
Neural Stem Cell Activation and the Role of Protein Synthesis
Abstract
Adult neural stem cells are generated at embryonic stages by entering a quiescent state that allows their retention into adulthood and thereby maintenance of life-long brain homeostasis. Thus, a tight balance between the quiescence and activation state is instrumental to meet the brain demands for a specific cell type at the correct numbers, at a given time and position. Protein synthesis is the most energy-consuming process within the cell and, not surprisingly, it occurs at low rates in quiescent stem cells. This way quiescent cells adjust to energy constraints and avoid their premature depletion. Stem cell activation is characterized by upregulation of protein synthesis followed by cell division and differentiation. The role of such upregulation as causative or rather a consequence of the activation remains elusive. Here we summarize recent findings connecting stem cell activation to the regulation of protein synthesis, particularly focusing on embryonic and adult neural stem cells of the ventricular zone.
INTRODUCTION
Adult neural stem cells (NSCs) are best characterized in two brain regions, the ventricular zone (VZ) of the lateral ventricles and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG). In the VZ, ventricle-contacting monociliated radial glia-like cells (RGLs) constantly produce olfactory bulb interneurons and their absence leads to olfactory bulb atrophy. In the SGZ, new granule neurons are generated by NSCs over the lifetime of an animal, but contrary to the VZ-NSCs, these cells have no replenishment function [1].
The fact that adult VZ-NSCs position, morphology and multipotent potential closely resemble their embryonic counterpart suggested their embryonic origin. Two recent studies show that in the embryonic brain a subset of neural progenitors become quiescent early during development to get reactivated postnatally [2, 3]. Of note, loss of quiescent NSCs at embryonic stages impairs the maintenance of NSCs into adulthood [3]. In adult life, quiescent (non-cycling) and activated (cycling) NSCs coexist both in the VZ [4, 5] and SGZ [6, 7].
NSCs of the VZ and SGZ can be characterized by their activation status, but so far only NSCs of the VZ were shown to display lineage heterogeneity relating to their restricted competence to generate certain neuronal subtypes in the olfactory bulb [8, 9]. Recently, this cellular heterogeneity could also be demonstrated on the molecular level using single cell transcriptomics [10]. Within the quiescent states, a dormant state is characterized by dependence on glycolysis and pentose phosphate pathway as energy source. These long-lived cells, which are in charge of providing differentiated progeny, require highest level of protection to their genomic DNA. The demand for minimal reactive oxygen species (ROS) production as well as general hypoxic environment of the neurogenic niche [11, 12] could be the prime reasons for quiescent NSCs to mainly rely on glycolysis and pentose phosphate in order to generate energy and intermediates for anabolic reactions and avoid excessive ROS [13, 14].
Single cell transcriptomes of NSCs also revealed that NSCs are not clearly separated into distinct populations of quiescent and activated NSCs, but rather transit through multiple intermediate stages before amplification and differentiation. One major hallmark of NSC activation is transcriptional upregulation of factors critical for the protein synthesis machinery (including multiple ribosomal proteins). Consequently, dormant NSCs (the most quiescent NSCs) show a particularly low level of protein translation, whereas progressive activation leads to increased global translation levels (Fig. 1). In hematopoietic stem cells, deletion of phosphatase and tensin homolog (PTEN), a negative regulator of phosphoinositide 3-kinase/protein kinase B (PI3K/PKB, rather known as PI3K/AKT) signaling, led to exhaustion of the stem cell pool and loss of stem cell-mediated reconstitution of the hematopoietic system [15]. Similarly, PTEN deletion in NSCs induced their terminal astrocytic differentiation and NSC depletion in the SGZ [6]. Altogether these data indicate that maintaining low protein synthesis is of key importance for stem cell function. As well, precise regulation of protein synthesis is instrumental for NSC activation and differentiation.
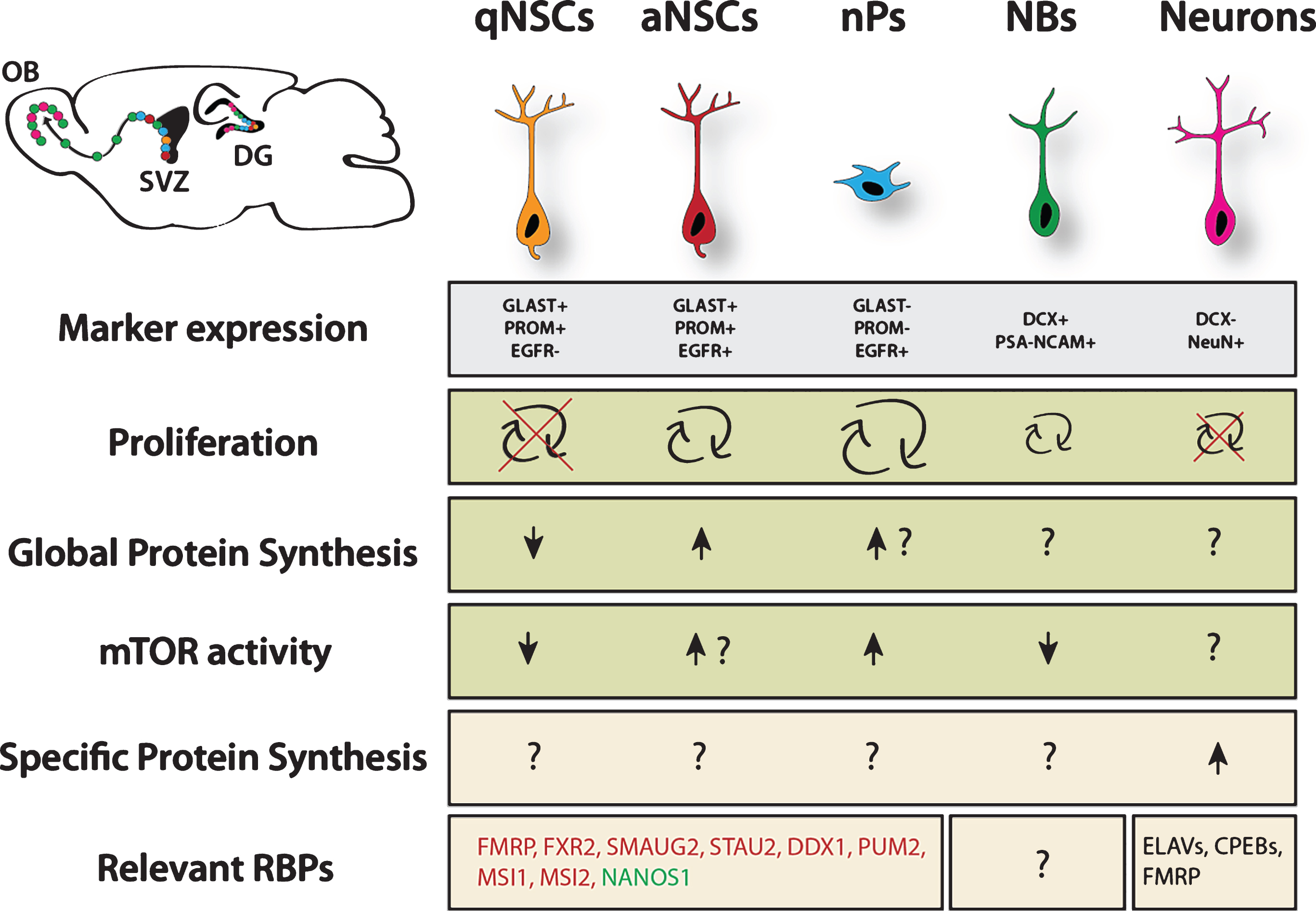
Fig.1
Overview of translational regulation taking place in the NSC lineage. Data was pooled from studies investigating cortical development, adult neurogenesis in the ventricular zone and hippocampus. Arrows represent relative estimation compared to other stages within the lineage. Question marks indicate unknown situation. Question marks next to arrows indicate likely scenario based on proliferation rate, however this is not experimentally proven. Green color marks proneurogenic factors, red color marks anti-neurogenic factors. qNSC = quiescent NSC, aNSC = active NSC, nP = neurogenic progenitor, NB = neuroblast. Major sources: Proliferation rates from Codega et al. (2014), neurons are known to be postmitotic. Marker expression and protein synthesis levels from Llorens-Bobadilla et al. (2015). mTOR activity from Paliouras et al. (2012). For more details, see the main text.
In this review we first discuss the necessity for tight control of protein synthesis in stem cells and introduce the multiple steps of translational control exemplified by studies in NSCs or related adult tissue stem cells. We further describe RNA binding proteins (RBPs) and posttranscriptional modifications of mRNAs, which are involved in NSC biology. Finally, we critically review current methods applied to investigate protein synthesis, discuss advantages and disadvantages over more widespread RNA-based techniques and suggest how to apply these methods to further extent our understanding of the complex regulation of NSCs dynamics via tight control of protein synthesis.
