
Sonic Hedgehog Signaling Rises to the Surface: Emerging Roles in Neocortical Development
Abstract
The mammalian neocortex is composed of a diverse population of neuronal and glial cells that are crucial for cognition and consciousness. Orchestration of molecular events that lead to the production of distinct cell lineages is now a major research focus. Recent studies in mammalian animal models reveal that Sonic Hedgehog (Shh) signaling plays crucial roles in this process. In this review, we will evaluate these studies and provide insights on how Shh signaling specifically influence cortical development, beyond its established roles in telencephalic patterning, by specifically focusing on its impact on cells derived from the cortical radial glial (RG) cells. We will also assess how these findings further advance our knowledge of neurological diseases and discuss potential roles of targeting Shh signaling in therapies.
THE EMBRYONIC NEOCORTEX GENERATES DIVERSE NEURONAL AND GLIAL CELL TYPES
The mature neocortex is crucial for the execution of complex tasks such as cognition, perception, and motor function. This unique function relies on the proper generation and positioning of neurons and glial cells, the principal cortical cell types, enabling the assembly of short- and long- range circuits. Two major classes of neurons are found in the neocortex, the excitatory projection neurons (PN) and inhibitory neurons or interneurons [1, 2]. PNs largely form long-range projections and originate from the neurogenic niches of the embryonic neocortex. On the other hand, interneurons form intra-cortical short-range projections and are generated in neurogenic regions of the embryonic ganglionic eminence located in the ventral forebrain. The generation of interneurons will not be discussed in this review, but we refer the reader to recent excellent reviews on this topic [3–5]. Oligodendrocytes and astrocytes are the major cortical glial cell types also largely generated in the embryonic neocortex. Oligodendrocytes and astrocytes contribute to neuronal function by myelinating axons and regulating neuronal networks.
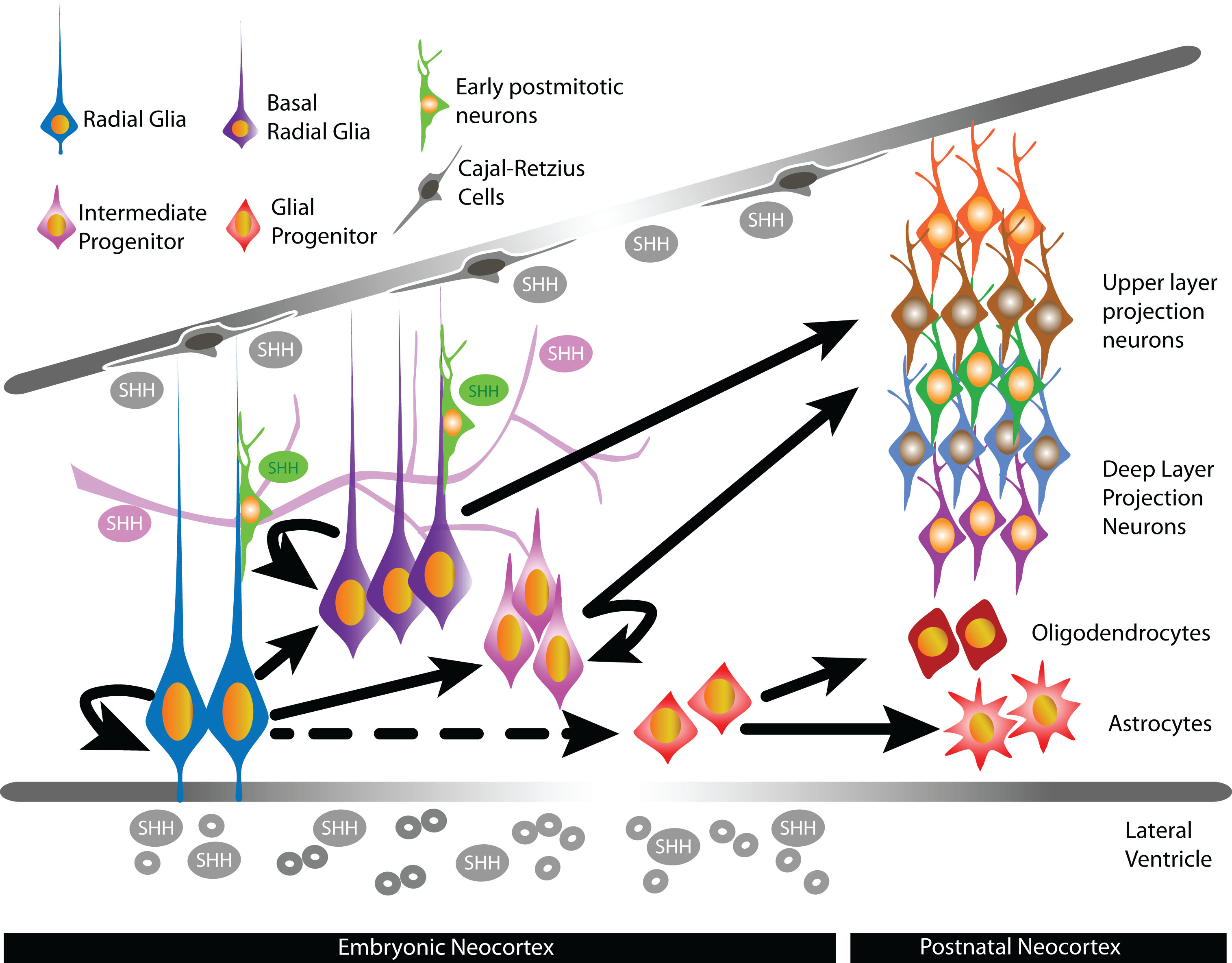
The germinal ventricular zone (VZ) of the embryonic neocortex is composed of radial glial cells (RG) that produce all PNs and the majority of glial cells that populate the mature neocortex. In the mouse, RGs initiate neurogenesis at embryonic day (E) 10.5 and continue until around E17 to produce PNs directly or indirectly via intermediate progenitors (IP) that occupy the adjacent subventricular zone (SVZ) [6]. Distinct PN lineages destined to specific cortical layers are typically produced in consecutive waves in an inside-out fashion, such that early born PNs migrate into the developing neocortex to settle in the deep layers (layers 5–6) while later born PNs form the upper layers (layers 2–4). Towards the end of cortical neurogenesis, gliogenesis commences with the generation of oligodendrocytes by E16.5 and followed by the generation of astrocytes by postnatal day (P) 1. RGs either generate oligodendrocyte (OPC) or astrocyte (APC) precursors, or transform into astrocytes themselves during gliogenesis [7]. As with PNs, recent studies have begun to uncover diverse features of oligodendrocyte and astrocyte subtypes that can have implications in the development and maintenance of neuronal circuits [8, 9]. To date, as with PNs, the mechanisms regulating glial subtype specification from cortical RG cells are largely unexplored.
