
Mitochondrial Metabolism-Mediated Regulation of Adult Neurogenesis
Abstract
The life-long generation of new neurons from radial glia-like neural stem cells (NSCs) is achieved through a stereotypic developmental sequence that requires precise regulatory mechanisms to prevent exhaustion or uncontrolled growth of the stem cell pool. Cellular metabolism is the new kid on the block of adult neurogenesis research and the identity of stage-specific metabolic programs and their impact on neurogenesis turns out to be an emerging research topic in the field. Mitochondrial metabolism is best known for energy production but it contains a great deal more. Mitochondria are key players in a variety of cellular processes including ATP synthesis through functional coupling of the electron transport chain and oxidative phosphorylation, recycling of hydrogen carriers, biosynthesis of cellular building blocks, and generation of reactive oxygen species that can modulate signaling pathways in a redox-dependent fashion. In this review, I will discuss recent findings describing stage-specific modulations of mitochondrial metabolism within the adult NSC lineage, emphasizing its importance for NSC self-renewal, proliferation of neural stem and progenitor cells (NSPCs), cell fate decisions, and differentiation and maturation of newborn neurons. I will furthermore summarize the important role of mitochondrial dysfunction in tissue regeneration and ageing, suggesting it as a potential therapeutic target for regenerative medicine practice.
INTRODUCTION
Most adult tissues retain a reservoir of self-renewing, multipotent stem cells that are capable of generating specific tissue components. It has been long thought that the brain is an exception to this rule and that the pool of neural stem cells (NSCs) is exhausted perinatally leading to a decrease in neurogenesis after birth. While this is certainly true for most brain regions, neurogenesis persists into adulthood in two specific micro-environments under physiological conditions: the subependymal zone (SEZ) of the lateral ventricle and the subgranular zone (SGZ) of the dentate gyrus (DG), leading to modifications of the olfactory bulb and hippocampal circuitry, respectively [1–3]. The continuous generation and integration of new neurons into existing circuitries is a key contribution to the adult’s brain plasticity. It allows for cellular and molecular remodeling in response to an individual’s interaction with the outside world and involves higher cognitive function such as memory processes as well as certain affective behaviors.
New neurons are generated at a considerable level also in the human hippocampus [4, 5]. From a clinical point of view adult neurogenesis is highly relevant as dysregulation is evolving as a significant contributor to neuropsychiatric symptoms in ageing and neurodegenerative diseases [6].Conversely, cumulative evidence from organs with high turnover suggests that ageing is a major contributor to loss or inactivation of stem cells [7] and to decreased neurogenesis [8, 9]. Consequently, there is an immense attention to both understanding the mechanisms of impaired neurogenesis as well as developing strategies to enhance neurogenesis duringageing.
Adult neurogenesis is a stereotypic sequence of distinct developmental steps: astrocytes with a highly characteristic radial morphology serve as quiescent NSCs. Upon activation, NSCs divide to generate actively proliferating intermediate progenitor cells (IPCs) that eventually give rise to neuronally committed neuroblasts (NBs). NBs in the hippocampus exit the cell cycle and enter the maturation phase during which dendrites are extended into the molecular layer and axons are sent to the CA3 region [2, 3]. In the SEZ, NBs migrate a long way through the rostral migratory stream to the olfactory bulb, where they eventually exit cell cycle and populate the glomerular and granular cell layer [10]. Despite a number of region-specific characteristics in the neurogenic lineage, both niches share basic principles, such as a stereotypic sequence of proliferation, differentiation, migration and maturation steps as well as embedding into a niche with specialized cellular cytoarchitecture, vascularization pattern, and extracellular matrix properties [1–3].
METABOLIC REGULATION OF NEUROGENESIS
The generation of a newborn neuron from a quiescent NSC is controlled by signals from the surrounding niche as well as intracellular pathways [1]. While tremendous effort has been taken in investigating the roles of signaling-, transcriptional-, and epigenetic pathways in controlling lineage progression [11], the discovery that a metabolic switch towards de novo lipogenesis is required for the proliferation of quiescent NSCs provided exciting first time evidence that hippocampal neurogenesis is functionally coupled to the activity of a specific metabolic program [12]. The role of lipid metabolism, including beta-oxidation, in the process of adult neurogenesis is reviewed in detail in this issue by MarlenKnobloch.
Differentiation from a quiescent radial glia-like NSC into a functional neuron requires coordinated remodeling of cell biological, biochemical and molecular pathways according to the changing demands of the developing neuron. It is appropriate to assume that this also comprises metabolic changes. While fundamental differences regarding the metabolic requirements between proliferative and differentiated cells have long been reported especially in the context of cancer cells [13], it is a fairly new concept that differential activity of metaboliccircuits - rather than being a passive response to cellular function - may represent a major driving force to attain or maintain a specific phenotype [14].
Metabolic differences may not only apply to proliferating versus non-proliferating cells, but also differentiated cells drastically differ in their metabolic requirements. Neurons have the highest energetic demands in the brain. Maintenance and restoration of ion gradients dissipated by the repeated generation of postsynaptic potentials and action potentials, and the neurotransmitter cycle are the main cause of neuronal energy need [15, 16]. Neurons display a high activity of oxidative metabolism and are metabolically tightly coupled to astrocytes. The recycling of neurotransmitters and metabolites in astrocytes is associated with significant metabolic costs. Astrocytes mostly satisfy their energetic demands via glycolysis. Being in close contact with the blood vessels, they take up glucose from the circulation, metabolize it into lactate via glycolysis and conversion of pyruvate, and release lactate into the extracellular space, from where it is taken up by neurons [17–19]. The profound differences in the prevalent metabolic circuits of highly specialized astrocyte-like NSCs (the virtual starting points of the neurogenic lineage) and postmitotic differentiated neurons (the outcome of the neurogenic lineage) suggest that metabolic adaption will be important during the course of adult neurogenesis for the generation and maturation of adult-bornneurons.
In the following I will give a r
METABOLIC REQUIREMENTS OF NEURAL STEM CELLS
Many quiescent stem cells rely on glycolysis [20]. It is hypothesized that this fits well with the lower energetic demands of the quiescent state and helps to limit oxidative metabolism-dependent generation of reactive oxygen species (ROS) in order to confine ROS-induced cellular damage and ensure life-long tissue renewal. Though glycolysis is much less efficient in terms of energy production, the generation of ATP does not appear to cause limitations in proliferating cells [21]. It may instead be advantageous for the generation of new daughter cells to maintain the pool of carbon intermediates required for biosynthesis of cellular building blocks by utilization of glycolysis [22]. Most precursors for essential biosynthetic pathways including purine and pyrimidine nucleotide, amino acids and triacylglycerols derive from intermediates of glycolytic and pentose phosphatase pathways [23].
Along with their glycolytic profile, stem cells often display an immature mitochondrial infrastructure characterized by immature spherical mitochondria with poorly developed cristae and low copy numbers of mitochondrial DNA [24]. Consequently, many stem cells display lower levels of oxidative respiration than their differentiated counterparts [25–31]. These observations also account for quiescent NSCs in the adult brain. Evidence from single cell transcriptome analysis of early stages of the hippocampal neurogenic lineage suggests beta-oxidation and glycolysis as the main energy-providing metabolic circuits in NSCs [32]. NSCs in the adult SEZ have been shown to mostly rely on fatty acid oxidation to produce energy and support their neurogenic activity [33], while metabolic analysis performed directly on NSC-containing embryonic cortical tissue suggested mostly (80–90%) non-mitochondrial ATP generation i.e. glycolysis [34]. The glycolytic profile of NSCs appears to be evolutionary conserved. Data from Jürgen Knoblich’s laboratory on lineage progression in the brain of Drosophila melanogaster suggest the interesting concept that induction of electron transport chain (ETC) and oxidative phosphorylation (oxPhos) in neuroblasts (as NSCs are called in Drosophila) is required to uncouple cell cycle from cell growth, leading to the terminal differentiation of stem cells. In that study, the authors further hypothesize that the metabolic switch from glycolysis to oxidative metabolism may be the cause and not just the consequence of differentiation [35]. Also neural progenitor cells in the Xenopus retina are less reliant on oxPhos than non-dividing differentiated neurons, and the transition from glycolysis to oxidative metabolisms is tightly coupled to neuronal differentiation [36]. In line with this, metabolic signature analysis of distinct stages within the programming process of human neural stem and progenitor cells (NSPCs) derived from embryonic stem cells or induced pluripotent stem cells (iPSCs) towards neurons suggests that the transition from aerobic glycolysis of NSCs toward neuronal oxPhos is marked by the downregulation of hexokinase and lactate dehydrogenase, essential components of the glycolytic pathway. Even more, the shut-off of glycolysis appeared to be essential for neuronal survival coupling neuronal differentiation with distinct metabolic reprogramming [37]. In the adult mouse hippocampus, electron microscopic studies showed that radial glia-like NSCs contained both tubular and spherical mitochondria with a significant smaller volume than differentiated neurons [38]. Furthermore cultured adult NSPCs have a significantly lower membrane potential than their neuronal counterparts indicative of low oxidative metabolisms. Intriguingly, disruption of ETC and oxPhos activity by conditionally deleting the mitochondrial transcription factor A (Tfam) in NSCs had no effect on NSC number. Even though proliferation and generation of new neurons were severely affected, the activation of NSCs remains intact, suggesting that a dysfunctional ETC and oxPhos machinery rather blocked neurogenesis at the level of actively proliferating IPC cells [38]. Hence, it appears as if NSCs live surprisingly well without oxidative metabolism as they meet most of their energetic demands and derive carbon intermediates from pathways others than ETC and oxPhos. It also seems as if upregulation of ETC and oxPhos activity plays a major role in further lineage progression, in exit from cell cycle and in differentiation as well as maturation of daughtercells.
