disease (AD) is the most common type of dementia. It is characterized pathologically by the presence of both extracellular neuritic plaques containing amyloid beta (Aβ) peptide and intracellular neurofibrillary tangles containing tau [17]. Clinically, AD results in a progressive loss of cognitive ability and eventually daily function activities [18, 19]. Currently approved therapies are only symptomatic, providing moderate improvements in memory without altering the progression of the disease pathology [20, 21]. Although a large number of clinical trials have been conducted in recent years with drug candidates designed to directly or indirectly reduce the amyloid plaque load, all of these trials have failed [2, 3].
Three different types of models have been used to study the possible beneficial effects of fisetin in AD: interventional, transgenic and sporadic. For the interventional studies, Aβ is injected directly into the cerebral ventricles in the brains of the mice (intracerebroventricular (icv) injection). There are numerous transgenic models of AD that are based on the mutations associated with the rare genetic form of the disease (familial AD or FAD). The models develop different degrees of cognitive impairment, levels of plaques and tangles, synaptic loss, gliosis and nerve cell death depending on the type and number of mutations [1–5] [22]. Although AD drug discovery has largely focused on these FAD models, this form of the disease accounts for only ∼ 1% of the total cases [23], and may be quite distinct from the much more prevalent, old age-associated, sporadic form of AD. Importantly, while many therapies directed against the amyloid pathway are effective in FAD transgenic mice, to date none has translated into the clinic [2]. Since old age is by far the greatest risk factor for AD [23, 24], animal models that incorporate aging into disease development may prove more useful for the development of therapies. One mouse model of aging that also develops characteristics of AD is the senescence-accelerated prone 8 (SAMP8) mouse that was developed in Japan by selective breeding of a rapidly aging phenotype. These mice exhibit a progressive, age-associated decline in brain function similar to human sporadic AD patients [reviewed in 25–27].
In all three types of AD models (icv Aβ injection, 2×FAD mice, SAMP8 mice), fisetin consistently prevented the loss of cognitive function [28–30]. Both oral administration in the food (500 mg/kg food; ∼45 mg/kg body weight (bw)/day) [28, 29] and intraperitoneal (ip) injection (20 mg/kg bw/day) [30] proved effective. In all of the models, fisetin maintained the levels of synaptic proteins and reduced markers of inflammation. It also reduced multiple markers of oxidative stress and particularly lipid peroxidation and activated the ERK pathway which is involved in both memory [31] and neurotrophic factor production and signaling [13]. However, the effects on the levels of soluble and insoluble Aβ varied between the different models suggesting that this may not be a critical target of fisetin in AD.
Although caused by dysfunction of the vascular endothelium, vascular dementia results in a similar loss of cognitive function as classical AD and is often a component of AD [32]. One approach to modeling vascular dementia is to treat rats with L-methionine which induces neurological changes similar to those seen in vascular dementia. Oral administration of fisetin (5–25 mg/kg bw/day) dose dependently reduced the impact of L-methionine treatment on cognitive function, vascular function and markers of neurodegeneration when administered to rats for 2 weeks during a 4 week treatment with L-methionine [33]. Thus, fisetin is effective at preventing loss of cognitive function and maintaining brain health in multiple, distinct models of dementia.
Fisetin and Parkinson’s disease
Parkinson’s disease (PD) is a chronic, progressive neurodegenerative disease and the second most common neurodegenerative disease after AD. The characteristic features of PD include resting tremor, bradykinesia (slowness of movement), rigidity and postural instability (for review see 34). PD is also associated with a variety of non-motor symptoms that contribute to disability. The majority of PD cases are sporadic with only about 10% of PD patients reporting a family history of the disease [35]. As with AD, age is the greatest risk factor for disease development. The pathological hallmark of PD is the degeneration of the dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) [34]. Since these neurons synapse with neurons in the striatum, their loss leads to the depletion of striatal dopamine. PD is also characterized by the presence of cytoplasmic protein aggregates, called Lewy bodies, in the remaining DA neurons of the SNc. Currently available treatments only improve symptoms but do not stop disease progression. Importantly, by the time that the first symptoms appear, striatal dopamine is reduced by ∼80% and ∼60% of the DA neurons of the SNc have died [36].
Modeling of PD in animals has most often involved treatment with a toxin such as a pesticide or other toxic compound that has been associated with PD in vivo. One of the most widely used models employs the toxin MPTP while chronic, low dose administration of the pesticide rotenone is gaining in popularity [37, 38]. However, neither of these models recapitulates all of the aspects of human PD [37, 38] and both are rapid onset as compared to the slow, age-dependent development of PD in human patients.
Several years ago, we tested fisetin in the MPTP model in mice [39]. Fisetin was administered orally at 10 and 25 mg/kg bw/day beginning one day before the MPTP treatment and continuing for 7 days after the treatment. At 25 mg/kg, fisetin increased striatal dopamine levels by slightly more than 2-fold and even at 10 mg/kg there was a 70% increase in striatal dopamine levels. Tyrosine hydroxylase (TH) immunoreactivity was also examined in the striatum, the site of DA nerve terminals that originate in the SN. MPTP caused a significant loss of TH immuno reactivity and this was largely prevented by fisetin treatment.
Very recently, fisetin was tested in the chronic, low dose rotenone model in rats using oral administration of 10 and 20 mg/kg bw/day beginning 10 days after the start of the rotenone treatment and continuing until the end of the treatment [40]. Both doses of fisetin improved motor function and reduced rotenone-mediated decreases in striatal dopamine levels and TH immuno reactivity although the higher dose was more effective. Both doses also improved mitochondrial function and markers of oxidative stress in the midbrain, the area affected in PD. Together these data indicate that fisetin is efficacious in two distinct rodent models and strongly support further testing in additional PD models.