The molecular players that regulate RG cell behavior, such as its proliferation, specification, and differentiation into diverse precursor populations and post-mitotic cell types are now beginning to be elucidated. Throughout this process, Sonic hedgehog (Shh) signaling is now emerging to be a key player in regulating various steps in neurogenesis and gliogenesis to ensure the proper generation of PNs and glial cells, specifically from RG cells in the germinal zones of the embryonic neocortex. In this review, we will discuss recent findings on the dynamic roles of Shh signaling, specifically in RG cells of the neocortex, to influence cortical development and its potential implications in the pathogenesis of neurological disorders.
SOURCES OF SHH LIGANDS AND EXPRESSION PATTERNS OF PATHWAY GENES DURING CORTICOGENESIS
The Shh signaling pathway is an evolutionarily conserved signaling network with broad roles in development, function, and disease. Activation of Shh signaling is initiated upon binding of extracellular Shh ligands to Patched (Ptch) transmembrane receptor and its co-receptors: Cell-adhesion-molecule-related/Downregulated by oncogenes (Cdon), Biregional Cdon-binding protein (Boc), and Growth Arrest Specific 1 (Gas1) [10–12]. Binding of Shh relieves the inhibitory binding of Ptch on the transmembrane receptor protein Smoothened (Smo) [13, 14]. Smo release from Ptch inhibition leads to a cascade of intracellular events resulting in the activation of the zinc-finger transcription factor, Gli, and its nuclear translocation and expression of Shh target genes to regulate Shh-dependent processes [15].
Shh ligands are secreted morphogens able to act in a paracrine or autocrine fashion by forming concentration gradients with short- and long- range transcriptional effects [16, 17]. In mammals, there are three known Shh ligands: sonic hedgehog (Shh), desert hedgehog (Dhh) and indian hedgehog (Ihh). Shh ligand is the best characterized and has been detected in the developing neocortex. Much less is known on the expression patterns of Ihh and Dhh in the developing neocortex, although Ihh has been detected in cultured cortical progenitors [18]. In the developing neocortex, expression of Shh has been detected in tangentially migrating interneurons, Cajal-Retzius cells localized in the marginal zones, the VZ/SVZ, and postmitotic neurons [19, 20]. Shh in the cerebrospinal fluid (CSF) is also a potential source of Shh throughout corticogenesis to influence the behavior of neural stem/progenitor cells lining the lateral ventricles. Indeed, expression of Shh mRNA is detectable in the choroid plexus of the lateral ventricle [21] and Shh has been detected in telencephalic CSF [22, 23]. Other potential sources of Shh are epithelial cells and the developing vascular system, which release Shh-containing microvesicles [24–26]. In the developing human fetal telencephalon, strong mRNA levels of Shh are detected in cells within the cortical plate and the subplate, and lower levels of Shh mRNA expression are observed in the VZ and SVZ [27]. The exact contributions of these potential sources in the developing neocortex and how varying levels of Shh throughout corticogenesis affect the generation of specific neuronal and glial lineages are unclear. In other tissues, extracellular levels of Shh exert differential effects on progenitor behavior to influence their differentiation into distinct cell types [28–30], and can therefore similarly affect the production of specific cell lineages from NSCs in the developing neocortex (Fig. 1).
Fig.1
In the developing neocortex, sources of Shh ligands include epithelial cells, blood vessels postmitotic neurons, Cajal-Retzius cells, and the cerebrospinal fluid of the lateral ventricles. The presence of Shh during corticogenesis have been shown or suggested to influence the proliferation, specification, and differentiation of specific cortical progenitors that lead to the generation of distinct neuronal and glial lineages.
The vertebrate Gli family of zinc-finger transcription factors, Gli1, Gli2, and Gli3, are the main effectors of Shh signaling [31]. Full-length Gli proteins act as transcriptional activators and transform into transcriptional repressors when cleaved. In the developing neocortex, Gli2 and Gli3 are predominantly expressed in their cleaved repressor forms (Gli2R and Gli3R, respectively) at early stages of corticogenesis and are prominently expressed in the proliferative VZ/SVZ during corticogenesis [20]. Gli2 expression is largely absent at later stages while expression of Gli3 persists particularly as a repressor [32]. Since Shh ligands are present in the developing neocortex, the prevalence of Gli repressors emphasize the importance of controlling Shh signaling activity. As we will discuss below, loss of Shh signaling in the developing neocortex has profound effects indicating that modulation of Shh signaling activity, rather than its complete deactivation, is an important regulatory step throughout corticogenesis. Indeed, Shh target genes include Ptch1 in order to induce a negative regulatory feedback mechanism to control the progression of the Shh signaling cascade.
Regulation of Shh signaling can be mediated upstream of the pathway at the level of Ptch1 expression. Patched 1 (Ptch1) mRNA is present at relatively low levels from E11.5 to E18.5 [21, 33] particularly in Nestin-expressing cells within the VZ of the embryonic neocortex [34]. As mentioned above, Ptch1 exerts its inhibitory effect upstream of Shh signaling through its repressive binding of Smo in the absence of Shh ligands. The exact mechanism of this inhibition has not been completely characterized in the developing neocortex and may include Smo conformational change from an active to inactive state, cellular membrane changes, activation of intracellular modulators, or prevention of Smo accumulation in the primary cilia which are essential Shh transduction sites in mammalian cells [14, 35, 36]. Additionally, the Shh co-receptors Boc and Cdon may contribute in facilitating Smo inhibition particularly since the expression of these proteins are spatially maintained at high levels in cortical progenitors in the VZ/SVZ of the developing neocortex [37–39].
