
PARP Inhibitors for Metastatic Urothelial Carcinoma: A Systematic Review of Efficacy and Safety
Abstract
BACKGROUND:
Poly (ADP-ribose) polymerase (PARP) inhibitors have activity in various cancers. Metastatic urothelial carcinoma (MUC) is platinum sensitive and a subset harbour DNA repair gene alterations.
OBJECTIVE:
To assess evidence for efficacy and safety of PARP inhibition for MUC.
METHODS:
This systematic review included randomised clinical trials (RCTs) evaluating PARP inhibitors as monotherapy, or in therapeutic combinations, compared to relevant comparators or best supportive care. The primary endpoint was progression free survival (PFS). We searched MEDLINE (Ovid), EMBASE, ClinicalTrials.gov and Cochrane Central Register of Controlled Trials from March 2013 to March 2023. Each study was appraised using the Cochrane Risk of Bias 2 Tool. Study results were synthesised descriptively. Registration: PROSPERO CRD42023403145.
RESULTS:
From 247 identified reports, we included three phase 2 RCTs including 252 patients. Two RCTs assessed PARP inhibition in unselected patient groups (one first line platinum ineligible, one post chemotherapy maintenance) and found no evidence of efficacy. All three RCTs assessed subgroups defined by biomarker selection for somatic DNA repair defects. Two of these identified PFS benefit with PARP inhibition compared to a relevant comparator (one first line in combination with immunotherapy, one maintenance monotherapy). Safety outcomes were consistent with prior experience of PARP inhibitors. The risk of bias across the outcomes was generally low.
CONCLUSIONS:
PARP inhibitors lack efficacy for unselected MUC patients. Phase 2 RCTs support further investigation of PARP inhibition within biomarker-selected patient subsets. The optimal biomarker is not yet determined. Limitations in the current evidence relate to small sample sizes and low statistical power.
INTRODUCTION
In 2020, 573,000 new cases of bladder cancer were diagnosed globally, with 213,000 deaths [1]. The predominant histology is urothelial carcinoma, which can occur throughout the urinary tract. Locally advanced or metastatic urothelial carcinoma (MUC) is an incurable disease with a poor prognosis. The optimal management approach has been typically with platinum based, combination, chemotherapy and PD-1/PD-L1 checkpoint-directed immunotherapy [2–6]. For patients fit enough to receive it, this would typically be with a cisplatin based chemotherapy combination, followed by maintenance avelumab immunotherapy. Even within this favourable prognosis group, median survival remains in the range of approximately 2 years [3]. Criteria for ‘cisplatin eligibility’, and more recent proposals to define ‘platinum eligibility’, provide a framework for clinical decision making for patients to receive chemotherapy safely [7, 8]. These criteria are an acknowledgment that some patients present, de novo, with a disease that may not be suitable for our current best options for treatment [3]. As such, there remains a substantial unmet need for improved therapeutic approaches and ideally predictive biomarker selection strategies to facilitate treatment recommendations and sequencing across the full spectrum of patient fitness.
Within this treatment setting, several molecularly defined therapeutic targets are under evaluation. The nectin-4 directed antibody drug conjugate enfortumab vedotin is an established treatment option for patients previously treated with both chemotherapy and immunotherapy [9]. Recently presented data have also demonstrated a survival advantage for a first line enfortumab vedotin combination with pembrolizumab immunotherapy that is likely to replace the current treatment pathway [10]. Other therapeutic targets, at varying stages of clinical development, include tumor-associated calcium signal transducer 2 (Trop-2), fibroblast growth factor receptors and HER2 [3].
