
Identification and Characterisation of cis-Regulatory Elements Upstream of the Human Receptor Tyrosine Kinase Gene MERTK
Abstract
BACKGROUND:
MERTK encodes a receptor tyrosine kinase that regulates immune homeostasis via phagocytosis of apoptotic cells and cytokine-mediated immunosuppression. MERTK is highly expressed in the central nervous system (CNS), specifically in myeloid derived innate immune cells and its dysregulation is implicated in CNS pathologies including the autoimmune disease multiple sclerosis (MS).
OBJECTIVE:
While the cell types and tissues that express MERTK have been well described, the genetic elements that define the gene’s promoter and regulate specific transcription domains remain unknown. The primary objective of this study was to define and characterise the human MERTK promoter region.
METHODS:
We cloned and characterized the 5’ upstream region of MERTK to identify cis-acting DNA elements that promote gene transcription in luciferase reporter assays. In addition, promoter regions were tested for sensitivity to the anti-inflammatory glucocorticoid dexamethasone.
RESULTS:
This study identified identified both proximal and distal-acting DNA elements that promote transcription. The strongest promoter activity was identified in an ∼850 bp region situated 3 kb upstream of the MERTK transcription start site. Serial deletions of this putative enhancer revealed that the entire region is essential for expression activity. Using in silico analysis, we identified several candidate transcription factor binding sites. Despite a well-established upregulation of MERTK in response to anti-inflammatory glucocorticoids, no DNA region within the 5 kb putative promoter was found to directly respond to dexamethasone treatment.
CONCLUSIONS:
Elucidating the genetic mechanisms that regulate MERTK expression gives insights into gene regulation during homeostasis and disease, providing potential targets for therapeutic modulation of MERTK transcription.
INTRODUCTION
MERTK (chr2 : 2q14.1) encodes a member of the TAM (TYRO3, AXL, MERTK) family of structurally and functionally related receptor tyrosine kinases that are broadly expressed in nervous, vascular and reproductive tissues [1–5]. Binding of the ligands growth-arrest-specific 6 (GAS6) or Protein S (PROS1) to MERTK initiates signaling and activation of a myriad of downstream pathways that modulate tissue repair, inflammation, cell survival and cell proliferation in many physiological systems [6–9]. This is primarily achieved via apoptotic cell engulfment, as well as interferon 1 and toll-like receptor (TLR) signaling [10–14].
Loss of MERTK signaling is linked to the development of autoimmunity, delayed apoptotic clearance and chronic inflammation with its disruption strongly implicated in the development of autoimmune and neurodegenerative diseases such as systemic lupus erythematosus (SLE), retinitis pigmentosa and MS [10, 13, 15–21]. Recently characterised polymorphisms within the gene have established MERTK as a novel risk locus for MS susceptibility [22, 23]. While loss or reduction of signaling is generally pathogenic, hypomorphic variation of MERTK has been observed to be protective in macrophage driven pathology of non-alcoholic steatohepatitis, further highlighting the complexity of MERTK expression and physiological function in different cellular contexts [24]. Conversely, overexpression of MERTK is associated with human cancers via potentiation of cellular proliferation, migration, invasion and survival [25–27]. The MERTK receptor can also be hijacked by enveloped viruses, whereby ectopic expression or overexpression of MERTK promotes viral entry and subsequent infection [28]. Therefore, the physiological maintenance of homeostasis mediated by MERTK is complex and dependent upon appropriate regulation of temporal and spatial expression profiles in specific tissues and cell types.
MERTK expression is enriched in CNS tissue, specifically in resident innate immune cells where it regulates several intersecting functions to support CNS plasticity and homeostasis. During development, MERTK expressing microglia and astrocytes actively participate in efferocytosis of immature neurons, synapses and myelin debris, helping to refine neuronal and synaptic architecture [18, 29, 30]. In response to inflammatory and pathogenic stimuli, MERTK expressing microglia, monocyte derived macrophages and dendritic cells phagocytose apoptotic cells and debris while releasing anti-inflammatory cytokines to promote tissue repair [10, 11, 31–33]. MERTK signaling in these cells has been shown to be essential for these functions and regulation of developmental and pathological CNS processes.
Despite the central importance of appropriate MERTK expression to the function of multiple cells in complex systems, transcriptional regulation of MERTK expression is not well understood [34]. To date, only one study has characterised the 5’ upstream promoter region of the MERTK gene and then only in the context of sertoli cells in mice [35]. In conjunction with upstream enhancer/suppressor elements, core gene promoters specify temporal and spatial gene expression patterns. Defining the promoter of human MERTK therefore presents a novel opportunity to further elucidate the genetic determinants of MERTK expression and the consequences of its regulation on cell function and CNS homeostasis.
A strong role in immune signaling and pathological outcomes makes MERTK an appealing drug target. Understanding the environmental and genetic factors that modulate MERTK expression would provide invaluable insight in realising this therapeutic potential. Glucocorticoids (GCs) represent one of the most effective clinical treatments for inflammatory disorders, enhancing the phagocytic and anti-inflammatory capacity of macrophages [36, 37]. The synthetic glucocorticoid dexamethasone upregulates MERTK expression in human monocyte derived macrophages and promotes clearance of apoptotic cells [15, 16, 38]. No studies have elucidated a specific mechanism (direct or indirect) of GC mediated upregulation of MERTK. GCs regulate transcription via the binding of a glucocorticoid receptor (GR) to highly conserved glucocorticoid receptor elements (GRE) in gene promoters [39, 40]. We posited that the MERTK promoter would contain cis-binding elements (GREs) that mediate its GC-induced upregulation.
In this study we have characterised the DNA elements that constitute the MERTK promoter and identified key regions that regulate gene transcription. To do this, we cloned and assayed a 5 kb region upstream of the MERTK transcription start site (TSS). Luciferase reporter assays in HEK293T cells identified minimal sequences in proximal and distal regions of the promoter capable of promoting gene expression. In silico analysis of these regions identified putative binding sites for both general and tissue specific transcription factors that may modulate gene expression, including the master myeloid factor CCAAT Enhancer Binding Protein Beta (C/EBPβ). We observed no further induction of reporter expression for any of the MERTK constructs following exposure to dexamethasone, suggesting that this promoter region does not contain cis-acting GREs that promote MERTK responsiveness to GC treatment. Overall, this work contributes to a holistic understanding of the transcriptional regulation of MERTK and its function in health and disease.
METHODS
Ethics statement
The DNA used in this study was sourced from a blood sample of a single healthy individual that was recruited based on previous genotyping and approved by the Melbourne Health Human Research Ethics committee (Project number: 2013.111) as a part of an ANZgene MS study [23]. The Research Ethics Committee which provided approval for this research are guided by national standards as outlined in the National Statement on Ethical Conduct in Human Research (https://www.nhmrc.gov.au/guidelines-publications/e72) issued by the National Health and Medical Research Council (Australia). The individual provided written consent for the use and storage of DNA samples.
Cloning of the 5’ upstream nucleotide region of MERTK
Seven overlapping fragments (each approximately 1 kb in size) that spanned a 5.735 kb region (chr2 : 112,651,004-112,656,738) from –5187 bp to +548 bp of the MERTK transcription start site were cloned using healthy control human DNA (see above) as a template. Primer pairs were designed using UCSC genome browser with the human reference genome (GRCh37/hg19) and were sourced from Integrated DNA technologies (Coralville, IA, USA) (Table 1). PCR reactions were carried out using 2 U recombinant Taq DNA polymerase recombinant (5 U/μl) (Invitrogen, CA, USA), 1× PCR reaction buffer (Invitrogen), 2 mM dNTPs, 1.5 mM MgCl2, 0.5μM forward and reverse primers and 100 ng of human DNA. PCR cycling conditions were as follows: 1 cycle of 95°C for 3 minutes; 35 cycles of 95°C for 30 seconds, annealing temperature (Individual Ta for each amplicon outlined in Table 1) for 30 seconds, 72°C for 2 minutes; followed by 1 cycle of 72°C for 10 minutes.
