
Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair
Abstract
Background:
Microglia are the resident macrophages of the central nervous system (CNS). In multiple sclerosis (MS) and related experimental models, microglia have either a pro-inflammatory or a pro-regenerative/pro-remyelinating function. Inhibition of Bruton’s tyrosine kinase (BTK), a member of the Tec family of kinases, has been shown to block differentiation of pro-inflammatory macrophages in response to granulocyte–macrophage colony-stimulating factor in vitro. However, the role of BTK in the CNS is unknown.
Methods:
Our aim was to investigate the effect of BTK inhibition on myelin repair in ex vivo and in vivo experimental models of demyelination and remyelination. The remyelination effect of a BTK inhibitor (BTKi; BTKi-1) was then investigated in LPC-induced demyelinated cerebellar organotypic slice cultures and metronidazole-induced demyelinated Xenopus MBP-GFP-NTR transgenic tadpoles.
Results:
Cellular detection of BTK and its activated form BTK-phospho-Y223 (p-BTK) was determined by immunohistochemistry in organotypic cerebellar slice cultures, before and after lysophosphatidylcholine (LPC)-induced demyelination. A low BTK signal detected by immunolabeling under normal conditions in cerebellar slices was in sharp contrast to an 8.5-fold increase in the number of BTK-positive cells observed in LPC-demyelinated slice cultures. Under both conditions, approximately 75% of cells expressing BTK and p-BTK were microglia and 25% were astrocytes. Compared with spontaneous recovery, treatment of demyelinated slice cultures and MTZ-demyelinated transgenic tadpoles with BTKi resulted in at least a 1.7-fold improvement of remyelination.
Conclusion:
Our data demonstrate that BTK inhibition is a promising therapeutic strategy for myelin repair.
INTRODUCTION
Bruton’s tyrosine kinase (BTK) is a member of the Tec family of non-receptor tyrosine kinases composed of five members, namely Tec, Btk, Itk/Emt/Tsk, Bmx, and Txk/Rlk [1]. BTK is expressed in cells of hematopoietic origin, including B cells, myeloid cells and platelets, but not T or NK cells [2, 3]. The function and role of BTK inhibition in human B cell development was demonstrated by its association with X-linked agammaglobulinemia, manifested by substantial reduction in B cells and immunoglobulins. BTK is important for B lymphocyte proliferation, maturation and antigen presentation.
In multiple sclerosis (MS), depletion of B cells with monoclonal antibodies targeting CD20, a 33-37 kDa trans-membrane protein [4, 5], reduces disease activity in relapsing-remitting MS (RRMS) patients [6]. Thus, inhibiting B cell function using a small molecule inhibitor of BTK may be a potential treatment for MS. Indeed, the BTK inhibitor evobrutinib recently met its primary endpoint in a phase 2 trial in MS patients [7].
In addition to its effects on B cells, BTK mediates signaling in monocytes and mast cells downstream of various receptors including Fc, integrin, Toll-like and chemokine receptors [8]. In rheumatoid arthritis (RA) and psoriatic arthritis, BTK expression was shown in monocytes, macrophages, and mast cells, in addition to B cells. Studies using X-linked agammaglobulinemia innate immune cells show that BTK is expressed in myeloid cell populations including monocytes and macrophages [9]. In myeloid cells, BTK was shown to function downstream of Fc-gamma, Fc-epsilon or TLR receptors [10]. Other studies indicate a role for BTK in migration of both B cells and myeloid cells downstream of integrin and chemokine receptors [11]. These data are based on genetic deficiency of BTK. Using pharmacologic inhibitors of BTK kinase activity, it was confirmed that BTK inhibition significantly inhibited macrophage IL-6 production induced by Fc receptor and CD40 ligation, as well as CD40L induced IL-8, TNF, and MMP production by RA synovial explants [12]. Both ibrutinib and CC292, which were used in the above-mentioned studies, inhibit numerous targets besides BTK while in contrast, evobrutinib as well as BTKi-1, the BTKi used here, are highly selective. Evobrutinib, strongly inhibits only 2 kinases (BTK and BMX) out of a panel of 267 kinases. Evobrutinib has been shown to increase the capacity of human pro-regenerative macrophages (M2-type) to take up apoptotic cells (unpublished data) and showed efficacy in preclinical models of lupus and RA [13]. Collectively, these results establish a crucial role for BTK in B cell function and antibody-mediated innate immune activation, and, therefore, in antibody-mediated autoimmune disease.
In contrast, little is known about the importance of BTK in myeloid cells in neurological diseases. Ibrutinib, a clinical BTK inhibitor used in cancer indications [3], inhibits maturation of IL-1ß in infiltrating macrophages and neutrophils in the infarcted area of ischemic brain [14]. Such involvement of BTK in NLRP3 (NOD-like receptor family, pyrin domain containing 3)-mediated inflammasome activation after ischemic brain injury might be of interest for other neurodegenerative or neuroinflammatory diseases like MS. Along this line, it has been reported that a mouse mutant lacking Btk, showed reduced inflammatory responses in experimental autoimmune encephalomyelitis (EAE)[15]. Evobrutinib was tested in rodent experimental autoimmune encephalomyelitis (EAE) models and showed profound efficacy, decreasing clinical score after disease induced with PLP 139–151 or MOG 35–55 peptide in SJL and C57/BL6 mouse strains [Boschert et al. ECTRIMS 2017. P678]. Of note, this efficacy was not mediated by B cell depletion as those models depend on T cell-mediated autoimmunity. The fact that BTK mediates signaling in myeloid cells and non-B cell dependent EAE suggests a benefit of BTK inhibitors beyond B cell inhibition, differentiating the BTK target from B cell depleting agents alone.
Our working hypothesis was therefore to explore the possibility that inhibition of BTK plays a role in myelin repair independently of any lymphocyte driven beneficial effect and raise the question of mode of action of BTK inhibitors in demyelination and remyelination. Is the beneficial effect mediated by a direct action on oligodendrocytes, or an indirect effect mediated by microglial cells or astrocytes, or both? Our first aim was to investigate the cellular pattern of expression of BTK in the mouse CNS, which was previously unknown. We then investigated the effect of BTKi-1, a highly specific BTK inhibitor on remyelination using an ex-vivo model involving mouse cerebellar slices, in which the remyelination process could be monitored after lysolecithin-induced demyelination. The third aim was to complement our ex vivo investigations by using an in vivo conditional demyelination model developed in Xenopus laevis—the transgenic MBP-GFP-NTR line — which has been shown to be well adapted to screen pro-remyelinating compounds [16].