In addition, MUC exhibits DNA repair gene alterations in a proportion of cases. Relevant genes include, BRCA1, BRCA2, ATM, RB1, PALB2, FANCC, FANCD2, and ERCC2. This raises the potential for a therapeutic vulnerability towards agents that interfere with DNA damage repair, of which the furthest developed are poly (ADP-ribose) polymerase (PARP) inhibitors [11–13]. MUC pre-clinical models are sensitive to PARP inhibition, and there is phenotypic overlap with gene alteration patterns that are known to predict for responsiveness to platinum based chemotherapy [14–20]. As such, clinical investigators have explored an over-arching hypothesis that agents designed to target cancer cell vulnerability to alterations in DNA repair, such as with PARP inhibition, might provide a novel approach for therapeutic development in this disease.
PARP inhibition is a standard of care approach in a variety of cancers including breast cancer, ovarian cancer and prostate cancer [11–13]. Across these diseases, there exist a multitude of treatment strategies, including monotherapy, combination or sequencing with platinum based chemotherapy or combination with other treatment modalities such as immunotherapy. An important consideration for PARP inhibitors is whether to treat ‘all comers’ or to select those with favourable predictive biomarkers. Where biomarkers are utilised, there are again multiple variations in practice including the use of germline and/or somatic alterations in BRCA1 and BRCA2, testing of broader DNA repair gene panels, the use of surrogate markers for a DNA repair deficient state, such as high genome-wide loss of heterozygosity (LOH), or clinical enrichment approaches such as through prior responsiveness to platinum-based chemotherapy.
PARP inhibition remains an experimental therapeutic approach for MUC. However, reports of phase I and II studies are emerging for this disease. We therefore undertook a systematic review of currently available clinical trial data for the use of PARP inhibition in MUC. We focussed on randomised comparisons for advanced disease, but also list data for non-randomised studies and other aspects of the treatment pathway for this disease.
MATERIALS AND METHODS
This study followed standard methods for conducting systematic reviews and meta-analyses and conforms to the 2020 Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [21].
Study objectives
Our objective was to determine the efficacy and safety of PARP inhibition, either as a monotherapy or in therapeutic combinations, compared to current standard therapy options, for adults with locally advanced or metastatic urothelial carcinoma.
Our chosen primary endpoint was progression free survival (PFS), defined as the time from randomized assignment to cancer progression or death from any cause. We also allowed for other definitions of progression free survival to be included, as necessary. Secondary endpoints included overall survival, response rates (which we anticipated might be more applicable to induction therapy rather than maintenance therapy), duration of response and adverse events. Data were collected for studies of both biomarker selected and unselected patient populations. For the purpose of this systematic review, biomarker positive status was treated as a binary distinction (positive or negative), regardless of the potential for variation in the methodology and composition of biomarkers between included studies.
Search strategy and selection criteria
We searched the following electronic bibliographic databases: MEDLINE (Ovid), EMBASE, ClinicalTrials.gov and Cochrane Central Register of Controlled Trials (CENTRAL). From this, we selected all randomised clinical trials published from March 2013 to March 2023. The search strategy included terms relating to, or describing, PARP inhibitors and MUC for patients of any age. Conference abstracts were included within our search from March 2020 onwards. Search strings included any PARP inhibitor used as a single agent or within combination treatment modalities and as either an induction or maintenance therapy. Standard care, as defined within each study, could include other systemic therapies such as chemotherapy and/or immunotherapy or supportive care alone. Language was restricted to English. Our search details are provided as supplementary data.
Titles and abstracts were screened by a minimum of two of the authors independently. Subsequent full text articles were screened by one person and checked by a minimum of one person. Any cases of disagreement were resolved by discussion among the reviewers and if necessary, referred to a third independent reviewer. Our search results included a study on which SJC was the principal investigator and lead author; he was excluded from the study selection decision making process and critical appraisal for that study [22].
In addition, we have listed non-randomised studies that have reported data for urothelial carcinoma.
Data extraction
Details of the study design, patient characteristics and eligibility criteria were extracted for each study, together with hazard ratios and associated confidence intervals for PFS, and data for other efficacy endpoints and adverse events extracted where available. Where multiple articles reported on a single study, these were assessed together to ensure that all available relevant data were considered. Data were extracted by one reviewer and checked by a second reviewer.