PROTEIN SYNTHESIS IN STEM CELLS – RELATION TO ENERGY METABOLISM
NSCs as well as other stem cells need to provide maximal protection to their genomic DNA over the life span of the host organism in order to give rise to non-mutated healthy progeny even at older stages. Reactive oxygen species (ROS) produced in a number of redox reactions appear to be the prime culprit responsible for accumulation of potentially malignant mutations [16, 17]. The main source of ROS production in the cell is oxidative phosphorylation in mitochondria, which could hint why NSCs and other stem cells predominantly rely on glycolysis and pentose phosphate pathway to generate energy and metabolic intermediates involved in maintenance of normal cell physiology [13, 18, 19]. Furthermore, NSCs need to adapt to the conditions of their hypoxic niche, which favors glycolysis over other mechanisms of energy production [11, 12]. Quiescent stem cells do not proliferate and differentiate, exhibiting minimal requirements for their metabolic rate. Therefore, it is not surprising that these cells feature a low efficiency of protein synthesis [10, 15, 20]. Since protein synthesis is the most highly energy consuming process in cells [21, 22], by keeping it at a minimal rate, NSCs can avoid intensive energy consumption.
Upon different stimulatory signals, including central nervous system (CNS) traumas and pathologies, NSCs become activated and begin to proliferate and differentiate in order to produce new neurons and glia cells, as well as to maintain their own pool [23]. Such activation immediately evokes intensive energy consumption and the acceleration of anabolic reactions to provide all necessary metabolites for cell growth and division. NSCs restructure their metabolism, intensifying oxidative phosphorylation in mitochondria as the most efficient metabolic pathway to generate energy, which results in the elevation of the intracellular ROS concentration [24–26]. Interestingly, a recent study demonstrated a direct effect of ROS on the activation of transcription factors involved in the regulation of lineage-related genes [14].
Together, low protein synthesis rate in stem cells correlates with their low cellular metabolism, which serves the purpose of longevity in a hierarchical system. Activation for proliferation and commitment to differentiate requires massive restructuring of cellular metabolism leading finally to considerable changes in energy production and consumption. This is in part reflected by the intensity of protein synthesis. Further studies are necessary to get into the details of mutual relation between stemness, energy metabolism and protein synthesis rates.
RIBOSOME BIOGENESIS AND GLOBAL TRANSLATIONAL CONTROL IN STEM CELLS
Ribosomes and other major components of the translation machinery are essential for even the most simple form of life [27]. Due to the high abundance and conservation over all species these factors are generally assumed to have mostly housekeeping function and not to be involved in making cellular decisions. However, recent evidence rather points into a direction where the control of ribosome biogenesis and their effect on global translation levels can actually trigger a certain behavior of cells. Particularly in the complex case of stem cells, which need to maintain a sensitive balance between quiescence, self-renewal and differentiation, this level of regulation is of utmost importance. Ribosomal DNA (rDNA) transcription produces ribosomal RNAs (rRNAs), which are the major component of ribosomes. A high level of rDNA transcription is a feature of stem cells and is downregulated in the progeny. Recently, a causal link between rDNA transcription and cellular behavior was shown in female drosophila germ line stem cells (GSCs). Here, manipulation of rDNA transcription by upregulation of POL1 delays differentiation, while reduction of rRNA production triggers cyst formation, a sign of differentiation [28].
Interestingly, translation rates in mammalian HSCs are lower than in any other more differentiated stage, independently of the rate of rDNA transcription [15]. This indicates that rRNA production might not be the only rate-limiting component of protein synthesis but rather the ratio of free rRNA molecules and ribosomal proteins predicts ribosome production and translational activity. Indeed, the study of adult neural stem cells at single-cell resolution showed that these cells, formerly treated as one population, can be actually grouped into four stages, which show varying levels of expression of ribosomal proteins [10, 29]. These four activation stages reveal progressive increase in the transcription of ribosomal genes and global protein synthesis levels. Since activation ultimately leads to cell division, the question arises whether higher protein synthesis levels actually trigger division or are rather the consequence of cellular decisions at a different level. In this line, Blanco et al. recently showed that skin stem cells, similar to NSCs and HSCs, synthesize less protein than their immediate progenitors [20]. The authors acknowledge that active cell cycle correlates with increasing translation rates. However, they conclude that proliferation does not drive translation in this system, since protein synthesis is highest in differentiating but non-cycling cells. It will be important to address the mechanisms which regulate protein synthesis since recent evidence points in a direction where translation is highly related to the differentiation status of stem cells. Consequently, stage-specific manipulation of related mechanisms could help making greater use of the regenerative potential of stem cells. Further, a direct link between the regulatory levels of transcription and translation was discovered in HSCs where loss of the runt-related transcription factor 1 (RUNX1) leads to decreased protein synthesis and higher stress response, outcompeting wild-typeHSCs [30].
Together, increased biogenesis of ribosomes by increased transcription of rRNA and/or ribosomal proteins often correlates with a proliferative status of stem cells (Fig. 1). However, non-cycling cells can also have high protein synthesis rates. Whether there is a causal link placing regulation of the translation machinery on top of the hierarchy of cell division decisions remains controversial and might vary between different stem cell systems.
GLOBAL CONTROL OF PROTEIN SYNTHESIS BY THE TRANSLATION MACHINERY AND UPSTREAM REGULATORS
NSCs underlie the same central dogma of biology as any other eukaryotic cell: gene regulation, both abundance and activity, is accomplished by precise control at the level of DNA, RNA and protein. Proteins are the major players of biological systems but their analysis on the global level is still challenging. Accordingly, the vast majority of recent studies focus on the analysis of total RNA by next generation sequencing. However, to which extent the abundance of individual mRNAs correctly mirrors the generation of corresponding functional proteins is still highly debated and is most likely dependent on the cellular context [31, 32].
When talking about “translational regulation”, we have to first distinguish two major mechanisms: the regulation of global translation by controlling key molecules involved in the process within the cells, including major factors of the translation machinery and the availability of ribosomes, which is believed to affect the vast majority of mRNAs equally, as well as specific regulation of certain transcripts. Thelatter is often based on sequence features recognized by a number of additional regulators including miRNAs, RBPs and more recently discovered circular RNAs (circRNAs), which are believed to compete with miRNAs for mRNA binding [33]. There is emerging evidence that mRNA methylation, particularly N6-methyladenosine (m6A) modification contributes significantly to post-transcriptional regulation (see later paragraph) [34]. Another exciting and currently evolving field explores to which level distinct composition of ribosomal proteins yields specialized ribosomes with tissue-specific regulatoryfunctions [35].
The following section focuses on mechanisms of global translation and introduces mammalian target of rapamycin (mTOR) signaling as a key regulatory component. The process of translation, responsible for the synthesis of all cellular proteins on corresponding mRNAs, recapitulates in NSCs the same canonical steps that have previously been described in detail for other eukaryotic cells (Fig. 2A) [36, 37]. The translation of each mRNA molecule starts with the formation of the 43S complex, comprising a 40S ribosomal subunit, associated eukaryotic initiation factors (eIFs) eIF1, eIF1A, eIF3, eIF5, and the so-called ternary complex eIF2-GTP-tRNAMeti. The 40S subunit could either directly be recycled from the stop codon of the same mRNA upon completion of the previous round of translation or generated de novo from the pool of free subunits. Initiation factors eIF1, 1A and 3, upon association with 40S subunits, stimulate the attachment of the ternary complex, thereby completing the 43S formation. Meanwhile, the mRNA destined to enter a new cycle of translation must prepare its 5’ end for the attachment of a pre-formed 43S complex. For that, the cap structure, present at 5’ ends of all cellular mRNAs, is recognized by the initiation factor eIF4F via its cap-binding subunit eIF4E. Two other constituents of eIF4F are the scaffold protein eIF4G and the RNA helicase eIF4A. eIF4G recruits available 43S complexes via direct association with its integral part, eIF3, and eIF4A can stimulate the 43S landing via unwinding any RNA structures in the vicinity of the cap that could negatively impact the 43S binding. Since start codons in eukaryotic mRNAs are generally located at some tens or even hundreds nucleotides away from the 5’ ends, the 43S complex has to scan such distance (called the 5’ untranslated region, 5’ UTR) till it encounters the start codon that is usually the first AUG in a proper context (so-called Kozak context: GCC(A/G)CCAUGG, start AUG underlined). The helicase eIF4A, stimulated by another initiation factor eIF4B, also provides the 5’-to-3’ direction to the scanning by preventing any backward movements of diffusion-based sliding 40S via a not yet fully deciphered mechanism. Upon the recognition of the correct AUG by the initiator tRNAMet anticodon, some structural rearrangements take place in 43S complexes resulting in the hydrolysis of eIF2-associated GTP and its following dissociation, accomplishing the formation of the 48S complex. Another GTPase eIF5B, also activated by the start codon recognition, is responsible for the joining of a 60S subunit to the 48S and the ultimate formation of the 80S complex that begins synthesis of the encoded protein in the process of elongation. Aminoacyl-tRNAs with anticodons complementary to corresponding codons in the A site of elongating ribosomes, are delivered by elongation factor eEF1A, and another elongation factor eEF2 stimulates ribosome translocation upon completion of the peptidyl-transferase reaction. The whole process of translation is concluded when ribosomes reach the stop codon, and two releasing factors eRF1 and eRF3 stimulate translation termination culminating in the complete dissociation of ribosomes from mRNAs. Recycled 40S subunits could again enter a new translation cycle at the 5’ end of the same or another mRNA.