MATERIALS & METHODS
Animals
Mice, either wild type (C57/Bl6) or our PLP-GFP transgenic line [17] were raised in our animal facility (agreement # A75-13-19). In the PLP-GFP transgenic line, oligodendrocytes were identified by the expression of the green fluorescent reporter. In addition, due to the presence in the construct of the PLP peptide signal upstream to the green fluorescent protein (GFP) coding sequence, the reporter is also inserted in the myelin sheath [18].
To test in vivo the effect of BTK inhibition on remyelination we used a conditional demyelination model, MBP-GFP-NTR, developed in Xenopus laevis, in which myelinating oligodendrocytes selectively express both the GFP reporter and the bacterial nitroreductase (NTR) enzyme, under the control of a portion of mouse myelin basic protein (MBP) regulatory sequence [19]. The NTR enzyme converts the nitro radical of prodrugs, such as metronidazole (MTZ), to a highly cytotoxic hydroxylamine derivative; therefore, introduction of MTZ into the swimming water of transgenic MBP-GFP-NTR tadpoles provokes an oligodendrocyte cell-death followed by demyelination [19]. After withdrawal of the demyelinating agent (MTZ), spontaneous remyelination occurs and this can be accelerated by addition of molecules such as retinoic acid, siponimod or clemastine [16, 20]. Xenopus tadpoles were staged according to Nieuwkoop and Faber (1994) [21]. Transgenic tadpoles were treated between stages NF 50 and 55, corresponding to pre-metamorphosis, a stage of ongoing myelination. Tadpoles of either sex were anesthetized in 0.05% MS-222 (ethyl-3-aminobenzoate methanesulfonate; Sigma-Aldrich) before quantification of GFP+ cells and returned to normal water to recover. Before brain and spinal cord dissection, tadpoles were euthanized in 0.5% MS-222. Animal care was in accordance with institutional and national guidelines. All animal procedures conformed to the European Community Council 1986 directive (86/609/EEC) as modified in 2010 (2010/603/UE) and have been approved by the ethical committee of the French Ministry of Higher Education and Research (APAFIS#5842-2016101312021965).
Preparation of BTKi
Stock solution (100 mM) of BTKi-1 (Merck KGaA, Darmstadt, Germany) was obtained by dissolving 200 mg in 4.49 mL of DMSO (Dimethyl sulfoxide D8418-50 ml Sigma-Aldrich). Further dilutions were either in culture medium for mouse cerebellar slices or water for Xenopus experiments.
Antibodies
Anti-BTK antibody was from BD Transduction Laboratories (Mouse Mab IgG2a, Ref 611117)(diluted 1/100) and the anti-p-BTK (p-TYR223) was from Novusbio (Rabbit polyclonal, NBP1-78295)(diluted 1/100). For double or triple immune labelling and secondary antibodies the following antibodies were used: Mab CC1 diluted 1/100 (mouse IgG1 anti-APC) was from Millipore (Ref 0p80); chicken anti-MBP Ab (diluted 1/500) was from Millipore (Ref AB9348); Mouse Mab IgG1 anti-S100ß clone 2720 was from Sigma (Ref. AMAB91038, diluted 1/100). Mouse microglial cells were labelled either with anti-IBa1 rabbit polyclonal antibodies (diluted 1/500) purchased from Abcam (Ref Ab5068), or with a cocktail of rat IgG anti F4-80(1/100), anti-CD11b (1/400) and anti-CD68 (1/400) from Biorad (ref MCA497, MCA74G,MCA1957). Goat anti-Mouse IgG2a Alexa Fluor 488 conjugated (green A21131) or 594 (red A21135), goat anti-rabbit IgG Alexa Fluor 488 conjugated (green A11034) or 594 (red A-11012), goat anti-chicken Ab Alexa Fluor 594 (red A-11042) were from InVitrogen (ThermoFisher Scientific, France) and all were used at a dilution of1/1000.
Isolectin B4 staining
Xenopus microglial cells were stained with Bandeiraea Simplicifolia isolectin B4 (IB4) on fixed tissue sections or whole tadpole brain and spinal cord prepared as above. Tissue sections were incubated overnight at 4°C with Alexa Fluor 647-conjugated IB4 (1 : 1000, Invitrogen) in PBT 0.1% supplemented with 1mM calcium, and then washed in PBT. For whole-mount staining, samples were incubated for 48 h with gentle shaking. When combined with immuno labeling, tissue sections or whole samples were incubated with fluorescent IB4 and washed before incubation in blocking solution and exposure to antibodies, as described above. Images were taken with Apotome microscope Zeiss 20X or 40X.
Mouse cerebellar slice preparation
P8–P10 mouse pups were euthanized and their brains dissected into ice-cold Hank’s Balanced Salt Solution. Sagittal slices of cerebellum (200–300 μm thick) were cut using a McIlwain tissue chopper [22, 23]. The slices were placed on Millipore Millicell-CM organotypic culture inserts in medium containing 50% MEM with Earle’s salts, 25% Earle’s Balanced Salt Solution, 25% heat-inactivated normal goat serum, glutamax-II supplement with penicillin–streptomycin, amphotericin B and 6.5 mg/ml glucose. Culture medium was changed every other day. After 6 days in culture, demyelination was induced by addition of 0.5 mg/ml lysophosphatidylcholine (lysolecithin, LPC, Sigma) to the medium for 16–17 h. Then slices were transferred back to normal medium. At DIV9 (the peak of demyelination), BTKi (1 μM) (BTKi-1) was added for 2 days and medium + BTKi was renewed at DIV11.
Quantification of myelin on mouse cerebellar slice preparation
Quantification of spontaneous remyelination (control) was compared to remyelination in the presence of BTKi at DIV13. Immunostainings were performed for PLP (myelin), Caspr (paranodes) and Calbindin (neuron) at DIV13 in order to quantify the number of myelinated segments and intact paranodal structures. Images were acquired using a Zeiss Axio-Imager-Apotome. Series of Z-sections were performed at 0.5 μm increment. Maximum orthogonal projection of images was carried out using Fiji software (NIH, Bethesda, Maryland). To analyze PLP expression (Fig. 1), images were quantified for labeled area fraction (expressed in %) by automated counting using Image J software. Briefly, images were normalized by subtracting background and an automatic thresholding (MaxEntropy) was applied. Mean values of area fraction (expressed in %) were obtained from images (n = 27) of cerebellum slices treated with LPC (n = 13) vs control (n = 14).