These data implicate that adaption of metabolic programs will become important for reprogramming protocols as already suggested by the work of laboratories of Magdalena Götz [39], Fred Gage and Tony Hunter [37]. Zheng et al., provide evidence that shut-off of glycolysis is critical for survival of neurons during neuronal reprogramming of NSPCs [37]. Recently, Gascon, Murenu et al. have shown that high levels of oxidative stress induces cell death and prevents the direct conversion of different cell types into neurons [39]. By inhibition of ferroptosis, an atypical form of cell death resulting from an iron-dependent accumulation of lipid ROS, the authors could significantly improve neuronal reprogramming. Furthermore, antioxidants treatment improved maturation of induced neurons in vitro and in vivo [39]. The latest findings underscore the significance of metabolic pathways in cellular reprogramming irrespective of cell types.
MITOCHONDRIAL METABOLISM IN ACTIVELY PROLIFERATING PROGENITOR CELLS
Recent work provides in vivo functional evidence supported by gene expression profiling that the adult hippocampal neurogenic lineage is critically dependent on mitochondrial complex function at the level of IPCs [38]. Transcriptional signature suggests that the transition of activated NSCs to IPCs is paralleled by upregulation of enzymes of the TCA cycle and components of mitochondrial complexes, most notably of genes related to complex V, the key complex for ATP production via oxPhos. In contrast, key enzymes of glycolysis are downregulated around the time of NSC activation, predicting a metabolic switch from glycolytic metabolism towards increased activity of ETC and oxPhos around the time of transition from activated NSCs to IPCs. The new energy demand of activated NSCs and IPCs might well be connected to the dramatic onset of translation [40] and the marked cell growth illustrated by Costa et al., [41]. Consistent with the single-cell transcriptome data, a severe loss of IPCs and subsequently of immature neuronal progeny upon conditional deletion of Tfam in NSCs was observed. Increased appearance of apoptotic cells in the hippocampal niche and massively increased cell death of proliferating NSPC in culture compared to BMP4-induced quiescent NSPCs suggest that impaired ETC and oxPhos activity is critical for viability of IPCs [38]. Thus, these data strongly support the emerging notion that stage-specific metabolic programs are functionally linked to distinct developmental steps within the NSC lineage [42–45].
Since high oxidative activity has up to now been mostly associated with termination of proliferation and neuronal differentiation [42–45], it was quite surprising that actively proliferating IPCs reacted in such dramatic way to ETC and oxPhos dysfunction. So, why is mitochondrial metabolism crucial for the highly proliferative IPCs in contrast to rarely proliferating NSCs? What is the specific metabolic need of IPCs that can be served by ETC and oxPhos? In general, ATP is not limiting in proliferating cells, and glycolysis as well as the pentose-phosphate pathway are better suited to meet the requirements for cellular building blocks including purine and pyrimidine nucleotides, amino acids and triacylglycerols. Two recently published papers provided a new and surprising perspective on the function of ETC in highly proliferating cancer cells [46, 47]. Although ETC is best known for its role in ATP synthesis, both groups showed independently that the central function of ETC in proliferating cells is the production of aspartate. These results solved a long-known mystery that cells lacking mtDNA failed to proliferate as a result of ETC dysfunction, which could be rescued by adding supraphysiological levels of pyruvate to the medium [48]. More than 25 years after this finding, Birsoy et al., and Sullivan et al., convincingly showed that ETC dysfunction impairs the redox balance of the cell and that pyruvate can restore redox homeostasis by serving as an electron acceptor. This regeneration of electron acceptors is required for the biosynthesis of the limiting amino acid aspartate in order to sustain high levels of proliferation [46, 47]. It will certainly be very interesting to investigate if hippocampal IPCs need ETC and aspartate to sustain their proliferative activity.
ROLE OF MITOCHONDRIA IN NEURONAL DIFFERENTIATION AND NEURITE GROWTH
Mitochondria play an important role in neuroplasticity and are involved in fundamental processes such as neural differentiation, growth of axons and dendrites, as well as formation of synapses and neurotransmitter release [49, 50]. First evidence for a role of mitochondria in neuronal differentiation came from the observation that differentiation of neurons is associated with an increase in mitochondrial mass [51, 52]. More recent findings show that differentiation from NSCs and other progenitor types generally involves increased aerobic activity, including upregulation of mitochondrial mass, mtDNA copy number, ETC capacity and activity of NADH-generating enzymes [49, 53]. The accumulation of mtDNA damage in NSCs deficient for the mitochondrial 8-oxoguanine DNA glycolsylase (OGG1) inhibits the normal maturation of mitochondria leading to inhibition of aerobic metabolism during differentiation [53]. This results in a preferential shift of ogg-/- NSCs in their differentiation direction towards an astrocytic lineage, while anti-oxidative treatment reverses the mtDNA damage accumulation and increase neurogenesis [54]. The underlying mechanism for this change in differentiation involves a redox shift that activates Sirt1 that in turn promote astrogliogenesis [50, 54, 55]. These results implicate that differentiation of progeny per se is not necessarily affected by dysfunction of mitochondrial metabolism as astrocyte generation and maturation appear normal. Instead it seems to be particularly important for neuronal differentiation and development. It is tempting to speculate if the switch to oxidative metabolism may even play an instructive role in driving differentiation toward the neuronal fate.
During axogenesis, mitochondria accumulate at the base of the developing neurites that are destined to become axons, and depletion of mitochondria at or before the stage of axogenesis prevents axon formation [56]. This indicates already that mitochondria play an important role in neural polarization and axonal outgrowth regulation. It has been further shown that mitochondria coordinate the sites of axon branching through localized intra-axonal protein synthesis [57]. Mitochondria are also required for dendritic development. Early studies found that mitochondria in dendrites are more active than those in the axons [58]. Disruption of mitochondrial protein translation in Drosophila olfactory projection neurons reduces dendritic arborization, while axonal morphology is relatively unaltered [59]. Moreover, mitochondrial distribution along dendrites is critical for synaptic support. Here, mitochondrial calcium buffering strongly influence synaptic plasticity as mice deficient for voltage-dependent anion channels (VDAC) located on the outer mitochondrial membrane show learning deficits and impairment of synaptic plasticity [60].
In the adult hippocampal neurogenic lineage, deletion of Tfam specifically in IPCs and neuroblasts by stereotactic injections of Cre-recombinase encoding mouse moloney leukemia virus in TfamloxP/loxP mice has a significant effect on neuronal maturation, in particular, dendritic growth. Tfam-ablated neurons display shortening of dendrites and decreased dendritic complexity [38]. Similar results have been obtained in mice lacking the cytoplasmic polyadenylation element binding protein 1 (CPEB1), which develop brain-specific dysfunctional mitochondria and a reduced ATP-production. This is due to defective polyadenylation-induced translation of ETC complex I protein NDUFV2 mRNA and results in defective dendritic morphogenesis of hippocampal neurons [61]. These studies indicate that functional ETC and oxPhos is required for the proper morphological development and growths of adult-bornneurons.