Risk of bias assessment
We used the Cochrane Risk of Bias Tool for randomised clinical trials (RCTs; version 2) to critically appraise included studies [23]. Risk of Bias 2 is an updated version of The Cochrane Collaboration’s tool for assessing risk of bias in RCTs, and has been widely used in both Cochrane and non-Cochrane systematic reviews. The tool includes five domains of bias (i.e. internal validity), focussing on different aspects of trial design, conduct, and reporting. A separate judgement (low risk/high risk/some concerns) is made for each study result included in this review, rather than for each study as a whole, to reflect risks of bias inherently specific to each result in each study. All studies were critically appraised by one reviewer and checked by a second. Any disagreement was resolved through discussion and if necessary, referred to a third reviewer to reach a decision. Risk of bias judgements were based on all available trial reports as well as study protocols/statistical analysis plans where these could be accessed. A summary of the bias judgments is presented in the Results section (Table 1). SJC was excluded from the risk of bias assessment for the study that he authored.
Table 1
Summary of risk of bias assessment
| Study | Randomization process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall judgement |
| Outcome: Progression-Free Survival (primary outcome for all studies) | ||||||
| ATLANTIS [22] | Low | Low | Low | Low | Low | Low |
| BAYOU [24] | Low | Low | Low | Low | Low | Low |
| Meet-URO12 [25] | Low | Low | Low | Low | Low | Low |
| Outcome: Overall Survival | ||||||
| ATLANTIS | Low | Low | Low | Low | Low | Low |
| BAYOU | Low | Low | Low | Low | Low | Low |
| Outcome: Response | ||||||
| ATLANTIS | Low | Low | Low | Low | Low | Low |
| BAYOU | Low | Low | Low | Low | Low | Low |
| Outcome: Adverse Events | ||||||
| ATLANTIS | Low | Low | Low | Low | Low | Low |
| BAYOU | Low | Low | Low | Low | Low | Low |
| MEET-URO 12 | Low | Low | Low | Low | Low | Low |
Strategy for data synthesis
According to the study protocol we aimed to quantitatively combine the studies in a meta-analysis for each outcome, subject to the studies being sufficiently homogeneous, i.e. having similar designs, settings and participant characteristics. We assessed the homogeneity of the studies by tabulating their study characteristics and populations and comparing these visually across the studies. If studies were deemed adequately homogeneous for meta-analysis we would synthesise their results for each outcome quantitatively following the analytical approach specified in the protocol. If substantial heterogeneity of trial populations or methods was detected we would compare the study results for each outcome in a descriptive data synthesis. As specified in the protocol, any risks of bias identified in the studies for each outcome would be considered when interpreting the data synthesis.
Study registration
We prospectively registered this systematic review on PROSPERO, an international prospective register of systematic reviews (CRD42023403145; https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42023403145).
RESULTS
Evidence synthesis
The search strategy identified 247 individual records (titles and abstracts) for screening, once duplicate records were removed. Of these, 41 potentially relevant records were selected for full text retrieval and further screening (Fig. 1). A total of 27 full text reports, representing 13 distinct studies (one a single patient case report), were subsequently excluded for not fully meeting the inclusion criteria. These included ongoing studies yet to report results, studies with non-randomised designs or studies in the neoadjuvant treatment setting. Three studies (with 14 reports) were identified which met the criteria for inclusion in this review, with a combined total of 252 patients [22, 24, 25].
Fig. 1
PRISMA flow chart for study identification and selection.
Risk of bias assessment
Table 1 provides a summary of the risk of bias judgements given to each study result included in this review. Generally, the risk of bias across the respective studies was considered low. A completed risk of bias template for each included study, containing the reviewers’ justification for their judgements, is available as supplementary material.