Table 1
Primers for PCR amplification of human MERTK promoter region
| MERTK promoter clone | Primer sequence | Length of Amplicon (bp) | Position (GRCh37/hg19) | Ta | |
| MERTK-1 | Fwd | GCAGGCCTGAAATTAGCTTG | 1329 | chr2 : 112651005-112652333 | 63.4°C |
| Rev | ACGTAGGGACCCCTGAGTCT | ||||
| MERTK-2 | Fwd | TTTTCTTACGGGCACCATTC | 1129 | chr2 : 112652086-112653214 | 63.4°C |
| Rev | AGGGAAGATTACCCCCACTG | ||||
| MERTK-3 | Fwd | CTGAAAGGGGTAGGGGACAG | 859 | chr2 : 112652959-112653817 | 67.0°C |
| Rev | GCCTTGGAGTAGCTGGAAGT | ||||
| MERTK-4 | Fwd | GCTAAATTTTGGCATTTGCAG | 988 | chr2 : 112653557-112654544 | 67.0°C |
| Rev | AAATCACAGGGCAGTCCAAC | ||||
| MERTK-5 | Fwd | TGACTTGGAGGAGAGCCAAC | 913 | chr2 : 112654304-112655216 | 67.0°C |
| Rev | AGCCAAATCATGTCCCTCTC | ||||
| MERTK-6 | Fwd | AGCCAAATCTACGGGAAGTG | 910 | chr2 : 112654995-112655904 | 69.6°C |
| Rev | GAGCCGGAAAGAGAAGTGAA | ||||
| MERTK-7 | Fwd | CGTTCATCTCCCTCCTTGAG | 1285 | chr2 : 112655455-112656739 | 62.0°C |
| Rev | TCCCAGACCAGACCTGCTAC | ||||
PCR amplicons and pGL3 enhancer vector (Promega, USA) were digested with appropriate restriction enzymes (NEB, USA) in 10× NEB cutsmart buffer (NEB). Each PCR amplicon (50 ng) was ligated into the reporter vector (50 ng) using 400 Units T4 DNA ligase (400 U/μl) (NEB). Plasmids were sequenced using Sanger sequencing (AGRF, VIC, AUS) to validate DNA sequence of cloned amplicons within pGL3 enhancer vector.
Site directed mutagenesis of MERTK-3
The Q5 Site-directed mutagenesis kit (NEB, USA) was used to generate 4 serial deletions (∼250 bp) of the MERTK-3 pGL3 construct. Primer pairs were designed using NEB Base changer (Table 2). PCR reaction cycling conditions were as follows: 1 cycle of 98°C for 30 seconds; 25 cycles of 98°C for 10 seconds, annealing temperature (Individual Ta for each primer pair outlined in Table 2) for 30 seconds, 72°C for 6 minutes and 1 cycle of 72°C for 12 minutes using 25 ng of MERTK-3 pGL3 vector as a template. Correct deletions were confirmed using Sanger sequencing (AGRF, VIC, AUS).
Table 2
Primers for mutagenesis of MERTK-3
| MERTK promoter clone | Primer sequence | Ta | |
| MERTK3- | Fwd | TTCCCTAGCCCTTAGATG | 61°C |
| ΔS1 | Rev | GGTACCTATCGATAGAGAAATG | |
| MERTK3- | Fwd | GGTCCGGGTTGTTTTTG | 59°C |
| ΔS2 | Rev | CATTGTGGTCACAATCTATC | |
| MERTK3- | Fwd | CACTTCTATCTTCAAGAGGTTG | 61°C |
| ΔS3 | Rev | ACTTACCTGGAACCCATTC | |
| MERTK3- | Fwd | CTCGAGATCTGCGATCTAAG | 61°C |
| ΔS4 | Rev | CTAAACTGTGAGATGTCAGC | |
Cell culture of HEK293T cells
HEK293T cells were cultured in DMEM with 4 mM Glutamine, 10% FCS, 100μg/ml penicillin streptomycin in 75 cm2 tissue culture flasks and incubated at 37°C/5% CO2. Cells were passaged every 3–4 days upon reaching confluency. Cells were treated with 0.25% trypsin for 5 minutes, pelleted by centrifugation at 1200 rpm for 5 minutes and subcultured (at a ratio of 1 : 8) in 10 ml media.
Lipofectamine LTX transfection of HEK293T cells
HEK293T cells were passaged as previously described and plated in a 24 well Nunclon delta surface plate at a density of 2×105 cells in 500μl media (as described above). Cells were incubated at 37°C/5% CO2 for 2 hours prior to transfection. Transfection reactions (one per well) were prepared as follows: 500 ng of pGL3 reporter plasmid (or an equimolar equivalent of a MERTK sequence containing plasmid) was added to 100μl of OPTI-MEM (1×) reduced serum medium. To control for variable levels of transfection between reactions, cells were co-transfected with 5 ng (1 : 100) of a pRL-TK Renilla luciferase control vector (Promega) and mixtures were incubated for 15 minutes at room temperature. Following incubation, 2μl of Lipofectamine LTX reagent (Invitrogen, CA) was added and mixtures were incubated at room temperature for a further 25 minutes. Mixtures were added to each well and cell lysates were harvested after 48 hours using the Promega Dual-Luciferase reporter 1000 assay system (Promega, USA) in accordance with manufacturer’s instructions. Quantification of firefly and renilla luciferase activity was measured with a POLARstar OMEGA microplate reader equipped with two injectors (BMG Labtech, Germany) in accordance with manufacturer’s instructions.
Statistical analysis
Relative luminescence was quantified as a ratio of firefly luciferase activity to the co-transfected renilla control. Data are expressed as means with error bars indicating standard error of mean (SEM) or standard deviation (SD). Results are displayed as a representative graph from the minimum of 3 independent experiments unless noted otherwise. Paired ratio t-tests and one-way ANOVA analyses with appropriate post hoc comparisons were used to identify statistically significant results.
Assessment of glucocorticoid responsiveness
To assess promoter constructs for GC response, transfections were completed as above except for the following: HEK293T cells were cultured in 2% FCS media and transfected with 300 ng pGL3 reporter plasmid, MERTK pGL3 construct or pGRE-Luc cis reporter plasmid (Agilent, CA, USA), 30 ng of human NR3C1 cDNA vector (pCMV6-XL5) (OriGene, MD, USA) and 3 ng of pRL-TK renilla. Cells were treated with 100 nM dexamethasone (Sigma, CA, USA) 24 hours prior to collection. Data collection and analysis was completed as described above.
Bioinformatics analysis of 5’ flanking region of MERTK/AXL
In silico analyses of the MERTK and AXL 5’ regulatory regions were undertaken using UCSC genome browser with the human reference genome (GRCh37/hg19 in humans, GRCm38/mm10 in mice). Reported transcription factor binding sites were obtained from ENCODE ChIP database [41]. TRANSFAC (http://genexplain.com/transfac/) and JASPAR (http://jaspar.genereg.net/) predictive software were used to characterise binding sites within regions of interest [42, 43]. The EMBOSS needle bioinformatics tool (EMBL-EBI, Cambridge, UK, https://www.ebi.ac.uk/) was used for pairwise sequence alignments [44].