Fig.2
Schematic representation of the process of protein biosynthesis in eukaryotes with main regulatory nodes. (A) Basic steps of translation beginning from the formation of the ternary complex transiting to the 43S complex, which, after loading onto an mRNA, scans the non-translatable region till the recognition of the initiation AUG resulting in the 48S complex assembly. After 60S subunit joining, newly formed 80S ribosomes proceed to elongation moving along the coding sequence until the stop codon appears in the acceptor A site of the ribosomes. This starts up the process of termination and recycling, releasing 40S and 60S ribosomal subunits for a new round of translation on the same or another mRNA molecule. (B) Regulation of the initiation of translation via the phosphorylation of factor eIF2 by stress-activated kinases HRI, PKR, GCN2, and PERK: phosphorylated eIF2 forms a very stable complex with the guanine exchange factor eIF2B exhausting the available pool of free eIF2B, thereby blocking the reaction of GDP-GTP exchange on eIF2. (C) mTORC1-mediated control including phosphorylation of 4E-BPs, S6K1/K2 and eEF2K. For more details, see the main text.
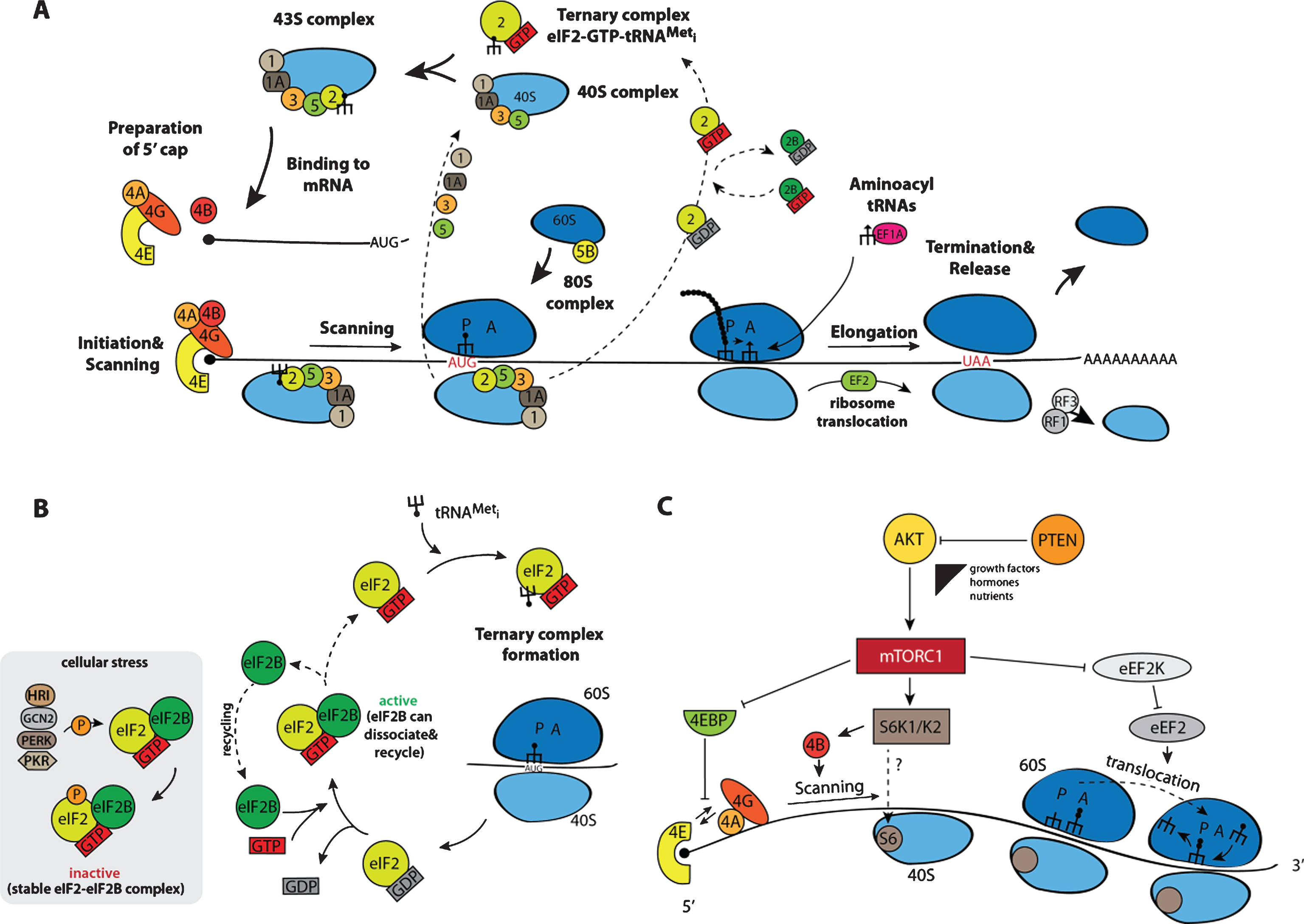
Not surprisingly, such complicated process is tightly regulated at several steps. As described in the introduction, protein biosynthesis is the most energy consuming cellular process and its regulation has a considerable impact on the total cellular metabolism. During their lifespan, particularly in multicellular organisms, cells are exposed to numerous stresses, which demand a tight translational control at the beginning of the process not only to save energy and adjust the set of actively translatable mRNAs, but also to prevent the synthesis of those unnecessary and even potentially deleterious proteins. Initiation factors eIF2 and eIF4E are two best described nexus regulators. Four kinases responsive to different stresses and specific to eIF2 have been described so far [38]: the heme-regulated inhibitor HRI, activated by heme-deficiency in erythroid cells as well as by oxidative stress and heat shock; the RNA-dependent protein kinase PKR, activated upon viral infections by double stranded RNAs and interferons; the general control non-derepressible-2 (GCN2), whose activity is induced by amino acid starvation and UV treatment; and the PKR-like endoplasmic reticulum (ER) resident protein kinase PERK, which is stimulated by UV and ER-stress upon accumulation of misfolded proteins. These four kinases specifically phosphorylate the α subunit of eIF2 at Ser51, and such modification highly elevates the affinity of p-eIF2 to the guanine exchange factor eIF2B, responsible for the conversion of post-initiation inactive GDP-eIF2 complexes into active GTP-eIF2 (Fig. 2B). Since the cellular concentration of eIF2 is much higher than that of eIF2B, even a partial phosphorylation of eIF2 is able to sequester all available eIF2B and, as a result, to block the GDP-GTP exchange. The phosphorylation of eIF2 negatively impacts the translation of most cellular mRNA, yet, in turn some mRNAs carrying short upstream open reading frames (uORFs) in their 5’UTRs get translated through a complex and not yet completely resolved mechanism. The proteins encoded by these mRNAs assist the affected cell for survival and recovery after the stress relieves. Accordingly, p-eIF2 is required for maintenance of global low translation levels in skeletal muscle cells, leading to accumulation of a considerable part of cellular mRNAs in stress-like cytoplasmic granules [39]. The p-eIF-mediated translation reduction is an integral component of a general program ensuring skeletal muscle cell quiescence. Alongside with the global translation repression, some mRNAs, whose protein products are important for sustaining the quiescent state, become activated upon eIF phosphorylation. Expression of a phosphorylation resistant form of eIF2 stimulates satellite cells’ metabolism and myogenic differentiation. Such p-eIF2-mediated control of quiescence has only been shown forskeletal muscle stem cells and needs to be addressed in other stem cell systems including NSCs.
