
DNA Methylation and Adult Neurogenesis
Abstract
The role of DNA methylation in brain development is an intense area of research because the brain has particularly high levels of CpG and mutations in many of the proteins involved in the establishment, maintenance, interpretation, and removal of DNA methylation impact brain development and/or function. These include DNA methyltransferase (DNMT), Ten-Eleven Translocation (TET), and Methyl-CpG binding proteins (MBPs). Recent advances in sequencing breadth and depth as well the detection of different forms of methylation have greatly expanded our understanding of the diversity of DNA methylation in the brain. The contributions of DNA methylation and associated proteins to embryonic and adult neurogenesis will be examined. Particular attention will be given to the impact on adult hippocampal neurogenesis (AHN), which is a key mechanism contributing to brain plasticity, learning, memory and mood regulation. DNA methylation influences multiple aspects of neurogenesis from stem cell maintenance and proliferation, fate specification, neuronal differentiation and maturation, and synaptogenesis. In addition, DNA methylation during neurogenesis has been shown to be responsive to many extrinsic signals, both under normal conditions and during disease and injury. Finally, crosstalk between DNA methylation, Methyl-DNA binding domain (MBD) proteins such as MeCP2 and MBD1 and histone modifying complexes is used as an example to illustrate the extensive interconnection between these epigenetic regulatory systems.
INTRODUCTION
The term epigenetics was first used by Conrad H Waddington in 1942, before the age of DNA, to define the “study of those processes by which genotype gives rise to phenotype” [1]. Later iterations of this definition required that epigenetic modifications lead to heritable changes in gene expression and function that are maintained across cell divisions or generations [2]. Thus, epigenetic regulation is particularly relevant in proliferative cells, such as those involved in AHN. Recent advances have shown that certain cell functions depend on dynamic regulation of epigenetic programs in addition to maintaining epigenetic programs through cell division. DNA methylation, one of the core epigenetic modifications along with histone modification, can be transmitted to daughter cells and was considered a largely permanent DNA modification until recent discovery of the Ten-Eleven Translocation (TET) enzymes in 2009 [3]. This discovery, coupled with the advent of more sophisticated techniques to measure and sequence DNA methylation and other derivatives has greatly expanded our knowledge of this epigenetic mark and the various roles it plays in the cell.
Likewise, the discovery and characterization of adult neurogenesis in humans and many other species over the past twenty years has caused a paradigm shift in the field of neuroscience: you are not born with all of the neurons you will ever have. In small regions of the brain stem cells continuously give rise to new neurons which play important roles in learning, memory and mood regulation. In addition, adult neurogenesis is also altered in many disease states— but can also be manipulated by pharmacological treatment and other interventions, making it a promising avenue for intervention. The role of DNA methylation in brain development is an intense area of research because the brain has particularly high levels of CpG and mutations in many of the proteins involved in the establishment, maintenance and interpretation of DNA methylation impact brain development and/or function. Epigenetic pathways, including DNA methylation, influence multiple aspects of neurogenesis from stem cell maintenance and proliferation, fate specification, neuronal differentiation and maturation, and synaptogenesis. In addition, DNA methylation during neurogenesis has been shown to be responsive to many extrinsic signals, both under normal conditions and during disease and injury.
NEUROGENESIS
The epigenetic networks regulating neurogenesis are highly connected with each other and other signaling pathways and regulatory networks. To date, embryonic cortical neurogenesis and adult hippocampal neurogenesis (AHN) are the best-studied systems. Although they have many similarities and are regulated by many of the same networks, there are also many differences that warrant investigation. Embryonic brain development must integrate growth, patterning and differentiation signals to generate neurons, astrocytes, and oligodendrocytes (and sub-types of cells) in a spatial and temporally-specific manner while still maintaining populations of stem cells. aNSCs also generate multiple cell types; moreover they are responsive to environmental inputs, pharmacological interventions, and are a major source of plasticity in the adult brain [4]. Adult neurogenesis contributes to the behavioral problems and learning deficits observed in many neurodevelopmental, neurodegenerative, and injury-based disorders [5, 6]. Animal models have provided an invaluable resource to evaluate adult neurogenesis and the mechanisms that regulate it. Several promising results indicate that it may be possible to improve function through treatment at post-natal and adult stages, even for disorders with partial developmental etiology, such as fragile X syndrome (FXS) and Rett syndrome (RTT) [7, 8]. Indeed, many anti-depressants and other medications are known to increase neural stem cell proliferation in animal models and humans. Because there is a distinct cellular progression of adult neurogenesis, and defined behavioral outcomes associated with altered neurogenesis, adult neurogenesis is an excellent model to study regulatory mechanisms, interventions, and functional outcomes.
Neurogenesis is defined as process that leads to the generation of new functional neurons. This process includes the proliferation and fate specification of neural stem cells and the differentiation and integration of newly generated neurons into the existing neural circuitry. Mammalian neurogenesis is divided into two phases: embryonic/developmental neurogenesis and adult neurogenesis. Embryonic neurogenesis encompasses the generation of neurons in the context of the formation of the central nervous system (CNS). In the adult brain, multipotent NSCs remain and continue to generate new neurons. There are two niches of the adult mammalian CNS confirmed to have ongoing neurogenesis: the subgranular zone (SGZ) of the dentate gyrus (DG) in the hippocampus and the SVZ bordering the lateral ventricles [9]. New neurons have also been observed in other regions of the brain, but these reports are more variable and may depend on the species and physiological or pathological state (reviewed in [10]). aNSCs in both the DG and the SVZ derive from embryonic NSCs. In mice, the DG begins to develop at E13.5 from NSCs located adjacent to the cortical hem and continues to develop until early postnatal stages [11]. Some NSCs retain their multi-potent properties and transform into radial glia like cells (RGLs), so named because their cell bodies reside in the subgranular zone (SGZ) and their radial processes extend through the DG molecular layer in manner reminiscent of RGs. RGLs, also known as type 1 cells, give rise to intermediate or transit-amplifying progenitors (type2a/b, type 3) which subsequently differentiate into neurons. A small number of astrocytes are generated from NSCs, but it is unclear if they arise directly from RGLs or if they pass through a non-committed transit amplifying intermediate [12, 13]. Cells in different stages of neurogenesis can be defined by a combination of morphology, cell-type specific markers, and proliferative capacity [14]. Throughout the process of neurogenesis, DNA methylation changes as cells proceed through different cell stages and respond to differentinputs.
Function of adult hippocampal neurogenesis
The hippocampus was recognized as being necessary for certain types of learning and memory based on cognitive studies of memory impairment in humans and hippocampal lesion studies in model animals [15]. Further studies determined that the hippocampus is required for spatial and contextual learning and memory [16]. Evidence for the importance of NSCs— and thus the formation of new neurons— in hippocampal learning and memory came from studies that ablated adult neural progenitors via genetic means, anti-proliferative drugs, or focal irradiation [17–22]. Hippocampal neurogenesis contributes to hippocampal plasticity and both play a role in hippocampal-dependent cognitive function.
AHN represents a unique source of plasticity in the brain. Plasticity, or the ability to respond and adapt to stimulus, is generated by AHN in the DG by two ways: 1) how many new cells are produced and 2) how these cell integrate into existing networks. First, increased NPC proliferation represents a major mechanism through which more neurons can be generated. The quiescence of RGLs, the cell cycle progression and differentiation of IPCs, and the maturation of new neurons are all tightly controlled by intricate molecular networks that consist of intrinsic genetic and epigenetic programs modulated by extrinsic physiological and pathological conditions [14, 23, 24]. The second mechanism driving adult neurogenesis-mediated plasticity is the integration of new cells to existing networks. These new neurons pass through a ‘critical period’ of enhanced synaptic and dendritic plasticity 3–6 weeks post-mitosis that is dependent upon the inputs cells receive [25–27]. Disruption of cells during this window of time disrupts hippocampal-dependent learning and memory [19, 21, 28, 29], revealing that cells in this stage are vital to the functional output of adult neurogenesis.
DNA METHYLATION
Epigenetic mechanisms, including DNA methylation, are critical for establishing the diverse cell fates present in the central nervous system. Different regions of the human brain (cerebral cortex, cerebellum, and pons) each have a characteristic DNA methylation signature [30]. And even within brain regions like the hippocampus, global methylation varies between neuronal subtypes [31]. There is good support from genome-wide methylation studies that DNA methylation globally represses neuronal genes in non-neuronal cells, supporting earlier studies of individual genes [32]. Multiple mechanisms likely contribute to gene repression by DNA methylation, including recruitment of repressive complexes by methyl-CpG binding proteins (MBPs) or through blocking the binding of pro-neuronal transcription factors (TFs). Though many studies have analyzed cell-type specific transcriptomes [33–37], the number of studies that combine transcriptome and genome-wide methylation is more limited. However, the growing feasibility of this type of study will allow researchers to ask questions about the role of DNA methylation in the cell. Such as, are DNA methylation patterns cell-type specific? Do they correspond to histone modifications or the binding patterns of other repressive complexes? Is methylation always associated with repression?
DNA methylation and DNMTs
DNA methylation is well known for its role in long-term gene silencing; it serves as the basis of imprinting, X chromosome inactivation, and the establishment of cell fate [38–40]. DNA methylation involves the covalent addition of a methyl group from the cofactor SAM (S-adenosyl-l- methionine) to C5 of cytosine in CG dinucleotides (also referred to as CpG, 5mC, or mCG). This addition is catalyzed by a family of DNA methyltransferases (DNMTs). DNMT3a and DNMT3b establish de novo methylation, whereas DNMT1 maintains methylation patterns in newly synthesized DNA by recognizing hemi-methylated DNA and methylating the unmodified strand [41].
In addition to CG methylation, other dinucleotide pairs containing cytosine can be methylated, referred to as CH or CpH methylation, where H = A/C/T. Recent studies have shown that CH methylation (mCH) is high in the brains of humans and mice [42, 43]. And within the brain, non-CG methylation is much more prevalent in neurons than non-neuronal cells and is estimated to account for 25–38% of total methylation [44–46]. CH methylation has been shown to accumulate dramatically in neurons but not astrocytes during postnatal development, a critical period of neuronal maturation and synaptogenesis [46]. There is evidence that DNMT3A is responsible for the deposition of mCH, and that non-CG methylation is also associated with gene repression [46]. The growing awareness of non-CG methylation has the potential toyield novel insights into the role of DNA methylation in regulating brain development and plasticity
Though many studies have analyzed cell-type specific transcriptomes [33–37], the number of studies that combine transcriptome and genome-wide methylation is more limited. However, the growing feasibility of this type of study will allow researchers to ask questions about the role of DNA methylation in the cell. Such as, are DNA methylation patterns cell-type specific? Do they correspond to histone modifications or the binding patterns of other repressive complexes? Is methylation always associated with repression?
TET proteins and demethylation
Until recently, methylation was thought to be a static DNA modification, with demethylation occurring only passively upon the reduction of DNMTs. However, the discovery of 5-hydroxymethyl cytosine (5hmC) and the subsequent elucidation of the cytosine demethylation pathway substantially changed the view of DNA methylation [47]. The regulation of DNA methylation and methylation derivatives is now known to be a dynamic and active process, thought the biological functions of this process are not yet entirely clear [48]. Active DNA demethylation is a multi-step process in which the methyl group is modified before the entire base is replaced via base excision repair (BER) pathways (reviewed in [3, 49]). First, members of the TET family of proteins, including TET1, TET2, and TET3, catalyze the conversion of methylated cytosine to 5hmC and subsequently to other derivatives such as 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) which are removed by BER glycosylases [50]. A second pathway, which is still controversial, involves deamination of 5hmC by AID/APOBEC to 5hmU, followed by base excision repair [51, 52]. Although this second pathway may be important in certain situations, such as neuronal activity induced demethylation (described below), it is considered unlikely that AID and APOBEC are generally involved in 5hmC-dependent demethylation [3].
Mounting evidence indicates that 5hmC methylation may have biological function beyond acting as a chemical demethylation intermediate. For example, 5hmC has a unique distribution pattern across the genome, leading to the question of how it is deposited and maintained. Compared to 5mC, 5hmC is relatively abundant at CG islands (CGIs), promoters, and within gene bodies (exons), but low in intergenic regions [53, 54]. In addition, 5hmC is relatively abundant in constitutively expressed exons and displays prominent 5hmC peak at the 5’splice site boundary [55, 56]. One of the key remaining questions is how 5hmC patterns are ‘read’ and interpreted by the cell. One possibility is through recruitment or exclusion of DNA-methyl binding proteins.
DNA methylation readers: MBPs
Three families of proteins are known to bind to methylated DNA, including the methyl binding domain (MBD) family, the zinc finger/Kaiso family, and SET and RING associated (SRA) domain family. In addition, recent work using quantitative proteomics has also allowed for the unbiased detection of proteins that interact with specific DNA sequences including methylated and hydroxymethylated sequences [57–59]. These methods have confirmed the binding properties of many MBPs and identified many novel methylated DNA binding proteins, such as RBP-J [58], a transcription factor within the Notch pathway. These findings highlight a growing appreciation of the contribution of DNA methylation to transcription factor binding, in addition to mediating the binding of the established MBP families. So far, only members of the MBD family have been implicated in human disorders affecting the brain [60], but many other MBPs are involved in cancer [61].