RESULTS
Identification of proximal and distal promoter regions 5’ of the MERTK TSS
The MERTK gene is located on chromosome 2, encoded by 19 canonical exons (plus a recently identified alternative final exon) and is distributed over a region of 130 kb [23]. As the human MERTK promoter has never been defined, we cloned 7 overlapping DNA fragments (spanning –5187 bp to +548 bp) into the pGL3 enhancer vector and assessed constructs for luciferase reporter expression in HEK293T cells (Fig. 1A). MERTK-3 had the strongest promoter activity of all clones, increasing reporter expression by 11.3 fold over the empty pGL3 vector (42.25±12.61 vs 3.75±0.53, p = 0.033) (Fig. 1A). This region, approximately 3 kb upstream of the MERTK TSS was nominated as a distal promoter element. MERTK-7 increased reporter expression 4.7 fold (17.44±2.71 vs 3.75±0.53, p = 0.023) over the empty pGL3 vector (Fig. 1A). This region spans a 1.2 kb region that overlaps with the MERTK TSS and was nominated as a proximal promoter region. Conversely, both MERTK-1 and MERTK-5 significantly reduced reporter gene expression, 12.5 fold (0.31±0.09 vs 3.75±0.53, p = 0.021) and 2.3 fold (1.60±0.22 vs 3.75±0.53, p = 0.020) respectively, when compared with the empty pGL3 vector (Fig. 1A). It is not surprising that the proximal element was capable of inducing transcription given its genomic location in relation to the TSS. In contrast, the discovery of the 850 bp region 3 kb upstream of MERTK was more novel, suggestive of a strongly acting putative enhancer of gene transcription.
Fig. 1
5’ upstream MERTK sequence contains regions which function as cis-acting promoter elements. Functional activity of generated MERTK promoter amplicons was assessed via luciferase reporter activity. A. Schematic representation showing the design of pGL3 reporters relative to the MERTK promoter region and their functional promoter activity. B. Schematic representation of a putative proximal promoter region relative to the MERTK gene with corresponding CpG islands. Numbers next to each CpG track represent the sum of consecutive CpG doublets for that island. Data obtained from UCSC Genome Browser (GRCh37/h19) C. Sequence alignment of MERTK transcription start site (+1) and the consensus sequence for a typical mammalian promoter initiator site with matching bases highlighted. Consensus symbols; Y = pyrimidine (C or T); N = any base; W = A or T. Luciferase expression was measured 48 hours following transfection in HEK293T cells. Relative luminescence represents ratio of firefly luciferase activity to the co-transfected renilla luciferase control. Data are expressed as means±SEM. Data are representative of 3 independent experiments (except when construct is denoted with a #, which is data representative of 1 experiment). Difference in activity of the pGL3 constructs was tested against the empty pGL3 enhancer vector using a paired ratio t test, * P < 0.05.
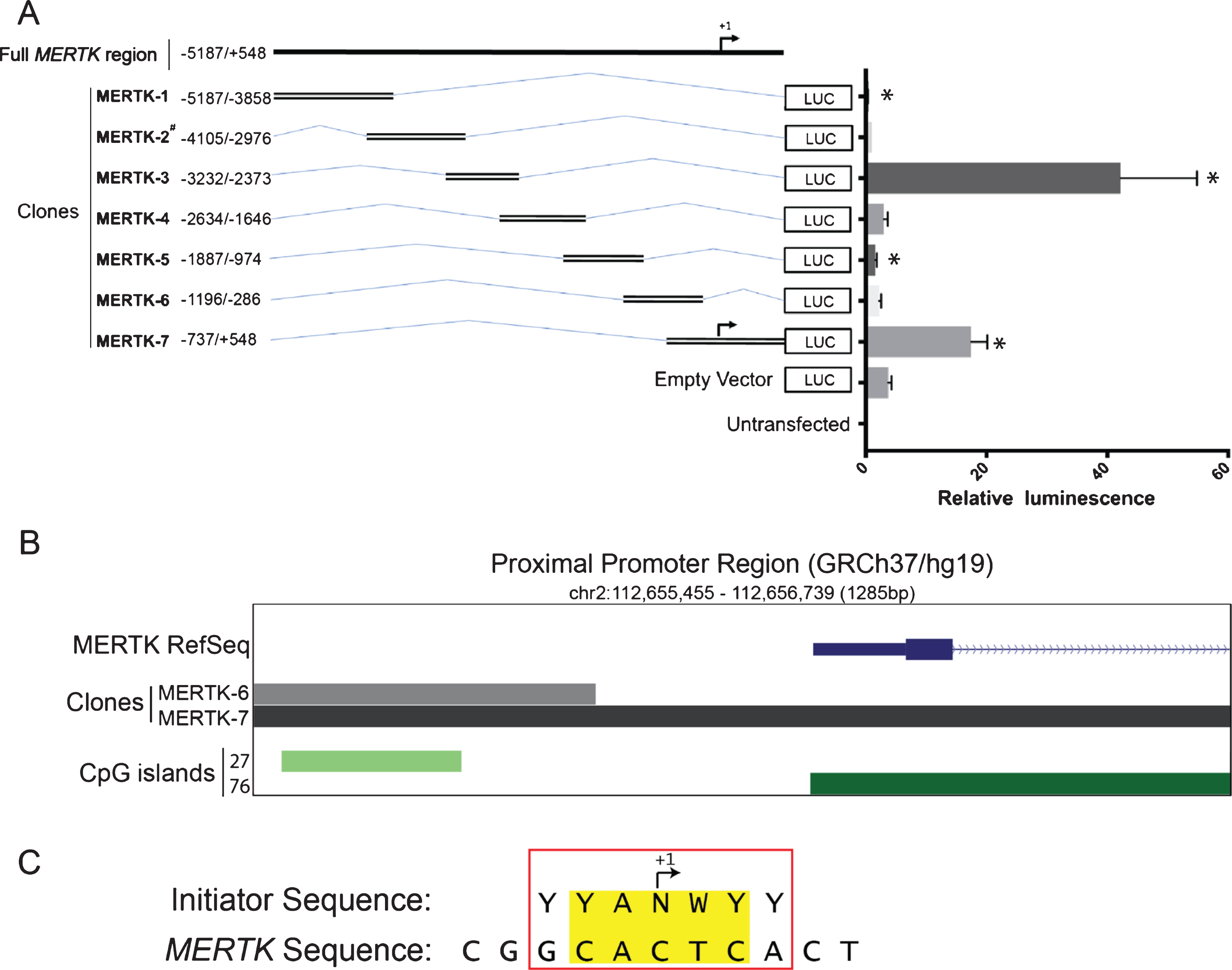
Proximal promoter region contains basal regulatory elements consistent with a TATA-less core gene promoter
To further characterise the defined proximal promoter element (MERTK-7, –737/+548), we undertook in silico analysis of the region using the UCSC genome browser (build hg19) and identified several putative regulatory elements. Analysis of core promoter elements revealed enrichment of CpG dinucleotides, the lack of a canonical TATA box and a 5/7 nucleotide sequence match with a core YYANWYY Initiator sequence at the +1 nucleotide (Fig. 1B and 1C). These data suggest that constitutive expression of human MERTK is driven by a core TATA-less CpG island promoter, the most highly represented class of mammalian promoters, present in 80% of human protein coding genes [45, 46].
In silico analysis reveals enrichment of SP1 and SP3 binding sites in proximal promoters of human/mouse MERTK and human AXL gene.
MERTK and its family TAM receptor AXL are broadly co-expressed in immune cells and both contribute to homeostatic and inflammatory processes in the CNS. Previously published proximal promoter analyses in both mouse Mertk and human AXL genes identified enrichment of binding motifs of the SP family of transcription factors [35, 47]. Binding of SP1 and SP3 were shown to be essential for basal expression of both TAM genes. We therefore wanted to determine whether a similar enrichment occurred in the human MERTK promoter. Using pairwise sequence alignment with EMBOSS needle we assessed sequence/motif conservation in the region of upstream DNA unique to the defined proximal promoter region that did not overlap the flanking (non-transcriptionally active) MERTK-6 clone (–300/+1). Pairwise alignment between human and mouse MERTK sequences revealed 52.1% identity for the region, with significant conservation of specific SP1/SP3 consensus binding sites, three of which were previously shown to bind and drive basal gene expression in mouse sertoli cells [35] (Fig. 2A). In addition, one other strongly conserved consensus site was predicted by JASPAR (Fig. 2A). Sequence alignment of MERTK and AXL revealed 44.6% identity but no strong conservation of specific binding motifs (data not shown). However, the proximal AXL promoter does contain SP1 and SP3 consensus sequences validated by Chromatin immunoprecipitation (ChIP) experiments in several human cell lines (Fig. 2B).