TRANSCRIPTION FACTOR- AND SIGNALING PATHWAY-MEDIATED MITOCHONDRIAL METABOLISM RESPONSE IN NEURONAL DEVELOPMENT
Mitochondria are hubs in which metabolism merges with transcription factor- and signaling pathway-mediated regulatory mechanisms. Cyclic AMP response element binding protein (CREB) is one such example of a neuronal activity-regulated transcription factor with a direct link to regulation of metabolic pathways in other cellular systems such as liver and lung. CREB-signaling is highly active in immature adult-generated neurons and CREB downregulation disturbs the maintenance of the neurogenic expression program and impairs dendritic development and survival of newborn neurons [62–66]. Additionally, CREB regulates the transcription of enzymes, which catalyze key steps of glucose metabolism [67] and impacts on mitochondrial biogenesis and mitochondrial respiratory chain function by directly controlling the expression of nuclear-encoded respiratory genes and through induction of expression of PGC1alpha and central transcriptional regulators of mitochondrial biogenesis, including Tfam [68].
Bit by bit, an increasing number of signaling functions for mitochondria are being discovered. Brain derived neurotrophic factor (BNDF) was shown to promote synaptic plasticity, in part, by enhancing mitochondrial energy production. In response to enhanced energy demands in cultured cortical neurons, BNDF increases glucose utilization and mitochondrial respiratory coupling at complex I [69, 70]. Also activity of the mammalian target of rapamycin (mTOR) mTOR has a direct links to the regulation of metabolic pathways in other cellular systems. By representing a metabolic rheostat mTOR integrates nutrient availability and growth factor signaling, and regulates mitochondrial biogenesis, and lipid and glucose metabolism through its transcriptional and translational targets [14]. The connection between mTOR-, BDNF and CREB-dependent signaling and adult-born neuron maturation and their regulatory function in metabolism strongly suggest that metabolic adaptation is closely linked to neuronal maturation in adult neurogenesis. But what about the converse scenario? Does neuronal activity impact on mitochondrial turnover? Do newborn neurons stimulated by local interneurons develop a mitochondrial metabolism different from that of unstimulatednewborn neurons? While certainly little is known, an interesting venue for future research is if and how neuronal activity regulates mitochondrial function.
MITOCHONDRIAL QUALITY CONTROL MECHANISMS IN NEURAL DEVELOPMENT
Along with the evolutionary process, mitochondria acquired an efficient system for quality control that detects and eliminates damaged mitochondria. Mitochondrial quality control involves both intra-mitochondrial and organellar mechanisms. The major role of intra-mitochondrial quality control is undoubtedly the maintenance of mtDNA. The organellar mitochondrial quality control system regulates mitochondrial integrity and dynamics involving mitochondrial membrane fusion and fission as well as selective mitophagy. Mitochondrial biogenesis and mitochondrial dynamics, i.e., the balance between mitochondrial fusion and fission processes, are major determinants of mitochondrial morphology. Rather than being generated de novo, mitochondria are generated by growth of preexisting mitochondria and subsequent fission [71, 72]. Mitochondrial fission also leads to mitochondrial fragmentation and facilitates the removal of damaged mitochondria through mitophagy. This type of autophagy or lysosomal degradation is crucial for the elimination of damaged mitochondrial organelles or content [73]. Mitophagy is associated with the control of organelle number, in response to developmental or physiological cues, resulting in removal of excessive mitochondria through activation of fission and blockage of fusion. Mitochondrial fusion enables exchange of genetic material and protects from mitochondrial damage accumulation [74]. Under cellular stress conditions, mitochondria form a hyperfused complex to prevent damage and to protect against mitophagy. This reaction ultimately promotes cell survival. Conditional deletion of the outer mitochondrial membrane fusion proteins MFN1/2 in adult NSCs causes severe mitochondrial fragmentation and lead to significant reduction in the number of Sox2+ NSCs in the SGZ of the hippocampus [34]. Recent work identified active mitophagy as a typical feature of early-stage mouse NSC differentiation. Attenuation of mitophagy through forced translocation of the apoptotic regulatory protein p53 to mitochondria increased neurogenic potential and neuriteoutgrowth [75].
The development of new neurons is coupled to extensive remodeling of the mitochondrial compartment [76]. During maturation of newborn neurons, mitochondria increase their mass and change their distribution and shape, characterized specifically by the appearance of round-shaped mitochondria in the dendritic arbor. Increased neurogenesis by voluntary exercise - the strongest pro-neural trigger of hippocampal neurogenesis [77, 78] - induces a profound increase in mitochondrial content, especially in the dendritic segments [76]. While mitochondrial fission would be essential to efficiently supply the dendritic arbor with new mitochondria [79], genetic inhibition of the mitochondrial fission factor dynamin-related protein-1 (Drp1) activity represses adult neurogenesis under basal and pro-neurogenic voluntary exercise conditions. In contrast, overexpression of Drp1 under exercise conditions promotes neuronal maturation even more than exercise alone [76]. Recently, Drp1 has been identified as a potential therapeutic target against Bisphenol A (BPA)-induced impairment of mitochondrial dynamics and neurodegeneration in the hippocampus [80]. BPA is a component of consumer plastic products and was shown to induce mitochondrial fragmentation and oxidative stress, which led to impaired NSC proliferation and differentiation and ultimately NSC apoptosis. BPA induces increased levels of Drp1 expression, and inhibition of Drp1, pharmacologically and genetically, reverses the BPA-induces mitochondrial dysfunction and ameliorates NSC defects and apoptosis [80]. Together, these results identify mitochondria not only as an essential component for the general neurogenesis process but also ascribed an active regulatory function to mitochondria during maturation of adult born neurons.
REACTIVE OXYGEN SPECIES AND THE ROLE OF CELLULAR REDOX STATE IN NEUROGENESIS
Reactive oxygen species (ROS) are produced in the cytosol, peroxisomes and the endoplasmatic reticulum, but the ETC, in particular complex I and to minor extent complex III, is still the major site of ROS production under physiological conditions. Here, instead of reduction of O2 to H2O, oxygen is prematurely and incompletely reduced to the superoxid anion radical (O - 2, SO), that can be further dismutated to hydrogen peroxid (H2O2). The redox state of a cell is defined by dynamic changes in the ration of the interconvertible oxidized and reduced form of specific redox couples such as NAD+/NADH, NAD phosphate (NADP+/NADPH), glutathione (GSSG/2GSH), superoxid/oxygen, peroxid/water. Accumulation of oxidizing molecules such as ROS shifts the intracellular redox environment towards a more oxidized state and promotes oxidation reactions. Like in a vicious circle, extensive ROS generation or impaired detoxification can result in pathological molecular modifications to proteins, nucleic acids, and lipids, and will on the long run inflict fatal damage on the cell.
ROS also plays a role as second messengers, regulating diverse cellular processes [50, 81]. Le Belle and colleagues have shown that proliferative NSCs have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependent manner [82]. Furthermore, induction of proliferation in NSCs results in a rapid increase in total mitochondrial numbers and overall ROS production, suggesting that oxidative stress is generated during a transient window of elevated neurogenesis that accompanies the normal neurogenesis process [83]. Recent findings suggest that changes in mitochondrial dynamics direct NSC fate through modification of complex-I-mediated ROS signaling triggering a dual program to suppress self-renewal and promote differentiation via nuclear factor erythroid 2-related factor 2 (NRF2)-mediated signaling [34]. Interestingly, NSCs in the embryonic mouse cerebral cortex exhibit intermittent spontaneous bursts of ROS generation that negatively regulate NSC self-renewal in the developing cerebral cortex [84]. Moreover, elevated ROS through loss of function of the tumor suppressor gene p53 leads to early neurogenesis, while restoration of p53 and anti-oxidative treatments partially reverse the phenotype [85]. These results suggest that p53-mediated regulation of physiological ROS levels by regulating oxidative stress gene expression may be required for the appropriate timing of neurogenesis in the developing telencephalon.
p53-mediated ROS is also important for cell fate decisions as shown in a study by Xavier and colleagues. Here, p53 reduces the levels of mitochondrial ROS, which leads to a shift in neural differentiation potential, favoring neuronal rather than astroglial conversion [75]. Along this line is the finding that the induction of mild oxidative stress triggers activation of the histone deacetylase sirtuin 1 (Sirt1), which subsequently leads to the inhibition of the proneural transcription factor Mash1, resulting in an increase in astrogliogenesis at the expense of the neuronal lineage [55]. These and other data implicate mitochondrial ROS (as well as ATP levels) as a rheostat to monitor cell fates and regulate cell cycle progression [75, 86]. Indeed, there is experimental evidence that mitochondrial dysfunction activates two signals resulting in cell cycle inhibition. The decrease in ATP production is usually followed by downregulation of cyclin E while increased ROS production induced up-regulation of p27 [87]. Regulation of neurogenesis is based on very complex and context-dependent mechanisms mediated by ROS. While the majority of studies have focused on the cellular toxicities of ROS, there is growing evidence that ROS (and ROS bursts) play a crucial role in regulation of proliferation, self-renewal and cell fate decisions of NSCs [82]. Nevertheless, the production of ROS has to be precisely regulated since ROS overload impairs adult neurogenesis during ageing, neuroinflammation and neurodegeneration [88] and may therefore have significant implications for brain injury, disease and repair.