MBD family
DNA methylation-induced gene repression is primarily mediated by proteins containing a methyl-CpG binding domain (MBD) [62]. Among MBPs, MBDs were discovered first and remain the best-studied to date. This family includes MBD1-6 and MeCP2 (Methyl CpG Binding Protein 2) (Fig. 1) [62]. In addition to the MBD domain, members of the MBD family possess diverse functional domains that enable them to bind to chromatin modifying proteins and other DNA-methylation specific proteins (Fig. 1). For example, MBD1 possesses a transcriptional repression domain (TRD) that enables binding to multiple histone-related proteins (Table 1). Both MBD1 and MeCP2 are highly expressed in the brain and play important roles in neurodevelopment and plasticity [60]. MBD1 Members of the MBD family play significant roles in the regulation of adult neurogenesis, which will be discussed below.
Fig.1
A schematic overview of the MBD family of methyl binding proteins (MBPs) including known protein domains in (MBD, methyl-CpG binding domain; TRD, transcriptional repression domain; AT hook; CXXC, zinc finger Cys-x-x-Cys domain; PWWP, Pro-Trp-Trp-Pro).
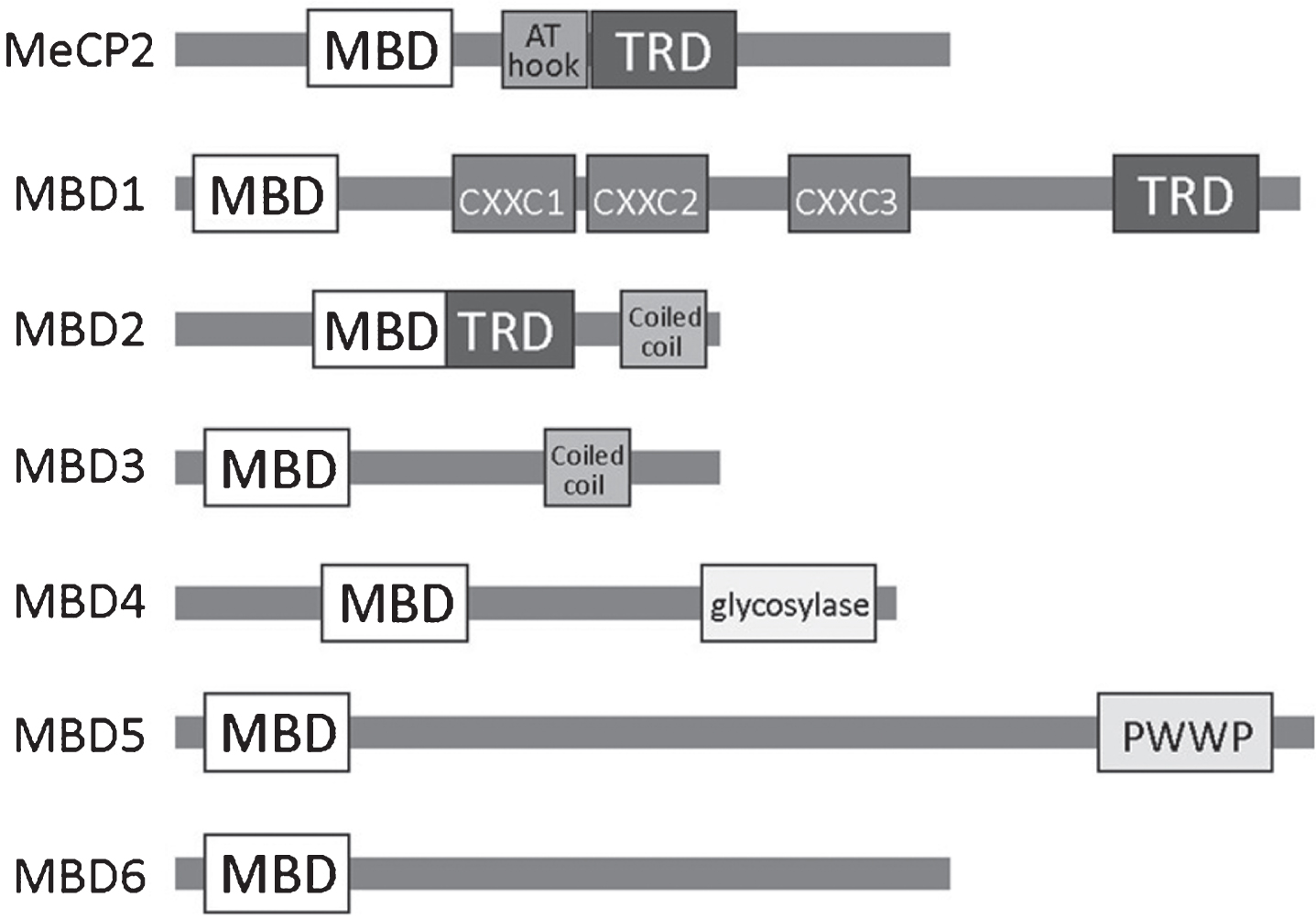
Table 1
MBD1 interacting proteins
| Reference | Interactor | Function | Methods | Required domains |
| Watanabe et al. [227] | MPG | DNA damage repair | Y2H (TRD) CoIP (HeLa) | TRD |
| Reese et al. [228] | CHAF1A | Histone octamer assembly on | Y2H CoIP (HeLa) | MBD |
| DNA during replication | ||||
| Fujita et al. [229] | MCAF1 | Transcriptional co-factor | Y2H | TRD |
| Sarraf et al. [230] | SETDB1 | H3K9 methyltransferase, PCR2 | Y2H | TRD |
| component | ||||
| CHAF1A | Y2H | MBD | ||
| Ichimura et al. [225] | MCAF2 | Transcriptional co-factor | GST pull-down Co-IP | TRD |
| SETDB1 | H3K9 methyltransferase, PCR2 | Co-IP | ||
| component | ||||
| SP1 | Zinc-finger TF, gene activation | Co-IP | ||
| HP1 | Pericentromic heterochromatin | Colocalization | ||
| Lyst et al. [222] | PIAS1/PIAS3 | E3 SUMO (small ubiquitin-like | Y2H See [230] | |
| modifier)-ligases | ||||
| Uchimura et al. [231] | MCAF1 | Transcriptional co-factor | Y2H | |
| Villa et al. [221] | HDAC3 | Histone deacetylase | CoIP | TRD |
| Sakamoto et al. [224] | HPC2 | PRC1 component | Y2H (Cxxc domain) | |
| Co-IP (HeLa) | ||||
| RING1B | PRC1 component | |||
| Xu et al. [232] | γH2AX | DNA-damage associated histone | Co-IP (PANC1) | |
| MDC1 | Cell cycle checkpoint | Co-IP (PANC1) |
CHAF1A, Chromatin Assembly Factor 1, Subunit A (P150), also known as CAF-1; γH2AX, phosphorylated Histone H2A.X; HDAC3, histone deacetylase 3; HPC2 also known as CBX2; MCAF1, MBD1-containing chromatin-associated factor 1, also known as AFT7IP1 or AM; HP1, heterochromatin protein 1; MDC1, Mediator of DNA damage checkpoint 1; MPG, N-methylpurine-DNA glycosylase; MCAF2, MBD1-containing chromatin-associated factor 2, also known as AFT7IP2; PIAS1/3, protein inhibitor of activated STAT-1/3; SETDB1, SET Domain, Bifurcated 1; SP1, specificity protein 1; RING1B, Ring Finger Protein 1B.
A critical step in understanding the function of MBDs is to identify their binding specificity and preferred sequences. The initial description of MeCP2 noted that it is localized to methylated DNA and pericentromic heterochromatin [63]. Since then, site-specific analysis has shown that these initial observations hold true: MeCP2 binds throughout the genome and tracks with mCG density [64]. Genomic mapping of the MBD proteins MeCP2 and MBD1-4 revealed that MBD binding correlates with DNA methylation, peaking at regions of dense methylation, such as methylated CG islands, except for MBD3 which does not bind to methylated DNA [65]. A comparison of genome-wide promoter methylation and RNA Pol II enrichment showed that active transcription is opposite to that of MeCP2, MBD1, MBD2, and MBD4, confirming the general role of these MBDs in gene repression [65]. MBD1 has multiple splice isoforms with the longest form containing both MBD domain that can bind to methylated CpG and a CXXC zinc finger domain that can bind to unmethylated CpG. A shorter version of MBD1, called MBD1v3, contains only MBD domain and 2 CXXC domains that do not bind to DNA [66]. Thus far, MBD5 and MBD6 have not been found to bind to methylated DNA, although they are localized to heterochromatin [67]. This localization may be mediate by co-factors such as the PR-DUB Polycomb complex [68].
The discovery of 5hmC in the brain led multiple groups to question whether or not known methylated DNA binding proteins also bound to 5hmC. Multiple labs have shown that the MBD domain is specific for 5mC and that the presence of 5hmC reduces binding of MeCP2 and MBD1 [59, 69–72]. There is some conflict over the ability of MeCP2 to bind to 5hmC [71]; however, some binding may be mediated by hemi-hydroxymethyled DNA as MeCP2 is already known to bind to hemimethylated DNA [73, 74]. It is likely that the in vivo binding affinity of MeCP2 and other MBDs may depend on the local chromatin environment or DNA sequence.
Based on the observation that mCH increases specifically in the brain during postnatal development, Gabel et al. investigated how MeCP2, which also exhibits high postnatal expression, binds to different methylated dinucleotide pairs [75]. They found that MeCP2 binds to mCG, mCA and 5hmCA with relatively high affinity and to mCC and mCT with low affinity. Moreover, they found that mCA was enriched in longer genes, longer genes are more enriched in the nervous system, and that changes in gene expression in the absence of MeCP2 corresponds with both of these factors [75]. The correlation between MeCP2 loss and upregulation of long genes has also been confirmed in expression profiles from various neuronal subtypes [76]. Another study found that genes that acquire more mCH methylation after birth are more likely to be differentially expressed in MeCP2 models, adding further support to the idea that binding of MeCP2 to non-CG methylation is critical for gene regulation [77]. In addition, both mCG and mCH appear to be associated with nucleosome positioning [44, 46, 78], indicating that non-CG methylation may be linked to other epigenetic mechanisms. These studies indicate that it is important to consider more than just CG methylation when evaluating MBP binding or changes in DNA methylation. DNA methylation is increasingly viewed as a dynamic modification that can be interpreted by the cell in a variety of ways to mediate gene regulation. Binding by MBPs to DNA is a major mechanism that cells use to interpret methylation status.
Zinc finger family
Kaiso (also known as ZBTB33) is a member of the BTB/POZ family of zinc-fingers (ZF) and contains three copies of a Krüppel-like C2H2 zinc finger. Kaiso was originally identified as a binding partner of p120 catenin [79]. It was later found to be part of the MeCP1 complex— a methylated DNA binding complex that also contains MBD2 and NuRD [80]. Kaiso is unique in that it binds to mCpG sites in a methylation-dependent manner but also binds to a specific sequence (TCCTGCNA) similar to a transcription factor [80–83]. Recent crystallization of the zinc-finger added insight into how two different types of binding are mediated by the same domain [84]. There are conflicting reports on Kaiso’s abilty to bind to hydroxymethylated DNA as some groups have found that it does not bind to 5hmC [85] while others have found that it binds to both 5mC and 5hmC [59]. Kaiso acts as a transcriptional repressor [80] and has been identified as a co-factor of N-CoR [83].
Other members in the Zinc finger/ Kaiso family include ZBTB38 (ZENON) and ZBTB4 (KIAA1538). The expression of ZBTB38 is restricted to the brain and specifically to differentiating neurons— it is highly expressed during development and continues in adult [86, 87]. ZBTB4 is expressed in multiple adult tissues, but not in embryonic stages [87]. Binding of both ZBTB38 and ZBTB4 is methylation dependent [87] and ZBTB4 also binds to the Kaiso non-methylated consensus sequence [88].
SRA domain family
UHRF1 and UHRF2 are ‘hub’ proteins that contain multiple epigenetic interaction domains including a ubiquitin-like (UBL) domain, tandem Tudor domain (TTD), plant homeodomain (PHD) finger domain, SET and RING associated (SRA) domain, and really interesting new gene (RING) finger domain [89]. The TTD and PHD domains are responsible for interactions with other epigenetic proteins such as DNMT1 and histones [90]. The SRA domain is responsible for recruitment to hemi-methylated DNA [91]. Crystalization of the SRA domain of UHRF1 revealed that binding of UHRF1 to DNA is not sequence dependent, as the SRA domain only makes contact with the hemimethylated CpG [92–94]. UHRF1 (also known as Np95) is essential in maintaining DNA methylation through cell division as it recruits DNMT1 to hemi-methylated sites [95, 96]. UHRF2, which is also known as Np95/ICBP90-like RING finger protein (NIRF), in contrast, does not maintain DNA methylation during DNA synthesis [97] but is involved in cell cycle progression and DNA damage repair. UHRF1 was shown to bind to 5mC and 5hmC with similar affinity [70]. This finding was confirmed by [59] who also showed that UHRF2 does not have any affinity for methylated DNA. Although further research is needed, this result is consistent with UHFR’s role in the DNA damage response which is connected to the demethylation and BER pathways.