Another well-established example of translational control is based on the regulation of the cap-binding protein eIF4E. This control involves the phosphorylation of a family of eIF4E-binding proteins 4E-BP1, 4E-BP2, and 4E-BP3 [40]. In a non-stimulated hypophosphorylated state, 4E-BP proteins bind eIF4E and prevent its interaction with the scaffold factor eIF4G, which, in turn, blocks the attachment of 43S complexes to the 5’ end of mRNAs. Upon activation of protein synthesis by extracellular stimuli including growth factors and nutrients, 4E-BPs become phosphorylated by the mechanistic/mammalian target of rapamycin kinase (mTOR). This kinase is present in two intracellular complexes mTORC1 and mTORC2 differing in their protein compositions, input control signals, and regulated processes [41]. mTORC1 is responsible for 4E-BP phosphorylation (Fig. 2C). Phosphorylated 4E-BPs lose their affinity for eIF4E and the latter can associate with eIF4G driving a normal translation process. As in the case of eIF2-mediated control (Fig. 2B), mRNAs have different sensitivity to the availability of eIF4E, and some of them are more sensitive to the 4E-BPs mediated repression than others [42, 43]. Apart of 4E-BPs, mTORC1 also phosphorylates and activates S6 kinase 1 (S6K1) and S6 kinase 2 (S6K2). S6K1/2 phosphorylate the ribosomal protein S6 (RPS6) and eIF4B, which stimulates general translation [44]. In addition, S6K1/2 phosphorylate and inactivate the eukaryotic elongation factor 2 kinase (eEF2K) that negatively affects protein synthesis via eEF2 phosphorylation [45]. Besides the phosphorylation of the proteins directly involved in translation, mTORC1 can stimulate the transcription of ribosomal RNAs and tRNAs via modification of transcription modulators initiation factors IA (TIF-IA) and IIIC (TF-IIIC), correspondingly [46, 47]. Even such a short description clearly highlights the exclusive role that mTORC1 plays in the regulation of translation.
mTORC1 is a node converging numerous signaling pathways stimulated by growth factors, nutrients, and metabolites. Thus, mTORC1 is an optimal master regulator of NSC activation by transducing signals from the niche. Most studies addressing potential roles of mTORC1 in NSCs use conditional knockout mouse models allowing for mTORC1 activation or repression specifically in NSCs and their progenies [48, 49]. In the embryonic brain, mTORC1 activity is critical for maintenance of proliferation anddifferentiation of early progenitor cells, and its inactivation results in microcephaly, impaired gliogenesis and neurogenesis [50–52]. Consistently, hyperactivation of mTORC1 stimulates neurogenic differentiation of NSCs at the expense of self-renewal, leading to premature exhaustion of the NSC pool [51]. The expression of an mTOR-resistant hypophosphorylated variant of 4E-BP reverses mTORC1-mediated massive NSC differentiation. In the adult neurogenic niche, mTORC1 becomes activated upon transition from NSCs into neurogenic intermediate progenitors, its activity is reduced in SVZ neuroblasts and gets activated in OB neuroblasts (Fig. 1) [53, 54]. Modulation of mTORC1 activity is often accomplished by manipulation of the PTEN phosphatase, an upstream negative regulator of the PI3K/AKT signaling pathway including mTORC. PTEN activity contributes to maintenance of a quiescent state in NSCs [6]. Conditional depletion of PTEN in quiescent NSCs forces their transition into the active states and subsequent differentiation into astrocytes and neurons. The involvement of mTORC1 activity in the transition from quiescent to active was recently demonstrated in muscle satellite cells [55]. However, the upstream regulators of mTORC1 during the transition from quiescence to NSCs activation, as well as the protein and RNA targets controlled by such stimulation remain to be addressed.
By the age of nine months the mouse brain shows considerable decline in the number of NSCs, which progressively continues with increasing age [56]. Notably, such decrease mostly occurs at the expense of active NSCs, hence accompanied by a similardrop of mTORC1 activity. Indeed, prolonged stimulation of mTORC1 with ketamin or premature inhibition by its specific inhibitor, rapamycin, either postpones or accelerates the age-related NSC decline, respectively. Summarizing, all these studies unambiguously underpin the pivotal role of mTORC1 in the timing of NSCs’ activation by switching-on anabolic processes, including protein biosynthesis. In support of the necessity to stimulate the process of translation for proper activation of quiescent NSCs, a recently published study from Miller’s and Kaplan’s labs found that many mRNAs encoding proneurogenic proteins such as neurod1, neurod4, neurogenin1, neurogenin2 could already be detected even in quiescent NSCs [57]. However, these mRNAs do not produce any protein since they are recruited into special cytoplasmic particles possessing characteristics of the processing bodies (P bodies). eIF4E bound to these mRNAs is associated with the negative regulator of translation 4E-transporter (4E-T) and such association stimulates the accumulation of the mRNAs in the P bodies (see next paragraph). Consequently, NSCs could be transcriptionally “personalized” to generate a great variety of NSCs upon activation. The authors speculate that differentiation-promoting extrinsic cues could stimulate the mRNA release from these particles and allow subsequent translation. The nature of such cues as well as the exact mechanism of translational stimulation remains a subject of future studies.
SPECIFIC TRANSLATION OF CELL FATE REGULATORS BY RBPS
Whereas the regulation of global translation levels for cell fate decisions is an emerging topic of interest, the translational control of specific cell fate modulators is a well-established concept for various molecules in multiple stem cell systems. Cells use a large repertoire of tools to post-transcriptionally control gene expression including RBPs and miRNAs.
RBPs are involved in a plethora of processes including alternative splicing, RNA processing, nuclear export, mRNA stability and translation [58]. Particularly in highly polarized cells like neurons, RBPs help to locally and spatially regulate translation efficiency and successfully integrate environmental signals [59]. Here we focus on a number of RBPs with reported significant contribution to cell fate decisions in the embryonic and adult brain.
FMRP
Fragile X mental retardation protein (FMRP) is a RBP with clear disease relevance. Fragile X syndrome is the most common inherited mental disorder, which results from the loss of FMRP. FMRP is known to bind specific mRNAs in order to inhibit their translation. Among its targets there are well-established regulators of the cell cycle including cyclin-dependent kinase 4 (CDK4) and cyclin D1, as well as members of the Wnt signaling pathway [60]. Accordingly, full knockout of FMRP leads to higher proliferation levels and altered fate decisions in the adult SGZ of the hippocampus. The closely related Fragile X relative protein 2 (FXR2) reduces the stability of Noggin mRNA [61]. Deficiency of FXR2 leads to increased Noggin expression and reduced BMP signaling, again resulting in increased proliferation of NSCs.
Musashi
The probably best-studied RBPs in the NSC field belong to the family of musashi proteins (MSI). Musashi1 (MSI1) and its homolog Musashi2 (MSI2) play an important role in neural development across vertebrates and invertebrates [62]. Both MSI proteins are highly expressed in the fetal and adult brain, highly enriched in the proliferative zones of the ventricular and subventricular zone. MSI1 is considered to be more restricted to multipotent stem cells and is often used as a NSC marker. Nonetheless, expression of the homologue proteins is actually largely overlapping indicating that they often act together. Accordingly, single knockout of MSI1 only has a minor NSC phenotype while double knockout of MSI1/MSI2 significantly decreases neurosphere-forming capacity of NSCs [63]. There are multiple ways of action described for Musashi proteins. However, the most canonical function is mediated over two RNA recognition motifs (RRMs). One of the most important targets is the Notch pathway component NUMB. Musashi binds at certain motifs in the 3’ UTR (usually enriched for GUAG or UAG), leading to downregulation of NUMB expression and subsequent activation of Notch signaling. Notably, Notch signaling is a well-known inhibitor of NSC differentiation [64] and pharmacological inhibition of Notch signaling in freshly isolated quiescent NSCs leads to activation and thereby increased protein synthesis [10].
Staufen and Pumilio
The balance between stem cell maintenance and differentiation can be also controlled by complexes of multiple RBPs and their target molecules. An RNA complex containing double-stranded RNA-binding protein Staufen homolog 2 (STAU2), Pumilio homolog 2 (PUM2) and ATP-dependent RNA helicase DDX1 was shown to be crucial for the correct development of radial glial precursors (RGPs) in the embryonic cortex [65]. STAU2 is apically localized in RPGs together with PUM2, DDX1 and target mRNAs including beta-actin, prospero and prox1. STAU2 and prox1 are asymmetrically enriched in some dividing precursors suggesting a stage specific role. Depletion of STAU2 was shown to increase genesis of neurons at expense of RPGs indicating that the STAU2 complex maintains the undifferentiated state. Knockdown of other complex members phenocopies STAU2 depletion, demonstrating a cooperative manner of regulation. Finally, it was shown that STAU2 RNA-binding domain is essential for proper RPG maintenance.