Data synthesis
The three RCTs included differed in several important respects. We therefore considered it inappropriate to combine the studies in a meta-analysis. Instead, we compare the studies’ results for each endpoint in a descriptive analysis below.
Patient and study characteristics
The characteristics of included studies are shown in Table 2. All three were prospective, randomised, phase 2 trials of adult patients with stage 4 urothelial carcinoma, with treatment given until disease progression, if tolerated. Two of these studies (ATLANTIS, Meet-URO12) tested PARP inhibitor monotherapy, with rucaparib and niraparib respectively, as a maintenance treatment, for patients who had gained stabilisation of their disease following first line, platinum-based, chemotherapy [22, 25]. Both had their recruitment halted, in 2020 and 2021 respectively, before reaching their planned sample size, following emergent data to support the use of maintenance avelumab immunotherapy in this treatment setting [5]. The global COVID-19 pandemic was also cited as a reason for recruitment discontinuation for ATLANTIS. Both study reports describe a process of prospective re-profiling of their statistical design for the subsequently reported study analysis based on the patients recruited to that point.
Table 2
Main characteristics of included studies
| ATLANTIS [22] | Meet-URO12 [25] | BAYOU [24] | |
| Study registration | ISRCTN25859465 | NCT03945084 | NCT03459846 |
| Study design | Prospective, randomised phase 2 | Prospective, randomised phase 2 | Prospective, randomised phase 2 |
| Clinical Setting | Maintenance therapy following platinum containing chemotherapy | Maintenance therapy following platinum containing chemotherapy | First line Platinum ineligible |
| Population | Stage IV urothelial carcinoma | Stage IV urothelial carcinoma | Stage IV urothelial carcinoma |
| Age restriction | ≥16 years | ≥18 years | ≥18 years |
| Intervention | Rucaparib | Niraparib | Durvalumab+olaparib |
| Comparator | Placebo | Best supportive care | Durvalumab+placebo |
| Biomarker selected | Yes (ITT population) | No (Reported as an exploratory subgroup analysis) | No (A planned secondary endpoint subgroup analysis) |
| Primary end point | Investigator assessed PFS | Investigator assessed PFS | Investigator assessed PFS |
| Number of patients in the ITT population | 40 | 58 | 154 |
| Randomisation ratio | 1 : 1 | 2 : 1 | 1 : 1 |
| Duration of follow up | 94.6 weeks (range, 11.4–148.9) | 8.5 months (interquartile range: 4.6–11.6) | 9.8 months (intervention arm; range, 0.0–29.0) 10.7 months (comparator arm; range, 1.0–29.0) |
| Stratification | Cisplatin vs non cisplatin based chemotherapy ECOG PS First line chemotherapy response Presence of visceral disease Presence of measurable disease Investigational site‘ | First line chemotherapy response ECOG PS | HRR mutant vs wild type Lymph node metastases vs other organ metastases ECOG performance status |
| Location | UK; multi centre | Italy; multi centre | Canada, South Korea, Russia, Spain, Taiwan, United States, Vietnam; multi centre |
ITT, intention to treat; PFS, progression free survival; ECOG PS, Eastern Cooperative Oncology Group performance status; HRR, Homologous recombination repair.
The third study (BAYOU) was undertaken in a treatment naive setting, for patients with platinum ineligible disease [24]. To our knowledge, this is the first prospective interventional clinical trial reported for this group of patients. Platinum ineligibility criteria were defined as the presence of at least one from a list of a creatinine clearance < 60 mL/min, grade≥2 audiometric hearing loss, grade≥2 peripheral neuropathy, New York Heart Association Class III heart failure or an Eastern Cooperative Oncology Group performance status (ECOG PS) of 2. BAYOU tested the PARP inhibitor olaparib in combination with durvalumab immunotherapy compared to durvalumab monotherapy. ATLANTIS and BAYOU were placebo controlled and double blinded whereas Meet-URO12 was an open label study.