Fig. 2
Comparison between proximal promoters of MERTK and AXL reveals similar enrichment for SP1 and SP3 binding sites. Partial nucleotide sequences (–300/+1) of 5’ flanking region of human and mouse MERTK and human AXL. A. Pairwise comparison of human and mouse sequences reveals conserved SP1 and SP3 consensus sequences (highlighted red) predicted by JASPAR and experimentally observed as binding sites in mouse sertoli cells [35]. Conserved binding sites predicted by JASPAR only are highlighted in blue. B. SP1 and SP3 binding sites in AXL proximal promoter have been experimentally observed in Rko and HeLa cells [47]. Sequence data obtained from UCSC Genome Browser (GRCh37/hg19 for human and GRCm38/mm10 for mouse).
![Comparison between proximal promoters of MERTK and AXL reveals similar enrichment for SP1 and SP3 binding sites. Partial nucleotide sequences (–300/+1) of 5’ flanking region of human and mouse MERTK and human AXL. A. Pairwise comparison of human and mouse sequences reveals conserved SP1 and SP3 consensus sequences (highlighted red) predicted by JASPAR and experimentally observed as binding sites in mouse sertoli cells [35]. Conserved binding sites predicted by JASPAR only are highlighted in blue. B. SP1 and SP3 binding sites in AXL proximal promoter have been experimentally observed in Rko and HeLa cells [47]. Sequence data obtained from UCSC Genome Browser (GRCh37/hg19 for human and GRCm38/mm10 for mouse).](https://content.iospress.com:443/media/bpl/2021/7-1/bpl-7-1-bpl200102/bpl-7-bpl200102-g002.jpg)
Distal promoter region (MERTK-3) contains binding sites for myeloid/cell type specific transcription factors
The distal promoter element (MERTK-3, –3232/–2373) exhibited the strongest promoter activity of all tested clones. Given that the proximal promoter region contains elements that usually drive constitutive gene expression, we next wanted to characterise and assess the distal promoter hypothesising that upstream regulatory elements could direct cell/tissue specificity of MERTK expression. Analysis of ENCODE ChIP data revealed a binding site for the myeloid specific C/EBPβ transcription factor (in HepG2 cells) in a region exclusive to the MERTK-3 clone (Fig. 3A). Binding sites for the general DNA binding proteins MAX, MAZ, FOXA2, TEAD4 and TAL1 (in K562 cells) were also observed in the overlapping region between MERTK-3 and its flanking clone MERTK-4 (Fig. 3A).
Fig. 3
MERTK-3 deletion constructs reveal entire 859 bp region is required for observed expression. Functional activity of MERTK-3 deletion constructs was assessed via luciferase reporter activity. A. Schematic representation of MERTK-3, flanking clones MERTK-2 and MERTK-4 and the corresponding region deleted by each MERTK-3 deletion construct (MERTK-3Δ S1–4) with accompanying ENCODE ChIP experimental data and TRANSFAC analysis of transcription factor binding sites. Data obtained from UCSC Genome Browser (GRCh37/hg19), ENCODE and TRANSFAC databases. B. Schematic representation showing the design of MERTK-3 deletion constructs relative to the full MERTK-3 vector and their functional promoter activity. Relative Luminescence represents ratio of firefly luciferase activity to the co-transfected renilla luciferase control. Data are expressed as means±SEM. Data are representative of 3 independent experiments. Data were analysed by a one-way ANOVA with post-hoc comparisons between MERTK-3 and each individual deletion construct. *p < 0.05, **p < 0.01, ***p < 0.001.
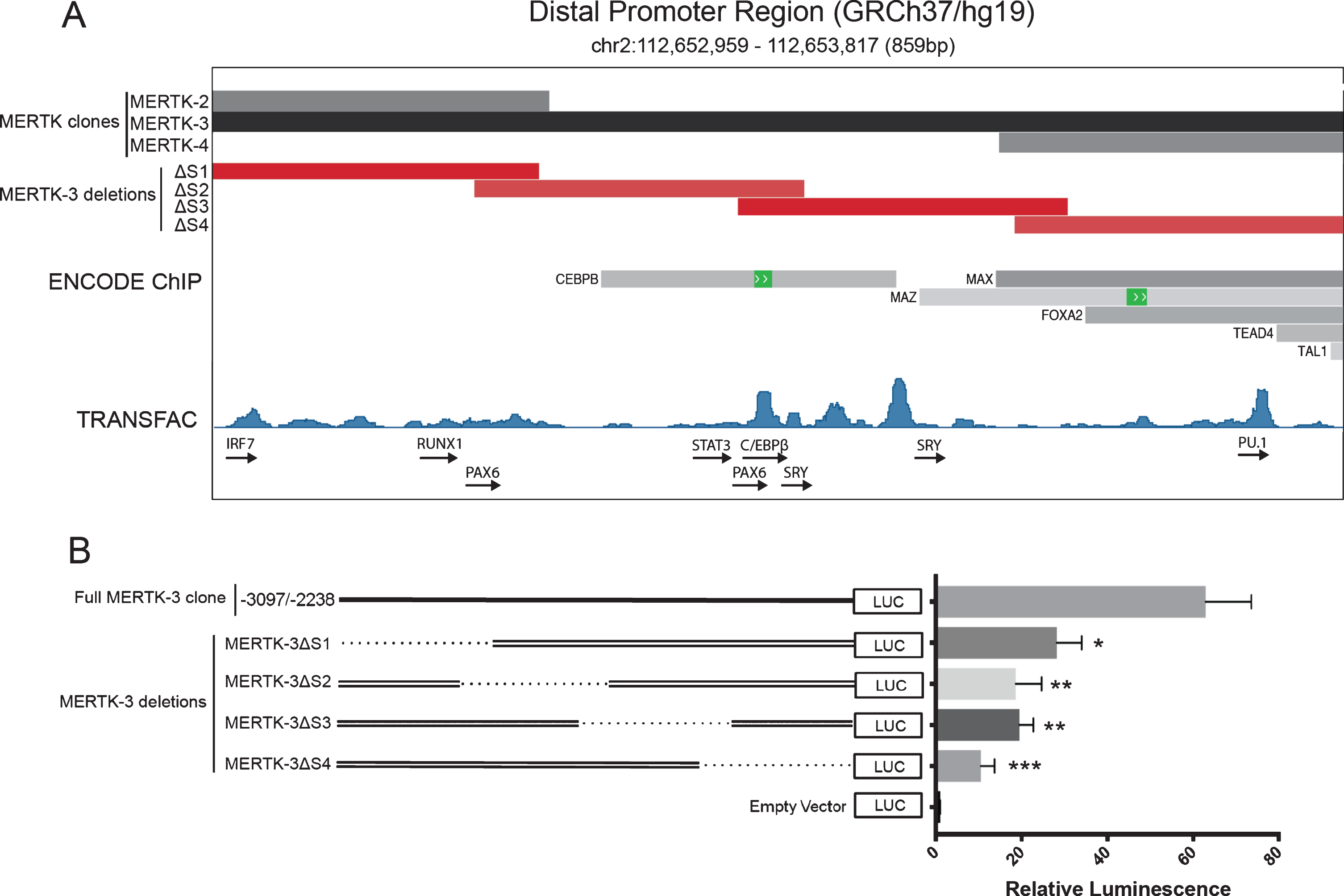
Further in silico analysis of MERTK-3 with TRANSFAC showed several peaks of predicted binding in MERTK-3 that supported the ENCODE data including C/EBPβ (Fig. 3A). MERTK-3 contains putative binding sites for the “master regulators” of myeloid lineage commitment PU.1 and RUNX1, as well as for SRY and PAX6 which are selectively expressed in MERTK enriched tissues including the testis and optic nerve (Fig. 3A). Putative binding sites for several transcription factors associated with inflammatory responses were also identified, including members of the IRF and STAT families (Fig. 3A).