A parallel study focuses on the asymmetric segregation of STAU2 during cell division [66]. Interestingly, STAU2 preferentially segregates to TBR2-positive intermediate progenitor cells (IPCs), a more differentiated cell than RGPs. Further, the authors identify STAU2-associated mRNAs by immunoprecipitation. These contain known regulators of asymmetric and basal cell fate including tripartite motif-containing protein 32 (TRIM32). These target mRNAs are also asymmetrically segregated during division. This data suggests that STAU2 ensures polarized distribution of cell fate regulators, which promote the IPC fate. Loss of STAU2 stimulates the RGP to intermediate-progenitor transition, most likely since intermediate-progenitor fate determining mRNAs are now not polarized to one but present in both daughter cells.
4E-T
In a hallmark study from 2014, Yang and colleagues describe a complex encompassing the well-known initiation factor eiF4E1, its binding partner 4E-T and multiple proneurogenic mRNAs, which regulates the generation of neurons in the embryonic brain [57]. The associated transcripts include the proneurogenic neurogenin- and neurod family members. This complex is dynamically assembled and disassembled during development in order to avoid premature production of neurons. Disruption of the complex causes enhanced neurogenesis and precursor depletion. It is hypothesized that 4E-T most likely identifies target mRNAs for recruitment to cellular storages including P body-like granules. However, since binding of 4E-T to the target mRNAs is not direct but instead requires an intermediate protein, the exact mechanism or recruitment remainsunclear [67].
SMAUG2 and NANOS1
Similar to aforementioned RBPs, the pair of SMAUG2 and NANOS1 is cooperatively controlling proper development of neural progenitors in the embryonic cortex by a bimodal translation switch where SMAUG2 inhibits and NANOS1 promotes neurogenesis [68]. Interestingly, SMAUG2 is silencing nanos1 mRNA by recruiting it into P body-like granules in association with the eiF4E binding partner 4E-T (see above) in order to keep neural precursors in a multipotent stage. On the contrary, NANOS1 protein promotes differentiation of precursors by repressing the precursor state. However, direct RNA targets of NANOS1, encoding likely mediators of precursor activation, still need to beidentified.
These examples illustrate that RBPs are usually negative regulators of NSC differentiation through repression of target transcripts, which is often driven by direct binding to regulatory motifs in the untranslated regions. Particularly in the embryonic brain, RBPs tune gene expression of factors critical for differentiation. This way, cells ensure proper balance between differentiated and undifferentiated cells and therefore correct expansion of the central nervous system. There is evidence that multiple RBPs have similar functions, just varying in their repertoire of target molecules. In fact, several of the aforementioned RBPs are found in P body like granules, which supports that RBP-mediated repression is the main and highly conserved mechanism. To which level RBPs need to act in complexes with other RBPs is poorly addressed. A cooperative function between STAU2, PUM2 and DDX1 is suggested by the fact that knockdown of PUM2 or DDX1 phenocopies the effect of STAU2 depletion. Further studies are necessary to fully uncover the regulatory networks of RBPs both in the developing and adult brain.
miRNAs are short, non-coding RNA molecules, which regulate the expression of target mRNAs by affecting stability and translation [69]. In contrast to the rather limited studies on RBPs in neurogenesis, miRNAs have been extensively studied, particularly in the setting of the developing brain [70, 71]. Deletion of Dicer, which is central to miRNA production, was shown to impair neurogenesis illustrating their crucial role in proper neuronal development [72]. MiRNA are highly abundant in the brain and subject of dynamic regulation. This is exemplified by miR-124, which is undetectable in progenitor cells and becomes expressed with differentiation and maturation [73].
Together we conclude that while the regulatory role of microRNAs in the mouse brain is very well established, RBPs only came recently to the fore. It will be important to further investigate RBPs role by carefully mapping binding sites and identifying target sequences, experiments that are challenging and highly dependent on the availability of excellent antibodies and bioinformatic tools.
EMERGING ROLE OF RNA METHYLATION
Methylation at the m6 position of adenosines in eukaryotic mRNAs represents another critical but not yet addressed level of regulation of thetransition between quiescent and active states in stem cells. The presence of methylated adenosines in RNAs was already reported half a century ago [74, 75]. However, only after the discovery of the enzymes responsible for such dynamic modification, m6A methylation got in the research spotlight. First, a multiprotein complex including the “m6A writers” methyltransferase like 3 (METTL3), methyltransferase like 14 (METTL14) and the regulatory wilms tumor 1 associated protein (WTAP) was purified and characterized [76, 77]. Later, two m6A demethylases (“m6A erasers”) were identified, which provide a dynamic regulation of the modification: fat mass and obesity associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) [78, 79]. Recent data shows that both m6A-marks and the proteins responsible for their establishment and recognition can participate in the control of almost all steps of mRNA metabolism in cells, including transcription, splicing, translation and degradation [80, 81]. Interestingly, this type of modification is among the earliest regulators of the transition between the self-renewal state of pluripotent embryonic stem cells to lineage-committed differentiated cells. Some mRNAs encoding pluripotency factors such as NANOG, sex determining region Y-box 2 (SOX2), and octamer-binding transcription factor 4 (OCT-4) undergo m6A methylation in a well-coordinated manner at the time of loss of pluripotency and onset of differentiation [82, 83]. One of the proposed mechanisms responsible for such timely-controlled regulation could be the degradation of corresponding mRNAs triggered by m6A-mediated binding of “m6A readers” such as YTH domain family 2 (YTHDF2), human antigen R (HuR) and others involved in mRNA stability regulation [84, 85]. The controlled stability of mRNAs on its own is among major factors affecting the final protein production in cells and its regulation is closely linked to the activity of mRNAs in translation [86]. So, it is not surprising that the same modification could modulate both processes. Indeed, apart from the direct regulation of mRNA degradation, two recent studies showed that m6A methylation in the 5’ UTR could direct cap-independent initiation of translation on substrate mRNAs adding another level of regulation whereby this modification controls stem cell fate [87, 88]. As previously discussed, stem cells undergo strict translational regulations upon their activation for proliferation and differentiation. Some mRNAs, which encode critical regulatory factors, could potentially avoid general control of protein biosynthesis via the controlled assembly of the eIF4F initiation complex at the capped 5’ end. This could equip cells with a fine-tuning level of regulation upon transition to differentiated cells, finally affecting the development and maintenance of the whole organism. Apart from the direct regulation of mRNA translation and stability via the methylation of adenines in mRNAs, this modification was detected in miRNA precursors (pri-miRNA) and long non-coding RNAs (lncRNA) [89, 90]. In the case of pri-miRNAs, m6A stimulates their processing allowing the formation of functional miRNAs. miRNAs are well-known effectors regulating mRNA translation and stability, and numerous data demonstrate their critical role in the defining stem cell fate and activation [91]. Further progress is required to get insight into the mechanism of m6A-mediated regulation of miRNAs and lncRNAs in stem cell metabolism and activation.
At the present moment, there are no available data delineating possible functions for the m6A modification in adult stem cells and in neural stem cells in particular. The generation of conditional knockout animals as well as scaling down those experimental biochemical approaches allowing to analyze the dynamic distribution of methylated adenosines in transcriptomes of few isolated cells should help to decipher the role of this modification duringneurogenesis.
APPLICATION OF NOVEL METHODS TO STUDY PROTEIN PRODUCTION IN VIVO
Aforementioned paragraphs illustrate that most gained knowledge on posttranscriptional control of NSC dynamics are based on studies focusing on single regulatory components. There is evidence that multiple elements act together to determine the cellular outcome. As much as this is emphasized about the cooperative function of transcription factors, the role of cooperativity of post-transcriptional regulators has been so far merely neglected, mostly due to technical limitations. Here we give a short overview of recent technical advancements, which will help identification of global regulation of posttranscriptional events in neurogenesis.
Global rates of protein synthesis have been characterized using sucrose gradient fractionation experiments, but also by usage of the translation inhibitor puromycin, a tRNA homologue that incorporates into nascent proteins [92]. Recently the Salic lab developed a modified puromycin to detect nascent proteins with key advantages over existing methods [93].O-propargyl-puromyin (OP-puro) is a modified version of conventional puromycin that bears a terminal alkyne group that can be efficiently detected by fluorescence-coupled azide molecules. OP-puro is easy to use, robust, sensitive and both applicable for in vitro and in vivo studies. OP-Puro massively extended our knowledge about protein synthesis levels in multiple stem cell systems both under homeostasis and stress conditions [10, 15, 20].
Traditionally, the translational efficiency of individual transcripts has been assessed by its loading onto multiple ribosomes (polysomes). Their separation by sucrose gradient centrifugation and analysis by cDNA arrays allowed for the first time the comparison of global changes in transcription versus translation [94]. This so-called translational state array analysis (TSAA) was applied to investigate the translational changes during differentiation of murine embryonic stem cells [95]. This data demonstrated that differentiation causes an anabolic switch that involves increasing transcript abundance, ribosome loading and global protein synthesis. Interestingly, some genes were only regulated on the translational level demonstrating that mRNA levels can be a misleading readout in certain cellular contexts.