Methylation-specific transcription factors
DNA methylation may also be interpreted/influenced by transcription factors that are sensitive to DNA methylation, or that require DNA methylation to bind. Because many TF display cell and developmental-stage specificity, they may represent a mechanism for fine-tuning the DNA methylation profile of different cell types. The relationship between TF and DNA methylation may function according to several different models (reviewed in [Reference 98]). Briefly, TFs can project specific sequences from DNA methylation. For example, deletion of the TF SP1, which binds to the consensus motif CCGCCC, has been shown to increase DNA methylation at a promoter with SP1 binding sites and to decrease gene expression [99]. Second, some TF complexes may promote DNA methylation. Although not a transcription factor itself, SETDB1, a H3K9 methyltransferase, interacts with DNMT3A and DNMT3B and is recruited to shared sites via TRIM28 and associated zinc-finger TFs [100, 101]. Third, there is some evidence that TF binding can reverse methylation. In REST knock out ES cells, sites surrounding REST binding motifs were methylated while they were not in WT cells, and reintroduction of REST into these cells removed the inappropriate methylation [102]. Finally, binding of TFs can reinforce repression of methylated regions. CpG methylation influences how E2F TFs bind to their recognition sites, which contain two possible methylation sites (T/CTTC/GG/CCGC/G); bi-methylation inhibits binding of all E2F TFs, but methylation of one site inhibits binding of E2F1 but not of E2F2-E2F5 [103]. E2F TFs are well known as regulators of the cell cycle, but during neurogenesis E2F3 and E2f4 bind to the promoters of many pathways involved in cell fate and differentiation such as members of the Notch, Wnt, and Fgf pathways [104].
DNA METHYLATION AND NEUROGENESIS
The epigenetic landscape of the brain is unique: it has a specific pattern compared to other tissues [105] characterized by high levels of non-CpG methylation and hydroxymethylation, which are exclusive to the brain [106]. In addition, many epigenetic factors, such as MBPs, are highly and/or uniquely expressed in the brain during development and beyond. These observations indicate that establishing, maintaining and interpreting DNA methylation serves an important function in the brain. These roles can be divided into two general processes: 1) to help establish the diverse cell fates found in the central nervous system and 2) to alter gene expression programs based on inputs received by the cell. These two processes occur in neurogenesis during development and in the adult. Technological advancements, such as the ability to detect DNA modifications beyond classical CpG methylation, have greatly expanded our knowledge of the function of DNA methylation in the brain.
Mounting evidence indicates that altered neurogenesis contributes to many pathological conditions [5, 6]. Adult neural stem/progenitor cells play a major role in normal brain functions and the brain’s response to injury and disease. Many neurodevelopmental, psychiatric, and neurodegenerative disorders are characterized by symptoms that have been linked to reduced adult neurogenesis, such as depressive behaviors and memory and learning deficits [107]. Knockout and conditional deletion models have been used to elucidate how the loss of DNA methylation-related proteins effects neurogenesis. Although the picture is not entirely complete, it is known that MBDs, DNMTs and TET proteins play a role in some stages of AHN ranging from stem cell maintenance and proliferation to neuronal differentiation and maturation (Fig. 2, Table 2).
Fig.2
Schematic drawing illustrates the two neurogenic regions in the adult rodent brains and the stages of neurogenesis regulated by DNA methylation-related epigenetic proteins. Top, Adult neurogenic niches in the brain include the dentate gyrus (DG, blue) and the subventricular zone (SVZ, purple). Bottom, the stages of DG neurogenesis and regulation by epigenetic proteins.
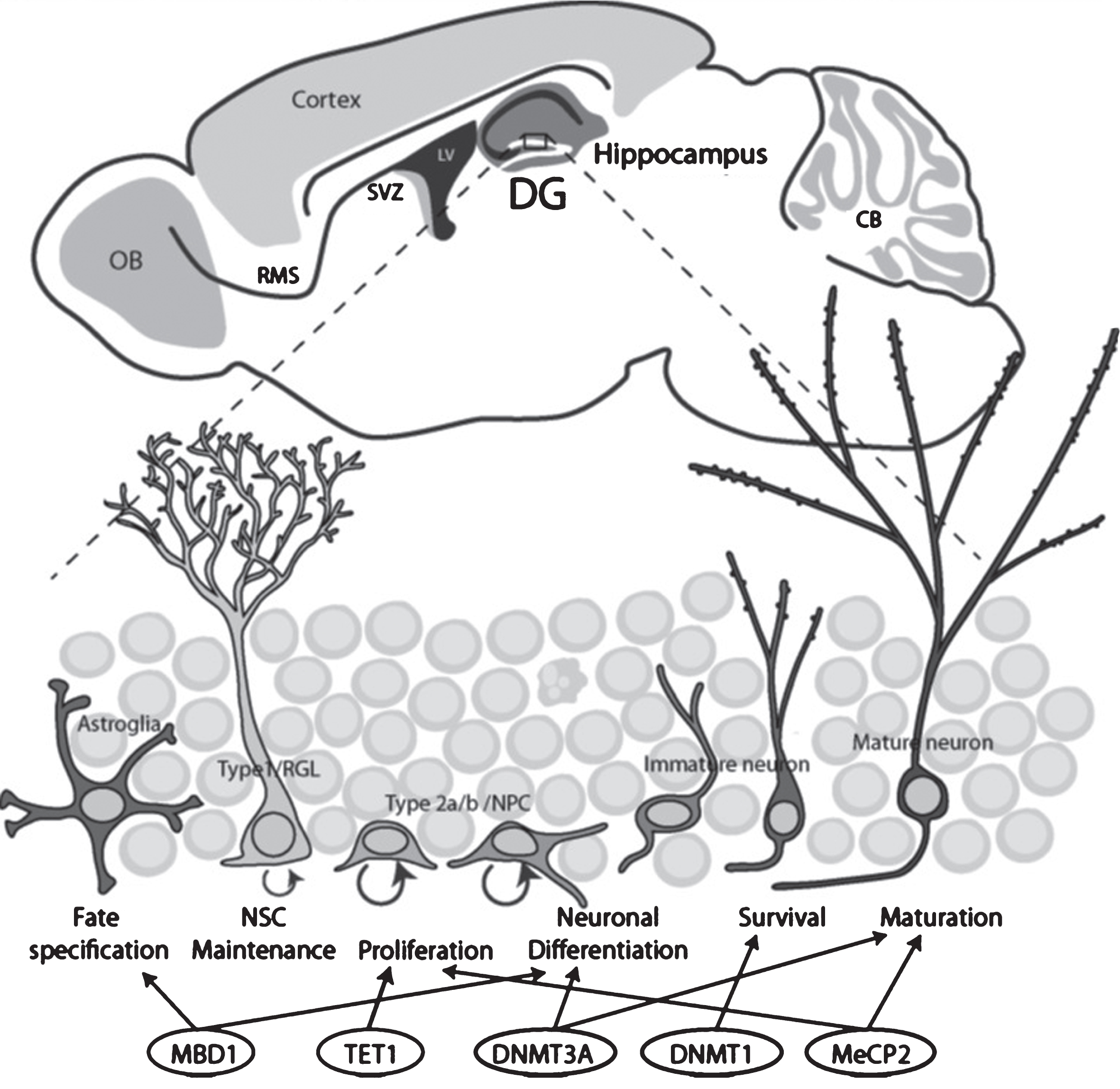
Table 2
DNA methylation related proteins in AHN
| Mouse model | AHN Stage | Direction | Details | Ref | |
| DNMT1 | Nestin-CreERT2 cKO | NSC maintenance | No change | Type 1 cells, 36 dpi Tamoxifen | [124] |
| Proliferation | No change | Ki67+, BrdU+(24 hrs), 36 dpi Tamoxifen | |||
| Survival | ↓ | BrdU+, 28 dpi BrdU | |||
| Synapsin1-Cre | Maturation | No change | DCX+ | ||
| DNMT1/DNMT3A | CamK-Cre dKO | Maturation | ↓ | Reduced hippocampal volume and impaired spatial memory | [126] |
| DNMT3A | KO, 4 week-old mice | Proliferation | No change | Ki67+ | [123] |
| Neuronal differentiation | ↓ | DCX+ | |||
| Neuronal survival | No change | TUNEL+ | |||
| TET1 | KO | NSC maintenance | No change | Type 1 cells | [141] |
| KO and Nestin-CreERT2 cKO | Proliferation | ↓ | Nestin-GFP+, BrdU+Ki67+ | ||
| Nestin-CreERT2 cKO | Neuronal differentiation | ↓ | 6 weeks pi Tamoxifen: DCX+BrdU+ (7 dpi), NeuN+BrdU+ (21 dpi) | ||
| KO | Behavior | ↓ | Learning and memory MWM | ||
| TET1 | KO | Neuronal diff/survival | No change | NeuN hippocampal density | [142] |
| Behavior | ↓ | Memory extinction in MWM and contextual fear conditioning | |||
| MeCP2 | Nestin-Cre cKO | Maturation | ↓ | Reduced neuronal cell body | [166] |
| MeCP2 | Retrovirus sh-MeCP2 | Maturation | ↓ | Reduced dendritic branching and spine development | [170] |
| MeCP2 | MeCP2S421A | Proliferation | ↓ | Ki67, BrdU (24 hrs) | [174] |
| Neuronal Differentiation | ↑ | NeuN+BrdU+ (4 weeks pi) | |||
| MBD1 | KO | Fate specification | ↓ neurons ↑ astrocytes | BrdU+NeuN+, BrdU+GFAP+(4 weeks pi) | [163] |
MWM, Morris Water Maze.
DNA methylation
Neurogenesis occurs before astrogenesis during embryonic brain development; therefore, suppressing astrocytic genes in NSCs is a vital step in the temporal fate specification of these cells. DNA methylation is a major mechanism through which this occurs. For example, during neurogenic phases the STAT3 binding sites in the promoters of Gfap (Glial Fibrillary Acidic Protein) and s100β are highly methylated, but at later stages that corresponding to astrogenesis they become demethylated, allowing STAT3 to bind [108, 109]. A similar phenomenon occurs in ESCs: the Gfap promoter is highly methylated but becomes demethylated in ESC-derived astrocytes [110, 111]. Binding of NFIA (Nuclear Factor I/A), a down-stream mediator of Notch and JAK/STAT activation, at the Gfap promoter has been shown to lead to DNA demethylation and gene activation via dissociation of DNMT1 [112].
The early embryo undergoes large-scale DNA methylation remodeling, but the changes that occur in DNA methylation during brain development are less clear. A large study that used in vitro neural differentiation showed that the DNA methylation state of a cell undergoes large changes as mouse embryonic stem cells (mESCs) transition to restricted neural progenitor cells and differentiated neurons [113]. Newly-methylated genes are associated with pluripotency, early embryonic development, or germ line development while the majority of the demethylated genes are brain-related, supporting the hypothesis that DNA methylation in general acts to repress genes. Another important finding of this study is that patterns of DNA methylation correlate more strongly with histone modifications than with the underlying genetic code, highlighting the interplay between these two systems [114].
DNMTs
The establishment of DNA methylation during development is essential as all DNMT null mutations (except DNMT2 and DNMT3L) are lethal in embryonic stages or shortly after birth [115]. DNMT1-KO mice develop neural tube defects (NTDs) and die by E9 [116]. Likewise, homozygous mutations in DNMT3B, which is highly expressed in the embryonic neural ectoderm, leads to NTDs and death by E9.5 in mice [117]. DNMT3B expression in the brain is high at embryonic day 10.5–13.5, corresponding with peak neurogenesis, but is not detectable after E13.5 [118]. In contrast, in human ESCs RNAi-mediated knockdown of DNMT3B resulted in accelerated maturation of the neuroepithelium and precocious expression of neuronal markers such as NEUROD1 coupled with loss of Polycomb repressive complex (PRC) component EZH2 at the promoters of early neuronal genes [119]. Does DNMT3B promote or suppress neuronal programs? Are the effects specific for developmental windows? DNMT3A-null mice appear normal at birth, but die at around 4 weeks of age [117]. The specific roles of DNA methyltransferases in development has been investigated using cell-type specific deletion models.
Deletion of DNMT1 in the entire CNS by E9-10 via crosses with Nestin-cre mice results in extensive hypomethylation in cells across the CNS [120]. These mice display precocious astrocytic differentiation linked to hypomethylation of astrocytic genes and activation of the JAK-STAT pathway [121]. This result mirrors the findings of the role of DNA methylation in astrocyte promoter activation and fate specification. Likewise, mice with conditional deletion of DNMT3A in nestin-expressing cells are apparently normal but lose DNA methylation at the Gfap promoter [122]. These mice die during young adult hood, most likely due to defects in the neuromuscular junctions of the diaphragm that cause respiratory failure [122].
DNMTs and adult neurogenesis
The observation that DNMT3A-null mice appear normal at birth but die by 4 weeks of age, suggests a role for DNMT3A in postnatal de novo methylation. A group that evaluated postnatal neurogenesis in the DG and SVZ found that the number of DCX+ immature neurons is greatly reduced, though there are no changes in proliferation [123]. This group found that DNMT3A promotes the transcription of a number of neurogenic genes by antagonizing PRC2-mediated repression. DNMT1 is also involved in adult neurogenesis: deletion of DNMT1 in DG aNSC does not change cell proliferation, but does reduce new neuron survival [124]. Retinal-specific Dnmt1 deletion from the onset of neurogenesis by Chx10-Cre also leads to defective neuronal differentiation and eventual wide-spread neuronal death [125]. These results indicate that DNA methylation in postnatal neurogenesis is required for neuronal differentiation and survival, as opposed to embryonic neurogenesis where DNA methylation contributes to cell fate specification. However, differences between in vivo and in vitro models may also contribute to some of these differences. Further exploration of how DNA methylation is altered in these mutants is needed to understand the mechanisms regulating neurogenesis.