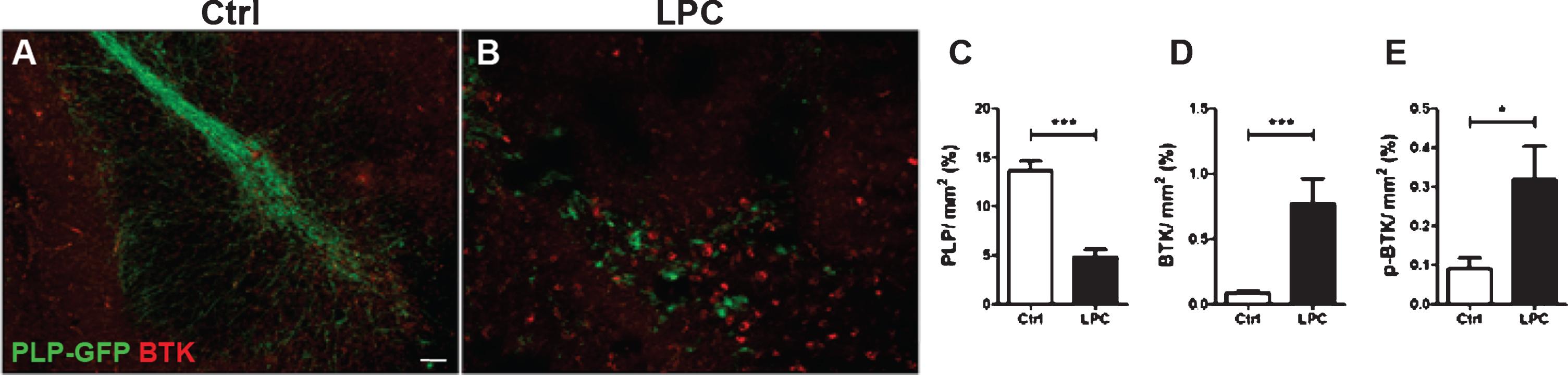
Fig. 1
Increased BTK signal upon demyelination. Cerebellar organotypic slices from P9 Plp-GFP mouse (green oligodendrocytes and myelin) were maintained in culture (A) or subjected to LPC treatment at 6 days in vitro (DIV) for 16–17 h (B) before being immunostained for BTK (red) or p-BTK at DIV9. LPC treatment induced a demyelination (loss of GFP signal B, C) and a concomitant 8.5-fold increase in BTK signal (B, D). Similarly, p-BTK was also increased after LPC demyelination (E). (*p < 0.01; ***p < 0.0001). Scale bar = 100 μm.
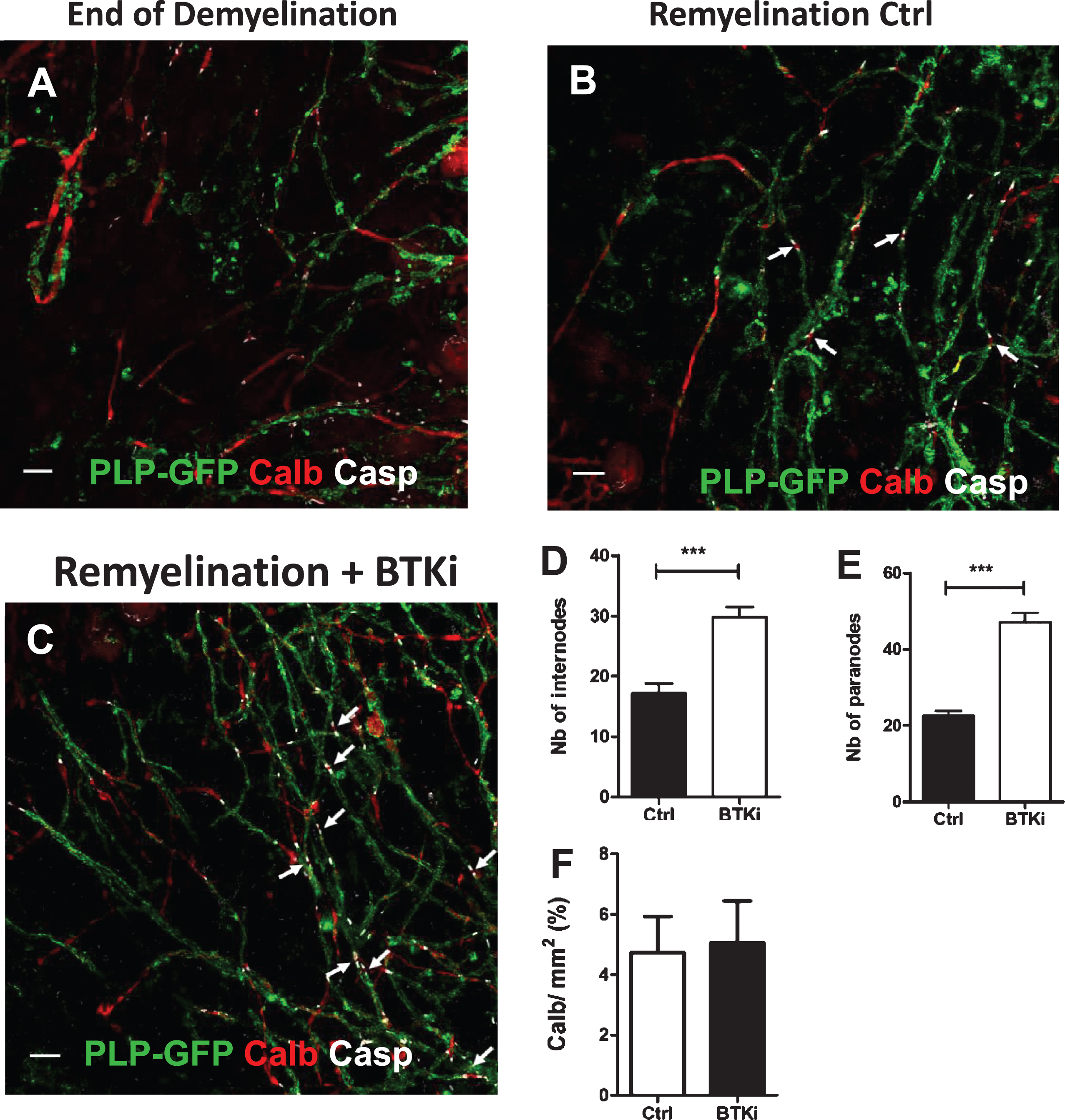
Using the same method of quantification, we analyzed the calbindin expression (Fig. 3). Mean values of Calb+ area fraction (expressed in %) were obtained from images (n = 8) of cerebellum slices treated with BTKi (n = 4) vs control (n = 4) and report that the axon density is the same in BTKi treated conditions compared to control conditions (Fig. 3F). To assess the remyelination in cerebellar organotypic slices after BTKi treatment, images were acquired using an Olympus FV-1200 Upright Confocal Microscope. Z-series were performed at 0.3 μm increment, and green, red, and far-red fluorescence were acquired sequentially. Z-projections of images were carried out using Fiji software (NIH, Bethesda, Maryland). Images were taken from 5 slices per condition, 4–6 images per slice. At least a coverage of 60–250 internodes per slice, identified by PLP+ myelinated segments were counted (Fig. 3D), and the number of paranodes was determined by counting the number of Caspr+ structures for at least a coverage of 105–380 paranodes per slice (Fig. 3E). Results were expressed as mean±SEM of at least three independent experiments.