Serial deletions within MERTK-3 reveal that the entire region is required for maximum transcriptional activity
To identify a minimal region capable of promoting gene expression, four overlapping 250 bp deletions were generated within MERTK-3 and individually assessed for luciferase expression. All MERTK-3 deletions resulted in the loss of at least half of the observed expression activity observed with the full MERTK-3 construct (Fig. 3B). MERTK-3ΔS4, which coincided with the highest number of observed ENCODE defined TF binding sites had the lowest expression, with an 83% reduction in activity when compared to the complete MERTK-3 region (10.44±3.24 vs 62.88±10.6, p = 0.0006) (Fig. 3A and 3B). MERTK-3ΔS2 and MERTK-3ΔS3 showed similar activity, reducing luciferase activity by 70% (18.65±6.00 vs 62.88±10.6, p = 0.0021) and 69% (19.46±3.27 vs 62.88±10.6, p = 0.0024) respectively (Fig. 3B). This near identical reduction suggests that deleted regions in both MERTK-3ΔS2 or MERTK-3ΔS3 are disrupting the same regulatory mechanism, the most likely candidate being C/EBPβ, given their overlap with the observed binding site. Additionally, although ENCODE ChIP data revealed no observed transcription factor binding in MERTK-3ΔS1, deletion of this region exhibited a 55% reduction in expression activity (28.22±5.80 vs 62.88±10.6, p = 0.016).
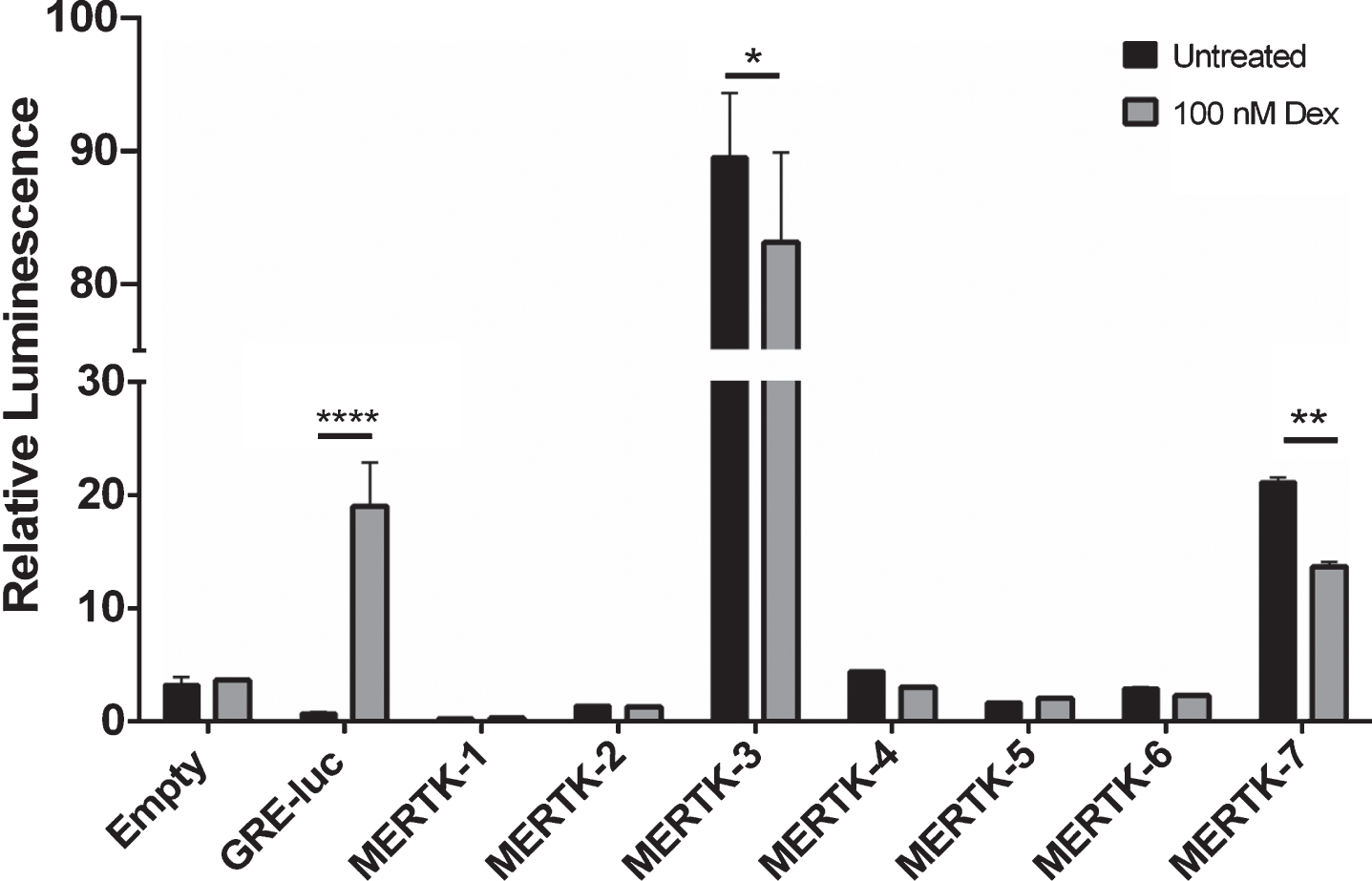
MERTK promoter activity is not enhanced by treatment with dexamethasone
As MERTK expression is known to be upregulated by GCs, we hypothesised that the MERTK promoter region would contain GRE elements strongly enhanced by GC treatment. To test this, MERTK pGL3 constructs were co-transfected with a GR (NR3C1) human cDNA clone and treated with 100 nM dexamethasone. None of the MERTK constructs upregulated luciferase expression following dexamethasone treatment, compared with a 25 fold induction observed using the GRE-luc control vector (18.97±3.85 vs. 0.75±0.07) (Fig. 4). Interestingly, both the distal and proximal promoter elements were both observed to statistically decrease reporter activity following treatment, 1.07 (89.43±4.8 vs. 83.07±6.70, p = 0.0145) and 1.55 (21.11±0.39 vs 13.65±0.40, p = 0.0025) fold respectively (Fig. 4).
Fig. 4
MERTK promoter activity is not enhanced following treatment with dexamethasone. Functional activity of MERTK promoter constructs was assessed via luciferase reporter activity following treatment with 100 nM dexamethasone. Each construct was co-transfected with a pCMV6-Xl5 vector encoding the full-length human cDNA for the glucocorticoid receptor NR3C1. The commercially available Pathdetect GRE-luc was used as a positive control for dexamethasone response. Relative Luminescence represents ratio of firefly luciferase activity to the co-transfected renilla luciferase control. Data are representative of one independent experiment and are expressed as means±SD. Data were analysed by a one-way ANOVA with post-hoc comparisons between treated and untreated constructs. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DISCUSSION
The MERTK receptor exhibits discrete expression profiles in specific cell types, including the myeloid cells of the innate immune system, where it contributes to homeostatic processes in the CNS. We and others have previously shown that MERTK expression is contextual, subject to environmental influence and genetic variation [22, 23, 48]. Given this background, it is fundamentally important to interrogate the MERTK promoter and identify extrinsic factors that regulate its expression. In the present study, we have systematically characterised the promoter of MERTK, identifying key regions, motifs and regulatory mechanisms that might drive both general and cell specific gene expression. Cloning and analysis of the 5’ upstream region of MERTK in HEK293T cells identified two distinct segments, a proximal (–737/+548) and distal (–3232/–2373) region relative to the TSS as DNA regions potentially capable of driving MERTK transcription. Regulation of mammalian gene transcription is accomplished via the involvement of both TSS-proximal promoter and TSS-distal regulatory elements and our identification of both in the 5’ region of MERTK provided a strong framework for further interrogation [49].