DNA methylation also likely contributes to neuronal maturation. In addition DNMT1 and DNMT3A may have overlapping functions in post-mitotic neurons. Only Camk2a-cre double KO, which affects neurons in the hippocampus, resulted in an observed phenotype [126]. These mice had smaller hippocampi, impaired spatial memory, increased expression of genes associated with synaptogenesis, and reduced DNA methylation [126]. DNMT-mediated methylation in post-mitotic neurons is also important in other regions of the brain. For example, Emx1-cre Dnmt1 conditional knockout mice fail to generate the correct structures in the somatosensory barrel cortex and do not establish long term potentiation (LTP) [127]. Deletion of Dnmt3a driven by Sim1-Cre (single-minded1) causes obesity and overgrowth phenotypes in mice [128]; SIM1 is expressed in a subpopulation of in neurons of the paraventricular hypothalamus (PVH) and amygdala that are known to contribute to energy homeostasis [129]. Because some phenotypes of DNMT3A overgrowth syndrome (discussed below) are recapitulated by loss of DNMT3A in SIM1 neurons, it suggests that de novo methylation may be more critical to the function of specific subpopulations of neurons which may contribute to the disease phenotype in different ways. Interestingly, deletion of MeCP2 in SIM1 expressing cells also causes obesity, highlighting the importance of both the establishment and interpretation of DNA methylation in this specific population of neurons [130]. In Purkinje neurons, DNMT3B is responsible for de-novo methylation of protocadherin genes that results in stochastic and monoallelic expression of different cadherin isoforms in individual neurons, a mechanism hypothesized to contribute to dendritic arborization by self-avoidance, synaptogenesis and circuit formation [131]. These studies indicate that de novo DNA methylation is required in post-mitotic neurons, thought the extent to which this is required for maturation or integration of input signals particularly in the hippocampus remains to be determined.
5hmC and TET
Multiple studies have shown that 5hmC is much more abundant in brain tissues relative to other tissues of the body [132–134]. In adult cells, 5hmC is abundant in the neurons relative to other cell types or cancer cells, constituting 0.6% of all nucleotides in Purkinje neurons and 0.2% in cerebellar granular neurons [135]. The abundance of 5hmC increases as neurons differentiate and mature in both embryonic and adult models [136, 137]. Multiple studies indicate that genes that gain or are enriched for 5hmC are important for neuronal differentiation [56, 136]. 5fC and 5caC, derivatives of 5hmC, also accumulate in differentiating human ESCs (hESCs), in mouse embryos during peak neurogenesis (E11.5-13.5), and during initial differentiation of aNPCs [138].
TET proteins have garnered significant interest in recent years for their role in activity-mediate demethylation, but they appear to be involved in early neuronal development as well. All TET proteins are expressed in the brain; TET3 is expressed at the highest level, followed by TET2 and TET1 [132]. About half of TET1-KO; TET2-KO mice survive to adulthood and are fertile; but those that die exhibit defects in head development, indicating that TET1 and TET2 are likely involved in neural development [139]. Deletion of TET3 in mice causes perinatal lethality early in development, but it is not known whether or not neurological defects contribute to this outcome [140]. Overexpression of TET2 or TET3 by in-utero electroporation enhanced cortical neuronaldifferentiation whereas knock-down via sh-RNA reduced the progression of neuronal differentiation and resulted in the accumulation of progenitor cells [136]. As described previously, other studies have found that DNA methylation is lost on neurogenic genes during neuronal differentiation, which is consistent with a role for TET proteins in this process.
TET and adult neurogenesis
Even though TET1 deletion is compatible with life, multiple groups have shown that its loss impacts adult neurogenesis. In TET1-KO mice there are no changes in the proliferation of RGLs but proliferation of nIPCs is reduced, leading to the generation of fewer new neurons [141]. TET1-KO animals also have learning and memory deficits [142]. Multiple studies indicate that TET1 controls the expression of neuronal-activity related genes such as Bdnf (Brain-derived neurotrophic factor) and c-Fos and that 5hmC is a key intermediate in this regulation [51, 142–144]. However, other labs have found that demethylation is not necessarily associated with increased transcription of neuronal genes during neuronal differentiation [136].
There is growing evidence that 5hmC, irrespective of its role in DNA demethylation, may be involved in transcriptional regulation, maintenance of bivalent chromatin states and neuronal differentiation. TET1 most likely does not bind to methylated DNA on its own: it likely recruits or is recruited by other factors. So far, three mechanisms have emerged that might couple 5hmC distribution, TET1 binding patterns, and gene expression. First, multiple reports have shown that TET1 and Polycomb (PcG) binding overlaps significantly genome-wide and that TET1 binds to PcG proteins targets [145–148]. PcG proteins play a critical role in regulating pluripotency genes and TET1 and TET2 knock-down leads to decreased expression of pluripotency genes [53], indicating that DNA demethylation may also be involved in resolving bivalent chromatin. Second, TET1 may be involved in recruiting the NURD (Nucleosome Remodeling and Deacetylase) repressive complex. There is evidence indicating that the NuRD complex is targeted to specific genomic loci by TET1 and 5hmC. MBD3, which is part of NuRD complex, co-localizes with TET1 across the genome, MBD3 knock down affects expression of genes with 5hmC, and localization of MBD3 is TET1 dependent [149]. PRC2 and the MBD1-NURD complex also co-regulate a set of common targets that are characterized by bivalent chromatin state (H3K27me3 and H3K4me3), highlighting the possible interplay of multiple epigenetic pathways [150]. Third, a recent report indicates that TET1 may be recruited to a portion of its genomic loci in by Lin28Aa protein which was previously thought of as an RNA binding protein [151]. Nor are the interacting partners of TET1 limited to the mechanisms described above. TET1 Co-IP identified methyl-CpG binding proteins MeCP2 and UHRF1, histone modifying proteins including several HDACs, SIN3A and LSD1 (lysine specific demethylase), and PCNA (proliferating cell nuclear antigen) [148]. Binding competition at 5mC may be one mechanism of TET1 regulation; one study found that MeCP2 inhibited TET1 activity [152]. In addition 5hmC levels in the mouse cerebellum have been correlated with MeCP2 gene dosage [153]. In summary, the precise role of TET proteins and DNA demethylation at different stages of development is still unclear. In addition, the crosstalk between TET proteins and MBPs in the context of neural development, whether it is facilitative (MBD3) or antagonistic (MeCP2), warrants further exploration.
MBDs
MeCP2, MBD1 and MBD5 are highly expressed in the brain. MeCP2 is highly expressed in neurons throughout the brain of adult rodents and primates and expression correlates with age [154–157]. MeCP2 is expressed in some nestin-positive precursor cells from the neuroepithelium [158] and its expression increases with neuronal differentiation [159]. Although initial reports focused on MeCP2 was expression in neurons [158, 159], subsequent reports have identified it in astrocytes and oligodendrocytes [160] and confirmed MeCP2 is not expressed in microglia [161]. MBD1 is expressed in the brain by P7 (postnatal day 7), but it is not known if it is expressed earlier [162]. In adult mice, MBD1 is expressed in neurons throughout the brain, though its expression is particularly high in the DG and CA1 of the hippocampus [163]. MBD5 is highly expressed in the brain relative to other tissues [67]. This expression pattern is strongly indicative of a role of MBDs in brain development and function.
MBDs and neurogenesis
MeCP2 performs dual complementary functions during embryonic fate specification of NPCs to neuronal and astrocytic fates. MeCP2 represses astrocyte genes during neurogenesis, and this repression must be released during astrogenesis. For example, during neurogenic phases, the s100β promoter is highly methylated, but at later stages that correspond to astrogenesis, it becomes demethylated, releasing MeCP2 during astrogenesis [109]. Other evidence suggests that the presence of MeCP2 supersedes other fate specification cues because the expression of MeCP2 in transplanted neuroepithelial progenitors promotes neuronal differentiation even when cells are transplanted into non-neurogenic brain regions [164]. In neurons, astrocyte induction cues were not sufficient to increase GFAP expression because the promoters of astrocytic genes Gfap and s100β were found to be highly methylated and repressed by MeCP2 [165]. It is still unclear if MeCP2 and other MBPs inhibit astrocyte genes directly, or if they also regulate the major pathways that promote gliogenesis. It is also possible that many of these experiments may be of limited biological relevance because the expression of MeCP2 is relatively low during embryonic development relative to its postnatal expression.
However, the majority of MeCP2 research involves the loss of MeCP2, which is more relevant for neuronal maturation rather than fate specification. NSCs derived from the cortex of E13.5 MeCP2-KO mice do not display altered fate specification for neurons, astrocytes or oligodendrocytes relative to controls [159]. Nor do MeCP2-null brains display obvious structural deficits associated with altered cell fate specification [166]. Rather, MeCP2-KO mice, similar to humans with RTT [167–169], have neurons with smaller nuclei, reduced dendritic branching and immature spines [170].
Both MBD1 and MeCP2 play a role in aNSC fate specification. In NPCs, oligodendrocytes, and astrocytes in vitro, both MeCP2 and MBD1 are localized to the RE1/NRSE region of the mGluR2 promoter, which is suppressed in non-neuronal cell-types [171]. This suggests that MBD1 and MeCP2 are involved in suppressing neuronal genes in astrocytes, which is distinct from MeCP2’s function in embryonic NSCs. A role for MBD1 in fate specification is supported by differentiation analysis of MBD1-KO animals injected with DNA analog BrdU (5-Bromo-2’deoxyuridine): 4 weeks after BrdU injection, the number of BrdU-labeled neurons decreases greatly, while the number of astrocytes increases slightly [163]. These mice also show signs of depression, and exhibit impaired spatial learning, and reduced long-term potentiation believed to be a result of decreased adult neurogenesis [172]. However, in the function of MBD1 in NSCs is unexplored and it is not clear what stage of NSC development leads to reduced neurogenesis. In summary, many questions remain about how MeCP2 and other MBDs regulate stem cell maintenance, proliferation, or fate commitment.
Although MeCP2 seems to play a more prominent role in neuronal maturation, it may also function in NSCs and other cell types. MeCP2 expression in astrocytes has recently been shown to have significant impact on neuronal function and RTT pathology [160, 173]. Since astrocyte-“specific” manipulations utilize the promoter of Gfap, a gene that is also expressed in NSCs, it is possible that some of these effects might be, at least in part, due to changes in NSCs. In addition, the phosphorylation-incompetent MeCP2 mutant S421A reduced proliferation of and enhanced neuronal differentiation in the adult dentate gyrus, indicating that MeCP2 function is important in NSC/NPCs [174].
MeCP2 has been shown to play an important role in translating neuronal activity and signaling cascades into epigenetic gene regulation. Because phosphorylation is a dynamic modification, MeCP2 phosphorylation can integrate activity-induced signaling and gene expression [175]. For example, Chen et al. 2003 found that depolarization of neurons led to phosphorylation of MeCP2 and decreased occupancy at the Bdnf promoter [176]. A complementary study by Martinowich et al. linked activity-dependent changes in DNA methylation with reduced MeCP2 binding at the Bdnf promoter which corresponded to increased BDNF expression [177]. BDNF signaling via its receptor, TRKB (Tropomyosin receptor kinase B), is also essential for AHN [178]. MeCP2 phosphorylation also integrates Notch signaling with neurogenesis: phosphorylation-incompetent MeCP2 mutants S421A exhibited deficits associated with reduced Notch signaling and could be rescued by overexpression of the Notch intra cellular domain (NICD) [174].
DNA METHYLATION IN DEVELOPMENT AND DISEASE
Given the unique methylation landscape in the brain and the contributions of epigenetics to brain development and function, connections between the factors responsible for the regulation of DNA methylation and human disease are of great interest. Indeed, mutations in a proportion of DNA methylation associated-genes have devastating impacts on brain development and function. Deficits in AHN may contribute to the etiology of some of these disorders [179]. However, mutations in others may be related to other brain regions and functions. But even in situations where there is no direct link between an epigenetic factor and AHN in humans, studies of AHN can be used to elucidate the function of a protein in different cells ranging from stem cells through new neurons. Moreover, AHN is a useful model system because aNSCs generate a relatively homogenous population of neurons that pass through well-defined developmental stages with consistent timing. Information gained from such studies may provide important mechanistic insights into disorders that effect the brain, but which are not primarily caused by defects in AHN, such as neurodegeneration caused by mutations in DNMT1 (discussed below). The scope of disorders linked to DNA methylation offers insight into the specific role of DNA methylation in the brain compared to other cell types and tissues.
DNMTs
Mutations in DNMT3A have recently been associated with human disease. Researchers searching for genes responsible for overgrowth disorders identified heterozygous de novo mutations DNMT3A in approximately 10% of overgrowth patients, all of whom also displayed moderate intellectual disability [180]. Mutations in DNMT3A are also frequently found in acute myeloid leukemia (AML) [181] and other hematological cancers like myelodysplastic syndromes (MDS) [182]. Mutations that cause DNMT3A overgrowth syndrome have been found in all functional domains of the protein and modeling suggests that many mutations interfere with histone binding [180]. Interestingly, other epigenetic proteins seem to play a dual role in growth syndromes and myeloid neoplasms. Mutations in EZH2, NSD1, and MLL (mixed lymphomic leukemia) cause Weaver, Sotos, and Wiedemann-Steiner syndromes, respectively, and are also associated with hematological cancers [180, 183]. This suggests that loss of de novo methylation or the inability to interpret the methylation state contributes to a cell’s oncogenic transformation.