Patient characteristics for each study are shown in Table 3 and reflect a typical MUC population in each case. Differences in characteristics seen between trials are consistent with the similar eligibility criteria for the ATLANTIS and Meet-URO12 post-chemotherapy maintenance studies, versus the first line, platinum ineligible, setting for BAYOU. In most regards, each were reasonably well balanced with respect to reported baseline characteristics and in the context of the modest respective sample sizes. Meet-URO12 had some imbalance between arms with respect to the proportion of those who had received prior cisplatin-based chemotherapy (62% intervention arm, 32% comparator).
Table 3
Baseline patient characteristics
| ATLANTIS [22] | Meet-URO12 [25] | BAYOU [24] | |||||
| Treatment arm | Rucaparib (N = 20) | Placebo (N = 20) | Niraparib (N = 39) | BSC (N = 19) | Durvalumab +Olaparib (N = 78) | Durvalumab +Placebo (N = 76) | |
| Median age, years | 69.5 | 71.5 | 71.0 | 68.4 | 79 | 72 | |
| Gender | Female | 5 (25%) | 2 (10%) | 0 | 0 | 22 (28%) | 21 (28%) |
| Male | 15 (75%) | 18 (90%) | 29 (74%) | 14(74%) | 56 (72%) | 55 (72%) | |
| ECOG PS | 0 | 11 (55%) | 10 (50%) | 26 (67%) | 12 (63%) | 12 (15%) | 14 (18%) |
| 1 | 9 (45%) | 10 (50%) | 13 (33%) | 7 (37%) | 30 (38%) | 34 (45%) | |
| 2 | 0 | 0 | – | – | 34 (44%) | 28 (37%) | |
| Missing | – | – | – | – | 2 (3%) | 0 | |
| Smoking | Current | 3 (15%) | 1 (5%) | NR | NR | 33 (42%) | 32 (42%) |
| Non | – | – | NR | NR | 42 (54%) | 43 (57%) | |
| Prior | 10 (50%) | 12 (60%) | NR | NR | – | – | |
| Never | 7 (35%) | 7 (35%) | NR | NR | – | – | |
| Missing | 0 | 0 | NR | NR | 3(4%) | 1(1%) | |
| Histology | Pure TCC | 19 (95%) | 20 (100%) | NR | NR | 72 (92%) | 68 (89%) |
| Mixed | 1 (5%) | 0 | NR | NR | 6 (8%) | 8 (11%) | |
| Disease extent | Node only | 12 (60%) | 10 (50%) | 7 (18%) | 6 (32%) | 26 (33%) | 28 (37%) |
| Visceral | 8 (40%) | 10 (50%) | 19 (49%) | 11 (58%) | 52 (67%) | 48 (63%) | |
| Prior platinum | Cisplatin | 13 (65%) | 12 (60%) | 24 (62%) | 6 (32%) | NA | NA |
| Carboplatin | 7 (35%) | 8 (40%) | 15 (38%) | 13 (68%) | NA | NA | |
| Chemotherapy | Stable disease | 2 (10%) | 2 (10%) | 17 (44%) | 9 (47%) | NA | NA |
| response | Partial | 12 (60%) | 12 (60%) | 19 (49%) | 10 (53%) | NA | NA |
| Complete | 6 (30%) | 6 (30%) | 3 (8%) | 0 | NA | NA | |
BSC, best supportive care; ECOG PS, Eastern Cooperative Oncology Group performance status; NR, not reported; TCC, transitional cell carcinoma; NA, not applicable.
Biomarker characteristics
Each study assessed biomarker status for a DNA repair defective phenotype, based on next generation sequencing analysis of archival, formalin fixed, paraffin embedded, tumour samples. The ATLANTIS rucaparib randomisation recruited solely from this biomarker-selected group of patients (from within the wider ATLANTIS platform study, which incorporated discrete parallel, biomarker selected, phase 2 randomisations, as previously described) [26]. Meet-URO12 and BAYOU recruited unselected patient populations for their stated primary endpoint analysis, but also undertook biomarker assessment of patient tumour samples. BAYOU incorporated this as a prospectively planned secondary endpoint analysis for PFS, and other stated secondary endpoints, within its biomarker positive subgroup. Whereas this subgroup analysis was described as exploratory for the Meet-URO12 study.