Immunolabelingof Xenopus tadpole brain sections
Tadpoles were fixed by immersion in 4% paraformaldehyde; fixation conditions varied according to the antibody used (see above). Fixed brains were rinsed in PBS (1×) and cryoprotected in sucrose (20% in PBS). Cryoprotected brains were embedded in OCT® (Tissue Tek). Cryosections (16 μm thick) were blocked in normal goat serum (10% in PBS) containing 0.1% Triton X-100 and incubated overnight at 4°C with primary antibodies. Slides were rinsed in PBT 0.1% and secondary antibody added. The slides were mounted with DAPI coated Fluoromount® (Sigma-Aldrich).
Metronidazole preparation and use
Metronidazole (MTZ, Fluka) was dissolved in filtered tap water containing 0.1% DMSO (Sigma Aldrich). MTZ was used at concentration of 10 mM with an exposure length of 10 days. Transgenic or non-transgenic sibling tadpoles were maintained in 600 ml of MTZ solution (maximum 10 tadpoles/600 ml) at 20°C in complete darkness (MTZ is light-sensitive) and the solution was changed daily throughout the duration of treatment. For regeneration experiments, MTZ-exposed animals were allowed to recover for 3 days in either normal water (control) or water containing BTKi (BTKi-1) at increasing concentrations in ambient laboratory lighting (12 h light/12 h dark).
Quantification of GFP+ cells
GFP was detected directly by fluorescence in live Xenopus embryos using an AZ100 Nikon Multizoom Macro-Microscope. The total number of GFP+ cells was counted in the optic nerve, from the emergence of the nerve (i.e., after the chiasm) to the retinal end. Of note, at stage 50 the number of myelinating oligodendrocytes in the optic nerve is very reproducible: mean value 18.5 ± 1. GFP+ cells were counted before (D0) and at the end of MTZ exposure (D10) and after 3 days of BTKi treatment (R3) at the concentration to be tested on the same embryos. Double-blind counts were performed independently by two researchers. Data were compared to control untreated animals of the same developmental stage. Data presented are the mean±SEM of number of GFP+ cells counted on at least 6 tadpoles per condition.
Statistical analysis
We used Prism v5 software for statistical analyses. Results were expressed as means±SEM. For organotypic slices experiments data obtained following BTK inhibition were compared to control using an unpaired two-tailed t-test. Data obtained in the Xenopus were compared using Student’s t-test. Statistical significance was set at p < 0.05.
RESULTS
BTK and p-BTK expression increased after demyelination
BTK and its phosphorylated activated form BTK-phospho-Y223 (p-BTK) were examined by immuno labeling of mouse PLP-GFP cerebellar slices prepared from P9 animals and maintained in culture for 6 days. Cultures were then demyelinated with LPC for 17 h before being immuno stained at DIV9with anti-BTK or anti-p-BTK antibodies. Treatment with LPC resulted in a dramatic demyelination as measured by the 3-fold decrease in the intensity of GFP fluorescence compare to control slices (Fig. 1A, B, C) and a concomitant 8.5-fold increase in BTK expression (Fig. 1A, B, D). Demyelination also resulted in a 3-fold increase in the level of p-BTK signaling (Fig. 1E).