In addition, DNMT3A was identified as an autism spectrum disorder candidate gene by genome-wide association from whole-genome sequencing [184]. One study suggests that DNMT3A overgrowth syndrome and possibly the intellectual disability associated with this disorder may be comorbid with autistic symptoms: a study that integrated two large copy number variation (CNV) data sets identified small de novo deletions of DNMT3A in 3 patients with autism who also had heights and weight characteristic of over-growth syndromes [185].
Mutations in DNMT3B results in ICF1 syndrome which is about 50–60% of the cases of ICF syndrome (immunodeficiency, centromere instability and facial abnormalities syndrome), a rare autosomal recessive disorder that leads to CG hypomethylation of pericentromic satellite regions of chromosomes 1, 9 and 16 [186-188]. About of half of ICF1 patients also display a high incidence of intellectual disability [189], indicating that de-novo methylation mediated by DNMT3B is important for proper brain function and development.
Mutations in DNMT1 have been linked to two rare neurodegenerative syndromes: autosomal dominant cerebellar ataxia-deafness and narcolepsy (ADCA-DN) and hereditary sensory neuropathy with dementia and hearing loss (HSN1E) [190, 191]. Mutations that cause HSN1E prevent binding of DNMT1 to heterochromatin during the G2 phase of the cell cycle, leading to global DNA hypomethylation and selective hypermethylation [190]. Whole genome bisulfite sequencing of HSN1E patients reveled that differentially methylated regions (DMR) are associated with neurological disorders and NAD+/NADH metabolism [192]. Defects in DNA methylation have been identified in many neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease and Huntington’s disease, reviewed in [193].
MBPs
Mutations in MBDs have been linked to multiple neurodevelopmental disorders that manifest with varying degrees of severity. A common thread between MBPs is autism-like symptoms, with disruptions to MeCP2 representing the most severe and MBD1 the most mild [194]. The similar yet distinct phenotypes associated with MBDs suggest that overlapping but distinct mechanisms contribute to disease etiology.
Mutations in MeCP2 are the cause of the neurodevelopmental disorder Rett syndrome (RTT), but they are also associated with a number of other neurological disorders, including cases of Angelman syndrome, Prader-Willi syndrome, autism, and non-syndromic mental retardation [195]. Mouse models have replicated key phenotypes of RTT and revealed that MeCP2 plays a critical role in neuronal maturation and synaptogenesis, in part through the regulation of dendritic morphology, synaptic transmission, and long-term plasticity [196, 197]. Both clinically and in mouse models of RTT, deletion or loss of function MeCP2 and duplication both result in similar pathology, highlighting the necessity for its precise control [195].
As detailed above, changes in adult neurogenesis have been described for MeCP2 mouse models [174, 198]. A potential exciting avenue of research is the modulation of adult neurogenesis by various pharmacological and non-pharmacological interventions. Because adult neurogenesis is responsive to a variety of inputs, it may be possible to counteract some behavioral deficits by increasing or normalizing adult neurogenesis. For example, treatment of FXS mice with a GSK3β inhibitor was capable of restoring hippocampal-dependent learning and rescuing neurogenesis in adult mice [8]. Reactivation of MeCP2 in post-natal neurons also partially rescues multiple pathologies observed in MeCP2-null mice [7]. These studies suggest that some aspects of neurodevelopmental-associated pathology may be reversible and may offer useful information in the development of therapies.
MBD5 was identified as the critical region contributing to 2q23.1 microdeletion syndrome [199, 200]. Microdeletions of that disrupt MBD5 cause a spectrum of neurodevelopmental phenotypes that share some behavioral, developmental, and neurologic features with autism, Rett syndrome, and other disorders [200]. In addition to being a hotspot for microdeletions, mutations in Mbd5 have been identified in ASD patients [185, 201–203]. Also, duplications of MBD5 have been identified in patients with similar features as those with the gene disruption, indicating that gene dosage of MBD5, like MeCP2, is critical for neural development [204, 205]. Mice with MBD5 deletions display craniofacial abnormalities and decreased motor function and altered social interaction [206]. The contribution of developmental or adult neurogenesis to these phenotypes has not been examined but may be affected.
In humans, mutations or polymorphisms in MBD1 have been identified in sporadic cases of autism spectrum disorder (ASD) [202, 207]. MBD1 is also contained within the region of del (18) (q12.2q21.1) syndrome, which has also been identified in some cases of atypical Rett syndrome [208, 209]. Mice with MBD1 deletion (MBD1-KO) exhibit behavioral deficits associated with ASD, including learning impairment, increased anxiety, reduced social interest, and impaired sensorimotor gating [163, 172]. These phenotypes are consistent with a role for MBD1 in the fate specification and differentiation of aNSCs.
So far, Kaiso has only been associated with cancer [210] and little is known about its function in the brain. Kaiso KO mice show no overt developmental phenotype, nor does its loss effect neural stem cell fate decision or stem cell maintenance [211]. However, another group that analyzed the behavior of the same mice found that the KO mice had increased locomotion, exploratory behavior and sensorimotor gating as well as shrunken ventricles [212]. A double KO of Kaiso and MeCP2 does not worsen the MeCP2-KO phenotype [213], indicating that Kaiso is not functionally redundant to MeCP2. It is unknown if ZBTB38 and ZBTB4 have a brain-specific function.
UHRF2 is expressed in the adult brain at comparable levels to other tissues in the body, while UHRF1 is expressed primarily in pluripotent and proliferative cells [214, 215]. Consistent with its function in DNA damage and repair, UHRF2 is frequently upregulated in cancer [89]. During embryogenesis, UHRF1 is highly expressed in proliferating NPCs around E11. In the adult hippocampus it is also expressed in proliferating NPCs, suggesting that it is involved in maintaining DNA methylation in this proliferating stem cell populations in the brain at multiple stages[216].
CROSSTALK BETWEEN EPIGENETIC PATHWAYS
DNA methylation is heavily interconnected with other epigenetic and regulatory systems such as histone modifying complexes, and transcription factor binding. The proteins that mediate the deposition, maintenance, interpretation and removal of DNA methylation are also highly connected with each other. For example, MeCP2 has been found to interact with diverse proteins including, but not limited to NCOR-SMRT (nuclear receptor co-repressor), HP1 (heterochromatin protein 1), SOX2, TET1, p300, and CREB1 (cAMP responsive element binding protein 1) [197]. In addition, it is becoming increasingly clear that signaling pathways and transcriptional regulation pathways are highly connected to epigenetic regulation. Understanding how disrupting one aspect of the network influences other pathways is vital in understanding the system as whole.
MBD1 is important in linking DNA methylation to the regulation of gene expression in combination with other epigenetic and regulatory pathways. MBD1 binds directly to the promoter and regulates the expression of Fgf-2, a molecule that promotes proliferation and is commonly used to expand NSC populations. Furthermore, a DNA methylation inhibitor blocked the effects of MBD1 in forebrain-derived aNSCs and increased Fgf-2 expression, underscoring the important role the methylation state of the promoter plays [217]. In part, MBD1 regulates neurogenesis by silencing miR-184; inhibiting miR-184 hinders proliferation and promotes the differentiation of hippocampal DG adult NSCs in vitro. In turn, miR-184 binds the 3’ UTR of Numblike (Numbl), a signaling protein required for differentiation in adult neurogenesis, and targets it for degradation [218]. Numbl is known to inhibit the Notch pathway [219], which has a significant impact on neurogenesis. Inhibition of miR-195, a microRNA repressed by MBD1, increases neuronal differentiation in vivo, similar to miR-184[220].
Similar to MeCP2, MBD1 has been found to interact with multiple epigenetic co-factors and it is highly likely that MBD1- mediated regulation of adult neurogenesis requires cooperation from other epigenetic machinery. MBD1-interactors have been identified primarily in cancer cell lines and it is not known if these interactions are relevant to MBD1 regulation in the context of adult neurogenesis (Table 1). HDAC3 can facilitate the regulation of gene expression by MBD1 in cancer cells [221] but it is not clear gene repression is mediated through the recruitment of HDACs by MBD1 as the HDAC inhibitor trichostatin A (TSA) does not always activate MBD1-repressed genes [222, 223]. However, MeCP2, MBD1, and HDACs have also been shown to target the RE1 REST binding site in a subset of neuronal genes, reflecting the possibility that both DNA methylation and histone remodeling are required to mediate gene repression [171]. In addition, MBD1 has been shown to interact with PRC1 components HPC2 and RING1B [224], and PRC2 component SETDB1 [225], indicating a possible role in the resolution of bivalent chromatin states. In addition, in the absence of MBD1, the levels of activating and repressive histone marks are altered at the promoter of miR-184 in aNSCs, suggesting that MBD1 can influence the recruitment of histone-modifying proteins [218]. However, exactly how MBD1 interacts with other epigenetic pathways remains unknown.
CONCLUSION
The brain is a hot-spot of epigenetic regulation: the generation of heterogeneous cell populations from shared progenitors requires many levels of generegulation. Many epigenetic factors are highly expressed or highly prevalent in the brain, such as many MBPs and DNA modifications. Recently, improved technologies for detecting DNA modifications beyond classical CpG methylation has greatly expanded our knowledge of the existence and possible function of mCH, 5hmC, other modifications, and demethylation pathways. Epigenetic pathways are increasingly found to be highly interconnected and DNA modifications are poised to be at the center of networks linking transcriptional regulation, chromatin states, and environmental inputs. The proteins that deposit, read and remove DNA methylation are vital for the function of the cell. The contributions of these proteins in the development of the nervous system are particularly interesting given how they are already known to regulate neurogenesis. However, there are many outstanding questions that remain regarding the function of DNA methylation in adult neurogenesis. The fields of epigenetics and AHN are starting to address what roles methylation-specific proteins play at each stage of neurogenesis (Fig. 2), but the functions in all cell types are not all known. One example is MBD1, which we have been studying for the past decade. Does MBD1 play a role in NSC maintenance and proliferation? Does it regulate neuronal maturation? What pathways and mechanisms control these possible functions? What are the cofactors or facilitators for MBD1 regulation of neurogenesis? Does MBD1 have a role in neurogenesis during juvenile period which may have important role in developing social ability [226]? Beyond neurogenesis, questions about the basic mechanisms of these proteins still remain. What is the binding specificity of MBDs in physiologically relevant cell types? How are MBDs recruited to methylated DNA? Do some MBDs have specific affinity for each type of non-CG methylation? Can MBDs compensate each other? Do MBDs interact with each other? As the field of adult neurogenesis moves increasingly towards an integrated network-based understand of regulatory networks and disease mechanisms, understanding the how epigenetics and other pathways intersect is becoming increasingly important.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH to X. Z. (MH080434, MH07897, R21NS095632), a center grant from the NIH to the Waisman Center (P30HD03352), and a NIH Molecular Biosciences Training Grant to E.M.J (MBTG: T32 GM07215).