The characteristics of the biomarkers used were prospectively defined in each study. They were substantially overlapping between studies, but minor differences existed, in terms of the gene lists utilised within each trial. ATLANTIS also designated patients as biomarker positive where their cancers exhibited high percentage, genome wide, LOH as a surrogate marker for DNA repair deficiency. The protocol also allowed for inclusion where a prior known germ line alteration in BRCA1 or BRCA2 existed (although no patients were actually included on this basis). Details of the biomarker composition within each trial are presented in Supplementary Table 1.
Primary endpoint: PFS
All three studies designated progression free survival as their primary endpoint.
Of note, ATLANTIS presented the data for its primary endpoint with 80% confidence intervals applied to the point estimates for median PFS, instead of confidence intervals at the conventional level of 95%. This prospectively determined approach was justified by the authors based on the relatively rare clinical setting (biomarker positive MUC patients) and within a ‘signal seeking’ study intended to inform a decision on progression to phase 3 investigation (rather than to directly change clinical practice). We have presented the data related to this outcome with the originally reported 80% confidence interval (in part, because the data were not available to recalculate to a conventional 95% interval) [22].
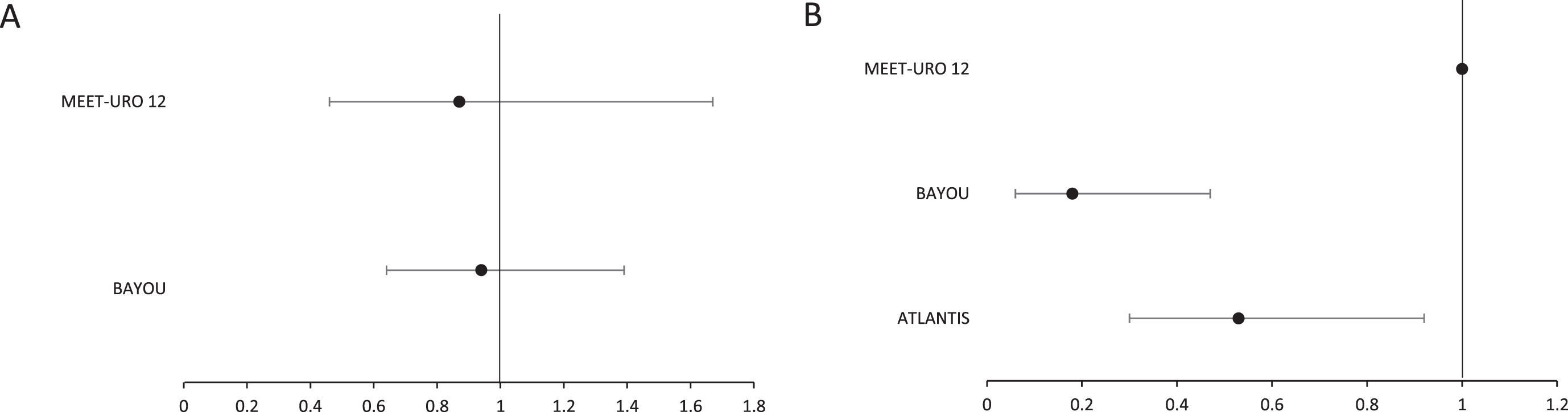
For unselected patient cohorts, no difference in PFS was demonstrated with the use of PARP inhibition in either the Meet-URO12 or BAYOU studies (Table 4, Fig. 2A). However, improvement in PFS was seen within biomarker selected patient cohorts, through the addition of PARP inhibition to standard care, in both ATLANTIS (primary endpoint) and BAYOU (planned secondary endpoint; Table 5, Fig. 2B). Analysis of PFS in biomarker selected patients was not pre-specified as an endpoint in the Meet-URO12 study, and only 47 (81%) of the intention to treat (ITT) population had samples available for biomarker analysis. However, point estimates for median PFS were reported, as an exploratory analysis, for subsets with alteration in pre-specified DNA repair genes for this study. The authors undertook this analysis for biomarker definitions with either ‘known’ pathogenic gene alterations (although this included only 6 randomised patients) and also a wider subset with ‘any’ gene alteration (including variants of unknown significance). No difference existed between treatment arms for median PFS for either of these biomarker definitions.