Fisetin and Huntington’s disease
Huntington’s disease is a late onset, progressive and fatal neurodegenerative disorder characterized by movement and psychiatric disturbances as well as cognitive impairment. There is, at present, no cure. HD is an autosomally dominant inherited disease that is caused by an unstable expansion of a trinucleotide repeat (CAG) that encodes an abnormally long polyglutamine tract in the huntingtin protein. The age at disease onset inversely varies with the CAG repeat number. The identification of the disease-causing mutation has allowed the development of a number of cellular and animal models of HD and these have been used to try to elucidate the mechanisms underlying disease development and progression [for reviews see 41, 42–44].
Fisetin was tested in the R6/2 mouse model of HD which is a transgenic line that has an aggressive disease phenotype and shortened lifespan [45]. Fisetin was fed to R6/2 mice and their wild type littermates in the food (500 mg/kg food; ∼45 mg/kg bw/day) beginning at ∼6 weeks of age. The mice were tested for motor function using the rotorod from ∼7–13 weeks of age and survival was followed. At the time of acquisition of the animals, rotorod performance was already impaired in the R6/2 mice as compared to their wild type littermates. Motor performance in the rotorod test declined significantly more rapidly in the R6/2 mice on the control diet as compared to those on the fisetin diet. Similarly, while the median life span of the R6/2 mice on the control diet was 104 days, that of fisetin-fed mice was increased by ∼30% to 139 days. The in vivo mechanisms underlying the effects of fisetin were not explored in this study. While encouraging, further studies are needed to support and extend these observations.
Fisetin and amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that is characterized by the loss of the motor neurons that control the voluntary movement of muscles resulting in paralysis and death, usually within 5 years of a diagnosis [46]. Approximately 10% of ALS cases are due to heritable gene mutations but different gene mutations are increasingly being found in patients with no family history of ALS suggesting that the genetic component is more complicated than originally thought [47]. Although there are three FDA-approved drugs for ALS, they all have very modest effects on survival (https://alsnewstoday.com/approved-treatments/).
The most commonly used mouse model of ALS is based on Cu/Zn superoxide dismutase 1 (SOD1), the first gene mutation that was shown to cause ALS [47]. SOD1 mutations are found in ∼20% of patients with the familial form of ALS. The most extensively used of these mouse models in the SOD1-G93A transgenic mouse which is characterized by abnormal protein aggregation, motor neuron loss, axonal denervation, progressive paralysis, and reduced lifespan [47].
Oral administration of fisetin (9 mg/kg/day) beginning at 2 months of age significantly delayed the development of motor deficits, reduced their rate of progression and increased lifespan [48] in the SOD1-G93A mouse. This correlated with a significant increase in the motor neuron count in the spinal cord. At the molecular level, fisetin increased the levels of phosphorylated and therefore activated ERK as well as the antioxidant protein heme oxygenase 1. Interestingly, fisetin also increased ERK phosphorylation in a transgenic AD model [28] and in HD flies [45] suggesting that this mechanism may at least partly contribute to its beneficial effects in these different models of neurodegenerative diseases. Further studies on fisetin and ALS are clearly warranted.
Fisetin and stroke
Ischemic stroke
Ischemic stroke occurs when the normal blood supply to the brain is disrupted due to artery blockage by a blood clot, thereby depriving the brain of oxygen and metabolic substrates and hindering the removal of waste products [for review see 49]. Ischemic stroke is believed to evolve in distinct phases. The initial ischemia results in immediate nerve cell death followed by an inflammatory response leading to secondary tissue damage after reperfusion [50]. There is good evidence that post-stroke inflammation contributes to secondary tissue damage. The nerve cell damage and death caused by cerebral ischemia results in functional impairment including cognitive impairment or death [51].
Fisetin has been tested in two different animal models of stroke. The rabbit small clot embolism model (SCEM) is a rigorous stroke model that recapitulates the initiating event in most cases of human stroke. For the SCEM, a suspension of small blood clots is injected into the carotid artery of a rabbit. To evaluate the quantitative relationship between clot dose and behavioral deficits, logistic S-shaped quantal analysis curves are fitted to the dose-response data as originally described by Waud [52] and adapted for stroke studies [53, 54]. A wide range of clot doses is used resulting in behaviorally normal and abnormal animals. In the absence of a neuroprotective agent, small numbers of microclots cause no grossly apparent neurologic dysfunction and large numbers of microclots invariably cause encephalopathy or death. Using a simple dichotomous rating system, each animal is rated by a blinded observer as either behaviorally normal or abnormal 24 hr post-embolization. Using quantal analysis, it is possible to detect behavioral changes following pharmacological intervention. A separate curve is generated for each treatment condition and a statistically significant increase in the P50 value or the amount of microclots that produce neurologic dysfunction in 50% of a group of animals compared to control is indicative of a behavioral improvement [53, 54]. To test the protective effect of fisetin in this model, fisetin (50 mg/kg bw) was injected intravenously at 5 min after microclot injection and behavioral analysis was conducted 24 hr after treatment. Fisetin significantly (p < 0.05) reduced stroke-induced behavioral deficits and increased the P50 value which directly correlated with an increase in the number of animals that were behaviorally “normal” [55].
Fisetin was also tested in the temporary middle cerebral artery occlusion (tMCAO) stroke model [56]. Temporary middle cerebral artery occlusion was induced in mice by the intraluminal filament method and maintained for 60 minutes. Animals were either injected intraperitoneally 20 min before or 180 min after the onset of ischemia with fisetin (25 or 50 mg/kg bw) or placebo. A significant, dose-dependent protective effect of fisetin on stroke size was observed. While animals injected immediately before the onset of ischemia with 25 mg/kg of fisetin showed a trend towards smaller infarcts, animals treated with the higher dose of fisetin (50 mg/kg) had significantly smaller infarcts. Even when injected three hours after the onset of ischemia, 50 mg/kg still retained its protective capabilities. A strong trend toward earlier recovery in both the low- and high-dose fisetin-treated animals compared to placebo following both the pre- and post-stroke treatment strategies was also seen. Importantly, fisetin prominently inhibited the infiltration of macrophages and dendritic cells into the ischemic hemisphere and suppressed intracerebral immune cell activation as measured by intracellular TNFα production [56].