Cellular expression of BTK and p-BTK
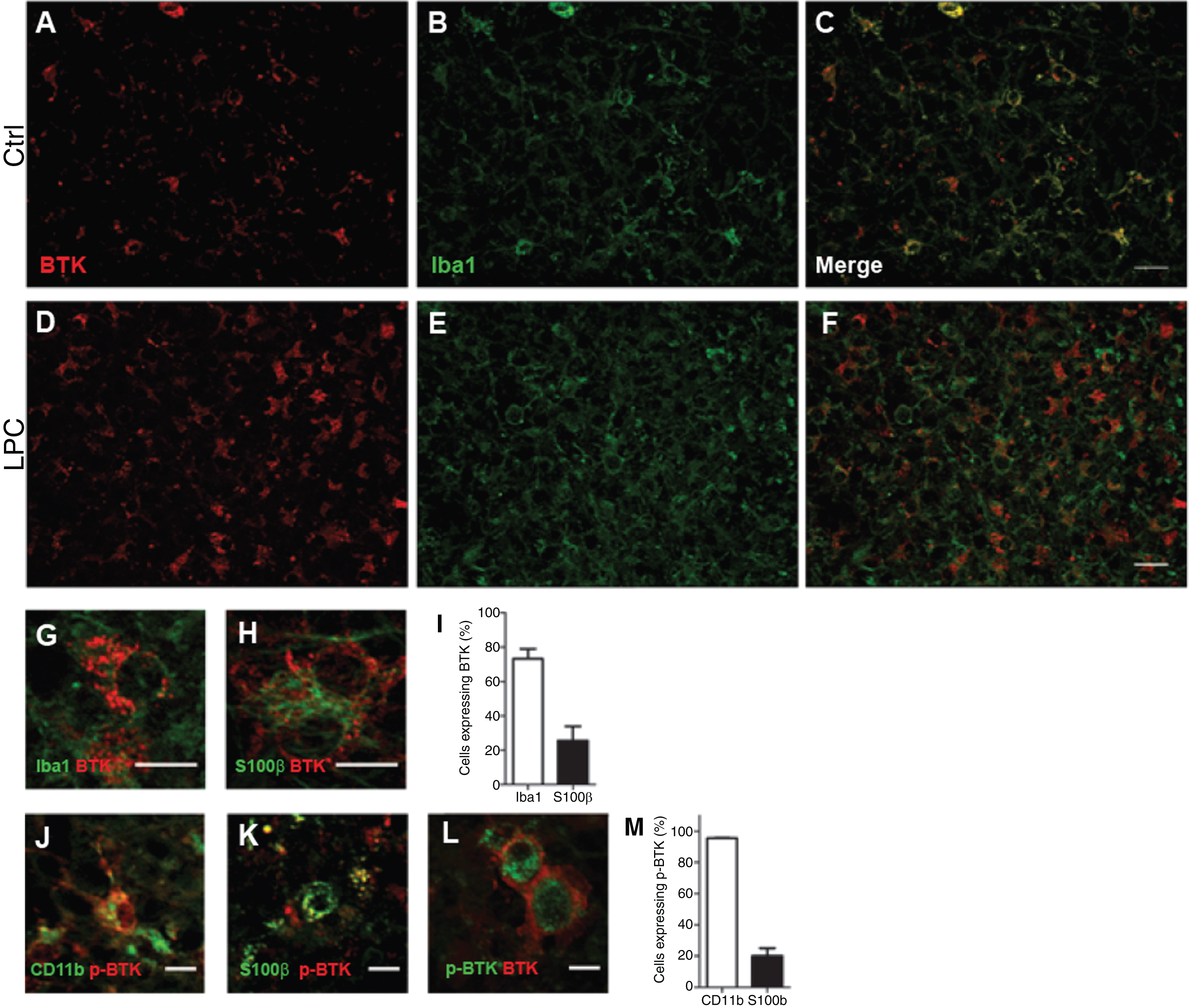
We then investigated the identity of cells expressing BTK and p-BTK (Fig. 2). Double-labeling experiments with anti-BTK and cell-specific markers showed that BTK-positive cells were rarely detected under control conditions (Fig. 2A–C). After LPC-induced demyelination, BTK was observed mainly in Iba1+ microglia (73.1%), and in 25.6% of S100ß+ astrocytes (Fig. 2 D–I). BTK was not detected in CC1+ oligodendrocytes, NeuN+ neurons, or Olig2+/CC1– oligodendrocyte precursor cells (Supplementary Figure 1). After LPC-induced demyelination of mouse cerebellar organotypic slices there was a substantial increase of p-BTK positive signal compared with control conditions; p-BTK was detected only in microglia (75.6%) and astrocytes (20.6%) (Fig. 2 J, K, M). Of note, 74.9% of BTK+ microglia were also p-BTK+ (Fig. 2L).
Fig. 2
Detection of BTK in microglia and astrocytes. Cerebellar organotypic slices from P9 mouse, were maintained in culture and submitted to LPC treatment at DIV6 for 16–17 h. At DIV9 (i.e., peak of demyelination) immuno staining was performed for BTK (Red) and Iba1 (microglia, green). A–F: Small magnification of control (A–C), and after LPC-induced demyelination (D–F). G,H,J, and K: Higher magnification illustrating co-localisation of BTK and p-BTK in microglia labeled with Iba1 (G) or CD11-F4/80-CD68 (J) and astrocyte S100ß+ (H, K).L: An example of two cells co-expressing BTK and p-BTK. Note that BTK is more at the surface while p-BTK is more intracytoplasmic. I and M: Quantification of microglia and astrocytes expressing BTK (I) or p-BTK (M). Scale bars: A–F = 20 μm; G,H, J,K, and L = 10 μm.
Remyelination in cerebellar organotypic slices after BTKi treatment
Cerebellar organotypic slices from P9 mice were maintained in culture and submitted to LPC treatment at DIV6 for 16–17 h. At DIV9 (i.e., peak of demyelination) (Fig. 3A), slices were treated or not (control) with BTKi (1 μM) before being triple-labeled at DIV13 for PLP, Caspr and Calbindin. Remyelination was quantified by counting the number of myelinated internodes labeled with the anti-PLP mAb and also the number of Caspr+ paranodal structures in the spontaneous remyelinating control and the BTKi treated cerebellar slices (Fig. 3B,C). At Day 4, BTKi treatment of demyelinated cerebellar slices significantly promoted remyelination as evaluated either by the number of myelinated internodes (1.7-fold increase) or the number of paranodal structures (2.1-fold increase) (Fig. 3D, E). Of note, compared to control condition, addition of BTKi in the demyelinated culture did not change the axonal density as measured by the intensity of Calbindin labeling (Fig. 3F).