Table 4
PFS outcomes in unselected patient populations
| Meet-URO12 [25] | BAYOU [24] | |||
| Treatment arm | Niraparib | Best supportive care | Durvalumab + olaparib | Durvalumab + placebo |
| n | 39 | 19 | 78 | 76 |
| Median PFS | 2.1 months (95% CI, 0.9–3.2) | 2.4 months (95% CI, 1.6–3.2) | 4.2 months (95% CI, 3.6–5.6) | 3.5 months (95% CI, 1.9–5.1) |
| Hazard Ratio | 0.87 (95% CI, 0.46–1.67, p = 0.7; adjusted for stratification factors) | 0.94 (95% CI, 0.64–1.39, p = 0.789; stratified by HRR status) | ||
PFS, progression free survival; CI, confidence interval; HRR, Homologous recombination repair.
Table 5
PFS outcomes in biomarker selected patient populations
| Outcomes | ATLANTIS [22] | Meet-URO12 [25] | BAYOU [24] | |||
| Rucaparib | Placebo | Niraparib | Best supportive care | Durvalumab +olaparib | Durvalumab +placebo | |
| n | 20 | 20 | 11 | 10 | 17 | 14 |
| Median PFS | 35.3 weeks (80% CI, 11.7–35.6) | 15.1 weeks (80% CI, 11.9–22.6) | 2.0 months (CI, NR) | 2.0 months (CI, NR) | 5.6 months (95% CI, 1.9–8.1) | 1.8 months (95% CI, 1.7–2.2) |
| Hazard Ratio | 0.53 (80% CI, 0.30 to 0.92; adjusted for minimisation factors) | NR | 0.18 (95% CI, 0.06 to 0.47; stratified by HRR status) | |||
PFS, progression free survival; CI, confidence interval; NR, not reported; HRR, Homologous recombination repair.
Fig. 2
Forest plot for hazard ratios and confidence intervals for selected studies for (A) unselected patients and (B) patients selected for the presence of a DNA repair gene defective phenotype. Note: the indicated confidence intervals are as reported for the respective trials. For Meet-URO12 and BAYOU this was set at 95%. For ATLANTIS this was set at 80%.
Secondary endpoints
Secondary endpoints relating to efficacy, as they were defined within the respective study protocols, are listed in Supplementary Table 2. No difference in overall survival was demonstrated between treatment arms in any of the three identified studies. Objective response rates were higher with the addition of olaparib to durvalumab in the first line BAYOU study (28.2% versus 18.4%) but not within the ATLANTIS study (response rates are not yet reported for Meet-URO12). Of note, 90% of patients in the ATLANTIS ITT population had already achieved an objective radiologic response to their prior first line chemotherapy indicating that, perhaps, further tumour response is not an informative secondary endpoint for a post chemotherapy maintenance treatment study. Median duration of response was actually shorter in the durvalumab plus olaparib treatment arm in BAYOU, compared to durvalumab plus placebo, with 32% versus 64% of patients in ongoing response at 12 months respectively. Other secondary endpoints within each study are either similar between treatment arms or not yet reported (Supplementary Tables 2 and 3).