The only currently FDA approved treatment for stroke is recombinant tissue plasminogen activator (rt-PA), a serine protease that degrades fibrin clots [57]. However, rt-PA has a narrow therapeutic time window with much poorer outcomes if administered outside of that window. A recent small scale (n = 192 patients) randomized, double-blind and placebo controlled clinical trial tested the ability of fisetin to enhance the effects of rt-PA when given either 0–3 hr after an ischemic stroke or 3–5 hr after an ischemic stroke [58]. When patients were tested at both 1 and 7 days after an ischemic stroke, co-treatment with fisetin (100 mg/day iv) did not provide additional benefits when rt-PA was given within 3 hr of the stroke. However, when rt-PA was given 3–5 hr after a stroke, then co-treatment with fisetin lead to significantly better outcomes as defined by the patients’ National Institutes of Health Stroke Scale (NIHSS) score. Moreover, there were strong linear correlations between the effects of fisetin on the NIHSS scores and its ability to lower serum levels of matrix metalloproteases 2 and 9 and the inflammation marker C reactive protein (CRP), all of which are thought to contribute to stroke severity. This is one of the first examples of a clinical trial with fisetin and it provides further support for the use of fisetin for treatment of a debilitating neurological disorder.
Hemorrhagic stroke
Hemorrhagic stroke results in a decrease in blood supply to the brain due to the rupture of a blood vessel (for review see 59). This type of stroke includes both intracerebral hemorrhage (ICH) which is due to bleeding within the brain and subarachnoid hemorrhage (SAH) which is due to bleeding in the subarachnoid space. While the majority of strokes are ischemic, ICH and SAH contribute to 10–15% of all strokes and are more likely to cause serious disability or death than ischemic strokes [59]. There are no good treatments for these types of strokes. Moreover, AD patients are at higher risk for hemorrhagic but not ischemic strokes [60].
The potential beneficial effects of fisetin have been studied in models of both ICH [61] and SAH [62]. To model ICH, collagenase, an enzyme that degrades the basement membrane and interstitial collagen, was injected into the striatum of mice. This results in rapid bleeding thereby mimicking both the spontaneous intraparenchymal bleeding as well as its expansion that is seen in human patients [63]. The authors used old (20–22 months) mice in this study because age is a significant risk factor for ICH [61]. Fisetin was administered by ip injection at doses ranging from 10–90 mg/kg bw/day for 72 hr after the induction of ICH by collagenase. Fisetin treatment dose-dependently reduced the neurologic severity score, brain edema and brain cell death observed in this model at 72 hr post-ICH with the greatest effects seen at 60 and 90 mg/kg bw. These results correlated with significant reductions in the brain levels of pro-inflammatory cytokines and glial activation.
To model SAH, autologous whole blood was sterotaxically injected into the subarachnoid space of rats [62]. Although this model of SAH does not fully recapitulate the situation in humans, it is the most widely used preclinical model of SAH and has translational relevance [64]. The rats were treated with 25 or 50 mg/kg bw of fisetin by ip injection 30 min after the induction of SAH and then examined 24 and 72 hr later [62]. Similar to the results in the ICH study, the high dose of fisetin significantly reduced both neurological deficits and brain edema at both 24 and 72 hr post-SAH. Also similar to the ICH study, fisetin reduced multiple markers of inflammation, including toll-like receptor 4, nuclear p65, TNF-α and IL-1β, in the cortex of the rats subjected to SAH. In addition, fisetin almost completely prevented the SAH-induced loss of ZO-1, a protein involved in the maintenance of the blood brain barrier which is damaged in this type of stroke. Thus, similar to ischemic stroke, fisetin has beneficial effects in two distinct animal models of hemorrhagic stroke suggesting that its actions in this type of stroke are worth further investigation.
Fisetin and traumatic brain injury
Traumatic brain injury (TBI) is caused by a bump, blow or jolt to the head or a penetrating head injury that disrupts the normal functioning of the brain [for reviews see 65, 66]. It is a growing cause of both disability and death. Similar to stroke, TBI also evolves in phases with the initial damage to axons and membranes eventually leading to an inflammatory response that causes further damage. Despite an increasing understanding of the underlying physiological changes that occur during TBI, there are still no good treatments.
There are multiple animal models of TBI including fluid percussive injury, controlled cortical impact injury, weight drop impact acceleration injury and blast injury and each has its advantages and disadvantages [66]. Fisetin was tested in the weight drop impact acceleration injury model using mice [67]. This model results in the development of injuries similar to those seen in motor vehicle or sports accidents [66]. The mice were injected ip with 25, 50 or 75 mg/kg bw fisetin 30 min after the induction of TBI and then evaluated 1, 3 and 7 days later [67]. Fisetin dose dependently improved the neurological score and motor function at 1 and 3 days and reduced brain edema, blood brain barrier disruption and lesion volume with 50 mg/kg bw showing the best effect. These beneficial physiological effects correlated with decreases in markers of oxidative stress and apoptosis and increases in the levels of nuclear Nrf2 as well as Nrf2 target proteins in the brains. To further evaluate the role of Nrf2 in the protective effects of fisetin against TBI, the same experiment was carried out in Nrf2 knockout mice. Interestingly, while fisetin failed to reduce markers of oxidative stress in the mice lacking Nrf2, it was still able to improve the neurological score and reduce the lesion volume as well as the markers of apoptosis. Thus, other targets of fisetin appear to be critical for many of its beneficial effects against TBI. However, given these promising results, further exploration of fisetin in additional models of TBI is clearly warranted.