Downstream of Smo, the cytoplasmic protein Suppressor of Fused (Sufu) regulates the activity of Gli transcription factors. Sufu exerts this role by regulating the stability, cytoplasmic-nuclear translocation, and proteosomal degradation or modification of Gli [40–42]. In the embryonic neocortex, high levels of Sufu mRNA expression are exclusively detected in the VZ/SVZ during corticogenesis. This expression pattern supports a role for Sufu in influencing the behavior of neural progenitors during corticogenesis by regulating the stability of Gli2 and Gli3 at early stages and the formation of Gli3R at later stages [43]. Overall, the presence of positive and negative regulatory mechanisms to modulate Shh signaling activity in the embryonic neocortex profoundly influences the behavior of cortical progenitors at various stages of corticogenesis.
MITOGENIC ROLES OF SHH SIGNALING IN CORTICAL PROGENITORS
Complete loss of Shh is deleterious in the developing embryo, emphasizing the critical roles of Shh signaling in developmental processes [44]. In the central nervous system (CNS), particularly in the developing forebrain, Shh signaling has prominent roles in the patterning of the dorsal and ventral axis [45]. This step establishes the molecular properties of the neurogenic zones in the dorsal and ventral forebrain, which is a crucial step in the generation of appropriate cell types: the ganglionic eminences of the ventral forebrain produce interneurons, whereas the VZ/SVZ of the neocortex produce glutamatergic PNs. Subsequently, the ventral forebrain maintains high levels of Shh signaling throughout corticogenesis, whereas Shh activity has been largely undetected in the developing neocortex. Studies in mice carrying conditional knockout alleles of pathway genes reveal that the mitogenic roles of Shh signaling extend to specific cortical progenitor populations during corticogenesis. Conditional deletion of Shh or Smo in Nestin-expressing cells of the neocortex by E12.5 cause microcephaly and result in lethality by postnatal day (P) 19 [46, 47]. Ablation of these genes at E12.5 does not disturb telencephalic patterning but profoundly affect proliferation by prolonging the cell cycle length of cortical progenitors within the VZ/SVZ at E13.5 [19]. These altered cell cycle kinetics result in a significant decrease in the numbers of Tbr2+ IP cells and consequently lead to the reduction of PNs in superficial layers 2–4. Milder phenotypes of Gli2 null mice (that are not exencephalic) show thinner neurogenic regions as a result of significantly fewer dividing cells at mid- to late- stages of corticogenesis, particularly in the SVZ [48]. Altogether, these studies point to a critical role for Shh signaling in regulating IP proliferation during corticogenesis.
The effect of Shh signaling on IP proliferation is sensitive to Shh levels. Shikata and colleagues used in utero electroporation to overexpress varying levels of Shh beginning at E13.5 and found differential effects on cortical development [49]. In areas with low to moderate levels of Shh overexpression (fewer than 20 cells transfected with Shh expression vector), significant increases in Tbr2+ IPs were evident. Interestingly, low levels of Shh expression resulted in higher numbers of IP cells, compared to moderate levels. Indeed, areas with high Shh levels (areas with over 20 cells transfected with Shh expression vector) did not show any increase in Tbr2+ cells. Rather, progenitor fate specification was compromised. These findings indicate that Shh levels critically affect IP cell behavior, in that a mild increase in Shh activity can enhance IP proliferation while a moderate to high increase in Shh activation alters IP identity.
Modulators are in place to control endogenous Shh signaling. Mice carrying hypomorphic or floxed alleles of Ptch1 displayed defects in the proliferation of both RG and IP cells [34, 49]. Removal of Ptch1 in Nestin+cortical progenitors at E11.5 resulted in extensive folding and decreased cortical thickness due to an increase in symmetrically dividing RG cells, at the expense of neurogenic division, thereby reducing IP cell production [34]. On the other hand, inactivation of Ptch1 in the neocortex of the E17-E18 mouse abolished RG self-renewal, indicating enhanced transition of RG progenitors to IP cells [49]. Similarly, deletion of Cdon, results in extensive cortical thinning partially due to reduced proliferation and differentiation of cortical progenitors [39]. Downstream of Shh receptors, Sufu and Gli have been shown to affect progenitor proliferation. Deletion of Sufu at E13.5 result in ectopic activation of Shh activity leading to an increase in the proliferation of IPs in the SVZ, and consequently, an increase in upper layer 4 PNs [50]. Similarly, reduction or complete removal of Gli3R accelerate the cell cycle [51] and ultimately lead to the failure of RG progenitors to self-renew [32]. Under these conditions, RGs prematurely exit the cell cycle and are unable to maintain the IP pool during the course of corticogenesis resulting in a decrease in the number of upper layer PNs in the postnatal neocortex.
Recent studies identified interesting new roles of Shh signaling in regulating specific progenitor subpopulations in the developing neocortex. Constitutive activation of Shh signaling achieved by expressing a constitutively active form of Smo (SmoM2; [52]) in cortical progenitors in mid-corticogenesis, results in an increase in the proliferation of Tbr2+ IPs and promotes the expansion of basal RGs (bRGs) [53]. bRGs are neural progenitors that exist in very small numbers in the mouse but are a predominant progenitor population in the developing human neocortex [54, 55]. bRGs undergo extensive symmetric and self-renewing asymmetric divisions to generate mostly upper layer PNs. Expansion of upper layer PNs are responsible for the large and folded neocortex present in primates and are thought to significantly contribute to the increased cortical size and functional complexity of the human brain [56]. Wang et al. observed that under high levels of Shh activation, bRGs and IPs remained in the cell cycle longer than controls, indicating an increase in self-renewal. This increase in bRGs and IPs consequently resulted in an increase in SatB2+ upper layer PNs and induced folding of the cingulate cortex. The findings by Wang et al. (2016) revealed a specific role for Shh signaling pathway in controlling bRG numbers in developing neocortex and provide insights on the mechanisms contributing to the evolution of the human neocortex.
ROLES OF SHH SIGNALING IN THE SPECIFICATION OF CORTICAL PROGENITORS
Cortical progenitors in the developing neocortex are highly heterogeneous and are distinguished according to unique transcriptional programs that remain dynamic during corticogenesis [57–60]. As a morphogen, Shh can trigger differential gene expression changes temporally and spatially that may influence progenitor heterogeneity and their ability to produce specific progenies throughout corticogenesis. Supporting this, ectopic activation of Shh signaling, as a result of removing Sufu or activating SmoM2 specifically in Emx1+ cortical progenitors using the Emx1-Cre driver [61] can alter the molecular identities of RG and IP progenitors [43]. RG and IP progenitors that lack Sufu ectopically express Dlx2 and Ascl1, markers of ventral forebrain progenitors, yet are still able to differentiate into cortical PNs that distinctly express PN-specific markers. However, these PNs are largely misspecified in that upper layer Cux1+ or Brn2+ cortical PNs also express markers for deep layer PNs such as Ctip2 [43]. These defects were only observed in mice in which Sufu deletion occurred at E10.5, but not at E13.5 [43, 53]. Moreover, Sufu deletion in Emx1+ cells at E10.5 did not cause defects in dorsoventral forebrain patterning. Thus, specification and production of molecularly distinct upper layer cortical PNs, which are largely generated at E14-E16, are dependent on the inhibition of Shh activity in cortical progenitors at earlier stages of corticogenesis.