REFERENCES
[1] | Hall BK . Waddington legacy in development and evolution. American Zoologist. (1992) ;32: (1):113–22. |
[2] | Yao B , Jin P . Unlocking epigenetic codes in neurogenesis. Genes Dev. (2014) ;28: (12):1253–71. |
[3] | Pastor WA , Aravind L , Rao A . TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. (2013) ;14: (6):341–56. |
[4] | Eisch AJ , Petrik D . Depression and hippocampal neurogenesis: A road to remission? Science. (2012) ;338: (6103):72–5. |
[5] | Kempermann G , Krebs J , Fabel K . The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr Opin Psychiatry. (2008) ;21: (3):290–5. |
[6] | Deng W , Aimone JB , Gage FH . New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nature Reviews Neuroscience. Nature Publishing Group. (2010) ;11: (5):339–50. |
[7] | Giacometti E , Luikenhuis S , Beard C , Jaenisch R . Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci USA. (2007) ;104: (6):1931–6. |
[8] | Guo W , Murthy AC , Zhang L , Johnson EB , Schaller EG , Allan AM , et al. Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet. (2012) ;21: (3):681–91. |
[9] | Zhao C , Deng W , Gage FH . Mechanisms and functional implications of adult neurogenesis. Cell. Elsevier; (2008) ;132: (4):645–60. |
[10] | Feliciano DM , Bordey A , Bonfanti L . Noncanonical Sites of Adult Neurogenesis in the Mammalian Brain. Cold Spring Harb Perspect Biol. (2015) ;7: (10):a018846. |
[11] | Li G , Pleasure SJ . Genetic regulation of dentate gyrus morphogenesis. Prog Brain Res. (2007) ;163: :143–52. |
[12] | Gebara E , Bonaguidi MA , Beckervordersandforth R , Sultan S , Udry F , Gijs P-J , et al. Heterogeneity of radial glia-like cells in the adult hippocampus. Stem Cells. (2016) ;34: (4):997–1010. doi: 10.1002/stem.2266. |
[13] | Bonaguidi MA , Wheeler MA , Shapiro JS , Stadel RP , Sun GJ , Ming G , et al. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. (2011) ;145: (7):1142–55. |
[14] | Kempermann G , Song H , Gage FH . Neurogenesis in the adult hippocampus. Cold Spring Harb Perspect Biol. (2015) ;7: (9):a018812. doi: 10.1101/cshperspect.a018812. page 1–14. |
[15] | Squire LR . Memory and the hippocampus: A synthesis from findings with rats, monkeys, and humans. Psychol Rev. (1992) ;99: (2):195–231. |
[16] | Jarrard LE . On the role of the hippocampus in learning and memory in the rat. Behav Neural Biol. (1993) ;60: (1):9–26. |
[17] | Barkho BZ , Munoz AE , Li X , Li L , Cunningham LA , Zhao X . Endogenous matrix metalloproteinase (MMP)-3 and MMP-9 promote the differentiation and migration of adult neural progenitor cells in response to chemokines. Stem Cells. (2008) ;26: (12):3139–49. |
[18] | Clelland CD , Choi M , Romberg C , Clemenson GD , Fragniere A , Tyers P , et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. (2009) ;325: (5937):210–3. |
[19] | Deng W , Saxe MD , Gallina IS , Gage FH . Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. (2009) ;29: (43):13532–42. |
[20] | Dupret D , Revest J-M , Koehl M , Ichas F , De Giorgi F , Costet P , et al. Spatial relational memory requires hippocampal adult neurogenesis. PLoS ONE. (2008) ;3: (4):e1959. |
[21] | Farioli-Vecchioli S , Saraulli D , Costanzi M , Pacioni S , Ciná I , Aceti M , et al. The timing of differentiation of adult hippocampal neurons is crucial for spatial memory. PLoS Biol. (2008) ;6: (10):e246. |
[22] | Imayoshi I , Sakamoto M , Ohtsuka T , Takao K , Miyakawa T , Yamaguchi M , et al. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. (2008) ;11: (10):1153–61. |
[23] | Xu L , Tang X , Wang Y , Xu H , Fan X . Radial glia, the keystone of the development of the hippocampal dentate gyrus. Mol Neurobiol. (2015) ;51: (1):131–41. |
[24] | Christian KM , Song H , Ming G . Functions and dysfunctions of adult hippocampal neurogenesis. Annu Rev Neurosci. (2014) ;37: :243–62. |
[25] | Ge S , Yang C-H , Hsu K-S , Ming G-L , Song H . A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. (2007) ;54: (4):559–66. |
[26] | Gu Y , Arruda-Carvalho M , Wang J , Janoschka SR , Josselyn SA , Frankland PW , et al. Optical controlling reveals time-dependent roles for adult-born dentate granule cells. Nat Neurosci. (2012) ;15: (12):1700–6. doi: 10.1038/nn.3260. |
[27] | Bergami M , Masserdotti G , Temprana SG , Motori E , Eriksson TM , Göbel J , et al. A critical period for experience-dependent remodeling of adult-born neuron connectivity. Neuron. (2015) ;85: (4):710–7. doi: 10.1016/j.neuron.2015.01.001. |
[28] | Kheirbek MA , Tannenholz L , Hen R . NR2B-dependent plasticity of adult-born granule cells is necessary for context discrimination. J Neurosci. (2012) ;32: (25):8696–702. |
[29] | Denny CA , Kheirbek MA , Alba EL , Tanaka KF , Brachman RA , Laughman KB , et al. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron. (2014) ;83: (1):189–201. |
[30] | Ladd-Acosta C , Pevsner J , Sabunciyan S , Yolken RH , Webster MJ , Dinkins T , et al. DNA methylation signatures within the human brain. Am J Hum Genet. (2007) ;81: (6):1304–15. |
[31] | Brown SE , Weaver ICG , Meaney MJ , Szyf M . Regional-specific global cytosine methylation and DNA methyltransferase expression in the adult rat hippocampus. Neurosci Lett. (2008) ;440: (1):49–53. |
[32] | Iwamoto K , Bundo M , Ueda J , Oldham MC , Ukai W , Hashimoto E , et al. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. (2011) ;21: (5):688–96. |
[33] | Zhang Y , Chen K , Sloan SA , Bennett ML , Scholze AR , O’Keeffe S , et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) ;34: (36):11929–47. |
[34] | Doyle JP , Dougherty JD , Heiman M , Schmidt EF , Stevens TR , Ma G , et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. (2008) ;135: (4):749–62. |
[35] | Lovatt D , Sonnewald U , Waagepetersen HS , Schousboe A , He W , Lin JH-C , et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. (2007) ;27: (45):12255–66. |
[36] | Rossner MJ , Hirrlinger J , Wichert SP , Boehm C , Newrzella D , Hiemisch H , et al. Global transcriptome analysis of genetically identified neurons in the adult cortex. J Neurosci. (2006) ;26: (39):9956–66. |
[37] | Cahoy JD , Emery B , Kaushal A , Foo LC , Zamanian JL , Christopherson KS , et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. (2008) ;28: (1):264–78. |
[38] | Edwards CA , Ferguson-Smith AC . Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol. (2007) ;19: (3):281–9. |
[39] | Klose RJ , Bird AP . Genomic DNA methylation: The mark and its mediators. Trends Biochem Sci. (2006) ;31: (2):89–97. |
[40] | Reik W . Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. (2007) ;447: (7143):425–32. |
[41] | Law JA , Jacobsen SE . Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics. Nature Publishing Group; (2010) ;11: (3):204–20. |
[42] | Xie W , Barr CL , Kim A , Yue F , Lee AY , Eubanks J , et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. (2012) ;148: (4):816–31. |
[43] | Varley KE , Gertz J , Bowling KM , Parker SL , Reddy TE , Pauli-Behn F , et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. (2013) ;23: (3):555–67. |
[44] | Guo JU , Su Y , Shin JH , Shin J , Li H , Xie B , et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. (2014) ;17: (2):215–22. |
[45] | Kozlenkov A , Roussos P , Timashpolsky A , Barbu M , Rudchenko S , Bibikova M , et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. (2014) ;42: (1):109–27. |
[46] | Lister R , Mukamel EA , Nery JR , Urich M , Puddifoot CA , Johnson ND , et al. Global epigenomic reconfiguration during mammalian brain development. Science. (2013) ;341: (6146):1237905. |
[47] | Tahiliani M , Koh KP , Shen Y , Pastor WA , Bandukwala H , Brudno Y , et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) ;324: (5929):930–5. |
[48] | Chen Z , Riggs AD . DNA methylation and demethylation in mammals. The Journal of Biological Chemistry. American Society for Biochemistry and Molecular Biology; (2011) ;286: (21):18347. |
[49] | Bhutani N , Burns DM , Blau HM . DNA Demethylation Dynamics. Cell. Elsevier. (2011) ;146: (6):866–72. |
[50] | Ito S , Shen L , Dai Q , Wu SC , Collins LB , Swenberg JA , et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. (2011) ;333: (6047):1300–3. |
[51] | Guo JU , Su Y , Zhong C , Ming G , Song H . Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. Elsevier; (2011) . |
[52] | Nabel CS , Jia H , Ye Y , Shen L , Goldschmidt HL , Stivers JT , et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. (2012) ;8: (9):751–8. |
[53] | Ficz G , Branco MR , Seisenberger S , Santos F , Krueger F , Hore TA , et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. (2011) ;473: (7347):398–402. |
[54] | Jin S-G , Wu X , Li AX , Pfeifer GP . Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. (2011) ;39: (12):5015–24. |
[55] | Wen L , Li X , Yan L , Tan Y , Li R , Zhao Y , et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. (2014) ;15: (3):R49. |
[56] | Khare T , Pai S , Koncevicius K , Pal M , Kriukiene E , Liutkeviciute Z , et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. (2012) ;19: (10):1037–43. |
[57] | Iurlaro M , Ficz G , Oxley D , Raiber E-A , Bachman M , Booth MJ , et al. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. (2013) ;14: (10):R119. |
[58] | Bartels SJJ , Spruijt CG , Brinkman AB , Jansen PWTC , Vermeulen M , Stunnenberg HG . A SILAC-based screen for methyl-CpG binding proteins identifies RBP-J as a DNA methylation and sequence-specific binding protein. PLoS ONE. Public Library of Science; (2011) ;6: (10):e25884. |
[59] | Spruijt CG , Gnerlich F , Smits AH , Pfaffeneder T , Jansen PWTC , Bauer C , et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. (2013) ;152: (5):1146–59. |
[60] | Guoping F , Hutnick L . Methyl-CpG binding proteins in the nervous system. Cell Research. Nature Publishing Group; (2005) ;15: (4):255–61. |
[61] | Parry L , Clarke AR . The roles of the methyl-CpG binding proteins in cancer. Genes & Cancer. SAGE Publications; (2011) ;2: (6):618–30. |
[62] | Defossez P-A , Stancheva I . Biological functions of methyl-CpG-binding proteins. Prog Mol Biol Transl Sci. (2011) ;101: :377–98. |
[63] | Lewis JD , Meehan RR , Henzel WJ , Maurer-Fogy I , Jeppesen P , Klein F , et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. (1992) ;69: (6):905–14. |
[64] | Skene PJ , Illingworth RS , Webb S , Kerr ARW , James KD , Turner DJ , et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. (2010) ;37: (4):457–68. |
[65] | Baubec T , Ivánek R , Lienert F , Schübeler D . Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. (2013) ;153: (2):480–92. |
[66] | Fujita N , Takebayashi S , Okumura K , Kudo S , Chiba T , Saya H , et al. Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol. (1999) ;19: (9):6415–26. |
[67] | Laget S , Joulie M , Le Masson F , Sasai N , Christians E , Pradhan S , et al. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE. (2010) ;5: (8):e11982. |
[68] | Baymaz HI , Fournier A , Laget S , Ji Z , Jansen PWTC , Smits AH , et al. MBD5 and MBD6 interact with the human PR-DUB complex through their methyl-CpG-binding domain. Proteomics. (2014) ;14: (19):2179–89. |
[69] | Valinluck V , Tsai H-H , Rogstad DK , Burdzy A , Bird A , Sowers LC . Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. (2004) ;32: (14):4100–8. |
[70] | Frauer C , Hoffmann T , Bultmann S , Casa V , Cardoso MC , Antes I , et al. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE. (2011) ;6: (6):e21306. |
[71] | Mellén M , Ayata P , Dewell S , Kriaucionis S , Heintz N . MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. (2012) ;151: (7):1417–30. |
[72] | Otani J , Arita K , Kato T , Kinoshita M , Kimura H , Suetake I , et al. Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J Biol Chem. (2013) ;288: (9):6351–62. |
[73] | Khrapunov S , Warren C , Cheng H , Berko ER , Greally JM , Brenowitz M . Unusual characteristics of the DNA binding domain of epigenetic regulatory protein MeCP2 determine its binding specificity. Biochemistry. (2014) ;53: (21):3379–91. |
[74] | Kimura H , Shiota K . Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. (2003) ;278: (7):4806–12. |
[75] | Gabel HW , Kinde B , Stroud H , Gilbert CS , Harmin DA , Kastan NR , et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. (2015) ;522: (7554):89–93. doi: 10.1038/nature14319. |
[76] | Sugino K , Hempel CM , Okaty BW , Arnson HA , Kato S , Dani VS , et al. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci. (2014) ;34: (38):12877–83. |
[77] | Chen L , Chen K , Lavery LA , Baker SA , Shaw CA , Li W , et al. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci USA. (2015) ;112: (17):5509–14. |
[78] | Chodavarapu RK , Feng S , Bernatavichute YV , Chen P-Y , Stroud H , Yu Y , et al. Relationship between nucleosome positioning and DNA methylation. Nature. (2010) ;466: (7304):388–92. |
[79] | Daniel JM , Reynolds AB . The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. (1999) :19: (5):3614–23. |
[80] | Prokhortchouk A , Hendrich B , Jørgensen H , Ruzov A , Wilm M , Georgiev G , et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. (2001) ;15: (13):1613–8. |
[81] | Daniel JM , Spring CM , Crawford HC , Reynolds AB , Baig A . The p120(ctn)-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. (2002) ;30: (13):2911–9. |