The proximal promoter region defined by MERTK-7 (–737/+548) encapsulates the MERTK TSS and its flanking DNA. In silico analysis of the region indicated that the MERTK TSS is regulated by a CpG rich core promoter with a consensus initiator sequence at the +1 nucleotide. Initiator sequences are prevalent in the human genome and regulate tight control of the TSS and recruitment of RNA Polymerase II [50, 51]. In general, this class of promoters is associated with non-specific transcriptional complexes that drive constitutive gene expression and thus must synergise with upstream regulatory elements to drive tissue-specific expression [52].
Previous analysis of the proximal promoter of mouse Mertk revealed that Sp1 and Sp3 were essential for gene expression in sertoli cells [35]. Members of the SP family encode zinc-finger proteins, defined by conserved C2H2 DNA binding domains that act through GC-box motifs and are closely associated with constitutive expression of genes with GC-rich promoters [53]. Our comparisons showed high conservation of specific SP1/SP3 binding motifs between mouse and human MERTK suggesting that the mechanics governing basal gene expression are similar between species. Furthermore, SP1/SP3 site conservation was also observed between MERTK and its family receptor AXL, which similarly mediates inflammatory and tolerogenic function in innate immune cells [16]. The AXL gene has been previously characterised as containing a CpG rich TATA-less core promoter regulated by the SP family of transcription factors [47]. Sequence alignment revealed low base similarity between the proximal promoters of human MERTK and AXL. However, general enrichment of SP1/SP3 sites in both suggests that regulatory elements in the core promoter of MERTK and AXL are conserved and may drive basal gene expression. Confirmation of SP1/SP3 binding to putative sites in vivo is required to accurately define specific mechanisms of MERTK transcriptional regulation.
Despite a broad co-regulation in innate immune cells, MERTK and AXL segregate into specific temporal niches of expression and immune function [16, 54]. MERTK is predominately expressed by microglia during steady state and promotes continuous cellular turnover and maintenance of a tolerogenic environment [16]. In contrast, AXL expression is more strongly expressed by dendritic cells where its phagocytic activity is stimulated specifically during inflammation [16]. This is supported by the observation that the two receptors exhibit reciprocal inductions to pro- and anti-inflammatory stimuli [16]. In addition, MERTK mediated efferocytosis has a defined role during CNS development driving neurogenesis, synaptic pruning and myelination [55, 56]. There is little evidence to suggest AXL contributes significantly to these processes, further supporting MERTK as being uniquely expressed within the CNS and having specific functional roles.
From an evolutionary perspective, Evans et al. have shown that the MERTK gene has significantly diverged from both its TAM family members AXL and TYRO3, having undergone significant positive selection in the exonic regions that code the signal peptide and transmembrane domains [54]. This study was confined to only the coding domain sequences of the TAM genes. As such, defining non-coding regulatory mechanisms that drive divergence in expression and function between MERTK and AXL is important for understanding combinatorial TAM signaling in health and disease. Here we have observed commonality between the proximal promoters of MERTK and AXL, suggesting that the specific niches of MERTK expression/function are potentially defined by regulatory elements located outside this core region. One potential candidate region for driving cell-specific expression of MERTK is the distal promoter that we identified in this study.
The distal promoter region defined by MERTK-3 (–3232/–2373) exhibited the strongest reporter gene expression, indicative of it being the predominant driver of MERTK transcription. ENCODE ChIP data revealed binding sites for the transcription factors MAX, MAZ, FOXA2 and TEAD4 in K562 cells. MAX and MAZ are constitutively expressed c-MYC associated binding proteins, TEAD4 is a broadly expressed transcriptional activator and FOXA2 is associated with priming regions of open chromatin [41, 57–61]. These transcription factors could therefore bind synergistically to enhance overall MERTK expression under permissive conditions.
ENCODE and TRANSFAC data identified several proteins binding to MERTK-3 that may drive cell/tissue specific expression of MERTK. These analyses collectively revealed a consensus site and observed protein binding for the transcription factor CCAAT/enhancer-binding protein β (C/EBPβ) in a region of DNA exclusive to MERTK-3 in HepG2 cells [41, 62]. C/EBPβ plays a critical role in myeloid cell development, differentiation and proliferation, and is linked to the activation of inflammatory and phagocytic pathways [63–66]. With such a prominent role in immune function, C/EBPβ is a promising candidate for a myeloid specific transcription factor that may be driving high expression of MERTK. Of note, it is also expressed in HEK293 cells [67]. TRANSFAC analysis also predicted the myeloid specific Runt-related transcription factor 1 (RUNX1) in MERTK-3, which could be responsible for enriching myeloid expression of MERTK. Given a strong role for MERTK in immune signaling, IRF and STAT binding sites within MERTK-3 may provide a mechanism for transcriptional modulation in response to inflammatory stimuli [69]. MERTK is also highly expressed in ocular (retinal pigment epithelial cells) and reproductive tissues (sertoli cells) [2, 70]. TRANSFAC analysis predicted binding sites for the testis-determining gene SRY and retinal gene PAX6 within MERTK-3 and suggests that the region may drive cell specific expression domains in a range of tissues.
It is plausible that binding of any one or a combination of these identified proteins could be driving the transcription observed to be enhanced by this region. Surprisingly, while many of these proteins were identified in silico to be binding to the regions that overlapped MERTK-3 and its flanking clones MERTK-2 and MERTK-4, neither of these regions in isolation were observed to strongly induce reporter expression. This suggests that either protein binding is occurring solely in the region specific to MERTK-3 or that expression activity depends on several cis-acting elements working in combination, some of which are not present in either MERTK-2 or MERTK-4. Our interrogation of a minimal response region via smaller deletions within MERTK-3 suggest that the latter is most likely.
Deletion of any 250 bp region within MERTK-3 resulted in a significant reduction in reporter expression, suggesting that several elements across the entire region are required for complete transcriptional activity. No deletion construct completely attenuated reporter expression which may indicate that isolated DNA elements within this region are sufficient to enhance some transcription, with elements working cumulatively to achieve the maximum response seen when assessing MERTK-3 as a whole. This result is most surprising for MERTK3-ΔS1, in which ENCODE ChIP experiments observed no protein binding. However, TRANSFAC analysis did predict putative transcription factor binding in this sub-region, including RUNX1 and PAX6. MERTK3-ΔS4 showed the lowest expression of all deletion constructs, suggesting that the observed transcription factors MAX, MAD, TEAD4, FOXA2 and TAL1 may play a critical role in ‘priming’ the region for transcription via chromatin modification and RNA Pol II recruitment while enabling cell/tissue specific factors such as C/EBPβ to access appropriate binding sites.
In addition to regions which enhanced expression, we also observed two other regions which modestly reduced reporter expression, MERTK-1 (–5187/–3858) and MERTK-5 (–1887/–974) which could therefore contain cis-acting suppressor elements. These elements require a native promoter to inhibit gene expression. The pGL3 enhancer vector contains no endogenous promoter elements and is lowly expressed and is therefore more suited to identifying DNA elements that can promote gene expression. Therefore, a different reporter environment would be more appropriate for formally identifying suppressing elements that may inhibit MERTK expression. This identification is important as suppression elements may assist restriction of MERTK expression to appropriate tissues.