Several pathways with well-known roles in self-renewal, lineage commitment and differentiation of NSCs - such as Notch and Wnt/β-catenin - may be modulated in a redox-dependent fashion [81]. Accumulating evidence suggest a crucial role for redox-sensitive molecules such as NAD+/NADH-dependent enzymes and NRF2 in spatiotemporal control of NSC fate decisions. NRF2 up-regulates genes involved in redox signaling such as thioredoxin as well as Notch and Jagged - two key proteins of Notch pathway [89–91]. Similar to other organs like liver and lung, NRF2 is essential for tissue regeneration in the brain. Here, it regulates neurogenesis and protects neural progenitor cells against ischemia-induced damage [92]. Overexpression of NRF2in vitro increases NSPC self-renewal under proliferating conditions and supports neuronal differentiation upon withdrawal of growth factors [92]. These results indicate that NRF2 may be involved in coordination of crucial stem cell- and lineage decisions potentially via interaction with the Notch-pathway.
NAD+/NADH is one of the most important redox pair due to the strong reducing capacity of NADH [93], and its activity is mainly defined by complex I of the ETC [94]. In addition to its function as an electron carrier, NAD+ has emerged as a critical co-regulator of signaling pathways, thereby providing a link between redox state and gene expression. Depletion of NAD+ in the murine brain is associated with a loss of proliferating NSPCs [95]. Deletion of nicotin-amide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in NAD+ biosynthesis,affects G1/S-phase transition, self-renewal capacity and maintenance of the stem cell pool. Nampt is crucial for fate-commitment of NSPCs into oligodendrocytes via activity of NAD+-dependent enzymes Sirt1 and Sirt2. Since Nampt expression is decreasing in the ageing brain, the authors suggest that Nampt-mediated biosynthesis may be a mediator of age-associated decline in NSPCs [95]. In line with this is a recent report showing that NAD+ improves mitochondrial activity and stem cell function in muscle stem cells leading to rejuvenation of muscle stem cells in aged mice [96]. The authors demonstrate that NAD+ precursor nicotinamide riboside delays senescence of neural and melanocyte stem cells and enhances life span in mice. Together these findings implicate NAD+ signaling as a potential strategy to reprogram senescent or dysfunctional stem cells in order to improve life span in mammals.
Box 1:
Mitochondria - more than just ATP synthesis
Most animal cells contain mitochondria. These organelles are surrounded by a double system of membranes conveniently described as the outer membrane and inner membrane. While the outer membrane is fairly similar in most cell membranes, the inner membrane is specialized due to particles that are involved in the ATP synthesis by oxidative phosphorylation (oxPhos) and are functionally associated with the respiratory chain or electron transport chain (ETC). The mitochondrial matrix contains enzymes of the beta-oxidation pathway and also most enzymes of the Krebs cycle or tricarboxylic acid (TCA) cycle (scheme in Fig. 1). Remarkably, mitochondria contain their own genome, which is maternally inherited. Mitochondrial DNA (mtDNA) is organized in a circular, covalently closed, double-stranded DNA. 37 genes are encoded by mtDNA; of these, 22 are for transfer RNA, 2 for the small and large subunit of ribosomal DNA, and 13 are encoding for components of ETC and oxPhos complex subunits.
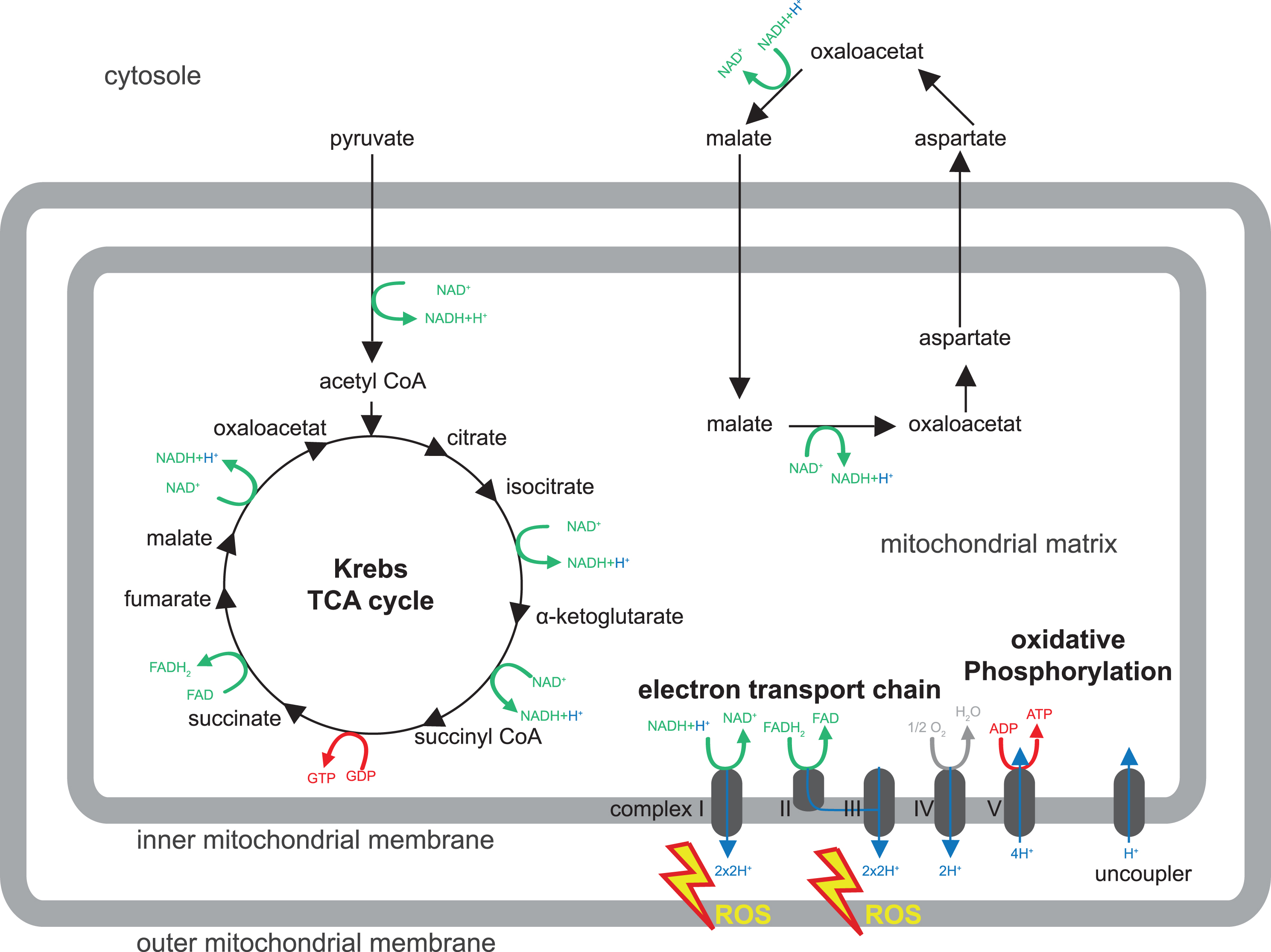
In all forms of life, ATP is used in innumerable metabolic reactions and physiological functions and it is vital to maintain a steady supply of ATP for a living cell to grow, divide, and response to challenges. In the presence of oxygen, oxPhos is by far the most important mechanism for generation of ATP. ATP synthesis via the mitochondrial respiratory chain is a result of two processes (electron transport and oxPhos) and inextricably coupled to oxidation of the reduced hydrogen carriers NADH (nicotinamide adenine dinucleotide) and FADH2 (Flavin adenine dinucleotide). Complex I - IV of the ETC operate the electron transport and involve oxidation (i.e., removal) of electrons from NADH or FADH2 coupled to pumping out protons from the mitochondrial matrix. This generates an electrochemical potential, which provides the energy for ATP synthesis by oxPhos conducted by complex V.