[82] | Arányi T , Faucheux BA , Khalfallah O , Vodjdani G , Biguet NF , Mallet J , et al. The tissue-specific methylation of the human tyrosine hydroxylase gene reveals new regulatory elements in the first exon. J Neurochem. (2005) ;94: (1):129–39. |
[83] | Yoon H-G , Chan DW , Reynolds AB , Qin J , Wong J . N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell. (2003) ;12: (3):723–34. |
[84] | Buck-Koehntop BA , Stanfield RL , Ekiert DC , Martinez-Yamout MA , Dyson HJ , Wilson IA , et al. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc Natl Acad Sci USA. (2012) ;109: (38):15229–34. |
[85] | Qin S , Zhang B , Tian W , Gu L , Lu Z , Deng D . Kaiso mainly locates in the nucleus in vivo and binds to methylated, but not hydroxymethylated DNA. Chin J Cancer Res. (2015) ;27: (2):148–55. |
[86] | Kiefer H , Chatail-Hermitte F , Ravassard P , Bayard E , Brunet I , Mallet J . ZENON, a novel POZ Kruppel-like DNA binding protein associated with differentiation and/or survival of late postmitotic neurons. Mol Cell Biol. (2005) ;25: (5):1713–29. |
[87] | Filion GJP , Zhenilo S , Salozhin S , Yamada D , Prokhortchouk E , Defossez P-A . A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol. (2006) ;26: (1):169–81. |
[88] | Sasai N , Nakao M , Defossez P-A . Sequence-specific recognition of methylated DNA by human zinc-finger proteins. Nucleic Acids Res. (2010) ;38: (15):5015–22. |
[89] | Mori T , Ikeda DD , Yamaguchi Y , Unoki M . NIRF/UHRF2 occupies a central position in the cell cycle network and allows coupling with the epigenetic landscape. FEBS Lett. (2012) ;586: (11):1570–83. |
[90] | Bronner C , Krifa M , Mousli M . Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem Pharmacol. (2013) ;86: (12):1643–9. |
[91] | Unoki M , Nishidate T , Nakamura Y . ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. (2004) ;23: (46):7601–10. |
[92] | Hashimoto H , Horton JR , Zhang X , Bostick M , Jacobsen SE , Cheng X . The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. (2004) ;455: (7214):826–9. |
[93] | Avvakumov GV , Walker JR , Xue S , Li Y , Duan S , Bronner C , et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. (2008) ;455: (7214):822–5. |
[94] | Arita K , Ariyoshi M , Tochio H , Nakamura Y , Shirakawa M . Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. (2008) ;455: (7214):818–21. |
[95] | Bostick M , Kim JK , Estève P-O , Clark A , Pradhan S , Jacobsen SE . UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. (2007) ;317: (5845):1760–4. |
[96] | Sharif J , Muto M , Takebayashi S , Suetake I , Iwamatsu A , Endo TA , et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. Nature Publishing Group; (2007) ;450: (7171):908–12. |
[97] | Zhang J , Gao Q , Li P , Liu X , Jia Y , Wu W , et al. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. (2011) ;21: (12):1723–39. |
[98] | Blattler A , Farnham PJ . Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. (2013) ;288: (48):34287–94. |
[99] | Lienert F , Wirbelauer C , Som I , Dean A , Mohn F , Schübeler D . Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. (2011) ;43: (11):1091–7. |
[100] | Li H , Rauch T , Chen Z-X , Szabó PE , Riggs AD , Pfeifer GP . The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. (2006) ;281: (28):19489–500. |
[101] | Schultz DC , Ayyanathan K , Negorev D , Maul GG , Rauscher FJ . SETDB A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. (2002) ;16: (8):919–32. |
[102] | Stadler MB , Murr R , Burger L , Ivanek R , Lienert F , Schöler A , et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. (2011) ;480: (7378):490–5. |
[103] | Campanero MR , Armstrong MI , Flemington EK . CpG methylation as a mechanism for the regulation of E2F activity. Proc Natl Acad Sci USA. (2000) ;97: (12):6481–6. |
[104] | Julian LM , Liu Y , Pakenham CA , Dugal-Tessier D , Ruzhynsky V , Bae S , et al. Tissue-specific targeting of cell fate regulatory genes by E2f factors. Cell Death Differ. (2015) ;1–11. |
[105] | Christensen BC , Houseman EA , Marsit CJ , Zheng S , Wrensch MR , Wiemels JL , et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. (2009) ;5: (8):e1000602. |
[106] | Kinde B , Gabel HW , Gilbert CS , Griffith EC , Greenberg ME . Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A. (2015) ;112: (22):6800–6 . |
[107] | Hsieh J , Eisch AJ . Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: Unraveling the genome to understand the mind. Neurobiology of Disease. Elsevier; (2010) ;39: (1):73–84. |
[108] | Takizawa T , Nakashima K , Namihira M , Ochiai W , Uemura A , Yanagisawa M , et al. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. (2001) ;1: (6):749–58. |
[109] | Namihira M , Nakashima K , Taga T . Developmental stage dependent regulation of DNA methylation and chromatin modification in a immature astrocyte specific gene promoter. FEBS Lett. (2004) ;572: (1-3):184–8. |
[110] | Hutnick LK , Golshani P , Namihira M , Xue Z , Matynia A , Yang XW , et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. (2009) ;18: (15):2875–88. |
[111] | Shimozaki K , Namihira M , Nakashima K , Taga T . Stage- and site-specific DNA demethylation during neural cell development from embryonic stem cells. J Neurochem. (2005) ;93: (2):432–9. |
[112] | Namihira M , Kohyama J , Semi K , Sanosaka T , Deneen B , Taga T , et al. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. (2009) ;16: (2):245–55. |
[113] | Mohn F , Weber M , Rebhan M , Roloff TC , Richter J , Stadler MB , et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Molecular Cell. Elsevier; (2008) ;30: (6):755–66. |
[114] | Meissner A , Mikkelsen TS , Gu H , Wernig M , Hanna J , Sivachenko A , et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. Nature Publishing Group; (2008) ;454: (7205):766–70. |
[115] | Bestor TH . The DNA methyltransferases of mammals. Hum Mol Genet. (2000) ;9: (16):2395–402. |
[116] | Lei H , Oh SP , Okano M , Jüttermann R , Goss KA , Jaenisch R , et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. (1996) ;122: (10):3195–205. |
[117] | Okano M , Bell DW , Haber DA , Li E . DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) ;99: (3):247–57. |
[118] | Feng J , Chang H , Li E , Fan G . Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res. (2005) ;79: (6):734–46. |
[119] | Martins-Taylor K , Schroeder DI , LaSalle JM , Lalande M , Xu R-H . Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics. (2012) ;7: (1):71–82. |
[120] | Fan G , Beard C , Chen RZ , Csankovszki G , Sun Y , Siniaia M , et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. (2001) ;21: (3):788–97. |
[121] | Fan G , Martinowich K , Chin MH , He F , Fouse SD , Hutnick L , et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. (2005) ;132: (15):3345–56. |
[122] | Nguyen S , Meletis K , Fu D , Jhaveri S , Jaenisch R . Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. (2007) ;236: (6):1663–76. |
[123] | Wu H , Coskun V , Tao J , Xie W , Ge W , Yoshikawa K , et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. (2010) ;329: (5990):444–8. |
[124] | Noguchi H , Kimura A , Murao N , Matsuda T , Namihira M , Nakashima K . Expression of DNMT1 in neural stem/precursor cells is critical for survival of newly generated neurons in the adult hippocampus. Neurosci Res. (2015) ;95: :1–11. |
[125] | Rhee K-D , Yu J , Zhao CY , Fan G , Yang X-J . Dnmt1-dependent DNA methylation is essential for photoreceptor terminal differentiation and retinal neuron survival. Cell Death Dis. (2012) ;3: :e427. |
[126] | Feng J , Zhou Y , Campbell SL , Le T , Li E , Sweatt JD , et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. (2010) ;13: (4):423–30. |
[127] | Golshani P , Hutnick L , Schweizer F , Fan G . Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus Relat Syst. (2005) ;3: (3):227–33. |
[128] | Kohno D , Lee S , Harper MJ , Kim KW , Sone H , Sasaki T , et al. Dnmt3a in Sim1 neurons is necessary for normal energy homeostasis. J Neurosci. (2014) ;34: (46):15288–96. |
[129] | Balthasar N , Dalgaard LT , Lee CE , Yu J , Funahashi H , Williams T , et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. (2005) ;123: (3):493–505. |
[130] | Fyffe SL , Neul JL , Samaco RC , Chao H-T , Ben-Shachar S , Moretti P , et al. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. (2008) ;59: (6):947–58. |
[131] | Toyoda S , Kawaguchi M , Kobayashi T , Tarusawa E , Toyama T , Okano M , et al. Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron. (2014) ;82: (1):94–108. |
[132] | Szwagierczak A , Bultmann S , Schmidt CS , Spada F , Leonhardt H . Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. (2010) ;38: (19):e181. |
[133] | Globisch D , Münzel M , Müller M , Michalakis S , Wagner M , Koch S , et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. (2010) ;5: (12):e15367. |
[134] | Münzel M , Globisch D , Brückl T , Wagner M , Welzmiller V , Michalakis S , et al. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem Int Ed Engl. (2010) ;49: (31):5375–7. |
[135] | Kriaucionis S , Heintz N . The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. (2009) ;324: (5929):930–1. |
[136] | Hahn MA , Qiu R , Wu X , Li AX , Zhang H , Wang J , et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. (2013) ;3: (2):291–300. |
[137] | Szulwach KE , Li X , Li Y , Song CX , Han JW , Kim SS , et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genetics. Public Library of Science; (2011) ;7: (6):e1002154. |
[138] | Wheldon LM , Abakir A , Ferjentsik Z , Dudnakova T , Strohbuecker S , Christie D , et al. Transient accumulation of 5-carboxylcytosine indicates involvement of active demethylation in lineage specification of neural stem cells. Cell Rep. (2014) ;7: (5):1353–61. |
[139] | Dawlaty MM , Breiling A , Le T , Raddatz G , Barrasa MI , Cheng AW , et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. (2013) ;24: (3):310–23. |
[140] | Gu T-P , Guo F , Yang H , Wu H-P , Xu G-F , Liu W , et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. (2011) ;477: (7366):606–10. |
[141] | Zhang R-R , Cui Q-Y , Murai K , Lim YC , Smith ZD , Jin S , et al. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. (2013) ;13: (2):237–45. |
[142] | Rudenko A , Dawlaty MM , Seo J , Cheng AW , Meng J , Le T , et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. (2013) ;79: (6):1109–22. |
[143] | Kaas GA , Zhong C , Eason DE , Ross DL , Vachhani RV , Ming G-L , et al. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. (2013) ;79: (6):1086–93. |
[144] | Ma DK , Jang M-H , Guo JU , Kitabatake Y , Chang M-L , Pow-Anpongkul N , et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. (2009) ;323: (5917):1074–7. |
[145] | Williams K , Christensen J , Pedersen MT , Johansen JV , Cloos PAC , Rappsilber J , et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. (2011) ;473: (7347):343–8. |
[146] | Wu H , D’Alessio AC , Ito S , Xia K , Wang Z , Cui K , et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. (2011) ;473: (7347):389–93. |
[147] | Wu H , D’Alessio AC , Ito S , Wang Z , Cui K , Zhao K , et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. (2011) ;25: (7):679–84. |
[148] | Cartron P-F , Nadaradjane A , Lepape F , Lalier L , Gardie B , Vallette FM . Identification of TET1 partners that control its DNA-demethylating function. Genes Cancer. (2013) ;4: (5-6):235–41. |
[149] | Yildirim O , Li R , Hung J-H , Chen PB , Dong X , Ee L-S , et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. (2011) ;147: (7):1498–510. |
[150] | Reynolds N , Salmon-Divon M , Dvinge H , Hynes-Allen A , Balasooriya G , Leaford D , et al. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. (2012) ;31: (3):593–605. |
[151] | Zeng Y , Yao B , Shin J , Lin L , Kim N , Song Q , et al. Lin28A binds active promoters and recruits tet1 to regulate gene expression. Mol Cell. (2016) ;61: (1):153–60. |
[152] | Hashimoto H , Liu Y , Upadhyay AK , Chang Y , Howerton SB , Vertino PM , et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. (2012) ;40: (11):4841–9. |
[153] | Szulwach KE , Li X , Li Y , Song C-X , Wu H , Dai Q , et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. (2011) ;14: (12):1607–16. |
[154] | Akbarian S , Chen RZ , Gribnau J , Rasmussen TP , Fong H , Jaenisch R , et al. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. (2001) ;8: (5):784–91. |
[155] | Shahbazian MD , Antalffy B , Armstrong DL , Zoghbi HY . Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. (2002) ;11: (2):115–24. |
[156] | Kishi N , Macklis JD . MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp Neurol. (2010) ;222: (1):51–8. |
[157] | Balmer D , Goldstine J , Rao YM , LaSalle JM . Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J Mol Med. (2003) ;81: (1):61–8. |
[158] | Jung BP , Jugloff DGM , Zhang G , Logan R , Brown S , Eubanks JH . The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol. (2003) ;55: (1):86–96. |
[159] | Kishi N , Macklis JD . MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. (2004) ;27: (3):306–21. |
[160] | Ballas N , Lioy DT , Grunseich C , Mandel G . Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. (2009) ;12: (3):311–7. |