In addition to cell and tissue restriction, MERTK transcription is known to alter in response to external stimuli. In particular, cell-based and genome wide assays have implicated MERTK as a target of GCs with this interaction central to the resolution of inflammatory insults [12, 71–73]. We did not, however, identify a cis-acting mechanism capable of upregulating MERTK expression in response to the synthetic glucocorticoid dexamethasone, which has been shown to enhance MERTK transcription [16, 73]. This suggests that either MERTK is indirectly upregulated by GCs, or that responsive GRE elements are located externally to the assayed 5 kb region. The latter is a strong possibility, as GREs have been reported to enhance target genes from several kilobases upstream and even downstream of the TSS [74]. Alternatively, GC binding could occur in this 5 kb region but is dependent on interactions with other proteins/complexes that are not present in the HEK293T cellular system. Both the proximal and distal MERTK constructs were observed to modestly downregulate luciferase expression following dexamethasone treatment. However, this most likely represents an unexpected interaction with the pGL3 vector backbone or unidentifiable in vitro effect rather than a biologically meaningful result.
Overall, these data suggest that transcription of MERTK is regulated by the combinatorial action of a proximal-core TSS promoter and a distal promoter regulatory element. The core promoter drives constitutive expression and recruitment of RNA Pol II, most likely through binding of general transcription factors such as SP1 and SP3. In contrast, tissue and environmental specific expression is specified by the strongly enhancing distal promoter element, with identification of putative binding sites for tissue specific regulatory elements including myeloid cell specific transcription factors (C/EBPβ, PU.1, RUNX1) and inflammatory transcriptional networks.
Identification of DNA regions capable of modulating MERTK expression and accompanying analysis has enabled a broad definition of the MERTK promoter in the context of in vitro HEK293T-based assays. However, refined in vivo experiments are required to confirm that these interactions occur in biologically relevant cell types/tissues. Precise regulation of MERTK expression is essential for its roles in both development and inflammation. In addition to a prominent role in innate immunity, MERTK is also a critical regulator of CNS plasticity, refining synapses, myelin and sculpting neuronal architecture. MERTK-mediated efferocytosis is central to its function in both contexts and so precise temporal and spatial regulation of its expression is essential for homeostasis. Consequently, dysregulation of MERTK expression is explicitly linked to outcomes of autoimmunity and neurodegeneration. Elucidating the specific genetic mechanisms that drive expression patterns of MERTK furthers our knowledge towards a holistic understanding of its transcriptional regulation and is an important step in informing new approaches to therapeutic intervention.
DATA AVAILABILITY
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
ACKNOWLEDGMENTS
The authors have no acknowledgements.
FUNDING
This work was supported by the National Multiple Sclerosis Society (US) Grant Number 6007629 to M.D.B. and T.J.K. The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. Australian Government Research Training Programme Scholarship to A.D.W. Florey Institute Alan and Elizabeth Finkel Award to AW. John A McKenzie / Selby Scientific Foundation Award to A.D.W. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICT OF INTEREST
The author(s) declare no competing interests.
REFERENCES
[1] | Prieto AL , Weber JL , Lai C . Expression of the receptor protein-tyrosine kinases Tyro-3, Axl, and mer in the developing rat central nervous system. J Comp Neurol. (2000) ;425: :295–314. |
[2] | Lu Q , et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. (1999) ;398: :723–728. doi:10.1038/19554. |
[3] | Lai C , Lemke G . An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron. (1991) ;6: :691–704. |
[4] | Gould WR , et al. Gas6 receptors Axl, Sky and Mer enhance platelet activation and regulate thrombotic responses. J Thromb Haemost. (2005) ;3: :733–741. doi:10.1111/j.1538-7836.2005.01186.x. |
[5] | Melaragno MG , et al. Gas6 inhibits apoptosis in vascular smooth muscle: role of Axl kinase and Akt. J Mol Cell Cardiol. (2004) ;37: :881–887. doi:10.1016/j.yjmcc.2004.06.018. |
[6] | Nagata K , et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. (1996) ;271: :30022–30027. |
[7] | Stitt TN , et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. (1995) ;80: :661–670. |
[8] | Lu Q , Lemke G . 306 (American Society for the Advancement of Science, 2001). |
[9] | Manfioletti G , Brancolini C , Avanzi G , Schneider C . The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol Cell Biol. (1993) ;13: :4976–4985. |
[10] | Scott RS , et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. (2001) ;411: :207–211. |
[11] | Rothlin CV , Ghosh S , Zuniga EI , Oldstone MBA , Lemke G . TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell. 2007;1124. |
[12] | Dransfield I , Michail K , Zagorska A , Lew ED , Lemke G . Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. CELL DEATH & DISEASE. 2015;6. |
[13] | Cohen PL , et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. (2002) ;196: :135–140. |
[14] | Barth ND , et al. Augmentation of Human Monocyte Responses to Lipopolysaccharide by the Protein S and Mer/Tyro3 Receptor Tyrosine Kinase Axis. J Immunol 2018. doi:10.4049/jimmunol.1800249. |
[15] | Endre Károly K , Gábor Z , László F . Role of transmembrane tyrosin kinases in dexamethasone induced enhanced phagocytosis of apoptotic cells. International Journal of Collaborative Research on Internal Medicine & Public Health. (2011) ;3: (3), 222–223. |
[16] | Zagorska A , Traves PG , Lew ED , Dransfield I , Lemke G . Diversification of TAM receptor tyrosine kinase function. Nature Immunology. 2014;920. doi:10.1038/ni.2986. |
[17] | Seitz HM , Camenisch TD , Lemke G , Earp HS , Matsushima GK . Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. Journal Of Immunology (Baltimore, Md.: 1950). (2007) ;178: :5635–5642. |
[18] | Healy LM , et al. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. Journal of Immunology. (2016) ;196: :3375–3384. doi:10.4049/jimmunol.1502562. |
[19] | Zizzo G , Hilliard BA , Cohen PL , Monestier M . Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. JOURNAL OF IMMUNOLOGY. (2012) ;189: :3508–3520. |
[20] | Rothlin CV , Carrera-Silva EA , Bosurgi L , Ghosh S . TAM Receptor Signaling in Immune Homeostasis. Annual Review of Immunology. (2015) ;33: :355–391. doi:10.1146/annurev-immunol-032414-112103. |
[21] | Gal A , et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. (2000) ;26: :270–271. doi:10.1038/81555. |
[22] | Ma GZM , Stankovich J , Kilpatrick TJ , Binder MD , Field J . Polymorphisms in the Receptor Tyrosine Kinase MERTK Gene Are Associated with Multiple Sclerosis Susceptibility. PLoS ONE. (2011) ;6: :1. |
[23] | Binder MD , et al. Common and Low Frequency Variants in MERTK Are Independently Associated with Multiple Sclerosis Susceptibility with Discordant Association Dependent upon HLA-DRB1*15:01 Status. PLoS Genetics. 2016. doi:10.1371/journal.pgen.1005853. |
[24] | Cai B , et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. (2020) ;31: :406–421.e407, doi:10.1016/j.cmet.2019.11.013. |
[25] | Wu YM , Robinson DR , Kung HJ . Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res. (2004) ;64: :7311–7320. doi:10.1158/0008-5472.CAN-04-0972. |
[26] | Linger RM , et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene. (2013) ;32: :3420–3431. doi:10.1038/onc.2012.355. |