Fisetin, memory and age-related changes in the brain
Similar to other organs, brain function declines with age. Human aging is associated with specific memory deficits including delayed recall of verbal information and declines in working memory, short-term recall, processing speed and spatial memory [for review see 68]. Although old age is not considered a disease, a decline in brain function can significantly impact the quality of life. In addition, age-related changes in brain function contribute to the development and progression of many of the neurological diseases/disorders described above.
In our first animal study with fisetin, we tested its effects on memory in young adult mice (3 month old male C57BL/6J) using the object recognition test [69]. In this test, during the training period, mice are presented with two identical objects, which they explore for a fixed time period. To test for long term memory (LTM), the mice are presented one day later with two different objects, one of which was presented previously during the training and is thus familiar to the mice; the other object is new to them. The better the mice remember the familiar object, the more time they will spend exploring the novel object. To test the effects of fisetin in this memory task, it was administered orally (5–25 mg kg bw) to the mice before the start of the training period [70]. Rolipram, administered by injection, was used as a positive control. In a dose dependent manner, fisetin significantly increased the time the mice spent exploring the novel object indicating a significant effect on LTM.
In order to understand the physiological mechanism underlying this effect of fisetin, we asked whether fisetin could affect long-term potentiation (LTP) in hippocampal slices from rat brains. LTP is an in vitro assay that is considered to be a good model of how memory is formed at the cellular level [71]. Furthermore, age-related changes in cognitive function have been shown to correlate with impaired induction and maintenance of LTP [68]. Although fisetin had no effect on basal synaptic responses in the CA1 area of rat hippocampal slices [70], it induced LTP in slices exposed to a weak tetanic stimulus (15 pulses at 100 Hz) that by itself failed to induce LTP. The facilitation of the induction of LTP by fisetin was dose dependent, with a maximal effect seen at 1μM and it persisted for at least 60 min.
Importantly, these results with hippocampal slices have recently been extended to in vivo studies in anesthetized rats [72]. Similar to the experiments with the slices, oral administration of fisetin (10 & 25 mg/kg bw) dose dependently facilitated the induction of LTP in vivo. In contrast, structurally related flavonoids including quercetin, myricetin and luteolin, did not have a significant effect on the facilitation of LTP when tested at the same doses [72].
More recently, the effects of fisetin on age-related changes in the brain were tested in 24 month old rats [73]. The animals were treated daily for 6 weeks with 15 mg/kg bw fisetin dissolved in 10% DMSO and administered orally. Fisetin reduced brain markers of oxidative stress, including lipid peroxidation, and increased the levels of several antioxidant enzymes relative to vehicle-treated old rats.
Consistent with these results, mice fed fisetin (500 mg/kg food) from 85 weeks of age until death showed significantly lower levels of age-related indicators of brain pathology as compared with mice fed a control diet [74] and this correlated with a significant increase in lifespan. Thus, fisetin may not only reduce the impact of neurological diseases/disorders themselves but also improve the overall environment in the brain so that these diseases/disorders are less likely to develop.
FISETIN MECHANISMS OF ACTION (Table 1)
Several reviews have highlighted many of the mechanisms by which fisetin may act to protect cells from various insults [16, 75–77]. These include fisetin’s antioxidant and transition metal chelating activity, indirect anti-oxidant activity via induction of the transcription factor Nrf2, anti-inflammatory activity, neurotrophic activity as well as its ability to modulate protein aggregation and stability. Thus, because these mechanisms have already been well covered in previous reviews, this review will focus on several other mechanisms through which fisetin may act in vivo that have received less attention but are likely to play critical roles in the beneficial effects of fisetin and deserve more investigation in the future.
Table 1
Mechanisms of Action of Fisetin and their Potential Relevance to Neurological Diseases
| Mechanism of Action | Potential Disease Relevance |
| Antioxidant and chelating activity | AD; PD; HD; ALS; stroke; TBI |
| Maintenance of GSH | AD; PD; stroke; TBI |
| Neurotrophic factor signaling pathways | AD; PD; HD; TBI |
| Anti-inflammatory activity | AD; PD; ALS; stroke; TBI |
| Modulation of protein aggregation and stability | AD; PD; HD; ALS |
| Inhibition of oxytosis/ferroptosis | AD; PD; HD; ALS; stroke; TBI |
| Modulation of gut microbiome | AD; PD; stroke; TBI |
| Senolytic activity | AD; PD; ALS |
Inhibition of oxytosis/ferroptosis
This type of regulated cell death was first described as being a form of oxidative stress-induced cell death that was initiated by the depletion of the major intracellular antioxidant glutathione (GSH) [10]. A reduction in GSH is seen in the aging brain and is accelerated in many neurological disorders/diseases [78]. Importantly, GSH loss in the brain is associated with impairments in cognitive function [78–80]. The depletion of GSH from cells leads to mitochondrial reactive oxygen species production, lipoxygenase activation, lipid peroxidation and calcium influx which initiates a form of cell death with features of both apoptosis and necrosis called oxytosis [10]. All of these changes are implicated in the nerve cell damage and death seen in neurological diseases [81]. Oxytosis appears to be very similar, if not identical, to another, recently described, form of cell death called ferroptosis [11]. We have found that most, if not all, compounds that inhibit oxytosis also inhibit ferroptosis [12] while the ferroptosis inhibitor ferrostatin-1 can also inhibit oxytosis [82]. This is of relevance because oxytosis/ferroptosis has been implicated in a number of pathological processes including neurodegenerative diseases [10, 12, 83, 84] and ICH [85]. Importantly, oxytosis/ferroptosis appears to be a process that can manifest itself over a lengthy time period before cells die, thereby offering a window for therapeutic intervention. Fisetin was first identified as a compound that could inhibit oxytosis in a nerve cell line [9] both through its ability to maintain GSH levels under conditions of stress and its ability to directly quench reactive oxygen species. Thus, at least some of the beneficial effects of fisetin seen in the context of neurological diseases are likely to be via its inhibition of oxytosis/ferroptosis.