The recent development of next generation sequencing techniques allows efficient production of sequencing libraries from minute amounts of mRNA at affordable rates [96]. Thus, TSAA studies are not any more limited to available probe sets of microarrays and offer much higher resolution. However, TSAA is still difficult to apply in vivo, since tissues harbor heterogeneous cell populations that hampers interpretation of results. The study of specific cell populations requires cell sorting that potentially introduces artifacts, since improper sample handling can easily cause ribosome dissociation.
In order to overcome these disadvantages, several mouse models have been established over the last years to investigate the ribosome-bound mRNA, called translatome. Translating ribosome affinity purification (TRAP) makes use of bacterial artificial chromosomes (BACs) introduced into a transgenic mouse model which expresses a GFP-tagged variant of the ribosomal protein L10a in a defined cell population [97, 98]. Using several TRAP mouse lines, where tagged L10 was expressed under different specific promoters, it could be shown that morphologically indistinguishable neuronal subtypes display massive differences in translated genes [97]. These studies provided a resource of 16 transgenic BAC mouse lines targeting 24 subtypes of neurons – a valuable dataset to study the wide molecular spectrum of neurons [98]. Over the last years additional TRAP lines were generated for various applications [99–101]. The TRAP methodology was originally generated to overcome technical limitations of neuronal cell sorting. Flow cytometry-mediated cell sorting of solid tissues can be difficult, especially in case of adult neurons, which are extremely connected in the brain and are highly myelinated. In order to compare the transcriptome to the TRAP-based translatome of adult brain neurons it will be necessary to establish improved protocols of high purity neuron isolation to obtain a reliable readout for translational efficiency. Application of TRAP to different stages of NSC activation would provide valuable insights into the post-transcriptional regulation during activation and differentiation, since the transcriptomes are well studied both on population- and single cell level [4, 10]. However, so far there are no published TRAP lines addressing the translatome of NSCs.
TRAP is majorly limited by the availability of mouse lines. For every cell type of interest, a new transgenic line has to be generated and characterized, where (in best case) an exclusive marker gene of this population is driving the expression of the ribosomal tag. This is laborious and a hinder to its application to study multiple cell types in vivo. A parallel approach makes use of preexisting Cre-recombinase driver lines. In Ribotag mice replacement of endogenous RPL22 protein, a ribosomal large subunit protein, with a HA-tagged variant is driven by cell-type specific Cre-recombinase expression and allows isolation of ribosome-associated mRNA [102]. Both TRAP and Ribotag have been mostly used to study rare types of neurons in the brain, which are challenging to isolate by conventional methods (see above). In fact, Ribotag was recently applied to study the axonal translatome of retinal cell axons of the developing and adult retinotectal projection, demonstrating localized translation in axons of the mammalian central nervous systems [103]. It will be crucial to apply these methods with NSC specific transgenic mouse lines in order to investigate, to which level the transcriptome correlates with actual protein expression, since local and temporal control of protein expression at posttranscriptional level might impact cell fate decisions. A comprehensive review on the correlation of transcriptome and proteome concluded that at steady-state mRNA levels match well to assessed protein levels. However at state transition as well as short-term adaptation periods the ratio of mRNA abundance to protein levels is highly modulated by posttranscriptional control mechanisms [104]. Thus, the process of neurogenesis, which involves transition of NSCs through multiple stages and requires adaptation to the changing microenvironment, might also be controlled at the translational level.
While TRAP and Ribotag offer the great advantage of targeting ribosome-associated transcripts of a defined cell population in vivo, these techniques cannot completely recapitulate ongoing translation since ribosomes can also “sit” on transcripts without active peptide synthesis. This ribosome stalling is considered to take place under certain conditions and can be addressed in great detail by ribosome profiling [105, 106]. Ribosome profiling is the deep sequencing of ribosome-protected mRNA fragments that are revealed by nuclease digestion. This method revolutionized the way we can look at ongoing translation allowing the identification of novel ORFs, the investigation of the number of ribosomes on certain transcripts and the speed at which they are moving along the transcript under different conditions [106, 107]. Due to the high demand of input material, application of ribosome profiling on mouse tissue has been challenging so far. Ribosome profiling of the hippocampus in different contextual paradigms demonstrated multiple repressive mechanisms during memory formation [108]. However, this study used total hippocampal tissue, not distinguishing the contribution of different cell types. Gonzalez and colleagues have further extended the methodology by doing ribosome profiling in combination with Ribotag mouse models [109]. This allowed detailed cell-type specific analysis of translational changes during glioma initiation. There are efforts to improve library preparation methods in order to do ribosome profiling from low input material, which will be crucial to understand ribosome behavior in specific subpopulations of cells in the brain [110].
Together, these methods offer great potential to further dissect both mechanisms of global and specific control of protein synthesis in stem cells. Their application on carefully characterized subpopulations can help to understand the key post-transcriptional events taking place during activation and differentiation.
CONCLUSIONS
Stemness features a population of cells that are present throughout the life of an animal with the ability to generate differentiated progeny. We now know that stem cell maintenance crucially depends on keeping a quiescent state, as generation of differentiated progeny depends on stem cell activation. Hence, a tight balance between quiescence and activation governs stem cell’s function. The switch between a quiescent and activated state crucially depends on repression or activation of protein synthesis, one of the most energy-demanding processes of the cell. Translation of mRNAs can be regulated at a more global level via ribosome availability. However, even repression of translation by factors presumably regulating the vast majority of mRNAs, like phosphorylation of eIF2 by stress-related kinases, can be evaded by a subset of transcripts -in case of eIF2, by mRNAs entailing short upstream ORFs. A more transcript-specific control of translation is imposed by the presence of motifs specifically recognized by RBPs, microRNA, circRNAs and others not yet defined factors; also posttranscriptional modification of mRNAs such as methylation dictates the life of a transcript. To date, most studies have focused on unraveling the role of specific translation-modulating factors by combining loss-of-function and gain-of-function studies with readouts of stem cell function. Fortunately, the forthcoming of next generation sequencing coupled to immunoprecipitation of ribosomal-associated, RBP-associated, dedicated ribosomal subunits, or specifically modified transcripts will expand our knowledge of global mechanism of translation governing the biology of stem cells in the different organs.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was supported by the University of Heidelberg and DKFZ (bridge-project ZMBH-DKFZ alliance), the DFG (SFB873) and the DKFZ. We apologize to authors whose work we were unable to cite due to space limitations.