ATP can be transported out of the mitochondrion via the ATP/ADP translocase and used otherwise. Hydrogen carriers are unable to cross the inner mitochondrial membrane. This requires active transfer of ‘reducing equivalents’ by two mechanisms: the malate/aspartate shuttle and the glycerol phosphate shuttle (particularly important in insects). The malate/aspartate shuttle transports malate from the cytosol to the mitochondrion, where it is transformed into aspartate, which subsequently leaves the mitochondrion again to restore malate in the cytosol. These steps are accompanied by regeneration of reducing equivalents NADH and FADH2. Also the TCA cycle is important for recycling of hydrogen carriers for the generation of ATP from glucose. Glucose is taken up by the cells and oxidized through glycolysis into two molecules of pyruvate. Pyruvate is fed into the TCA cycle where it is completely oxidized into carbon dioxide. In this process, the hydrogen carriers NAD+ and FAD become reduced to NADH and FADH2, and carry hydrogen to the respiratory chain. In summary, ATP is synthesized in the inner mitochondria membrane through sequential metabolic reactions that crucially involve the recycling of hydrogen carriers in the TCA cycle and the malate/aspartate shuttle, which are needed for the respiratory chain and oxPhos (Fig. 1).
Fig.1
Mitochondrial metabolism. Shown is a schematic drawing of the major mitochondrial metabolism pathways discussed in this review (for β-oxidation please refer to Knobloch, 2017 in this issue). For details, please refer to Box 1. Pyruvate is transported from the cytosol to the mitochondrion and enters the Krebs or tricarboxylic (TCA) cycle. TCA and malate/aspartate shuttle serve to recycle hydrogen carriers required for electron transport in the inner mitochondrial membrane (complex I - IV) to generate an electrochemical potential, which provides energy for ATP synthesis by oxidative phosphorylation (complex V). Reactive oxygen species (ROS) are produced by complex I and III of the electron transport chain.
Sirtuins, an evolutionary conserved family of deacetylase, in particular Sirt1, play a key role in redox/metabolism-dependent control of NSPC fate. Sirt1 expression and activity is tightly regulated by changes in the NAD+/NADH redox state [97] and, the other way around, sirtuins are associated with a variety of cellular functions connected to redox state [98, 99]. Sirtuins act as cellular anti-oxidative defense mechanisms, and Sirt1, 2, and 3 reduce ROS damage by FoxO3-mediated activation of superoxid dismutase 2 and catalase [100–102]. During CNS development, Sirt1 is expressed at early stages and Sirt1-/- mice exhibit severe cognitive defects associated with reduced synaptic plasticity and altered activation of genes involved in synaptic function, lipid metabolism and myelination [103]. During caudal neural tube development, Sirt1 supports NSPC maintenance by de-acetylating Pax3, subsequent induction of Hes1 and inhibition of Neurogenin2 [104]. Overall, cellular redox-state critically determines NSC maintenance, self-renewal, proliferation, and differentiation program and may also play an instructive role for distinct developmental steps of the neurogenic lineage. It will therefore be crucial to determine how subcellular alterations in NAD+/NADH ration might drive cell type-specific programs i.e. through Sirt1-dependent changes in gene expression.
MITOCHONDRIAL METABOLISM IN AGEING
Mitochondrial dysfunction is postulated to be one of the hallmarks contributing to organismal ageing [105, 106]. mtDNA is the major target for age-associated mutations, most likely due to the oxidative microenvironment of mitochondria and the limitations in mtDNA protection and repair mechanisms compared to nuclear DNA [107]. While the causal implications of mtDNA mutation are controversially debated, it is certain that the mutational load of single-cells in ageing significantly increases [108]. The other way around, data from human multisystem disorders caused by mtDNA mutation partially phenocopy ageing [109]. Moreover, mice deficient for mtDNA polymerase γ exhibit aspects of premature ageing and reduced life-span [110–112]. The reduced mitochondrial bioenergetics upon ageing may be caused by a variety of mechanisms, including accumulation of mutations and deletions in mtDNA, oxidation of respiratory proteins, destabilization of the respiratory supercomplexes, changes in the lipid composition of mitochondrial membranes, imbalance of fission and fusion events resulting in alterations of mitochondrial dynamics, and limited quality control by defective mitophagy [106, 113].
Mitochondrial dysfunction was previously proposed to contribute significantly to age-associated neurogenesis deficits [49, 83, 114, 115]. Aged iPSCs from human fibroblasts fail to properly undergo neurogenesis. This is accompanied by an increase in mitochondria with aberrant mitochondrial membrane potential and altered expression of genes related to mitochondrial biogenesis [83]. Adult-derived human iPSCs accumulate mtDNA defects in an age-dependent manner, which can lead to respiratory defects. These results implicate that for therapeutical application, genetic integrity of iPSCs, especially those derived from older patients, should be monitored for their metabolic status [116]. NSPCs isolated from aged mouse forebrains display an aberrant metabolic phenotype characterized by decreased mitochondrial mass, lower oxygen consumption rates, and increased resistance to mitochondrial inhibitors [115, 117], suggesting that mitochondrial function during aging contributes to impaired neurogenesis. In line with this are recent results from our laboratory showing that deletion of Tfam in adult NSCs recapitulates hallmarks of ageing in the hippocampal neurogenic lineage, including proliferation deficits, decreased generation of new neurons, and maturation delay of newborn neurons [38]. While it is clear that mitochondrial function has a profound effect on ageing processes, it is less clear whether improving mitochondrial function can ameliorate the ageing phenotypes. In order to test if neurogenesis can be improved by boosting mitochondrial function, we administered the nootropic compound piracetam to aged animals [38]. Piracetam is used for treatment of age-related cognitive decline and dementia [118] and has been found to enhance mitochondrial respiration, ETC activity and ATP production only in cells with impaired mitochondrial functions [119, 120], while it appears to have no effect on healthy young cells. Intriguingly, administration of piracetam to aged mice significantly improved proliferation, generation and maturation of newborn neurons in vivo [38]. These findings identify mitochondrial function as a potential target to ameliorate the age-associated decline in hippocampal neurogenesis.
CONCLUSIONS
Over the last years, mitochondria turned out to be key players in regulating the balance of stem cell self-renewal, proliferation and differentiation in various tissues, including the brain. Mitochondria act through controlling energy and redox states of a cell, by providing hydrogen carriers and electron acceptors, and by generating intermediates that serve as cellular building blocks. Furthermore, mitochondria unite metabolic and signaling pathways. There is growing evidence for ROS- and redox state-mediated regulation of neurogenic pathways and gene expression on one hand, and control of mitochondrial function and dynamics through well-known neurogenic regulators like CREP and BDNF on the other hand. Mitochondrial dynamics and distribution play a crucial role in the differentiation and maturation of newborn neurons, while proliferation of progenitor cells may be dependent on biosynthesis of aspartate through regeneration of electron acceptors by ETC. Experimental findings from various organisms and from reprogramming of iPSCs to neurons support the hypothesis that a metabolic switch from glycolytic to mitochondrial oxidative metabolism may be instructive for initiation of neuronal differentiation and indispensable of the survival of newly generated neurons (Fig. 2).
Fig.2
Mitochondrial metabolism-mediated regulation of adult neurogenesis. Schematic drawing summarizing how mitochondrial metabolism influences the distinct developmental steps of adult neurogenesis: neural stem cells (NSCs) generate actively proliferating intermediate progenitor cells (IPCs). IPCs give rise to neuronally committed neuroblasts (NBs) that differentiated into mature neurons. NSCs also generate new astrocytes. Impact of mitochondrial functions on NSC self-renewal, NSCs and IPCs proliferation, differentiation and maturation of progeny is illustrated.