Modulation of the gut microbiome
The gut microbiome has come under increasing attention as a potential contributor to multiple human diseases including some neurological disorders [86]. There is strong evidence that the gut microbiota is important for normal brain development and the maintenance of brain function. Thus, it is perhaps not surprising that animal and a few human studies have provided data suggesting that dysregulation of the gut microbiota contributes to PD, AD and the susceptibility to stroke and TBI [86]. However, since it is still not clear exactly what constitutes a healthy gut microbiota, how it can be altered to improve outcomes will require a great deal more study.
Most studies on the gut microbiome use DNA analysis of fecal material. However, evidence for changes in the gut microbiota can also be obtained from plasma metabolomic studies as some metabolites found in the plasma can only be made by bacteria [87, 88]. We analyzed the effects of fisetin on the plasma metabolome of 10 month old age-accelerated SAMP8 mice, an age where the mice show significant levels of cognitive dysfunction, following 7 months of fisetin treatment in the diet (500 mg/kg diet) [29]. Although fisetin only altered 11 metabolites relative to untreated 10 month old SAMP8 mice, most of these changes resulted in a prevention of the changes that occurred with aging in these mice [29]. Interestingly, 27% of these metabolites (4-hydroxyphenylacetate, indoleacetate and taurourso deoxycholate) are mainly derived from the gut microbiome [87, 88] suggesting that some of the beneficial effects of fisetin may be mediated by alterations to the gut microbiota.
More direct evidence for an effect of fisetin on the gut microbiome was obtained in a recent study on fisetin in a model of premature ovarian failure (POF) in mice [89]. POF was induced chemically over 4 weeks in the absence or presence of fisetin (100 ng/kg bw, ip). Analysis of fecal samples at the end of the study found that fisetin significantly altered the distribution and diversity of the gut microbiome in POF mice as well as the metabolic pathways associated with the microbiome. These results correlated with a significant improvement in markers of POF and a reduction in inflammatory markers in the blood. Clearly, the effects of fisetin on the microbiome is an area of research that deserves further attention.
Senolytic activity
Cellular senescence is caused by the stable arrest of the cell cycle and results in an altered cellular phenotype (for reviews see 90, 91, 92). Acute senescence plays an important role during development and in tissue repair while chronic senescence is associated not only with a failure of the arrested cell to perform its normal functions but also an increase in both local and systemic inflammation. Chronically senescent cells normally increase during aging and even higher numbers have been found in a large number of age-related diseases [90, 91] including some neurological disorders such as AD, PD and ALS [92]. Studies have shown that the removal of senescent cells can be beneficial in the context of both aging and disease [92–94]. Compounds that can selectively eliminate senescent cells are termed senolytics [91] and there is evidence that fisetin is one of these compounds.
A study published several years ago [95] showed that fisetin was much more effective at killing senescent as compared to proliferating human umbilical vein endothelial (HUVEC) cells. In contrast, fisetin was equally toxic to senescent and proliferating human preadipocytes.
These studies were subsequently extended to other cell lines as well as mouse models of senescence [74]. Using a progeroid mouse model carrying a luciferase tagged version of p16INK4a, a marker for senescent cells, fisetin treatment in food (500 mg/kg diet) for 10 weeks beginning at 10 weeks of age not only reduced p16INK4a expression but also other markers of senescence as well as markers of inflammation and oxidative stress in multiple tissues. Interestingly, fisetin did not need to be continuously fed to the animals to suppress markers of senescence consistent with a senolytic mode of action which would result in the elimination of senescent cells.
CONCLUSIONS AND OUTLOOK
There are few effective treatments for neurological disorders in general and no effective treatments for age-dependent neurodegenerative diseases. To address this major public health crisis, one or more effective drugs are required. Based on the evidence highlighted in this review, it seems time that fisetin, either alone or in combination with other compounds, is taken seriously as a possible treatment for neurological diseases/disorders (Fig. 2). Fisetin is already being tested in several short-term clinical trials for effects on senescence-related musculo-skeletal and kidney disorders (https://clinicaltrials.gov/ct2/results?cond=&term=fisetin&cntry=&state=% city=&dist=). Moreover, as described in this review, it has recently been tested in China in a double blind, randomized, placebo controlled clinical trial as an adjuvant to rtPA therapy for ischemic stroke where it significantly improved the therapeutic outcome relative to rtPA alone at longer treatment times [58]. However, it is not currently being directly tested for the treatment of any neurological disorder/disease in the US. While it is clear that the companies that have the funds to carry out clinical trials do not see a financial benefit to testing fisetin due to multiple factors including a lack of patent protection [96], alternative approaches to financing need to be tried to determine if fisetin could be useful to treat these intractable health problems.
Fig. 2
FUNDING
This study was supported by grants from the NIH (RO1 AG046153, RF1 AG054714 and R41 AI104034) and the Edward N. & Della Thome Memorial Foundation.
CONFLICT OF INTEREST
The author has no conflicts to report.
ACKNOWLEDGMENTS
The author has no acknowledgements.