REFERENCES
[1] | Bond AM , Ming G-L , Song H . Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell. (2015) ;17: (4):385–95. |
[2] | Fuentealba LC , Obernier K , Alvarez-Buylla A . Adult neural stem cells bridge their niche. Cell Stem Cell. (2012) ;10: (6):698–708. |
[3] | Furutachi S , Miya H , Watanabe T , Kawai H , Yamasaki N , Harada Y , et al. Slowly dividing neural progenitors are an embryonic origin of adult neural stem cells. Nat Neurosci. (2015) ;18: (5):657–65. |
[4] | Codega P , Silva-Vargas V , Paul A , Maldonado-Soto AR , Deleo AM , Pastrana E , et al. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. (2014) ;82: (3):545–59. |
[5] | Calzolari F , Michel J , Baumgart EV , Theis F , Götz M , Ninkovic J . Fast clonal expansion and limited neural stem cell self-renewal in the adult subependymal zone. Nat Neurosci. (2015) ;18: (4):490–2. |
[6] | Bonaguidi MA , Wheeler MA , Shapiro JS , Stadel RP , Sun GJ , Ming G-L , et al. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. (2011) ;145: (7):1142–55. |
[7] | Encinas JM , Michurina TV , Peunova N , Park J-H , Tordo J , Peterson DA , et al. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. (2011) ;8: (5):566–79. |
[8] | Merkle FT , Mirzadeh Z , Alvarez-Buylla A . Mosaic Organization of Neural Stem Cells in the Adult Brain. Science. (2007) ;317: (5836):381–4. |
[9] | Merkle FT , Fuentealba LC , Sanders TA , Magno L , Kessaris N , Alvarez-Buylla A . Adult neural stem cells in distinct microdomains generate previously unknown interneuron types. Nat Neurosci. (2014) ;17: (2):207–14. |
[10] | Llorens-Bobadilla E , Zhao S , Baser A , Saiz-Castro G , Zwadlo K , Martin-Villalba A . Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. (2015) ;17: (3):329–40. |
[11] | Erecińska M , Silver IA . Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. (2001) ;128: (3):263–76. |
[12] | Mazumdar J , O’Brien WT , Johnson RS , LaManna JC , Chavez JC , Klein PS , et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nature Cell Biology. (2010) ;12: (10):1007–13. |
[13] | Candelario KM , Shuttleworth CW , Cunningham LA . Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor-1alpha expression. J Neurochem. (2013) ;125: (3):420–9. |
[14] | Khacho M , Clark A , Svoboda DS , Azzi J , MacLaurin JG , Meghaizel C , et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell. (2016) ;19: (2):232–47. |
[15] | Signer RAJ , Magee JA , Salic A , Morrison SJ . Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. (2014) ;509: (7498):49–54. |
[16] | Dröge W . Free radicals in the physiological control of cell function. Physiological Reviews. (2002) ;82: (1):47–95. |
[17] | Signer RAJ , Morrison SJ . Mechanisms that regulate stem cell aging and life span. Cell Stem Cell. (2013) ;12: (2):152–65. |
[18] | Wanet A , Arnould T , Najimi M , Renard P . Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. (2015) ;24: (17):1957–71. |
[19] | Folmes CDL , Terzic A . Energy metabolism in the acquisition and maintenance of stemness. Semin Cell Dev Biol. (2016) ;52: :68–75. |
[20] | Blanco S , Bandiera R , Popis M , Hussain S , Lombard P , Aleksic J , et al. Stem cell function and stress response are controlled by protein synthesis. Nature. (2016) ;534: (7607):335–40. |
[21] | Buttgereit F , Brand MD . A hierarchy of ATP-consuming processes in mammalian cells. Biochem J. (1995) ;312: (Pt 1):163–7. |
[22] | Rolfe DF , Brown GC . Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiological Reviews. (1997) ;77: (3):731–58. |
[23] | Arvidsson A , Collin T , Kirik D , Kokaia Z , Lindvall O . Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. (2002) ;8: (9):963–70. |
[24] | Le JE , Orozco NM , Paucar AA , Saxe JP , Mottahedeh J , Pyle AD , et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. (2011) ;8: (1):59–71. |
[25] | Kokovay E , Wang Y , Kusek G , Wurster R , Lederman P , Lowry N , et al. VCAM1 is essential to maintain the structure of the SVZ niche and acts as an environmental sensor to regulate SVZ lineage progression. Cell Stem Cell. (2012) ;11: (2):220–30. |
[26] | Walton NM , Shin R , Tajinda K , Heusner CL , Kogan JH , Miyake S , et al. Adult neurogenesis transiently generates oxidative stress. PLoS one. (2012) ;7: (4):e35264. |
[27] | Hutchison CA , Chuang R-Y , Noskov VN , Assad-Garcia N , Deerinck TJ , Ellisman MH , et al. Design and synthesis of a minimal bacterial genome. Science. (2016) ;351: (6280):aad6253. |
[28] | Zhang Q , Shalaby NA , Buszczak M . Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science. (2014) ;343: (6168):298–301. |
[29] | Shin J , Berg DA , Zhu Y , Shin JY , Song J , Bonaguidi MA , et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. (2015) ;17: (3):360–72. |
[30] | Cai X , Gao L , Teng L , Ge J , Oo ZM , Kumar AR , et al. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell. (2015) ;17: (2):165–77. |
[31] | Schwanhäusser B , Busse D , Li N , Dittmar G , Schuchhardt J , Wolf J , et al. Global quantification of mammalian gene expression control. Nature. (2011) ;473: (7347):337–42. |
[32] | Li JJ , Bickel PJ , Biggin MD . System wide analyses have underestimated protein abundances and the importance of transcription in mammals. Peer J. (2014) ;2: :e270. |
[33] | Chen L-L . The biogenesis and emerging roles of circular RNAs. Nature Publishing Group. (2016) ;17: (4):205–11. |
[34] | Fu Y , Dominissini D , Rechavi G , He C . Gene expression regulation mediated through reversible m6A RNA methylation. Nature Reviews Genetics. (2014) ;15: (5):293–306. |
[35] | Shi Z , Barna M . Translating the Genome in Time and Space: Specialized Ribosomes, RNA Regulons, and RNA-Binding Proteins. Annu Rev Cell Dev Biol. (2015) ;31: (1):31–54. |
[36] | Hinnebusch AG , Lorsch JR . The mechanism of eukaryotic translation initiation: New insights and challenges. Cold Spring Harb Perspect Biol. (2012) ;4: :a011544. |
[37] | Dever TE , Green R . The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol. (2012) ;4: (7):a013706. |
[38] | Koromilas AE . Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochim Biophys Acta. (2015) ;1849: (7):871–80. |
[39] | Zismanov V , Chichkov V , Colangelo V , Jamet S , Wang S , Syme A , et al. Phosphorylation of eIF2α Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell. (2016) ;18: (1):79–90. |
[40] | Pause A , Belsham GJ , Gingras AC , Donzé O , Lin TA , Lawrence JC , et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature. (1994) ;371: (6500):762–7. |
[41] | Laplante M , Sabatini DM . mTOR signaling in growth control and disease. Cell. (2012) ;149: (2):274–93. |
[42] | Graff JR , Konicek BW , Carter JH , Marcusson EG . Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. (2008) ;68: (3):631–4. |
[43] | Morita M , Gravel S-P , Chénard V , Sikström K , Zheng L , Alain T , et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. (2013) ;18: (5):698–711. |
[44] | Holz MK , Ballif BA , Gygi SP , Blenis J . mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. (2005) ;123: (4):569–80. |
[45] | Wang X , Li W , Williams M , Terada N , Alessi DR , Proud CG . Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. The EMBO Journal. (2001) ;20: (16):4370–9. |
[46] | Mayer C , Zhao J , Yuan X , Grummt I . mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes & Development. (2004) ;18: (4):423–34. |
[47] | Wei Y , Tsang CK , Zheng XFS . Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. The EMBO Journal. (2009) ;28: (15):2220–30. |
[48] | Lee DY . Roles of mTOR Signaling in Brain Development. Exp Neurobiol. (2015) ;24: (3):177–85. |
[49] | Tee AR , Sampson JR , Pal DK , Bateman JM . The role of mTOR signalling in neurogenesis, insights from tuberous sclerosis complex. Semin Cell Dev Biol. (2016) ;52: :12–20. |
[50] | Cloëtta D , Thomanetz V , Baranek C , Lustenberger RM , Lin S , Oliveri F , et al. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci. (2013) ;33: (18):7799–810. |
[51] | Hartman NW , Lin TV , Zhang L , Paquelet GE , Feliciano DM , Bordey A . mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Reports. (2013) ;5: (2):433–44. |
[52] | Mahoney C , Feliciano DM , Bordey A , Hartman NW . Switching on mTORC1 induces neurogenesis but not proliferation in neural stem cells of young mice. Neurosci Lett. (2016) ;614: :112–8. |
[53] | Paliouras GN , Hamilton LK , Aumont A , Joppe SE , Barnabe-Heider F , Fernandes KJL . Mammalian Target of Rapamycin Signaling Is a Key Regulator of the Transit-Amplifying Progenitor Pool in the Adult and Aging Forebrain. J Neurosci. (2012) ;32: (43):15012–26. |
[54] | Zhu G , Chow LML , Bayazitov IT , Tong Y , Gilbertson RJ , Zakharenko SS , et al. Pten deletion causes mTorc1-dependent ectopic neuroblast differentiation without causing uniform migration defects. Development. (2012) ;139: (18):3422–31. |
[55] | Rodgers JT , King KY , Brett JO , Cromie MJ , Charville GW , Maguire KK , et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. (2014) ;510: (7505):393–6. |
[56] | Romine J , Gao X , Xu X-M , So KF , Chen J . The proliferation of amplifying neural progenitor cells is impaired in the aging brain and restored by the mTOR pathway activation. Neurobiol Aging. (2015) ;36: (4):1716–26. |