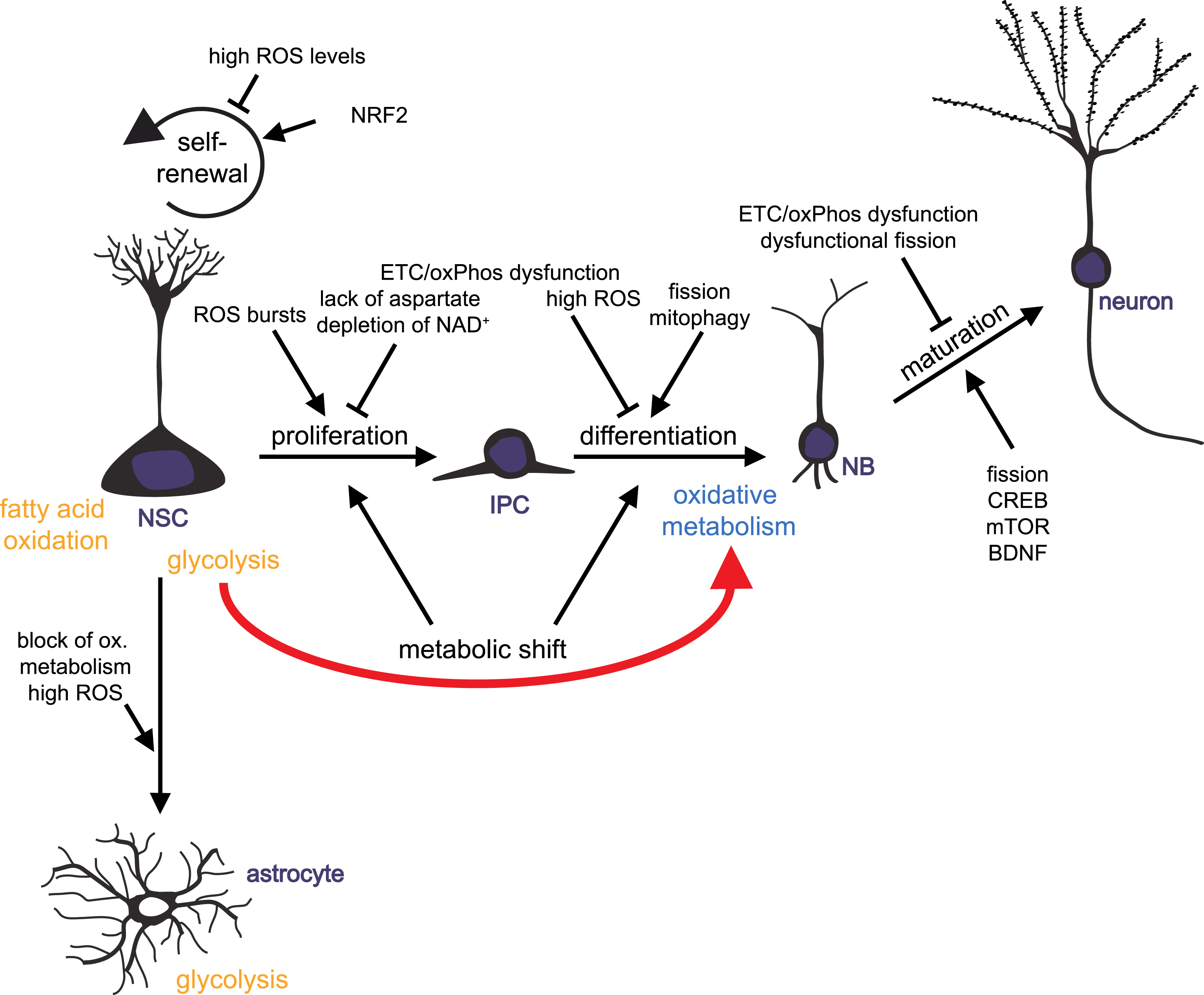
At the recent state of knowledge, it is crucially important to understand how distinct metabolic profiles and shifts within metabolic pathways are linked to distinct cellular stages, to cell fate decisions and to differentiation processes in order to understand what might be altered in neurological disorders. Disturbances in mitochondrial function and signaling lead to impaired neuroplasticity, ageing and neurodegeneration, while enhancing of mitochondrial activity can have beneficial effects on the generation of new neurons. Therapeutic strategies that efficiently preserve or improve mitochondrial function may represent a novel direction for numerous mitochondrial-implicated, age-related disorders.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
ACKNOWLEDGMENTS
I would like to thank Alexander Bonk for carefully revising the manuscript. This work is funded by the Deutsche Forschungsgemeinschaft (BE 5136/1-1) and the research training group 2162 “Neurodevelopment and Vulnerability of the Central Nervous System” of the Deutsche Forschungsgemeinschaft (DFG GRK2162/1).
REFERENCES
[1] | Bond AM , Ming GL , Song H . Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell. (2015) ;17: (4):385–95. |
[2] | Kempermann G , Song H , Gage FH . Neurogenesis in the Adult Hippocampus. Cold Spring Harbor Perspectives in Medicine. (2015) ;5: (7):a018812. |
[3] | Ming GL , Song H . Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron. (2011) ;70: (4):687–702. |
[4] | Spalding KL , Bergmann O , Alkass K , Bernard S , Salehpour M , Huttner HB , et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. (2013) ;153: (6):1219–27. |
[5] | Bergmann O , Spalding KL , Frisen J . Adult Neurogenesis in Humans. Cold Spring Harbor Perspectives in Biology. (2015) ;7: (7):a018994. |
[6] | Winner B , Kohl Z , Gage FH . Neurodegenerative disease and adult neurogenesis. The European Journal of Neuroscience. (2011) ;33: (6):1139–51. |
[7] | Yun MH . Changes in Regenerative Capacity through Lifespan. International Journal of Molecular Sciences. (2015) ;16: (10):25392–432. |
[8] | Lee SW , Clemenson GD , Gage FH . New neurons in an aged brain. Behavioural Brain Research. (2012) ;227: (2):497–507. |
[9] | Seib DR , Martin-Villalba A . Neurogenesis in the Normal Ageing Hippocampus: A Mini-Review. Gerontology. (2015) ;61: (4):327–35. |
[10] | Gotz M , Nakafuku M , Petrik D . Neurogenesis in the Developing and Adult Brain-Similarities and Key Differences. Cold Spring Harbor Perspectives in Biology. (2016) ;8: (7):a018853. |
[11] | Aimone JB , Li Y , Lee SW , Clemenson GD , Deng W , Gage FH . Regulation and function of adult neurogenesis: From genes to cognition. Physiological Reviews. (2014) ;94: (4):991–1026. |
[12] | Knobloch M , Braun SM , Zurkirchen L , von Schoultz C , Zamboni N , Arauzo-Bravo MJ , et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. (2013) ;493: (7431):226–30. |
[13] | Ward PS , Thompson CB . Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell. (2012) ;21: (3):297–308. |
[14] | Mihaylova MM , Sabatini DM , Yilmaz OH . Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell. (2014) ;14: (3):292–305. |
[15] | Attwell D , Laughlin SB . An energy budget for signaling in the grey matter of the brain. Journal of cerebral blood flow and metabolism: Official journal of the International Society of Cerebral Blood Flow and Metabolism. (2001) ;21: (10):1133–45. |
[16] | Alle H , Roth A , Geiger JR . Energy-efficient action potentials in hippocampal mossy fibers. Science. (2009) ;325: (5946):1405–8. |
[17] | Pellerin L , Magistretti PJ . Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proceedings of the National Academy of Sciences of the United States of America. (1994) ;91: (22):10625–9. |
[18] | Rouach N , Koulakoff A , Abudara V , Willecke K , Giaume C . Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. (2008) ;322: (5907):1551–5. |
[19] | Belanger M , Allaman I , Magistretti PJ . Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metabolism. (2011) ;14: (6):724–38. |
[20] | Folmes CD , Terzic A . Energy metabolism in the acquisition and maintenance of stemness. Seminars in Cell & Developmental Biology. (2016) ;52: :68–75. |
[21] | Guppy M , Greiner E , Brand K . The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. European Journal of Biochemistry / FEBS. (1993) ;212: (1):95–9. |
[22] | Vander Heiden MG , Cantley LC , Thompson CB . Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. (2009) ;324: (5930):1029–33. |
[23] | Lunt SY , Vander Heiden MG . Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annual Review of Cell and Developmental Biology. (2011) ;27: :441–64. |
[24] | Sun X , St John JC . The role of the mtDNA set point in differentiation, development and tumorigenesis. The Biochemical Journal. (2016) ;473: (19):2955–71. |
[25] | Folmes CD , Nelson TJ , Martinez-Fernandez A , Arrell DK , Lindor JZ , Dzeja PP , et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metabolism. (2011) ;14: (2):264–71. |
[26] | Cho YM , Kwon S , Pak YK , Seol HW , Choi YM , Park DJ , et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochemical and Biophysical Research Communications. (2006) ;348: (4):1472–8. |
[27] | Chung S , Dzeja PP , Faustino RS , Perez-Terzic C , Behfar A , Terzic A . Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nature Clinical Practice Cardiovascular Medicine. (2007) ;4: (Suppl 1):S60–7. |
[28] | Kondoh H , Lleonart ME , Nakashima Y , Yokode M , Tanaka M , Bernard D , et al. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxidants & Redox Signaling. (2007) ;9: (3):293–9. |
[29] | Simsek T , Kocabas F , Zheng J , Deberardinis RJ , Mahmoud AI , Olson EN , et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. (2010) ;7: (3):380–90. |
[30] | Chen CT , Shih YR , Kuo TK , Lee OK , Wei YH . Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. (2008) ;26: (4):960–8. |
[31] | Turner WS , Seagle C , Galanko JA , Favorov O , Prestwich GD , Macdonald JM , et al. Nuclear magnetic resonance metabolomic footprinting of human hepatic stem cells and hepatoblasts cultured in hyaluronan-matrix hydrogels. Stem Cells. (2008) ;26: (6):1547–55. |
[32] | Shin J , Berg DA , Zhu Y , Shin JY , Song J , Bonaguidi MA , et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. (2015) ;17: (3):360–72. |
[33] | Stoll EA , Makin R , Sweet IR , Trevelyan AJ , Miwa S , Horner PJ , et al. Neural Stem Cells in the Adult Subventricular Zone Oxidize Fatty Acids to Produce Energy and Support Neurogenic Activity. Stem Cells. (2015) ;33: (7):2306–19. |
[34] | Khacho M , Clark A , Svoboda DS , Azzi J , MacLaurin JG , Meghaizel C , et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell. (2016) ;19: (2):232–47. |
[35] | Homem CC , Steinmann V , Burkard TR , Jais A , Esterbauer H , Knoblich JA . Ecdysone and mediator change energy metabolism to terminate proliferation in Drosophila neural stem cells. Cell. (2014) ;158: (4):874–88. |
[36] | Agathocleous M , Love NK , Randlett O , Harris JJ , Liu J , Murray AJ , et al. Metabolic differentiation in the embryonic retina. Nature Cell Biology. (2012) ;14: (8):859–64. |
[37] | Zheng X , Boyer L , Jin M , Mertens J , Kim Y , Ma L , et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife. (2016) ;5. |