REFERENCES
[1] | Collaborators GN . Global, regional, and national burden of neurological disorders, 1990–2016: a systemic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) ;18: :459–80. |
[2] | Gold M . Phase II clinical trials of anti-amyloid β antibodies: When is enough, enough? Alzheimer’s & Dementia. (2017) ;3: :402–9. |
[3] | Cummings J , Lee G , Ritter A , Zhong K . Alzheimer’s drug development pipeline: 2018. Alzheimers Dement. (2018) ;4: :195–214. |
[4] | Schubert D , Currais A , Goldberg J , Finley K , Petrascheck M , Maher P . Geroneuroprotectors: Effective geroprotectors for the brain. Trends Pharmacol Sci. (2018) ;39: :1004–7. |
[5] | Dias DA , Urban S , Roessner U . A historical overview of natural products in drug discovery. Metabolites. (2012) ;2: :303–36. |
[6] | Yun B-W , Yan Z , Amir R , Hong S , Jin Y-W , Lee E-K , et al. Plant natural products: history, limitations and the potential of cambial meristematic cells. Biotech Gen Engineer Rev. (2013) ;28: :47–60. |
[7] | Beking K , Vieira A . Flavonoid intake and disability-adjuested life years due to Alzheimer’s and related dementias: a population-based study involving twenty-three developed countries. Public Health Nutrition. (2010) ;13: :1403–9. |
[8] | Commenges D , Scotet V , Renaud S , Jacqmin-Gadda H , Barberger-Gateau P , Dartigues JF . Intake of flavonoids and risk of dementia. Eur J Epidemiol. (2000) ;16: :357–63. |
[9] | Ishige K , Schubert D , Sagara Y . Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med. (2001) ;30: :433–46. |
[10] | Tan S , Schubert D , Maher P . Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. (2001) ;1: :497–506. |
[11] | Dixon SJ , Lemberg KM , Lamprecht MR , Skouta R , Zaitsev EM , Gleason CE , et al. Ferroptosis: An iron-dependent form of non-apoptotic cell death. Cell. (2012) ;149: :1060–72. |
[12] | Lewerenz J , Ates G , Methner A , Conrad M , Maher P . Oxytosis/ferroptosis-(Re-)emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases on the central nervous system. Front Neurosci. (2018) ;12: :214. |
[13] | Sagara Y , Vahnnasy J , Maher P . Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J Neurochem. (2004) ;90: :1144–55. |
[14] | Arai Y , Watanabe S , Kimira M , Shimoi K , Mochizuki R , Kinae N . Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL and cholesterol concentration. J Nutri. (2000) ;130: :2243–50. |
[15] | Hendler SS . PDR for Nutritional Supplements, 2nd edition: Thomson Reuters; (2008) . |
[16] | Maher P . How fisetin reduces the impact of age and disease on CNS function. Front Biosci. (2015) ;7: :58–82. |
[17] | Goedert M , Spillantini MG . A century of Alzheimer’s disease Science. (2006) ;314: :777–81. |
[18] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM . Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Humans Services Task Force on Alzheimer’s disease. Neurol. (1984) ;34: :939–44. |
[19] | McKeith I , Cummings JL . Behavioral changes and psychological symptoms in dementia disorders. Lancet Neurol. (2005) ;4: :735–42. |
[20] | Haas C . Strategies, development and pitfalls of therapeutic options for Alzheimer’s disease. J Alzheimer’s Dis. (2012) ;28: :241–81. |
[21] | Rafil MS , Aisen PS . Recent developments in Alzheimer’s disease therapeutics. BMC Med. (2009) ;7: :7. |
[22] | https://www.alzforum.org/research-models/alzheimers-disease. |
[23] | Swerdlow RH . Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol Aging. (2007) ;28: :1465–80. |
[24] | Herrup K . The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. (2015) ;18: :794–9. |
[25] | Pallas M . Senescence-accelerated mice P8: A tool to study brain aging and Alzheimer’s disease in a mouse model. ISRN Cell Biol. (2012) ;2012: :917167. |
[26] | Morley JE , Armbrecht HJ , Farr SA , Kumar VB . The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim Biophys Acta. (2012) ;1822: :650–6. |
[27] | Cheng XR , Zhou WX , Zhang YX . The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res Rev. (2014) ;13: :13–37. |
[28] | Currais A , Prior M , Dargusch R , Armando A , Ehren J , Schubert D , et al. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell. (2014) ;13: :379–90. |
[29] | Currais A , Farrokhi C , Dargusch R , Armando A , Quehenberger O , Schubert D , et al. Fisetin reduces the impact of aging on behavior and physiology in the rapidly aging SAMP8 mouse. J Gerentol A Biol Sci Med Sci. (2018) ;73: :299–307. |
[30] | Ahmad A , Ali T , Park HY , Badshah H , Rehman SU , Kim MO . Neuroprotective effect of fisetin against amyloid-beta-induced cognitive/synaptic dysfunction, neuroinflammation and neurodegeneration in mice. Mol Neurobiol. (2017) ;54: :2269–85. |
[31] | Sweatt JD . Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. (2004) ;14: :311–7. |
[32] | O’Brien JT , Thomas A . Vascular dementia. Lancet. (2015) ;386: :1698–706. |
[33] | Kumar BH , Reddy RA , Kumar JM , Kumar BD , Diwan PV . Effects of fisetin on hyperhomocysteinemia-induced experimental endothelial dysfunction and vascular dementia. Can J Physiol Pharmacol. (2017) ;95: :32–42. |
[34] | Weintraub D , Comella CL , Horn S . Parkinson’s disease. Am J Manag Care. (2008) ;14: :S40–S69. |