Gli transcription factors, particularly Gli3R, influence the specification of neuronal subtypes in the developing neocortex. The fate of cortical progenitors is dependent on Gli3 since deletion of Gli3 in Emx1+ cortical progenitors at early stages of corticogenesis prolongs the production of deep layer Ctip2+ PNs at the expense of upper layer Cux1+ PNs [32]. The repressor function of Gli3 is especially important in this process since restoration of Gli3R levels reversed this phenotype. In correlation to these observations, the absence of Sufu at early stages of corticogenesis destabilized full-length Gli3, which likely prevented the proteolytic cleavage and formation of Gli3R [43]. Curiously, mice that lacked Sufu or expressed constitutively active Smo in Emx1+ progenitors displayed more severe defects in PN specification compared to Gli3 mutant cortical progenitors at similar timepoints. Additionally, Amaniti et al., (2013) reported that specification of callosal PNs located in upper cortical layers are also unaffected when Gli3 is inactivated in Emx1+ progenitors [62]. Whether the requirement for Gli3R is partially compensated by an increase in Gli2 expression in cortical progenitors in which Gli3 is absent is plausible. It is also possible that additional downstream targets of Shh signaling, aside from those targeted by Gli3, may influence the specification of cortical progenitors. Whether Sufu affects the expression of these yet unidentified targets, independent of its role as a regulator of Gli transcription factors, is also a likely possibility.
The ectopic expression of Dlx2 and Ascl1 observed in cortical progenitors lacking Sufu [43] may have implications in the specification of human cortical progenitors. Dlx2 and Ascl1 are markers for ventral forebrain progenitors that eventually generate GABAergic interneurons [63]. Thus, whether a subset of mispecified cortical progenitors also continued on to generate GABAergic interneurons are not known. In humans and likely other primate species, Dlx2 and Ascl1 are highly expressed in the fetal proliferative zones of the neocortex and may generate a subpopulation of GABAergic interneurons [64–67]. Given the expansion of bRG progenitors in SmoM2-expressing cortical progenitors, human cortical progenitors could have also acquired the ability to generate GABAergic interneurons partially due to increasing Shh signaling activity observed in the fetal human neocortex [53]. Indeed, detectable Shh mRNA is observed in the proliferative outer SVZ (where bRGs are largely localized), RG progenitors, and early post-mitotic neurons in the human fetal neocortex at mid-gestation (at ∼20 gestational weeks), a time of active cortical neurogenesis [53, 68, 69]. Furthermore, cultured human RG cells are Shh-responsive, and in the presence of exogenous Shh, can differentiate into Nkx2.1 lineage GABAergic interneurons [70]. These studies, although conducted in vitro, provide further insights on the evolutionary divergent mechanisms underlying the formation of a highly complex human neocortex.
ROLE FOR SHH SIGNALING IN CORTICAL GLIOGENESIS
Shh signaling has prominent roles in the regulation of gliogenic fates in the developing spinal cord and the ventral forebrain [18, 71, 72]. Surprisingly however, there are very limited studies on the roles of Shh signaling in glial production from cortical RG progenitors, which generate the vast majority of cortical oligodendrocytes populating the neocortex [72–74]. These studies are largely in vitro based but hint at potentially significant roles for Shh signaling in glial precursor proliferation and specification. For example, upon addition of Shh, cultured E13 or E15 rodent cortical RG progenitors can generate large numbers of oligodendrocytes and astrocytes but not neuronal progenies [75, 76] indicating that RG progenitors in the glial lineage at mid- to late- stage corticogenesis are highly responsive to Shh signaling. Similarly, cultured dorsal forebrain RG cells derived from human fetal brain are highly responsive to Shh, and significantly expand the number of Olig2+ OPCs [77]. Since sources of Shh ligands increase during the course of corticogenesis, this will likely influence the behavior of a growing number of Shh-responsive cortical progenitors. Furthermore, recent findings indicate that Shh signaling play significant roles in oligodendrocyte proliferation and specification in the postnatal forebrain, much of which are generated by neural stem cells (NSC) derived from embryonic cortical RG progenitors [6, 78–80]. With the recent characterization of molecularly diverse oligodendrocyte and astrocyte lineages in the forebrain [8, 9], it would be interesting to determine whether Shh signaling is involved in the specification of distinct oligodendroglial and astroglial lineages from RG progenitors during cortical development.
IMPLICATIONS OF SHH SIGNALING IN NEUROLOGICAL DISORDERS
Abnormalities in neuronal number, differentiation, and connectivity in the neocortex commonly cause a spectrum of neurodevelopmental and cognitive disorders. In most cases, the cause of these disorders is attributed to known genetic changes. Inactivating mutations in genes associated with the Shh signaling pathway, for example, severely disrupt brain development and lead to malformations of the neocortex. Nonsyndromic holoprosencephaly, a condition in which the brain fails to form two distinct hemispheres, is commonly caused by inactivation of Shh [81]. Mutations in Shh receptors, Ptch1 and CDON, have also been reported in a number of human cases of holoprosencephaly [82, 83]. Mutations in Gli3 cause Greig cephalopolysyndactyly syndrome or acrocallosal syndrome, both are rare conditions characterized by polydactyly and severe brain abnormalities such as macrocephaly and callosal agenesis [84, 85]. Moderate to severe developmental delays and intellectual disabilities, as well as seizures, are typical in patients with these syndromes.