[161] | Song C , Feodorova Y , Guy J , Peichl L , Jost KL , Kimura H , et al. DNA methylation reader MECP Cell type- and differentiation stage-specific protein distribution. Epigenetics Chromatin. (2014) ;7: :17. |
[162] | Wood KH , Johnson BS , Welsh SA , Lee JY , Cui Y , Krizman E , et al. Tagging methyl-CpG-binding domain proteins reveals different spatiotemporal expression and supports distinct functions. Epigenomics. (2016) ;8: (4):455–73. |
[163] | Zhao X , Ueba T , Christie BR , Barkho B , McConnell MJ , Nakashima K , et al. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci U S A. (2003) ;100: (11):6777–82. |
[164] | Tsujimura K , Abematsu M , Kohyama J , Namihira M , Nakashima K . Neuronal differentiation of neural precursor cells is promoted by the methyl-CpG-binding protein MeCP2. Exp Neurol. (2009) ;219: (1):104–11. |
[165] | Setoguchi H , Namihira M , Kohyama J , Asano H , Sanosaka T , Nakashima K . Methyl-CpG binding proteins are involved in restricting differentiation plasticity in neurons. J Neurosci Res. (2006) ;84: (5):969–79. |
[166] | Chen RZ , Akbarian S , Tudor M , Jaenisch R . Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. (2001) ;27: (3):327–31. |
[167] | Armstrong DD . Neuropathology of Rett syndrome. J Child Neurol. (2005) ;20: (9):747–53. |
[168] | Bauman ML , Kemper TL , Arin DM . Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology. (1995) ;45: (8):1581–6. |
[169] | Armstrong D , Dunn JK , Antalffy B , Trivedi R . Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. (1995) ;54: (2):195–201. |
[170] | Zhou Z , Hong EJ , Cohen S , Zhao W-N , Ho H-YH , Schmidt L , et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. (2006) ;52: (2):255–69. |
[171] | Kuwabara T , Hsieh J , Nakashima K , Taira K , Gage FH . A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell. Elsevier; (2004) ;116: (6):779–93. |
[172] | Allan AM , Liang X , Luo Y , Pak C , Li X , Szulwach KE , et al. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum Mol Genet. (2008) ;17: (13):2047–57. |
[173] | Lioy DT , Garg SK , Monaghan CE , Raber J , Foust KD , Kaspar BK , et al. A role for glia in the progression of Rett’s syndrome. Nature. (2011) ;475: (7357):497–500. |
[174] | Li H , Zhong X , Chau KF , Santistevan NJ , Guo W , Kong G , et al. Cell cycle-linked MeCP2 phosphorylation modulates adult neurogenesis involving the Notch signalling pathway. Nat Commun. (2014) ;5: :5601. |
[175] | Li H , Chang Q . Regulation and function of stimulus-induced phosphorylation of MeCP2. Front Biol (Beijing). (2014) ;9: (5):367–75. |
[176] | Chen WG , Chang Q , Lin Y , Meissner A , West AE , Griffith EC , et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. (2003) ;302: (5646):885–9. |
[177] | Martinowich K , Hattori D , Wu H , Fouse S , He F , Hu Y , et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. (2003) ;302: (5646):890–3. |
[178] | Li Y , Luikart BW , Birnbaum S , Chen J , Kwon C-H , Kernie SG , et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. (2008) ;59: (3):399–412. |
[179] | Fitzsimons CP , van Bodegraven E , Schouten M , Lardenoije R , Kompotis K , Kenis G , et al. Epigenetic regulation of adult neural stem cells: Implications for Alzheimer’s disease. Mol Neurodegener. (2014) ;9: :25. |
[180] | Tatton-Brown K , Seal S , Ruark E , Harmer J , Ramsay E , Del Vecchio Duarte S , et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. (2014) ;46: (4):385–8. |
[181] | Ley TJ , Ding L , Walter MJ , McLellan MD , Lamprecht T , Larson DE , et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. (2010) ;363: (25):2424–33. |
[182] | Nikoloski G , van der Reijden BA , Jansen JH . Mutations in epigenetic regulators in myelodysplastic syndromes. Int J Hematol. (2012) ;95: (1):8–16. |
[183] | Jones WD , Dafou D , McEntagart M , Woollard WJ , Elmslie FV , Holder-Espinasse M , et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet. (2012) ;91: (2):358–64. |
[184] | Jiang Y , Yuen RKC , Jin X , Wang M , Chen N , Wu X , et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. (2013) ;93: (2):249–63. |
[185] | Sanders SJ , He X , Willsey AJ , Ercan-Sencicek AG , Samocha KE , Cicek AE , et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. (2015) ;87: (6):1215–33. |
[186] | Hansen RS , Wijmenga C , Luo P , Stanek AM , Canfield TK , Weemaes CM , et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. (1999) ;96: (25):14412–7. |
[187] | Xu GL , Bestor TH , Bourc’his D , Hsieh CL , Tommerup N , Bugge M , et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. (1999) ;402: (6758):187–91. |
[188] | Weemaes CMR , van Tol MJD , Wang J , vanOstaijen-ten Dam MM , van Eggermond MCJA , Thijssen PE , et al. Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur J Hum Genet. (2013) ;21: (11):1219–25. |
[189] | Smeets DF , Moog U , Weemaes CM , Vaes-Peeters G , Merkx GF , Niehof JP , et al. ICF syndrome: A new case and review of the literature. Hum Genet. (1994) ;94: (3):240–6. |
[190] | Klein CJ , Botuyan M-V , Wu Y , Ward CJ , Nicholson GA , Hammans S , et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. (2011) ;43: (6):595–600. |
[191] | Winkelmann J , Lin L , Schormair B , Kornum BR , Faraco J , Plazzi G , et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. (2012) ;21: (10):2205–10. |
[192] | Sun Z , Wu Y , Ordog T , Baheti S , Nie J , Duan X , et al. Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics. (2014) ;9: (8):1184–93. |
[193] | Lardenoije R , Iatrou A , Kenis G , Kompotis K , Steinbusch HWM , Mastroeni D , et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. (2015) ;131: :21–64. |
[194] | Castro J , Mellios N , Sur M . Mechanisms and therapeutic challenges in autism spectrum disorders: Insights from Rett syndrome. Curr Opin Neurol. (2013) ;26: (2):154–9. |
[195] | Chahrour M , Zoghbi HY . The story of Rett syndrome: From clinic to neurobiology. Neuron. (2007) ;56: (3):422–37. |
[196] | Smrt RD , Zhao X . Epigenetic regulation of neuronal dendrite and dendritic spine development. Front Biol (Beijing). (2010) ;5: (4):304–23. |
[197] | Lyst MJ , Bird A . Rett syndrome: A complex disorder with simple roots. Nat Rev Genet. (2015) ;16: (5):261–75. |
[198] | Szulwach KE , Li X , Smrt RD , Li Y , Luo Y , Lin L , et al. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. (2010) ;189: (1):127–41. |
[199] | Talkowski ME , Maussion G , Crapper L , Rosenfeld JA , Blumenthal I , Hanscom C , et al. Disruption of a large intergenic noncoding RNA in subjects with neurodevelopmental disabilities. Am J Hum Genet. (2012) ;91: (6):1128–34. |
[200] | Hodge JC , Mitchell E , Pillalamarri V , Toler TL , Bartel F , Kearney HM , et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry. 2014;19: (3):368–79. doi: 10.1038/mp.2013.42. |
[201] | Talkowski ME , Mullegama SV , Rosenfeld JA , van Bon BWM , Shen Y , Repnikova EA , et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. (2011) ;89: (4):551–63. |
[202] | Cukier HN , Rabionet R , Konidari I , Rayner-Evans MY , Baltos ML , Wright HH , et al. Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics. (2010) ;11: (3):291–303. |
[203] | Turner TN , Hormozdiari F , Duyzend MH , McClymont SA , Hook PW , Iossifov I , et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. (2016) ;98: (1):58–74. |
[204] | Chung BHY , Mullegama S , Marshall CR , Lionel AC , Weksberg R , Dupuis L , et al. Severe intellectual disability and autistic features associated with microduplication 2q23.1.. Eur J Hum Genet. (2012) ;20: (4):398–403. |
[205] | Mullegama SV , Rosenfeld JA , Orellana C , van Bon BWM , Halbach S , Repnikova EA , et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet. (2014) ;22: (1):57–63. |
[206] | Camarena V , Cao L , Abad C , Abrams A , Toledo Y , Araki K , et al. Disruption of Mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome. EMBO Mol Med. (2014) ;6: (8):1003–15. |
[207] | Li H , Yamagata T , Mori M , Yasuhara A , Momoi MY . Mutation analysis of methyl-CpG binding protein family genes in autistic patients. Brain Dev. (2005) ;27: (5):321–5. |
[208] | Gustavsson P , Kimber E , Wahlström J , Annerén G . Monosomy 18q syndrome and atypical Rett syndrome in a girl with an interstitial deletion (18)(q21.1q22.3). Am J Med Genet. (1999) ;82: (4):348–51. |
[209] | Imataka G , Ohwada Y , Shimura N , Yoshihara S , Arisaka O . Del(18)(q12.2q21.1) syndrome: A case report and clinical review of the literature. Eur Rev Med Pharmacol Sci. (2015) ;19: (17):3241–5. |
[210] | Koh D-I , Han D , Ryu H , Choi W-I , Jeon B-N , Kim M-K , et al. KAISO, a critical regulator of p53-mediated transcription of CDKN1A and apoptotic genes. Proc Natl Acad Sci USA. (2014) ;111: (42):15078–83. |
[211] | Prokhortchouk A , Sansom O , Selfridge J , Caballero IM , Salozhin S , Aithozhina D , et al. Kaiso-deficient mice show resistance to intestinal cancer. Mol Cell Biol. (2006) ;26: (1):199–208. |
[212] | Kulikov AV , Korostina VS , Kulikova EA , Fursenko DV , Akulov AE , Moshkin MP , et al. Knockout Zbtb33 gene results in an increased locomotion, exploration and pre-pulse inhibition in mice. Behav Brain Res. (2016) ;297: :76–83. |
[213] | Martín Caballero I , Hansen J , Leaford D , Pollard S , Hendrich BD . The methyl-CpG binding proteins Mecp2, Mbd2 and Kaiso are dispensable for mouse embryogenesis, but play a redundant function in neural differentiation. PLoS ONE. (2009) ;4: (1):e4315. |
[214] | Pichler G , Wolf P , Schmidt CS , Meilinger D , Schneider K , Frauer C , et al. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J Cell Biochem. (2011) ;112: (9):2585–93. |
[215] | Hopfner R , Mousli M , Jeltsch JM , Voulgaris A , Lutz Y , Marin C , et al. ICBP90, a novel human CCAAT binding protein, involved in the regulation of topoisomerase IIalpha expression. Cancer Res. (2000) ;60: (1):121–8. |
[216] | Murao NTMHNHKMN , Nakashima K . Characterization of Np95 expression in mouse brain from embryo to adult: A novel marker for proliferating neural stem/precursor cells. Neurogenesis. (2014) ;1: :e976026. |
[217] | Li X , Barkho BZ , Luo Y , Smrt RD , Santistevan NJ , Liu C , et al. Epigenetic regulation of the stem cell mitogen Fgf-2 by Mbd1 in adult neural stem/progenitor cells. J Biol Chem. (2008) ;283: (41):27644–52. |
[218] | Liu C , Teng Z-Q , Santistevan NJ , Szulwach KE , Guo W , Jin P , et al. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell. (2010) ;6: (5):433–44. |
[219] | Petersen PH , Tang H , Zou K , Zhong W . The enigma of the numb-Notch relationship during mammalian embryogenesis. Dev Neurosci. (2006) ;28: (1-2):156–68. |
[220] | Liu C , Teng Z-Q , McQuate AL , Jobe EM , Christ CC , von Hoyningen-Huene SJ , et al. An epigenetic feedback regulatory loop involving microRNA-195 and MBD1 governs neural stem cell differentiation. PLoS ONE. (2013) ;8: (1):e51436. |
[221] | Villa R , Morey L , Raker VA , Buschbeck M , Gutierrez A , De Santis F , et al. The methyl-CpG binding protein MBD1 is required for PML-RARalpha function. Proc Natl Acad Sci USA. (2006) ;103: (5):1400–5. |
[222] | Lyst MJ , Nan X , Stancheva I . Regulation of MBD1-mediated transcriptional repression by SUMO and PIAS proteins. The EMBO Journal. Nature Publishing Group; (2006) ;25: (22):5317–28. |
[223] | Ng HH , Jeppesen P , Bird A . Active repression of methylated genes by the chromosomal protein MBD1. Mol Cell Biol. (2000) ;20: (4):1394–406. |
[224] | Sakamoto Y , Watanabe S , Ichimura T , Kawasuji M , Koseki H , Baba H , et al. Overlapping roles of the methylated DNA-binding protein MBD1 and polycomb group proteins in transcriptional repression of HOXA genes and heterochromatin foci formation. J Biol Chem. (2007) ;282: (22):16391–400. |
[225] | Ichimura T , Watanabe S , Sakamoto Y , Aoto T , Fujita N , Nakao M . Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J Biol Chem. (2005) ;280: (14):13928–35. |
[226] | Wei L , Meaney MJ , Duman RS , Kaffman A . Affiliative behavior requires juvenile, but not adult neurogenesis. The Journal of Neuroscience. Society for Neuroscience; (2011) ;31: (40):14335–45. |
[227] | Watanabe S , Ichimura T , Fujita N , Tsuruzoe S , Ohki I , Shirakawa M , et al. Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc Natl Acad Sci USA. (2003) ;100: (22):12859–64. |
[228] | Reese BE , Bachman KE , Baylin SB , Rountree MR . The methyl-CpG binding protein MBD1 interacts with the p150 subunit of chromatin assembly factor 1. Molecular and Cellular Biology. Am Soc Microbiol. (2003) ;23: (9):3226. |
[229] | Fujita N , Watanabe S , Ichimura T , Ohkuma Y , Chiba T , Saya H , et al. MCAF mediates MBD1-dependent transcriptional repression. Mol Cell Biol. (2003) ;23: (8):2834–43. |
[230] | Sarraf SA , Stancheva I . Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. (2004) ;15: (4):595–605. |
[231] | Uchimura Y , Ichimura T , Uwada J , Tachibana T , Sugahara S , Nakao M , et al. Involvement of SUMO modification in MBD1- and MCAF1-mediated heterochromatin formation. J Biol Chem. (2006) ;281: (32):23180–90. |
[232] | Xu J , Zhu W , Xu W , Cui X , Chen L , Ji S , et al. Silencing of MBD1 reverses pancreatic cancer therapy resistance through inhibition of DNA damage repair. Int J Oncol. (2013) ;42: (6):2046–52. |