[35] | Klein C , Westenberger A . Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. (2012) ;2: :a008888. |
[36] | Schulz JB , Falkenburger BH . Neuronal pathology in Parkinson’s disease. Cell Tissue Res. (2004) ;318: :135–47. |
[37] | Tieu K . A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb Perspect Med. (2011) ;1: :a009316. |
[38] | Blesa J , Przedborski S . Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat. (2014) ;8: :155. |
[39] | Maher P . Protective effects of fisetin and other berry flavonoids in Parkinson’s disease. Food Func. (2017) ;8: :3033–42. |
[40] | Alikatte K , Palle S , Kumar JR , Pathakala N . Fisetin improved rotenone-induced behavioral deficits, oxidative changes and mitochondrial dysfunctions in rat model of Parkinson’s disease. J Dietary Suppl. 2020;DOI:10.1080/19390211.2019.1710646. |
[41] | Borrell-Pages M , Zala D , Humbert S , Saudou F . Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. (2006) ;63: :2642–60. |
[42] | Ramaswamy S , Shannon KM , Kordower JH . Huntington’s disease: Pathological mechanisms and therapeutic strategies. Cel Transplant. (2007) ;16: :301–12. |
[43] | Imarisio S , Carmichael J , Korolchuk V , Chen C-W , Saiki S , Rose C , et al. Huntington’s disease: from pathology and genetics to potential therapeutics. Biochem J. (2008) ;412: :191–209. |
[44] | Gil JM , Rego AC . Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. (2008) ;27: :2803–20. |
[45] | Maher P , Dargusch R , Bodai L , Gerard P , Purcell JM , Marsh JL . ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Gen. (2011) ;20: :261–70. |
[46] | Riancho J , Gil-Bea FJ , Santurtan A , Lopez de Munain A . Amytotrophic lateral sclerosis: a complex syndrome that needs an integrated research approach. Neural Regen Res. (2019) ;14: :193–6. |
[47] | Lutz C . Mouse models of ALS. Brain Res. (2018) ;1693: :1–10. |
[48] | Wang TH , Wang SY , Wang XD , Jiang HQ , Yang YQ , Wang Y , et al. Fisetin exerts antioxidant and neuroprotective effects in mulitple mutant hSOD1 modles of amyotrophic lateral sclerosis by activating ERK. Neuroscience. (2018) ;379: :152–66. |
[49] | Lapchak PA , Araujo DM . Advances in ischemic stroke treatment: neuroprotective and combination therapies. Expert Opin Emerg Drugs. (2007) ;12: :97–112. |
[50] | Gelderblom M , Leypoldt F , Steinback K , Behrens D , Choe C-U , Siler DA , et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. (2009) ;40: :1849–57. |
[51] | Cumming TB , Marshall RS , Lazar RM . Stroke, cognitive deficits and rehabilitation: still an incomplete picture. Int J Stroke. (2013) ;8: :38–45. |
[52] | Waud DR . On biological assays involving quantal responses. J Pharmacol Exper Theory. (1972) ;183: :577–607. |
[53] | Zivin JA , Waud DR . Quantal bioassay and stroke. Stroke. (1992) ;23: :767–73. |
[54] | Lapchak PA , Araujo DM , Song D , Wei J , Zivin J . Neuroprotective effects of the spin trap agent disodium-[tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (generic NXY-059) in a rabbit small clot embolic stroke model: combination studies with the thrombolytic tissue plasminogen activator. Stroke. (2002) ;33: :1411–5. |
[55] | Maher P , Salgado KF , Zivin JA , Lapchak PA . A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. (2007) ;1173: :117–25. |
[56] | Gelderblom M , Leypoldt F , Lewerenz J , Birkenmayer G , Orozco D , Ludewig P , et al. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J Cereb Blood Flow Metab. (2012) ;32: :835–43. |
[57] | Christophe BR , Mehta SH , Garton AL , Sisti J , Connolly ES . Current and future perspectives on the treatment of cerebral ischemia. Expert Opin Pharmacother. (2017) ;18: :573–80. |
[58] | Wang L , Cao D , Wu H , Jia H , Yang C , Zhang L . Fisetin prolongs therapy window of brain ischemic stroke using tissue plasminogen activator: A double blind randomized placebo-controlled clinical trial. Clin ApplThromb Hemost. (2019) ;25: :1–8. |
[59] | Grysiewicz RA , Thomas K , Pandey DK . Epidemiology of ischemic and hemorrhagic stroke: Incidence, prevalence, mortality and risk factors. Neurol Clin. (2008) ;26: :871–95. |
[60] | Waziry R , Chibnik LB , Bos D , Ikram MK , Hofman A . Risk of hemorrhagic and ischemic stroke in patients with Alzheimer’s disease: A synthesis of the literature. Neurol. (2020) ;94: :265–72. |
[61] | Chen C , Yao L , Cui J , Liu B . Fisetin protects against hemorrhage-induced neuroinflammation in aged mice. Cerebrovasc Dis. (2018) ;45: :154–61. |
[62] | Zhou C-H , Wang C-X , Xie G-B , Wu L-Y , Wei Y-X , Wang Q , et al. Fisetin alleviates early brain injury following experimental subarachnoid hemorrhage in rats possibly by suppressing TLR 4/NF-kB signaling pathway. Brain Res. (2015) ;1629: :250–9. |
[63] | Ma Q , Khatibi N , Chen H , Tang J , Zhang JH . History of preclinical models of intracerebral hemorrhage. Acta Neurochir Suppl. (2011) ;111: :3–8. |
[64] | Leclerc JL , Garcia JM , Diller MA , Carpenter A-M , Kamat PK , Hoh BL , et al. A comparison of pathophysiology in humans and rodent models of subarachnoid hemorrhage. Front Mol Neurosci. (2018) ;11: :71. |
[65] | Capizzi A , Woo J , Verduzco-Gutierrez M . Traumatic Brain Injury. Med Clin N Am. (2020) ;104: :213–38. |