[38] | Beckervordersandforth R , Ebert B , Schaffner I , Moss J , Fiebig C , Shin J , et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron. (2017) ;93: (3):560–573 e6. |
[39] | Gascon S , Murenu E , Masserdotti G , Ortega F , Russo GL , Petrik D , et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell. (2016) ;18: (3):396–409. |
[40] | Llorens-Bobadilla E , Zhao S , Baser A , Saiz-Castro G , Zwadlo K , Martin-Villalba A . Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. (2015) ;17: (3):329–40. |
[41] | Costa MR , Ortega F , Brill MS , Beckervordersandforth R , Petrone C , Schroeder T , et al. Continuous live imaging of adult neural stem cell division and lineage progression in vitro. Development. (2011) ;138: (6):1057–68. |
[42] | Knobloch M , Jessberger S . Metabolism and neurogenesis. Current Opinion in Neurobiology. (2016) ;42: :45–52. |
[43] | Rafalski VA , Brunet A . Energy metabolism in adult neural stem cell fate. Progress in Neurobiology. (2011) ;93: (2):182–203. |
[44] | Yeo H , Lyssiotis CA , Zhang Y , Ying H , Asara JM , Cantley LC , et al. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. The EMBO Journal. (2013) ;32: (19):2589–602. |
[45] | Beckervordersandforth R , Häberle BM and Lie DC . Metabolic regulation of adult stem cell-derived neurons. Frontier of Biology. (2015) (10):107–16. |
[46] | Sullivan LB , Gui DY , Hosios AM , Bush LN , Freinkman E , Vander Heiden MG . Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. (2015) ;162: (3):552–63. |
[47] | Birsoy K , Wang T , Chen WW , Freinkman E , Abu-Remaileh M , Sabatini DM . An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. (2015) ;162: (3):540–51. |
[48] | King MP , Attardi G . Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. (1989) ;246: (4929):500–3. |
[49] | Cheng A , Hou Y , Mattson MP . Mitochondria and neuroplasticity. ASN Neuro. (2010) ;2: (5):e00045. |
[50] | Xavier JM , Rodrigues CM , Sola S . Mitochondria: Major Regulators of Neural Development. The Neuroscientist: A review Journal Bringing Neurobiology, Neurology and Psychiatry. (2016) ;22: (4):346–58. |
[51] | Moyes CD , Mathieu-Costello OA , Tsuchiya N , Filburn C , Hansford RG . Mitochondrial biogenesis during cellular differentiation. The American Journal of Physiology. (1997) ;272: (4 Pt 1):C1345–51. |
[52] | Vayssiere JL , Cordeau-Lossouarn L , Larcher JC , Basseville M , Gros F , Croizat B . Participation of the mitochondrial genome in the differentiation of neuroblastoma cells. In vitro Cellular & Developmental Biology: Journal of the Tissue Culture Association. (1992) ;28A: (11-12):763–72. |
[53] | Wang W , Osenbroch P , Skinnes R , Esbensen Y , Bjoras M , Eide L . Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells. (2010) ;28: (12):2195–204. |
[54] | Wang W , Esbensen Y , Kunke D , Suganthan R , Rachek L , Bjoras M , et al. Mitochondrial DNA damage level determines neural stem cell differentiation fate. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2011) ;31: (26):9746–51. |
[55] | Prozorovski T , Schulze-Topphoff U , Glumm R , Baumgart J , Schroter F , Ninnemann O , et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nature Cell Biology. (2008) ;10: (4):385–94. |
[56] | Mattson MP . Establishment and plasticity of neuronal polarity. Journal of Neuroscience Research. (1999) ;57: (5):577–89. |
[57] | Spillane M , Ketschek A , Merianda TT , Twiss JL , Gallo G . Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Reports. (2013) ;5: (6):1564–75. |
[58] | Overly CC , Rieff HI , Hollenbeck PJ . Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. Journal of Cell Science. (1996) ;109: (Pt 5):971–80. |
[59] | Chihara T , Luginbuhl D , Luo L . Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nature Neuroscience. (2007) ;10: (7):828–37. |
[60] | Levy M , Faas GC , Saggau P , Craigen WJ , Sweatt JD . Mitochondrial regulation of synaptic plasticity in the hippocampus. The Journal of Biological Chemistry. (2003) ;278: (20):17727–34. |
[61] | Oruganty-Das A , Ng T , Udagawa T , Goh EL , Richter JD . Translational control of mitochondrial energy productionmediates neuron morphogenesis. Cell Metabolism. (2012) ;16: (6):789–800. |
[62] | Nakagawa S , Kim JE , Lee R , Chen J , Fujioka T , Malberg J , et al. Localization of phosphorylated cAMP response element-binding protein in immature neurons of adult hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2002) ;22: (22):9868–76. |
[63] | Merz K , Herold S , Lie DC . CREB in adult neurogenesis–master and partner in the development of adult-born neurons? The European Journal of Neuroscience. (2011) ;33: (6):1078–86. |
[64] | Herold S , Jagasia R , Merz K , Wassmer K , Lie DC . CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Molecular and Cellular Neurosciences. (2011) ;46: (1):79–88. |
[65] | Jagasia R , Steib K , Englberger E , Herold S , Faus-Kessler T , Saxe M , et al. GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2009) ;29: (25):7966–77. |
[66] | Giachino C , De Marchis S , Giampietro C , Parlato R , Perroteau I , Schutz G , et al. cAMP response element-binding protein regulates differentiation and survival of newborn neurons in the olfactory bulb. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2005) ;25: (44):10105–18. |
[67] | Altarejos JY , Montminy M . CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nature Reviews Molecular Cell Biology. (2011) ;12: (3):141–51. |
[68] | Spiegelman BM . Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. (2007) ;287: :60–3; discussion 3-9. |
[69] | Burkhalter J , Fiumelli H , Allaman I , Chatton JY , Martin JL . Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2003) ;23: (23):8212–20. |
[70] | Markham JA , Greenough WT . Experience-driven brain plasticity: Beyond the synapse. Neuron Glia Biology. (2004) ;1: (4):351–63. |
[71] | Attardi G , Schatz G . Biogenesis of mitochondria. Annual Review of Cell Biology. (1988) ;4: :289–333. |
[72] | Osman C , Voelker DR , Langer T . Making heads or tails of phospholipids in mitochondria. The Journal of Cell Biology. (2011) ;192: (1):7–16. |
[73] | Mishra P , Chan DC . Mitochondrial dynamics and inheritance during cell division, development and disease. Nature Reviews Molecular Cell Biology. (2014) ;15: (10):634–46. |
[74] | Chen H , Chan DC . Emerging functions of mammalian mitochondrial fusion and fission. Human Molecular Genetics. (2005) ;14 Spec No. 2: :R283–9. |
[75] | Xavier JM , Morgado AL , Sola S , Rodrigues CM . Mitochondrial translocation of p53 modulates neuronal fate by preventing differentiation-induced mitochondrial stress. Antioxidants & Redox Signaling. (2014) ;21: (7):1009–24. |
[76] | Steib K , Schaffner I , Jagasia R , Ebert B , Lie DC . Mitochondria modify exercise-induced development of stem cell-derived neurons in the adult brain. The Journal of Neuroscience: The official Journal of the Society for Neuroscience. (2014) ;34: (19):6624–33. |
[77] | van Praag H , Christie BR , Sejnowski TJ , Gage FH . Running enhances neurogenesis, learning, and long-term potentiation in mice. Proceedings of the National Academy of Sciences of the United States of America. (1999) ;96: (23):13427–31. |
[78] | van Praag H , Kempermann G , Gage FH . Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nature Neuroscience. (1999) ;2: (3):266–70. |
[79] | Li Z , Okamoto K , Hayashi Y , Sheng M . The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. (2004) ;119: (6):873–87. |