[57] | Yang G , Smibert CA , Kaplan DR , Miller FD . An eIF4E1/4E-T Complex Determines the Genesis of Neurons from Precursors by Translationally Repressing a Proneurogenic Transcription Program. Neuron. (2014) ;84: (4):723–39. |
[58] | Ye J , Blelloch R . Regulation of pluripotency by RNA binding proteins. Cell Stem Cell. (2014) ;15: (3):271–80. |
[59] | Jung H , Yoon BC , Holt CE . Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci. (2012) ;13: (5):308–24. |
[60] | Luo Y , Shan G , Guo W , Smrt RD , Johnson EB , Li X , et al. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. (2010) ;6: (4):e1000898. |
[61] | Guo W , Zhang L , Christopher DM , Teng Z-Q , Fausett SR , Liu C , et al. RNA-binding protein FXR2 regulates adult hippocampal neurogenesis by reducing Noggin expression. Neuron. (2011) ;70: (5):924–38. |
[62] | Okano H , Kawahara H , Toriya M , Nakao K , Shibata S , Imai T . Function of RNA-binding protein Musashi-1 in stem cells. Experimental Cell Research. (2005) ;306: (2):349–56. |
[63] | Sakakibara S-I , Nakamura Y , Yoshida T , Shibata S , Koike M , Takano H , et al. RNA-binding protein Musashi family: Roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci USA. (2002) ;99: (23):15194–9. |
[64] | Pierfelice T , Alberi L , Gaiano N . Notch in the vertebrate nervous system: An old dog with new tricks. Neuron. (2011) ;69: (5):840–55. |
[65] | Vessey JP , Amadei G , Burns SE , Kiebler MA , Kaplan DR , Miller FD . An asymmetrically localized Staufen2-dependent RNA complex regulates maintenance of mammalian neural stem cells. Cell Stem Cell. (2012) ;11: (4):517–28. |
[66] | Kusek G , Campbell M , Doyle F , Tenenbaum SA , Kiebler M , Temple S . Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell. (2012) ;11: (4):505–16. |
[67] | Kamenska A , Lu W-T , Kubacka D , Broomhead H , Minshall N , Bushell M , et al. Human 4E-T represses translation of bound mRNAs and enhances microRNA-mediated silencing. Nucleic Acids Research. (2014) ;42: (5):3298–313. |
[68] | Amadei G , Zander MA , Yang G , Dumelie JG , Vessey JP , Lipshitz HD , et al. A Smaug2-Based Translational Repression Complex Determines the Balance between Precursor Maintenance versus Differentiation during Mammalian Neurogenesis. J Neurosci. (2015) ;35: (47):15666–81. |
[69] | Bartel DP . MicroRNAs: Target recognition and regulatory functions. Cell. (2009) ;136: (2):215–33. |
[70] | Greve TS , Judson RL , Blelloch R . microRNA Control of Mouse and Human Pluripotent Stem Cell Behavior. Annu Rev Cell Dev Biol. (2013) ;29: (1):213–39. |
[71] | Shi Y , Zhao X , Hsieh J , Wichterle H , Impey S , Banerjee S , et al. MicroRNA Regulation of Neural Stem Cells and Neurogenesis. J Neurosci. (2010) ;30: (45):14931–6. |
[72] | Bernstein E , Kim SY , Carmell MA , Murchison EP , Alcorn H , Li MZ , et al. Dicer is essential for mouse development. Nat Genet. (2003) ;35: (3):215–7. |
[73] | Deo M , Yu J-Y , Chung K-H , Tippens M , Turner DL . Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn. (2006) ;235: (9):2538–48. |
[74] | Iwanami Y , Brown GM . Methylated bases of ribosomal ribonucleic acid from HeLa cells. Arch Biochem Biophys. (1968) ;126: (1):8–15. |
[75] | Desrosiers R , Friderici K , Rottman F . Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. (1974) ;71: (10):3971–5. |
[76] | Bokar JA , Rath-Shambaugh ME , Ludwiczak R , Narayan P , Rottman F . Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. (1994) ;269: (26):17697–704. |
[77] | Liu J , Yue Y , Han D , Wang X , Fu Y , Zhang L , et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) ;10: (2):93–5. |
[78] | Jia G , Fu Y , Zhao X , Dai Q , Zheng G , Yang Y , et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) ;7: (12):885–7. |
[79] | Zheng G , Dahl JA , Niu Y , Fedorcsak P , Huang C-M , Li CJ , et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell. (2013) ;49: (1):18–29. |
[80] | Yue Y , Liu J , He C . RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes & Development. (2015) ;29: (13):1343–55. |
[81] | Cao G , Li H-B , Yin Z , Flavell RA . Recent advances in dynamic m6A RNA modification. Open Biol. (2016) ;6: :160003. |
[82] | Batista PJ , Molinie B , Wang J , Qu K , Zhang J , Li L , et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. (2014) ;15: (6):707–19. |
[83] | Geula S , Moshitch-Moshkovitz S , Dominissini D , Mansour AA , Kol N , Salmon-Divon M , et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. (2015) ;347: (6225):1002–6. |
[84] | Wang X , Lu Z , Gomez A , Hon GC , Yue Y , Han D , et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) ;505: (7481):117–20. |
[85] | Wang Y , Li Y , Toth JI , Petroski MD , Zhang Z , Zhao JC . N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nature Cell Biology. (2014) ;16: (2):191–8. |
[86] | Roy B , Jacobson A . The intimate relationships of mRNA decay and translation. Trends Genet. (2013) ;29: (12):691–9. |
[87] | Meyer KD , Patil DP , Zhou J , Zinoviev A , Skabkin MA , Elemento O , et al. 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell. (2015) ;163: (4):999–1010. |
[88] | Zhou J , Wan J , Gao X , Zhang X , Jaffrey SR , Qian S-B . Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. (2015) ;526: (7574):591–4. |
[89] | Alarcón CR , Lee H , Goodarzi H , Halberg N , Tavazoie SF . N6-methyladenosine marks primary microRNAs for processing. Nature. (2015) ;519: (7544):482–5. |
[90] | Liu N , Parisien M , Dai Q , Zheng G , He C , Pan T . Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. (2013) ;19: (12):1848–56. |
[91] | Shenoy A , Blelloch RH . Regulation of microRNA function in somatic stem cell proliferation and differentiation. Nature Publishing Group. (2014) ;15: (9):565–76. |
[92] | Nathans D . Puromycin inhibition of protein synthesis: Incorporation of puromycin into peptide chains. Proc Natl Acad Sci USA. (1964) ;51: :585–92. |
[93] | Liu J , Xu Y , Stoleru D , Salic A . Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci USA. (2012) ;109: (2):413–8. |
[94] | Zong Q , Schummer M , Hood L , Morris DR . Messenger RNA translation state: The second dimension of high-throughput expression screening. Proc Natl Acad Sci USA. (1999) ;96: (19):10632–6. |
[95] | Sampath P , Pritchard DK , Pabon L , Reinecke H , Schwartz SM , Morris DR , et al. A Hierarchical Network Controls Protein Translation during Murine Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell. (2008) ;2: (5):448–60. |
[96] | Adiconis X , Borges-Rivera D , Satija R , DeLuca DS , Busby MA , Berlin AM , et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Meth. (2013) ;10: (7):623–9. |
[97] | Heiman M , Schaefer A , Gong S , Peterson JD , Day M , Ramsey KE , et al. A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell. (2008) ;135: (4):738–48. |
[98] | Doyle JP , Dougherty JD , Heiman M , Schmidt EF , Stevens TR , Ma G , et al. Application of a Translational Profiling Approach for the Comparative Analysis of CNS Cell Types. Cell. (2008) ;135: (4):749–62. |
[99] | Brichta L , Shin W , Jackson-Lewis V , Blesa J , Yap E-L , Walker Z , et al. Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat Neurosci. (2015) ;18: (9):1325–33. |
[100] | Zhou P , Zhang Y , Ma Q , Gu F , Day DS , He A , et al. Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc Natl Acad Sci USA. (2013) ;110: (38):15395–400. |
[101] | Zhang KX , Tan L , Pellegrini M , Zipursky SL , McEwen JM . Rapid Changes in the Translatome during the Conversion of Growth Cones to Synaptic Terminals. CellReports. (2016) ;14: (5):1258–71. |
[102] | Sanz E , Yang L , Su T , Morris DR , McKnight GS , Amieux PS . Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA. (2009) ;106: (33):13939–44. |
[103] | Shigeoka T , Jung H , Jung J , Turner-Bridger B , Ohk J , Lin JQ , et al. Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell. (2016) ;166: (1):181–92. |
[104] | Liu Y , Beyer A , Aebersold R . On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. (2016) ;165: (3):535–50. |
[105] | Ingolia NT , Ghaemmaghami S , Newman JRS , Weissman JS . Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. (2009) ;324: (5924):218–23. |
[106] | Ingolia NT , Lareau LF , Weissman JS . Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. (2011) ;147: (4):789–802. |
[107] | Ingolia NT , Brar GA , Stern-Ginossar N , Harris MS , Talhouarne GJS , Jackson SE , et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. (2014) ;8: (5):1365–79. |
[108] | Cho J , Yu N-K , Choi J-H , Sim S-E , Kang SJ , Kwak C , et al. Multiple repressive mechanisms in the hippocampus during memory formation. Science. (2015) ;350: (6256):82–7. |
[109] | Gonzalez C , Sims JS , Hornstein N , Mela A , Garcia F , Lei L , et al. Ribosome Profiling Reveals a Cell-Type-Specific Translational Landscape in Brain Tumors. J Neurosci. (2014) ;34: (33):10924–36. |
[110] | Hornstein N , Torres D , Sharma Das S , Tang G , Canoll P , Sims PA . Ligation-free ribosome profiling of cell type-specific translation in the brain. Genome Biol. Genome Biology. (2016) 1–15. |