A number of cortical defects are caused by changes in the activity of Shh signaling independent of mutations in genes associated with the canonical Shh signaling pathway. This can occur in the presence of defects in the primary cilia, a microtubule-based organelle present in cortical progenitors that functions to integrate and transduce extracellular signals, including Shh signaling [86]. Mutations in genes that encode ciliary proteins, such as Kif3a, Ift88, and Katnb1, significantly disrupt Shh signaling, and contribute to a number of human ciliopathies with defects in cortical development [51, 87–89]. Altered Shh signaling is also thought to contribute to defects in neurogenesis leading to microcephaly in Down syndrome (DS), the most common genetic cause of mental retardation. Human fetal brains with DS and the Tg65DN mouse model of DS show high levels of Ptch1 overexpression in the VZ/SVZ of the developing forebrain, thereby repressing Shh signaling activity, as a result of the increase in transcriptional activity of Amyloid precursor protein intracellular domain [90]. Although the effect of Shh agonists in cortical progenitors have not been examined, it could be a promising therapy particularly since the Shh agonist SAG1.1 increased the proliferation of Shh-responsive cerebellar granule cell precursors in Ts65DN mice [91]. Altogether, these studies indicate that alterations in the activity of Shh signaling, particularly during corticogenesis, can have profound impact in cortical development and cause neurological disorders.
As recent studies in mammalian systems uncover important roles of Shh signaling in neuronal and glial specification in the developing neocortex, there is a need to investigate the potential contributions of Shh signaling in complex neurodevelopmental diseases that are polygenic in nature. Notably, upper layer PNs are critical for a number of higher associative cognitive functions and are prominently affected in developmental psychiatric disorders such as schizophrenia and autism. Studies point to aberrant control of cortical progenitor behavior as the cause of abnormal cortical circuitry in these disorders, including callosal projections formed by upper layer PNs [92–94]. We discussed in this review that alterations in Shh signaling specifically affect the proliferation and specification of cortical IP or bRG cells, which are major sources of upper layer PNs in mice and humans, respectively. Yet, studies that examine Shh signaling in the developing neocortex of mouse models of these diseases are largely lacking. This could be due to the largely absent bRG population in the developing mouse neocortex. It may be useful to utilize in vitro models of human cortical development such as human cerebral organoids formed from patient-derived induced pluripotent stem (IPS) cells [95]. Furthermore, whether variations in Shh signaling activity during corticogenesis affect maturation and connectivity of upper layer PNs produced by IPs and bRGs should be investigated. These inquiries can provide critical insights on how Shh signaling in human cortical progenitors contributes to the pathogenesis of neurological diseases or be utilized in the development of regenerative therapies.
CONCLUSIONS
Novel roles of Shh signaling in regulating cortical progenitor behavior, beyond forebrain patterning, are now beginning to be unraveled. With the availability of pharmacological modulators of Shh signaling, this pathway could be a promising treatment target for neurological disorders caused by defects in cortical development. Thus, further elucidation of the mechanisms controlled by Shh signaling in corticogenesis can lead to a better understanding of the etiology and treatment of complex neurological diseases.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health to S.J.P. (NINDS R01MH077694) and O.R.Y. (NCI K01CA201068).
REFERENCES
[1] | Greig LC , Woodworth MB , Galazo MJ , Padmanabhan H , Macklis JD . Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. (2013) ;14: :755–69. doi:10.1038/nrn3586 |
[2] | Lodato S , Shetty AS , Arlotta P . Cerebral cortex assembly: Generating and reprogramming projection neuron diversity. Trends Neurosci. (2014) ;38: :117–25. doi:10.1016/j.tins.2014.11.003 |
[3] | Hu JS , Vogt D , Sandberg M , Rubenstein JL . Cortical interneuron development: A tale of time and space. Development. (2017) ;144: :3867–78. doi:10.1242/dev.132852 |
[4] | Bandler RC , Mayer C , Fishell G . Cortical interneuron specification: The juncture of genes, time and geometry. Curr Opin Neurobiol. (2017) ;42: :17–24. doi:10.1016/j.conb.2016.10.003 |
[5] | DeBoer EM , Anderson SA . Fate determination of cerebral cortical GABAergic interneurons and their derivation from stem cells. Brain Res. (2017) ;1655: :277–82. doi:10.1016/j.brainres.2015.12.031 |
[6] | Kriegstein A , Alvarez-Buylla A . The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. (2009) ;32: :149–84. doi:10.1146/annurev.neuro.051508.135600 |
[7] | Noctor SC , Martínez-Cerdeño V , Kriegstein AR . Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol. (2008) ;508: :28–44. doi:10.1002/cne.21669 |
[8] | John Lin C-C , Yu K , Hatcher A , Huang T-W , Lee HK , Carlson J , et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci. (2017) ;20: :396–405. doi:10.1038/nn.4493 |
[9] | Dimou L , Simons M . Diversity of oligodendrocytes and their progenitors. Curr Opin Neurobiol. (2017) ;47: :73–9. doi:10.1016/j.conb.2017.09.015 |
[10] | Allen BL , Song JY , Izzi L , Althaus IW , Kang J-S , Charron F , et al. Overlapping Roles and Collective Requirement for the Coreceptors GAS1, CDO, and BOC in SHH Pathway Function. Dev Cell. (2011) ;20: :775–87. doi:10.1016/j.devcel.2011.04.018 |