[80] | Agarwal S , Yadav A , Tiwari SK , Seth B , Chauhan LK , Khare P , et al. Dynamin-related Protein 1 Inhibition Mitigates Bisphenol A-mediated Alterations in Mitochondrial Dynamics and Neural Stem Cell Proliferation and Differentiation. The Journal of Biological Chemistry. (2016) ;291: (31):15923–39. |
[81] | Prozorovski T , Schneider R , Berndt C , Hartung HP , Aktas O . Redox-regulated fate of neural stem progenitor cells. Biochimica et Biophysica Acta. (2015) ;1850: (8):1543–54. |
[82] | Le Belle JE , Orozco NM , Paucar AA , Saxe JP , Mottahedeh J , Pyle AD , et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. (2011) ;8: (1):59–71. |
[83] | Walton NM , Shin R , Tajinda K , Heusner CL , Kogan JH , Miyake S , et al. Adult neurogenesis transiently generates oxidative stress. PLoS One. (2012) ;7: (4):e35264. |
[84] | Hou Y , Ouyang X , Wan R , Cheng H , Mattson MP , Cheng A . Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells. (2012) ;30: (11):2535–47. |
[85] | Forsberg K , Wuttke A , Quadrato G , Chumakov PM , Wizenmann A , Di Giovanni S . The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. The Journal of Neuroscience: The official Journal of the Society for Neuroscience. (2013) ;33: (36):14318–30. |
[86] | Maryanovich M , Gross A . A ROS rheostat for cell fate regulation. Trends in Cell Biology. (2013) ;23: (3):129–34. |
[87] | Owusu-Ansah E , Yavari A , Mandal S , Banerjee U . Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nature Genetics. (2008) ;40: (3):356–61. |
[88] | Yuan TF , Gu S , Shan C , Marchado S , Arias-Carrion O . Oxidative Stress and Adult Neurogenesis. Stem Cell Reviews. (2015) ;11: (5):706–9. |
[89] | Wakabayashi N , Skoko JJ , Chartoumpekis DV , Kimura S , Slocum SL , Noda K , et al. Notch-Nrf2 axis: Regulation of Nrf2 gene expression and cytoprotection by notch signaling. Molecular and Cellular Biology. (2014) ;34: (4):653–63. |
[90] | Im JY , Lee KW , Woo JM , Junn E , Mouradian MM . DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Human Molecular Genetics. (2012) ;21: (13):3013–24. |
[91] | Kim JH , Thimmulappa RK , Kumar V , Cui W , Kumar S , Kombairaju P , et al. NRF2-mediated Notch pathway activation enhances hematopoietic reconstitution following myelosuppressive radiation. The Journal of Clinical Investigation. (2014) ;124: (2):730–41. |
[92] | Karkkainen V , Pomeshchik Y , Savchenko E , Dhungana H , Kurronen A , Lehtonen S , et al. Nrf2 regulates neurogenesis and protects neural progenitor cells against Abeta toxicity. Stem Cells. (2014) ;32: (7):1904–16. |
[93] | Unden G , Bongaerts J . Alternative respiratory pathways of Escherichia coli: Energetics and transcriptionalregulation in response to electron acceptors. Biochimica et Biophysica Acta. (1997) ;1320: (3):217–34. |
[94] | Mimaki M , Wang X , McKenzie M , Thorburn DR , Ryan MT . Understanding mitochondrial complex I assembly in health and disease. Biochimica et Biophysica Acta. (2012) ;1817: (6):851–62. |
[95] | Stein LR , Imai S . Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO Journal. (2014) ;33: (12):1321–40. |
[96] | Zhang H , Ryu D , Wu Y , Gariani K , Wang X , Luan P , et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. (2016) ;352: (6292):1436–43. |
[97] | Lin SJ , Ford E , Haigis M , Liszt G , Guarente L . Calorie restriction extends yeast life span by lowering the level of NADH. Genes & Development. (2004) ;18: (1):12–6. |
[98] | Webster BR , Lu Z , Sack MN , Scott I . The role of sirtuins in modulating redox stressors. Free Radical Biology & Medicine. (2012) ;52: (2):281–90. |
[99] | Radak Z , Koltai E , Taylor AW , Higuchi M , Kumagai S , Ohno H , et al. Redox-regulating sirtuins in aging, caloric restriction, and exercise. Free Radical Biology & Medicine. (2013) ;58: :87–97. |
[100] | Alcendor RR , Gao S , Zhai P , Zablocki D , Holle E , Yu X , et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circulation Research. (2007) ;100: (10):1512–21. |
[101] | Brunet A , Sweeney LB , Sturgill JF , Chua KF , Greer PL , Lin Y , et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. (2004) ;303: (5666):2011–5. |
[102] | Wang F , Nguyen M , Qin FX , Tong Q . SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging cell. (2007) ;6: (4):505–14. |
[103] | Michan S , Li Y , Chou MM , Parrella E , Ge H , Long JM , et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2010) ;30: (29):9695–707. |
[104] | Ichi S , Boshnjaku V , Shen YW , Mania-Farnell B , Ahlgren S , Sapru S , et al. Role of Pax3 acetylation in the regulation of Hes1 and Neurog2. Molecular Biology of the Cell. (2011) ;22: (4):503–12. |
[105] | Lopez-Otin C , Galluzzi L , Freije JM , Madeo F , Kroemer G . Metabolic Control of Longevity. Cell. (2016) ;166: (4):802–21. |
[106] | Lopez-Otin C , Blasco MA , Partridge L , Serrano M , Kroemer G . The hallmarks of aging. Cell. (2013) ;153: (6):1194–217. |
[107] | Linnane AW , Marzuki S , Ozawa T , Tanaka M . Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. (1989) ;1: (8639):642–5. |
[108] | Khrapko K , Bodyak N , Thilly WG , van Orsouw NJ , Zhang X , Coller HA , et al. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Research. (1999) ;27: (11):2434–41. |
[109] | Wallace DC . A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annual Review of Genetics. (2005) ;39: :359–407. |
[110] | Kujoth GC , Prolla TA . Evolving insight into the role of mitochondrial DNA mutations in aging. Experimental Gerontology. (2008) ;43: (1):20–3. |
[111] | Trifunovic A , Larsson NG . Mitochondrial dysfunction as a cause of ageing. Journal of Internal Medicine. (2008) ;263: (2):167–78. |
[112] | Trifunovic A , Wredenberg A , Falkenberg M , Spelbrink JN , Rovio AT , Bruder CE , et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. (2004) ;429: (6990):417–23. |
[113] | Wang K , Klionsky DJ . Mitochondria removal by autophagy. Autophagy. (2011) ;7: (3):297–300. |
[114] | Ahlqvist KJ , Hamalainen RH , Yatsuga S , Uutela M , Terzioglu M , Gotz A , et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metabolism. (2012) ;15: (1):100–9. |
[115] | Stoll EA , Cheung W , Mikheev AM , Sweet IR , Bielas JH , Zhang J , et al. Aging neural progenitor cells have decreased mitochondrial content and lower oxidative metabolism. The Journal of Biological Chemistry. (2011) ;286: (44):38592–601. |
[116] | Kang E , Wang X , Tippner-Hedges R , Ma H , Folmes CD , Gutierrez NM , et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell Stem Cell. (2016) ;18: (5):625–36. |
[117] | Perry SW , Norman JP , Barbieri J , Brown EB , Gelbard HA . Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Bio Techniques. (2011) ;50: (2):98–115. |
[118] | Waegemans T , Wilsher CR , Danniau A , Ferris SH , Kurz A , Winblad B . Clinical efficacy of piracetam in cognitive impairment: A meta-analysis. Dementia and Geriatric cognitive disorders. (2002) ;13: (4):217–24. |
[119] | Kurz C , Ungerer I , Lipka U , Kirr S , Schutt T , Eckert A , et al. The metabolic enhancer piracetam ameliorates the impairment of mitochondrial function and neurite outgrowth induced by beta-amyloid peptide. British Journal of Pharmacology. (2010) ;160: (2):246–57. |
[120] | Leuner K , Kurz C , Guidetti G , Orgogozo JM , Muller WE . Improved mitochondrial function in brain aging and Alzheimer disease - the new mechanism of action of the old metabolic enhancer piracetam. Frontiers in Neuroscience. (2010) ;4: :pii:44. |