Fig. 3
Inhibition of BTK favors remyelination. A–C: Cerebellar organotypic slices from P9 mouse, were exposed to LPC at DIV6 for 16–17 h. At DIV 9, i.e., peak of demyelination, myelinated internodes were no longer observed and only PLP + myelin debris were detected A). At the end of LPC-induced demyelination, cultures were treated with BTKi (BTKi-1) (1 μM) from DIV 9 before triple labeling with anti-PLP (myelinated internodes, green), anti-Caspr (paranodes, white) and anti-Calbindin (axons, red) antibodies. Small white arrows in control (Remyelinated-Ctrl B) and BTKi treated (Remyelinated-BTKi C) cultures point to some complete nodal structures with the Caspr+ paranode on each side of the Calbindin+ naked axon. D–F: Remyelination was evaluated by counting either PLP+ myelinated internodes (D) or Caspr+ paranodal structures (E) and axonal density as measured by the intensity of Calbindin labeling (F) (see M & M section for details). Scale bar = 10 μm
Cellular expression of BTK in Xenopus brain
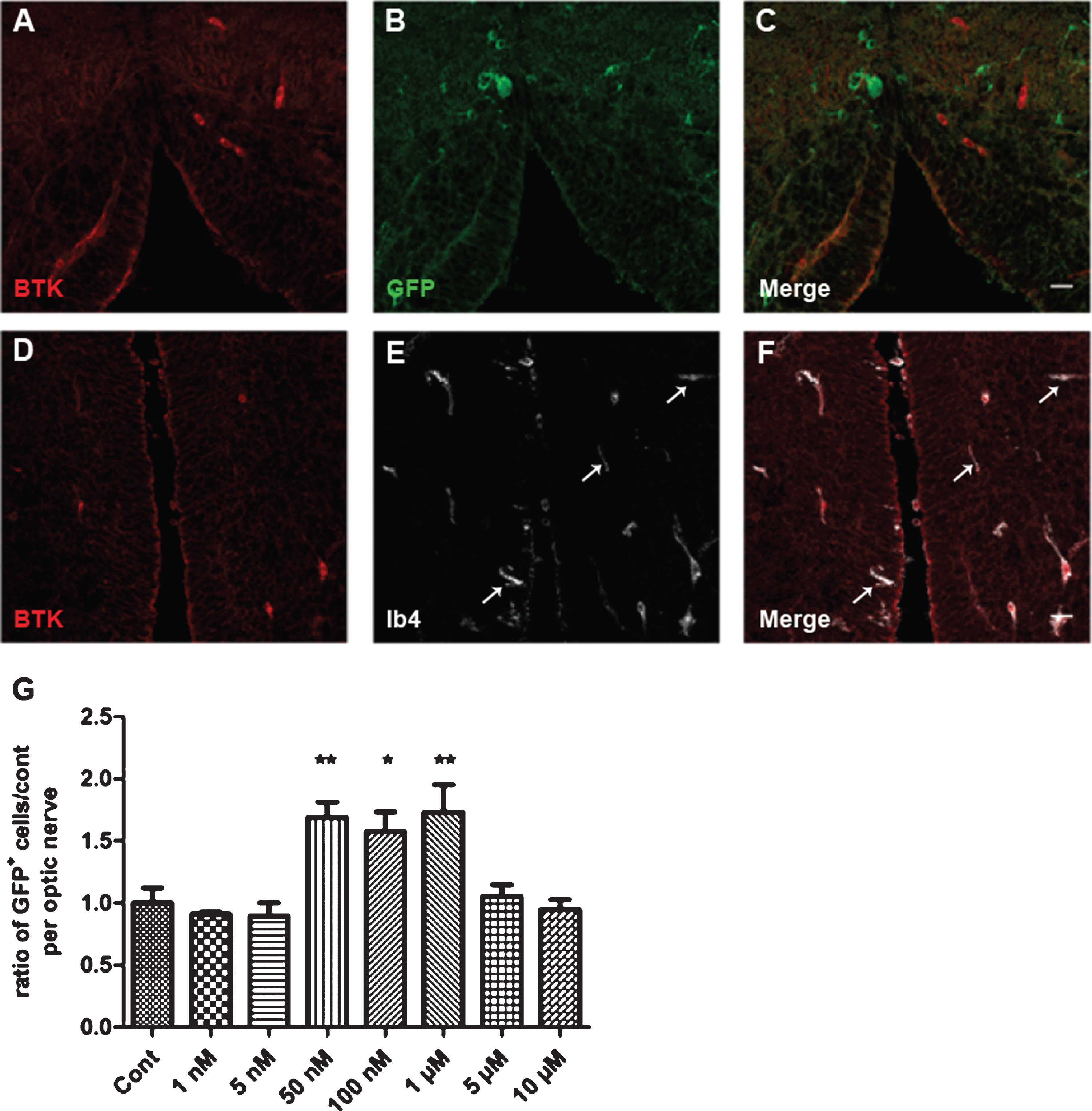
To determine the cellular expression of BTK in the Xenopus brain, tissue sections across the brain stem of transgenic MBP-GFP-NTR Xenopus tadpoles (stage 52–53) were immuno stained with anti-BTK antibodies, and either anti-GFP antibodies or pre-labeled with fluorescent-conjugated isolectin IB4. Co-immunostaining with anti-GFP showed a complete exclusion of BTK cells with GFP+ oligodendrocytes (Fig. 4 A–C). In contrast, BTK was detected only in IB4 microglial cells. Of note, while all BTK+ cells were microglial, a few IB4 microglia were BTK-(Fig. 4D–F).
Fig. 4
Cellular expression of BTK in Xenopus and dose–response of remyelination by BTKi (BTKi-1). A–F: Immunodetection of BTK in microglial cells, but not in oligodendrocytes. Coronal (A–C) or horizontal (D–F) tissue sections across the brain stem of stage 52–53 MBP-GFP-NTR tadpoles double-labeled with anti-BTK (A, C, D, F, red) and anti-GFP (B, C, oligodendrocytes, green) antibodies or isolectin IB4 (E, F, microglia, white). Note in A–C the complete exclusion of the two labels, illustrating absence of BTK in oligodendrocytes. (D–F) In contrast, all BTK+ cells were also IB4+. However, some IB4+ cells (white arrows) were not BTK+. G: Demyelination of stage 52–53 MBP-GFP-NTR tadpoles was achieved by 10 days exposure to metronidazole (10 mM) in the swimming water. Tadpoles were then returned to normal water or water containing increasing concentration of BTKi (BTKi-1) for 3 days. Remyelination was assayed by counting the number of GFP+ cells per optic nerve on day 3 of the repair period. Treatment of tadpoles with BTKi (BTKi-1) at concentrations ranging between 50 nM and 1 μM improved remyelination up to 1.7-foldcompared to spontaneous recovery (control) set as 1. (*p < 0.01; **p < 0.001). Scale bar in A–F = 20 μm.
BTKi treatment increases remyelination in vivo
To examine whether BTKi would also favor remyelination in vivo, we took advantage of our MBP-GFP-NTR transgenic Xenopus laevis model, which permits live imaging of demyelination and remyelination by conditionally triggering apoptotic ablation of myelin forming oligodendrocytes [19]. In MTZ-exposed transgenic MBP-GFP-NTR tadpoles demyelination is restricted to the CNS, without axonal damage. At the end of MTZ exposure spontaneous remyelination occurs and can already be evaluated after 3 days by counting the number of GFP+ oligodendrocyte per optic nerve of live animal, which we have previously shown to be a reliable index of demyelination and remyelination [19, 16].
Transgenic MBP-GFP-NTR Xenopus tadpoles (stage 52–53) were treated for 10 days with MTZ (10 mM) then returned to either fresh water (controls) or water containing increasing concentrations of BTKi. The number of GFP+ cells per optic nerve was counted in vivo before (D0), at the end of MTZ treatment (D10), and then on day 3 (R3) of the repair period. Treatment of demyelinated tadpoles with BTKi (1 μM) improved remyelination by a factor of 1.7-fold in comparison to control. The dose–response of BTKi efficiency to accelerate remyelination showed a bell-shaped curve with a maximum remyelination effect at concentrations of BTKi ranging between 50 nM and 1 μM; the effect was not observed at lower (1 nM and 5 nM) or higher concentrations (5 μM or 10 μM) (Fig. 4G).