[11] | Stone DM , Hynes M , Armanini M , Swanson TA , Gu Q , Johnson RL , et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. (1996) ;384: :129–34. doi:10.1038/384129a0 |
[12] | Marigo V , Davey RA , Zuo Y , Cunningham JM , Tabin CJ . Biochemical evidence that Patched is the Hedgehog receptor. Nature. (1996) ;384: :176–9. doi:10.1038/384176a0 |
[13] | Murone M , Rosenthal A , de Sauvage FJ . Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr Biol. (1999) ;9: :76–84. |
[14] | Taipale J , Cooper MK , Maiti T , Beachy PA . Patched acts catalytically to suppress the activity of Smoothened. Nature. (2002) ;418: :892–6. doi:10.1038/nature00989 |
[15] | Rimkus T , Carpenter R , Qasem S , Chan M , Lo H-W . Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel). (2016) ;8: :22. doi:10.3390/cancers8020022 |
[16] | Fuccillo M , Joyner AL , Fishell G . Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. (2006) ;7: :772–83. doi:10.1038/nrn1990 |
[17] | Balaskas N , Ribeiro A , Panovska J , Dessaud E , Sasai N , Page KM , et al. Gene Regulatory Logic for Reading the Sonic Hedgehog Signaling Gradient in the Vertebrate Neural Tube. Cell. (2012) ;148: :273–84. doi:10.1016/j.cell.2011.10.047 |
[18] | Tekki-Kessaris N , Woodruff R , Hall AC , Gaffield W , Kimura S , Stiles CD , et al. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development. (2001) ;128: :2545–54. |
[19] | Komada M , Saitsu H , Kinboshi M , Miura T , Shiota K , Ishibashi M . Hedgehog signaling is involved in development of the neocortex. Development. (2008) ;135: :2717–27. doi:10.1242/dev.015891 |
[20] | Dahmane N , Sanchez P , Gitton Y , Palma V , Sun T , Beyna M , et al. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. (2001) ;128: :5201–12. |
[21] | The Developing Mouse Brain. Allen Brain Dev Mouse Brain Atlas [Internet] n.d. http://developingmouse.brain-maorg. |
[22] | Chau KF , Springel MW , Broadbelt KG , Park H-Y , Topal S , Lun MP , et al. Progressive Differentiation and Instructive Capacities of Amniotic Fluid and Cerebrospinal Fluid Proteomes following Neural Tube Closure. Dev Cell. (2015) ;35: :789–802. doi:10.1016/j.devcel.2015.11.015 |
[23] | Lun MP , Johnson MB , Broadbelt KG , Watanabe M , Kang Y-J , Chau KF , et al. Spatially heterogeneous choroid plexus transcriptomes encode positional identity and contribute to regional CSF production. J Neurosci. (2015) ;35: :4903–16. doi:10.1523/JNEUROSCI.3081-14.2015 |
[24] | Choe Y , Huynh T , Pleasure SJ . Epithelial cells supply Sonic Hedgehog to the perinatal dentate gyrus via transport by platelets. Elife. (2015) ;4. doi:10.7554/eLife.07834 |
[25] | Benameur T , Soleti R , Porro C , Andriantsitohaina R , Martínez MC . Microparticles Carrying Sonic Hedgehog Favor Neovascularization through the Activation of Nitric Oxide Pathway in Mice. PLoS One. (2010) ;5: :e12688. doi:10.1371/journal.pone.0012688 |
[26] | Soleti R , Martínez MC . Microparticles harbouring Sonic Hedgehog: Role in angiogenesis regulation. Cell Adh Migr n.d. 3: :293–5. |
[27] | Tichy J , Zinke J , Bunz B , Meyermann R , Harter PN , Mittelbronn M . Expression Profile of Sonic Hedgehog Pathway Members in the Developing Human Fetal Brain. Biomed Res Int. (2015) ;2015: :494269. doi:10.1155/2015/494269 |
[28] | Cohen M , Briscoe J , Blassberg R . Morphogen interpretation: The transcriptional logic of neural tube patterning. Curr Opin Genet Dev. (2013) ;23: :423–8. doi:10.1016/j.gde.2013.04.003 |
[29] | Kugler MC , Joyner AL , Loomis CA , Munger JS . Sonic hedgehog signaling in the lung. From development to disease. Am J Respir Cell Mol Biol. (2015) ;52: :1–13. doi:10.1165/rcmb.2014-0132TR |
[30] | Zhulyn O , Li D , Deimling S , Vakili NA , Mo R , Puviindran V , et al. A switch from low to high Shh activity regulates establishment of limb progenitors and signaling centers. Dev Cell. (2014) ;29: :241–9. doi:10.1016/j.devcel.2014.03.002 |
[31] | Hui C , Angers S . Gli Proteins in Development and Disease. Annu Rev Cell Dev Biol. (2011) ;27: :513–37. doi:10.1146/annurev-cellbio-092910-154048 |
[32] | Wang H , Ge G , Uchida Y , Luu B , Ahn S . Gli3 is required for maintenance and fate specification of cortical progenitors. J Neurosci. (2011) ;31: :6440–8. doi:10.1523/JNEUROSCI.4892-10.2011 |
[33] | Carpenter D , Stone DM , Brush J , Ryan A , Armanini M , Frantz G , et al. Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc Natl Acad Sci U S A. (1998) ;95: :13630–4. |
[34] | Dave RK , Ellis T , Toumpas MC , Robson JP , Julian E , Adolphe C , et al. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS One. (2011) ;6: :e14860. doi:10.1371/journal.pone.0014680 |
[35] | Ingham PW , Nystedt S , Nakano Y , Brown W , Stark D , van den Heuvel M , et al. Patched represses the Hedgehog signalling pathway by promoting modification of the Smoothened protein. Curr Biol. (2000) ;10: :1315–8. doi:10.1016/S0960-9822(00)00755-7 |
[36] | Rohatgi R , Milenkovic L , Scott MP . Patched1 regulates hedgehog signaling at the primary cilium. Science. (2007) ;317: :372–6. doi:10.1126/science.1139740 |
[37] | Mulieri PJ , Kang J-S , Sassoon DA , Krauss RS . Expression of the boc gene during murine embryogenesis. Dev Dyn. (2002) ;223: :379–88. doi:10.1002/dvdy.10063 |
[38] | Mulieri PJ , Okada A , Sassoon DA , Mcconnell SK , Krauss RS . Developmental expression pattern of thecdo gene. Dev Dyn. (2000) ;219: :40–9. doi:10.1002/1097-0177(2000)9999:9999<::AID-DVDY1032>3.0.CO;2-M |