[66] | Galgano M , Toshkezi G , Qiu X , Russell T , Chin L , Zhao L-R . Traumatic brain injury: Current treatment strategies and future endeavors. Cell Transplantation. (2017) ;26: :1118–30. |
[67] | Zhang L , Wang H , Zhou Y , Zhu Y , Fei M . Fisetin alleviates oxidative stress after traumatic brain injury via the Nrf2-ARE pathway. Neurochem Int. (2018) ;118: :304–13. |
[68] | Yankner BA , Lu T , Loerch P . The aging brain. Annu Rev Pathol Mech Dis. (2008) ;3: :41–66. |
[69] | Bevins RA , Besheer J . Object recognition in rats and mice: a non-trial non-matching-to-sample learning task to study ‘recognition memory’. Nature Protocols. (2006) ;1: :1306–11. |
[70] | Maher P , Akaishi T , Abe K . Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc Natl Acad Sci USA. (2006) ;103: :16568–73. |
[71] | Bliss TVP , Collingridge GL . A synaptic model of memory: long-term potentiation in the hippocampus. Nature. (1993) ;361: :31–9. |
[72] | He WB , Abe K , Akaishi T . Oral administration of fisetin promotes the induction of hippocampal lonr-term potentiation in vivo. J Pharmacol Sci. (2018) ;136: :42–5. |
[73] | Singh S , Singh AK , Garg G , Rizvi SI . Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci. (2018) ;193: :171–9. |
[74] | Yousefzadeh MJ , Zhu Y , McGowan SJ , Angelini L , Fuhrmann-Stroissnigg H , Xu M , et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. (2018) ;36: :18–28. |
[75] | Nabavi SF , Braidy N , Habtemariam S , Sureda A , Manayi A , Nabavi SM . Neuroprotective effects of fisetin in Alzheimer’s and Parkinson’s diseases: From chemistry to medicine. Curr Top Med Chem. (2016) ;16: :1910–5. |
[76] | Lall RK , Adhami VM , Mukhtar H . Dietary flavonoid fisetin for cancer prevention and treatment. Mol Nutr Food Res. (2016) ;60: :1396–405. |
[77] | Kashyap D , Sharma A , Sak K , Tuli HS , Buttar HS , Bishayee A . Fisetin: A bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci. (2018) ;194: (75-87). |
[78] | Currais A , Maher P . Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid Redox Signal. (2013) ;19: :813–22. |
[79] | Feng W , Rosca M , fan Y , Hu Y , Feng P , Lee H-G , et al. Gclc deficiency in mouse CNS causes mitochondrial damage and neurodegeneration. Hum Mol Gen. (2017) ;26: :1376–90. |
[80] | Fernandez-Fernandez S , Bobo-Jimenez V , Requejo-Aguilar R , SGonzalez-Fernandez S , Resch M , Carabias-Carrasco M , et al. Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol. (2018) ;38: :5415–28. |
[81] | Maher P . The potential of flavonoids for the treatment of neurodegenerative diseases. Int J Mol Sci. (2019) ;20: :3056. |
[82] | Kang Y , Tiziani S , Park G , Kaul M , Paternostro G . Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nature Commun. (2014) ;5: :3672. |
[83] | Schubert D , Piasecki D . Oxidative glutamate toxicity can be a part of the excitotoxicity cascade. J Neurosci. (2001) ;21: :7455–62. |
[84] | Xie Y , Hou W , Song X , Yu Y , Huang J , Sun X , et al. Ferroptosis: process and function. Cell Death Differ. (2016) ;23: :369–79. |
[85] | Zille M , Karuppagounder SS , Chen Y , Gough PJ , Bertin J , Finger J , et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. (2017) ;48: :1033–43. |
[86] | Cryan JF , O’Riordan KJ , Sandhu K , Peterson V , Dinan TG . The gut microbiome in neurological disorders. Lancet Neurol. (2020) ;19: :179–94. |
[87] | Nicholson JK , Holmes E , Kinross J , Burcelin R , Gibsonb G , Jia W , et al. Host-gut microbiota metabolic interactions. Science. (2012) ;336: :1262–7. |
[88] | Guo L , Milburn MV , Ryals JA , Lonergan SC , Mitchell MW , Wulff JE , et al. Plasma metabolomic profiles enhance precision medicine for volunteers of normal health. Proc Natl Acad Sci U S A. (2015) ;2015: :E4901–E10. |
[89] | Lin J , Nie X , Xiong Y , Gong Z , Chen J , Cehn C , et al. Fisetin regulates gut microbiota to decrease CCR9+/CXCR3+/CD4+ T lymphocytes count and IL-12 secretion to alleviate premature ovarian failure in mice. Am J Transl Res. (2020) ;12: (203-247). |
[90] | de Keizer PLJ . The fountain of youth by targeting senescent cells? Trends Mol Med. (2017) ;23: :6–17. |
[91] | Gerdes EOW , Zhu Y , Tchkonia T , Kirkland JL . Discovery, development and future application of senolytics: theories and predictions. FEBS J. 2020. doi:10.1111/febs.15264. |
[92] | Martinez-Cue C , Rueda N . Cellular senescence in neurodegnerative diseases. Front Cell Neurosci. (2020) ;14: :16. |
[93] | Musi N , Valentine JM , Sickora KR , Baeuerle E , Thompson CS , Shen Q , et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. (2018) ;17: :e12840. |
[94] | Zhang P , Kishimoto Y , Grammatikakis I , Gottimukkala K , Cutler RG , Zhang S-Q , et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nature Neurosci. (2019) ;22: :719–28. |
[95] | Zhu Y , Doornebal EJ , Pirtskhalava T , Giorgadze N , Wentworth M , Fuhrmann-Stroissnigg H , et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging. 2017;9: 955-63. |
[96] | Paller CJ , Denmeade SR , Carducci MA . Challenges of conducting clinical trials of natural products to combat cancer. Clin. Adv. Hematol. Oncol. (2016) ;14: :447–55. |