DISCUSSION
Here we report that the vast majority of CNS cells expressing BTK are microglial cells and, to a lesser extent, astrocytes, and that the level of BTK expression increased after demyelination. Our key finding was that inhibition of BTK favors remyelination. This was illustrated using both ex vivo (mouse cerebellar slices) and in vivo (transgenic Xenopus laevis), two complementary experimental models of demyelination independent of adaptive immunity. Our data complement preclinical and clinical findings with BTKi, which have been shown to affect B cell proliferation and survival (for review see [24, 25]). Within the limitations of our study, we cannot conclude whether the action of BTKi on remyelination was mediated by microglia, astrocytes, or both. In this respect, it would be of interest to investigate whether inhibition of BTK alters astrocyte morphology, and/or affects microglia/astrocyte cell number and/or microglia phagocytosis. The molecular target and cellular mechanism favoring remyelination following inhibition of BTK also remains to be determined.
In a different model using rodent cell lines and primary cultures, ibrutinib, another BTKi, significantly reduced LPS-induced increases in pro-inflammatory cytokine levels in microglial cells, but not in astrocytes [26]. It has recently been reported that blocking BTK function ex vivo in acute rat brain slices reduced microglial phagocytosis and maintained numbers of resting microglia [27]. This observation is in apparent contradiction with our finding since it is well established that phagocytosis of myelin debris is an important step to favor remyelination in rodents [28–31], as well as in Xenopus [32]. Both ibrutinib and CC292, which were used in the above-mentioned studies, inhibit numerous targets besides BTK while, in contrast, the BTKi used here is highly selective. Furthermore, evobrutinib, another highly selective BTKi, has been shown to increase the capacity of human M2 macrophages to take up apoptotic cells (unpublished data).
Although activation of microglia has often been viewed as detrimental in the context of neurological disorders of the CNS, an increasing number of reports have revealed its neuroprotective and regenerative functions, in particular in demyelinating diseases such as MS. The mechanisms by which microglia are involved in remyelination are not completely understood [33, 34]. The TAM family of receptors is a different family of protein tyrosine kinases comprising three known members, Tyro3, Axl, and Mer. These receptors are widely expressed in the nervous system, including in oligodendrocytes and microglial cells. Growth arrest gene 6 (Gas 6), the principle cognate ligand of TAM receptors, has been shown to promote oligodendrocyte survival and to increase migration and proliferation of microglia/macrophages in the cuprizone model of demyelination [35]. A similar mechanism explaining the remyelination potency of BTKi acting both directly and indirectly on oligodendrocytes is unlikely since we have shown that BTK is expressed by less than 1% of oligodendrocytes.
A pan-kinase inhibitor, tyrphostin AG 126, has been described as exerting neuroprotection in EAE. The anti-inflammatory potential and especially interference with Toll-like receptor signaling was shown to involve AG126 hydrolysis and conversion of its dinitrile side chain to malononitrile [36]. The authors concluded that the beneficial effects of AG126 in EAE rely on a dual hit on adaptive and innate immune functions that builds on BTK inhibition and AG126 conversion to malononitrile as a novel TLR response modifier.
In our conditional Xenopus demyelination model, the dose–response of BTKi showed a bell-shaped curve, with a maximum effect observed between 50 nM and 1 μM, and no effect at either lower or higher concentrations. It is interesting that the higher doses of the BTK inhibitor failed to promote any remyelination rather than showing a plateau of effect. One possible interpretation of this effect could be related to the fact that the BTKi used in our study (BTKi-1) is first solubilized in DMSO before being diluted in the water vehicle. At higher concentration, it is likely that BTKi-1 is under a micelle form, which may mask the real concentration, a phenomenon well described by lipid enzymologists [37, 38].
Together, our data show that BTK inhibition has a net positive effect on remyelination both ex vivo (rodent) and in vivo (Xenopus), which suggests that BTK inhibition is a promising new therapeutic strategy to promote remyelination by targeting microglia. This activity would be complementary to the well-established inhibition of B cell function and the recently described promotion of anti-inflammatory macrophage differentiation.
FUNDING
This work was supported by Inserm, CNRS, UPMC, the program “Investissements d’Avenir” ANR-10-IAIHU-06 and NeurATRIS, and ANR grant BRECOMY to BZ. The study was partially funded by a research grant from Merck-Serono to BZ.
CONFLICT OF INTEREST
R. Grenningloh is an employee of EMD Serono, Billerica, MA, USA (a business of Merck KGaA, Darmstadt, Germany). U. Boschert is an employee of Merck Serono S.A., Eysin, Switzerland.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/BPL-200100.