[39] | Zhang W , Yi M-J , Chen X , Cole F , Krauss RS , Kang J-S . Cortical Thinning and Hydrocephalus in Mice Lacking the Immunoglobulin Superfamily Member CDO. Mol Cell Biol. (2006) ;26: :3764–72. doi:10.1128/MCB.26.10.3764-3772.2006 |
[40] | Humke EW , Dorn KV , Milenkovic L , Scott MP , Rohatgi R . The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. (2010) ;24: :670–82. doi:10.1101/gad.1902910 |
[41] | Liu X , Wang X , Du W , Chen L , Wang G , Cui Y , et al. Suppressor of fused (Sufu) represses Gli1 transcription and nuclear accumulation, inhibits glioma cell proliferation, invasion and vasculogenic mimicry, improving glioma chemo-sensitivity and prognosis. Oncotarget. (2014) ;5: :11681–94. |
[42] | Chen M-H , Wilson CW , Li Y-J , Lo Law KK , Lu C-S , Gacayan R , et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. (2009) ;23: :1910–28. doi:10.1101/gad.1794109 |
[43] | Yabut OR , Fernandez G , Huynh T , Yoon K , Pleasure SJ . Suppressor of Fused Is Critical for Maintenance of Neuronal Progenitor Identity during Corticogenesis. Cell Rep. (2015) ;12: :2021–34. doi:10.1016/j.celre2015.08.031 |
[44] | Chiang C , Litingtung Y , Lee E , Young KE , Corden JL , Westphal H , et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. (1996) ;383: :407–13. doi:10.1038/383407a0 |
[45] | Rash BG , Grove EA . Patterning the dorsal telencephalon: A role for sonic hedgehog? J Neurosci. (2007) ;27: :11595–603. doi:10.1523/JNEUROSCI.3204-07.2007 |
[46] | Machold R , Hayashi S , Rutlin M , Muzumdar MD , Nery S , Corbin JG , et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. (2003) ;39: :937–50. |
[47] | Xu Q , Wonders CP , Anderson SA . Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon. Development. (2005) ;132: :4987–98. doi:10.1242/dev.02090 |
[48] | Palma V , Ruiz i Altaba A . Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. (2004) ;131: :337–45. doi:10.1242/dev.00930 |
[49] | Shikata Y , Okada T , Hashimoto M , Ellis T , Matsumaru D , Shiroishi T , et al. Ptch1-mediated dosage-dependent action of Shh signaling regulates neural progenitor development at late gestational stages. Dev Biol. (2011) ;349: :147–59. doi:10.1016/j.ydbio.2010.10.014 |
[50] | Yabut O , Ng H , Fernandez G , Yoon K , Kuhn J , Pleasure S . Loss of Suppressor of Fused in Mid-Corticogenesis Leads to the Expansion of Intermediate Progenitors. J Dev Biol. (2016) ;4: :29. doi:10.3390/jdb4040029 |
[51] | Wilson SL , Wilson JP , Wang C , Wang B , McConnell SK . Primary cilia and Gli3 activity regulate cerebral cortical size. Dev Neurobiol. (2012) ;72: :1196–212. doi:10.1002/dneu.20985 |
[52] | Jeong J , Mao J , Tenzen T , Kottmann AH , McMahon AP . Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. (2004) ;18: :937–51. doi:10.1101/gad.1190304 |
[53] | Wang L , Hou S , Han Y-G . Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat Neurosci. (2016) . doi:10.1038/nn.4307 |
[54] | Hansen DV , Lui JH , Parker PRL , Kriegstein AR . Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. (2010) ;464: :554–61. doi:10.1038/nature08845 |
[55] | Wang X , Tsai J-W , LaMonica B , Kriegstein AR . A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci. (2011) ;14: :555–61. doi:10.1038/nn.2807 |
[56] | Molnár Z , Métin C , Stoykova A , Tarabykin V , Price DJ , Francis F , et al. Comparative aspects of cerebral cortical development. Eur J Neurosci. (2006) ;23: :921–34. doi:10.1111/j.1460-9568.2006.04611.x |
[57] | Telley L , Govindan S , Prados J , Stevant I , Nef S , Dermitzakis E , et al. Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science. (1443) ;351: :1443–6. doi:10.1126/science.aad8361 |
[58] | Johnson MB , Wang PP , Atabay KD , Murphy EA , Doan RN , Hecht JL , et al. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci. (2015) ;18: :637–46. doi:10.1038/nn.3980 |
[59] | Pollen AA , Nowakowski TJ , Chen J , Retallack H , Sandoval-Espinosa C , Nicholas CR , et al. Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell. (2015) ;163: :55–67. doi:10.1016/J.CELL.2015.09.004 |
[60] | Thomsen ER , Mich JK , Yao Z , Hodge RD , Doyle AM , Jang S , et al. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat Methods. (2016) ;13: :87–93. doi:10.1038/nmeth.3629 |
[61] | Gorski JA , Talley T , Qiu M , Puelles L , Rubenstein JLR , Jones KR . Cortical Excitatory Neurons and Glia, But Not GABAergic Neurons, Are Produced in the Emx1-Expressing Lineage. J Neurosci. (2002) ;22: :6309–14. |
[62] | Amaniti E-M , Fu C , Lewis S , Saisana M , Magnani D , Mason JO , et al. Expansion of the Piriform Cortex Contributes to Corticothalamic Pathfinding Defects in Gli3 Conditional Mutants. Cereb Cortex. (2013) :bht244. doi:10.1093/cercor/bht244 |
[63] | Letinic K , Zoncu R , Rakic P . Origin of GABAergic neurons in the human neocortex. Nature. (2002) ;417: :645–9. doi:10.1038/nature00779 |
[64] | Al-Jaberi N , Lindsay S , Sarma S , Bayatti N , Clowry GJ . The Early Fetal Development of Human Neocortical GABAergic Interneurons. Cereb Cortex. (2015) ;25: :631–45. doi:10.1093/cercor/bht254 |
[65] | Yu X , Zecevic N . Dorsal Radial Glial Cells Have the Potential to Generate Cortical Interneurons in Human But Not in Mouse Brain. J Neurosci. (2413) ;31: :2413–20. doi:10.1523/JNEUROSCI.5249-10.2011 |
[66] | Zecevic N , Hu F , Jakovcevski I . Interneurons in the developing human neocortex. Dev Neurobiol. (2011) ;71: :18–33. doi:10.1002/dneu.20812 |
[67] | Jakovcevski I , Mayer N , Zecevic N . Multiple Origins of Human Neocortical Interneurons Are Supported by Distinct Expression of Transcription Factors. Cereb Cortex. (1771) ;21: :1771–82. doi:10.1093/cercor/bhq245 |
[68] | Kang HJ , Kawasawa YI , Cheng F , Zhu Y , Xu X , Li M , et al. Spatio-temporal transcriptome of the human brain. Nature. (2011) ;478: :483–9. doi:10.1038/nature10523 |