[27] | Schlegel J , et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J Clin Invest. (2013) ;123: :2257–2267. doi:10.1172/JCI67816. |
[28] | Bhattacharyya S , et al. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe. (2013) ;14: :136–147. doi:10.1016/j.chom.2013.07.005. |
[29] | Paolicelli RC , et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) ;333: :1456–1458. doi:10.1126/science.1202529. |
[30] | Schafer DP , et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) ;74: :691–705. doi:10.1016/j.neuron.2012.03.026. |
[31] | Healy LM , et al. MerTK-mediated regulation of myelin phagocytosis by macrophages generated from patients with MS. Neurology(R) Neuroimmunology & Neuroinflammation. (2017) ;4: :e402–e402. doi:10.1212/NXI.0000000000000402. |
[32] | Behrens EM , et al. The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur J Immunol. (2003) ;33: :2160–2167. doi:10.1002/eji.200324076. |
[33] | Zhang Y , et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. JOURNAL OF NEUROSCIENCE. (2014) ;34: :11929–11947. |
[34] | Lemke G , Rothlin CV . Immunobiology of the TAM receptors. Nat Rev Immunol. (2008) ;8: :327–336. doi:10.1038/nri2303. |
[35] | Wong CCS , Lee WM . The proximal cis-acting elements Sp1, Sp3 and E2F regulate mouse mer gene transcription in Sertoli cells. European Journal Of Biochemistry. (2002) ;269: :3789–3800. |
[36] | Aisleen M , Sylwia M , Ian D , Adriano GR . Effects of Glucocorticoids on Apoptosis and Clearance of Apoptotic Cells. The Scientific World Journal. (2007) ;7: :1165–1181. 1165, doi:10.1100/tsw.2007.224. |
[37] | Heasman SJ , et al. Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. The Journal Of Endocrinology. (2003) ;178: :29–36. |
[38] | McColl A , et al. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. Journal Of Immunology (Baltimore, Md.: 1950). (2009) ;183: :2167–2175. doi: 10.4049/jimmunol.0803503. |
[39] | La Baer J , Yamamoto KR . Analysis of the DNA-binding affinity, sequence specificity and context dependence of the glucocorticoid receptor zinc finger region. Journal Of Molecular Biology. (1994) ;239: :664–688. |
[40] | Chandler VL , Maler BA , Yamamoto KR . DNA sequences bound specifically by glucocorticoid receptor in vitro render a heterologous promoter hormone responsive in vivo. Cell. (1983) ;33: :489–499. |
[41] | The Encode Project Consortium, a. The ENCODE (ENCyclopedia of DNA Elements) Project. Science. 2004;636. |
[42] | Sandelin A , Alkema W , Engstrom P , Wasserman WW , Lenhard B . JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. (2004) ;32: :D91–94. doi:10.1093/nar/gkh012. |
[43] | Matys V , et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. (2006) ;34: :108–D110. doi:10.1093/nar/gkj143. |
[44] | Chojnacki S , Cowley A , Lee J , Foix A , Lopez R . Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res. (2017) ;45: :W550–W553. doi:10.1093/nar/gkx273. |
[45] | Kim TH , et al. A high-resolution map of active promoters in the human genome. Nature. (2005) ;436: :876–880. doi: 10.1038/nature03877. |
[46] | Cross SH , Bird AP . CpG islands and genes. Curr Opin Genet Dev. (1995) ;5: :309–314. |
[47] | Mudduluru G , Allgayer H . The human receptor tyrosine kinase Axl gene-promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. BIOSCIENCE REPORTS. (2008) ;28: :161–176. |
[48] | Fourgeaud L , et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016;240. |
[49] | Kim TK , Shiekhattar R . Architectural and Functional Commonalities between Enhancers and Promoters. Cell. (2015) ;162: :948–959. doi:10.1016/j.cell.2015.08.008. |
[50] | Javahery R , Khachi A , Lo K , Zenzie-Gregory B , Smale ST . DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol Cell Biol. (1994) ;14: :116–127. |
[51] | Yang C , Bolotin E , Jiang T , Sladek FM , Martinez E . Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene. (2007) ;389: :52–65. doi:10.1016/j.gene.2006.09.029. |
[52] | Schug J , et al. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. (2005) ;6: :R33. doi:10.1186/gb-2005-6-4-r33. |
[53] | Suske G . The Sp-family of transcription factors. Gene. (1999) ;238: :291–300. |
[54] | Evans AL , et al. Antagonistic Coevolution of MER Tyrosine Kinase Expression and Function. Molecular Biology And Evolution. (2017) ;34: :1613–1628. doi:10.1093/molbev/msx102. |
[55] | Hagemeyer N , et al. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathologica. (2017) ;134: :441. |
[56] | Chung WS , et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. (2013) ;504: :394–400. doi:10.1038/nature12776. |
[57] | Bossone SA , Asselin C , Patel AJ , Marcu KB . MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proceedings of the National Academy of Sciences of the United States. 1992;7452. |
[58] | Grandori C , Cowley SM , James LP , Eisenman RN . The Myc/Max/Mad network and the transcriptional control of cell behavior. Annual Review of Cell and Developmental Biology. (2000) ;16: :653–699. |
[59] | Kyrmizi I , et al. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes & Development. (2006) ;20: :2293–2305. |
[60] | Porcher C , Chagraoui H , Kristiansen MS . SCL/TAL a multifaceted regulator from blood development to disease. Blood. (2017) ;129: :2051–2060. doi:10.1182/blood-2016-12-754051. |
[61] | Shi Z , et al. DNA-binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene. (2017) ;36: :4362–4369. doi:10.1038/onc.2017.24. |
[62] | Osada S , Yamamoto H , Nishihara T , Imagawa M . DNA binding specificity of the CCAAT/enhancer-binding protein transcription factor family. The Journal Of Biological Chemistry. (1996) ;271: :3891–3896. |
[63] | Landschulz WH , Johnson PF , Adashi EY , Graves BJ , McKnight SL . Isolation of a recombinant copy of the gene encoding C/EBP. Genes & Development. (1988) ;2: :786–800. |
[64] | Ramji D , Foka P . CCAAT/enhancer-binding proteins: structure, function and regulation. Biochemical Journal. (2002) ;365: :561. |
[65] | Huber R , Pietsch D , Panterodt T , Brand K . Regulation of C/EBPβ and resulting functions in cells of the monocytic lineage. Cellular Signalling. (2012) ;24: :1287–1296. doi:10.1016/j.cellsig.2012.02.007. |
[66] | Tsai VW , et al. CCAAT/enhancer binding protein-delta expression by dendritic cells regulates CNS autoimmune inflammatory disease. J Neurosci. (2011) ;31: :17612–17621. doi:10.1523/JNEUROSCI.3449-11.2011. |
[67] | Uhlén M , et al. Proteomics. Tissue-based map of the human proteome. Science (New York, N.Y.). (2015) ;347: :1260419–1260419. doi:10.1126/science.1260419. |
[68] | Speck NA , Gilliland DG . Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. (2002) ;2: :502–513. doi:10.1038/nrc840. |
[69] | Chiang HS , Liu HM . The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Front Immunol. (2018) ;9: :3086. doi:10.3389/fimmu.2018.03086. |
[70] | Prasad D , et al. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. (2006) ;33: :96–108. doi:10.1016/j.mcn.2006.06.011. |
[71] | Carter BS , Meng F , Thompson RC . Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiol Genomics. (2012) ;44: :1188–1200. doi:10.1152/physiolgenomics.00097.2012. |
[72] | Garabuczi E , Szondy Z , Sarang Z . Glucocorticoids enhance prolonged clearance of apoptotic cells by upregulating liver X receptor, peroxisome proliferator-activated receptor-delta and UCP2. BIOCHIMICA ET BIOPHYSICA ACTA-MOLECULAR CELL RESEARCH. (2015) ;1853: :573–582. |
[73] | Zahuczky G , Kristóf E , Majai G , Fésüs L . Differentiation and Glucocorticoid Regulated Apopto- Phagocytic Gene Expression Patterns in Human Macrophages. Role of Mertk in Enhanced Phagocytosis. PLoS ONE. (2011) ;6: :1–10. doi:10.1371/journal.pone.0021349. |
[74] | Yick-Lun So A , Chaivorapol C , Bolton EC , Hao L , Yamamoto KR . Determinants of Cell- and Gene-Specific Transcriptional Regulation by the Glucocorticoid Receptor. PLoS Genetics. (2007) ;3: :e94–0938. doi: 10.1371/journal.pgen.0030094. |