REFERENCES
[1] | Mano H . Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. déc (1999) ;10: (3-4):267–80. |
[2] | Hendriks RW . Drug discovery: New Btk inhibitor holds promise. Nat Chem Biol. janv(2011) ;7: (1):4–5. |
[3] | Hendriks RW , Yuvaraj S , Kil LP . Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. avr (2014) ;14: (4):219–32. |
[4] | Gasperini C , Haggiag S , Ruggieri S . Drugs in clinical development for multiple sclerosis: focusing on anti-CD20 antibodies. Expert Opin Investig Drugs. oct (2013) ;22: (10):1243–53. |
[5] | Barun B , Bar-Or A . Treatment of multiple sclerosis with anti-CD20 antibodies. Clin Immunol Orlando Fla. janv (2012) ;142: (1):31–7. |
[6] | Kappos L , Li D , Calabresi PA , O’Connor P , Bar-Or A , Barkhof F , et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet Lond Engl. 19 nov (2011) ;378: (9805):1779–87. |
[7] | Montalban X , Arnold DL , Weber MS , Staikov I , Piasecka-Stryczynska K , Willmer J , et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N Engl J Med. 20 (2019) ;380: (25):2406–17. |
[8] | López-Herrera G , Vargas-Hernández A , González-Serrano ME , Berrón-Ruiz L , Rodríguez-Alba JC , Espinosa-Rosales F , et al. Bruton’s tyrosine kinase–an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. févr (2014) ;95: (2):243–50. |
[9] | Marron TU , Martinez-Gallo M , Yu JE , Cunningham-Rundles C . Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines. J Allergy Clin Immunol. janv (2012) ;129: (1):184–190.e1-4. |
[10] | Whang JA , Chang BY . Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov Today. août (2014) ;19: (8):1200–4. |
[11] | Block H , Zarbock A . The role of the tec kinase Bruton’s tyrosine kinase (Btk) in leukocyte recruitment. Int Rev Immunol. avr (2012) ;31: (2):104–18. |
[12] | Hartkamp LM , Fine JS , van Es IE , Tang MW , Smith M , Woods J , et al. Btk inhibition suppresses agonist-induced human macrophage activation and inflammatory gene expression in RA synovial tissue explants. Ann Rheum Dis. août (2015) ;74: (8):1603–11. |
[13] | Haselmayer P , Camps M , Liu-Bujalski L , Nguyen N , Morandi F , Head J , et al. Efficacy and Pharmacodynamic Modeling of the BTK Inhibitor Evobrutinib in Autoimmune Disease Models. J Immunol Baltim Md 15 (2019) ;202: (10):2888–906. |
[14] | Ito M , Shichita T , Okada M , Komine R , Noguchi Y , Yoshimura A , et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 10 juin (2015) ;6: :7360. |
[15] | Mangla A , Khare A , Vineeth V , Panday NN , Mukhopadhyay A , Ravindran B , et al. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 15 août (2004) ;104: (4):1191–7. |
[16] | Mannioui A , Vauzanges Q , Fini JB , Henriet E , Sekizar S , Azoyan L , et al. The Xenopus tadpole: An in vivo model to screen drugs favoring remyelination. Mult Scler Houndmills Basingstoke Engl. (2018) ;24: (11):1421–32. |
[17] | Spassky N , Olivier C , Cobos I , LeBras B , Goujet-Zalc C , Martínez S , et al. The early steps of oligodendrogenesis: insights from the study of the plp lineage in the brain of chicks and rodents. Dev Neurosci. (2001) ;23: (4-5):318–26. |
[18] | Wight PA , Duchala CS , Readhead C , Macklin WB . A myelin proteolipid protein-LacZ fusion protein is developmentally regulated and targeted to the myelin membrane in transgenic mice. J Cell Biol. oct (1993) ;123: (2):443–54. |
[19] | Kaya F , Mannioui A , Chesneau A , Sekizar S , Maillard E , Ballagny C , et al. Live imaging of targeted cell ablation in Xenopus: a new model to study demyelination and repair. J Neurosci Off J Soc Neurosci. 12 sept (2012) ;32: (37):12885–95. |
[20] | Mannioui A , Zalc B . Conditional Demyelination and Remyelination in a Transgenic Xenopus laevis. Methods Mol Biol Clifton NJ. (2019) ;1936: :239–48. |
[21] | Normal Table of Xenopus Laevis (Daudin): A Systematical & Chronological Survey of the Development from the Fertilized Egg till the End of Metamorphosis [Internet]. CRC Press. [cité 10 janv 2020]. Disponible sur: https://www.crcpress.com/Normal-Table-of-Xenopus-Laevis-Daudin-A-Systematical–Chronological/Faber-Nieuwkoop/p/book/9780815318965 |
[22] | Birgbauer E , Rao TS , Webb M . Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J Neurosci Res. 15 oct (2004) ;78: (2):157–66. |
[23] | Zhang H , Jarjour AA , Boyd A , Williams A . Central nervous system remyelination in culture–a tool for multiple sclerosis research. Exp Neurol. juill (2011) ;230: (1):138–48. |
[24] | Pal Singh S , Dammeijer F , Hendriks RW . Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 19 (2018) ;17: (1):57. |
[25] | Corneth OBJ , Klein Wolterink RGJ , Hendriks RW . BTK Signaling in B Cell Differentiation and Autoimmunity. Curr Top Microbiol Immunol (2016) ;393: :67–105. |
[26] | Nam HY , Nam JH , Yoon G , Lee J-Y , Nam Y , Kang H-J , et al. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J Neuroinflammation. 19 sept (2018) ;15: (1):271. |
[27] | Keaney J , Gasser J , Gillet G , Scholz D , Kadiu I . Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol. sept (2019) ;14: (3):448–61. |
[28] | Bao Z , Hao J , Li Y , Feng F . Promotion of microglial phagocytosis by tuftsin stimulates remyelination in experimental autoimmune encephalomyelitis. Mol Med Rep. déc (2019) ;20: (6):5190–6. |
[29] | He Y , An J , Yin J-J , Sui R-X , Miao Q , Ding Z-B , et al. Ethyl pyruvate enhances spontaneous remyelination by targeting microglia phagocytosis. Int Immunopharmacol. déc (2019) ;77: :105929. |
[30] | Lloyd AF , Davies CL , Holloway RK , Labrak Y , Ireland G , Carradori D , et al. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat Neurosci. (2019) ;22: (7):1046–52. |
[31] | Karamita M , Barnum C , Möbius W , Tansey MG , Szymkowski DE , Lassmann H , et al. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 20 avr (2017) ;2: (8). |
[32] | Sekizar S , Mannioui A , Azoyan L , Colin C , Thomas J-L , Du Pasquier D , et al. Remyelination by Resident Oligodendrocyte Precursor Cells in a Xenopus laevis Inducible Model of Demyelination. Dev Neurosci. (2015) ;37: (3):232–42. |
[33] | Schwartz M . Macrophages and microglia in central nervous system injury: are they helpful or harmful? J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. avr (2003) ;23: (4):385–94. |
[34] | Lloyd AF , Miron VE . The pro-remyelination properties of microglia in the central nervous system. Nat Rev Neurol. août (2019) ;15: (8):447–58. |
[35] | Binder MD , Cate HS , Prieto AL , Kemper D , Butzkueven H , Gresle MM , et al. Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J Neurosci Off J Soc Neurosci. 14 mai (2008) ;28: (20):5195–206. |
[36] | Menzfeld C , John M , van Rossum D , Regen T , Scheffel J , Janova H , et al. Tyrphostin AG126 exerts neuroprotection in CNS inflammation by a dual mechanism. Glia. juin (2015) ;63: (6):1083–99. |
[37] | Gatt S , Gazit B , Barenholz Y . Effect of bile salts on the hydrolysis of gangliosides, glycoproteins and neuraminyl-lactose by the neuraminidase of Clostridium perfringens. Biochem J. 1 janv (1981) ;193: (1):267–73. |
[38] | Azum N , Kumar D . Kinetic study of the metal-dipeptide complex with ninhydrin facilitated by gemini (m-s-m) surfactant micelles. Sci Rep. 5 mars (2020) ;10: (1):4088. |