[69] | Miller JA , Ding S-L , Sunkin SM , Smith KA , Ng L , Szafer A , et al. Transcriptional landscape of the prenatal human brain. Nature. (2014) ;508: :199–206. doi:10.1038/nature13185 |
[70] | Radonjić NV , Memi F , Ortega JA , Glidden N , Zhan H , Zecevic N . The Role of Sonic Hedgehog in the Specification of Human Cortical Progenitors In Vitro. Cereb Cortex. (2016) ;26: :131–43. doi:10.1093/cercor/bhu183 |
[71] | Lu QR , Yuk D , Alberta JA , Zhu Z , Pawlitzky I , Chan J , et al. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron (2000) ;25: :317–29. |
[72] | Kessaris N , Fogarty M , Iannarelli P , Grist M , Wegner M , Richardson WD . Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. (2006) ;9: :173–9. doi:10.1038/nn1620 |
[73] | Tripathi RB , Clarke LE , Burzomato V , Kessaris N , Anderson PN , Attwell D , et al. Dorsally and ventrally derived oligodendrocytes have similar electrical properties but myelinate preferred tracts. J Neurosci. (2011) ;31: :6809–19. doi:10.1523/JNEUROSCI.6474-10.2011 |
[74] | Crawford AH , Tripathi RB , Richardson WD , Franklin RJM . Developmental Origin of Oligodendrocyte Lineage Cells Determines Response to Demyelination and Susceptibility to Age-Associated Functional Decline. Cell Rep. (2016) ;15: :761–73. doi:10.1016/j.celre2016.03.069 |
[75] | Araujo GLL , Araujo JAM , Schroeder T , Tort ABL , Costa MR . Sonic hedgehog signaling regulates mode of cell division of early cerebral cortex progenitors and increases astrogliogenesis. Front Cell Neurosci. 77 ((2014) ;8: doi:10.3389/fncel.2014.00077 |
[76] | Murray K , Calaora V , Rottkamp C , Guicherit O , Dubois-Dalcq M . Sonic Hedgehog Is a Potent Inducer of Rat Oligodendrocyte Development from Cortical Precursors in Vitro. Mol Cell Neurosci. (2002) ;19: :320–32. doi:10.1006/mcne.2001.1079 |
[77] | Ortega JA , Radonjić NV , Zecevic N . Sonic hedgehog promotes generation and maintenance of human forebrain Olig2 progenitors. Front Cell Neurosci. (2013) ;7: :254. doi:10.3389/fncel.2013.00254 |
[78] | Tong CK , Fuentealba LC , Shah JK , Lindquist RA , Ihrie RA , Guinto CD , et al. A Dorsal SHH-Dependent Domain in the V-SVZ Produces Large Numbers of Oligodendroglial Lineage Cells in the Postnatal Brain. Stem Cell Reports. (2015) ;5: :461–70. doi:10.1016/j.stemcr.2015.08.013 |
[79] | Furutachi S , Miya H , Watanabe T , Kawai H , Yamasaki N , Harada Y , et al. Slowly dividing neural progenitors are an embryonic origin of adult neural stem cells. Nat Neurosci. (2015) ;18: :657–65. doi:10.1038/nn.3989 |
[80] | Pozniak CD , Langseth AJ , Dijkgraaf GJP , Choe Y , Werb Z , Pleasure SJ . Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing Suppressor of Fused expression. Proc Natl Acad Sci U S A. (2179) ;107: :5–800. doi:10.1073/pnas.1016485107 |
[81] | Nanni L , Ming JE , Bocian M , Steinhaus K , Bianchi DW , Die-Smulders C , et al. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. (1999) ;8: :2479–88. |
[82] | Ming JE , Kaupas ME , Roessler E , Brunner HG , Golabi M , Tekin M , et al. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. (2002) ;110: :297–301. doi:10.1007/s00439-002-0695-5 |
[83] | Bae G-U , Domené S , Roessler E , Schachter K , Kang J-S , Muenke M , et al. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet. (2011) ;89: :231–40. doi:10.1016/j.ajhg.2011.07.001 |
[84] | Elson E , Perveen R , Donnai D , Wall S , Black GCM . De novo GLI3 mutation in acrocallosal syndrome: Broadening the phenotypic spectrum of GLI3 defects and overlap with murine models. J Med Genet. (2002) ;39: :804–6. |
[85] | Démurger F , Ichkou A , Mougou-Zerelli S , Le Merrer M , Goudefroye G , Delezoide A-L , et al. New insights into genotype-phenotype correlation for GLI3 mutations. Eur J Hum Genet. (2015) ;23: :92–102. doi:10.1038/ejhg.2014.62 |
[86] | Sasai N , Briscoe J . Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip Rev Dev Biol. (2012) ;1: :753–72. doi:10.1002/wdev.43 |
[87] | Hu WF , Pomp O , Ben-Omran T , Kodani A , Henke K , Mochida GH , et al. Katanin p80 Regulates Human Cortical Development by Limiting Centriole and Cilia Number. Neuron. (1240) ;84: :1470–57. doi:10.1016/j.neuron.2014.12.017 |
[88] | Willaredt MA , Hasenpusch-Theil K , Gardner HAR , Kitanovic I , Hirschfeld-Warneken VC , Gojak CP , et al. A Crucial Role for Primary Cilia in Cortical Morphogenesis. J Neurosci. (1288) ;28: :7–900. doi:10.1523/JNEUROSCI.2084-08.2008 |
[89] | Youn YH , Han Y-G . Primary Cilia in Brain Development and Diseases. Am J Pathol. (2018) ;188: :11–22. doi:10.1016/j.ajpath.2017.08.031 |
[90] | Trazzi S , Mitrugno VM , Valli E , Fuchs C , Rizzi S , Guidi S , et al. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum Mol Genet. (1560) ;20: :1560–73. doi:10.1093/hmg/ddr033 |
[91] | Das I , Park J-M , Shin JH , Jeon SK , Lorenzi H , Linden DJ , et al. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci Transl Med. (2013) ;5: :201ra120. doi:10.1126/scitranslmed.3005983 |
[92] | Wegiel J , Kuchna I , Nowicki K , Imaki H , Wegiel J , Marchi E , et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. (2010) ;119: :755–70. doi:10.1007/s00401-010-0655-4 |
[93] | Fame RM , MacDonald JL , Macklis JD . Development, specification, and diversity of callosal projection neurons. Trends Neurosci. (2011) ;34: :41–50. doi:10.1016/j.tins.2010.10.002 |
[94] | Sacco R , Cacci E , Novarino G . Neural stem cells in neuropsychiatric disorders. Curr Opin Neurobiol. (2018) ;48: :131–8. doi:10.1016/j.conb.2017.12.005 |
[95] | Di Lullo E , Kriegstein AR . The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. (2017) ;18: :573–84. doi:10.1038/nrn.2017.107 |