Each study reported adverse events according to the Common Terminology Criteria for Adverse Events version 4.03 within a defined safety population of patients that had received at least one dose of study medication (for study arms containing either an active treatment or a placebo). Rates of adverse events are shown in Supplementary Table 4. Broadly, these indicate anaemia, fatigue, nausea and anorexia as the most common treatment emergent adverse events that differed between treatment arms of the respective studies. Furthermore, these were predominantly of low grade and remained consistent with the established adverse event profiles for PARP inhibition in other clinical settings. No new safety signals emerged for the use of PARP inhibition through these studies.
Identified studies not meeting criteria for systematic review inclusion
Other PARP inhibitor trials have been undertaken in MUC, or are in progress, but did not meet our prospectively defined criteria for this systematic review, primarily due to lack of randomised treatment assignment, and in some cases also their treatment setting. Those that have reported data, comprising 172 patients in total, are listed in Table 6.
Table 6
Non-randomised studies of PARP inhibitors that include urothelial carcinoma patients
| Study | PARP inhibitor | Number of patients* | Setting | Design | Comments |
| ATLAS [28] | Rucaparib | 97 | Second or third line, MUC | Single arm, phase 2 | No significant clinical activity in unselected patients |
| BISCAY [29] | Olaparib (+durvalumab) | 29 | Second or subsequent line, MUC | Non randomised, biomarker selected, phase 1b | Outcomes did not appear qualitatively different compared with durvalumab monotherapy (not randomised) |
| Case report [30] | Olaparib | 1 | Post-immunotherapy and chemotherapy, MUC | Single patient case report | Clinical and radiologic response in a patient with a BRCA homozygous deletion |
| ARIANES [31] | Rucaparib (+atezolizumab) | 16 | ‘Platinum sensitive’ MUC | Multi-cancer ‘basket study’, phase 2 | 2 of 16 patients achieved an objective response |
| NEODURVARIB [32] | Olaparib (+durvalumab) | 29 | Neoadjuvant, MIBC | Single arm, phase 2 | Pathological complete response rate 50% (secondary endpoint) |
*Representing the ITT population for MUC patients allocated to receive a PARP inhibitor (some studies had non-PARP inhibitor or non-MUC components). MUC, metastatic urothelial carcinoma; MIBC, muscle invasive bladder cancer.
DISCUSSION
Outcomes for MUC remain inadequate and improvements to therapeutic options are required. The current use of chemotherapy, immunotherapy, and nectin-4 directed therapy with enfortumab vedotin, is based on data in unselected patient populations. However, it is likely that future therapeutic advance will require patient selection strategies through the development of predictive biomarkers for treatment benefit. Recent phase 3 data, demonstrating superior overall survival with the FGFR inhibitor erdafitinib compared to chemotherapy, in a pre-treated MUC subset with alteration in either FGFR2 or FGFR3, provides an example of this principle now entering routine practice. This example is based on a biomarker that is said to be present in approximately 20% of cases in the advanced disease setting [27].
MUC is a platinum chemotherapy responsive disease that also harbours DNA repair gene alterations in a subset of patients. As such, clinical evaluation of PARP inhibition has biological plausibility and it is also reasonable to hypothesise that phenotypic overlap between platinum sensitivity and PARP inhibitor sensitivity would exist. We investigated the current state of evidence for efficacy and safety for PARP inhibition in MUC through systematic review. The randomised data that exist are limited to phase 2 clinical trials and the number of patients incorporated into these is modest in total. As such, we view the current data as signal seeking in nature and routine clinical use of PARP inhibition would not be supported currently.
In unselected patient groups, there is no current evidence for clinical effectiveness for PARP inhibitors, at least in the treatment settings that the studies identified here have evaluated. As such, further development of PARP inhibition without biomarker selection is not now supported by the randomised data available. However, two trials, ATLANTIS and BAYOU have found evidence to support potential clinical effectiveness in biomarker selected patients. A third trial (Meet-URO12) did not show efficacy within patients with biomarker positive cancers. However, the number of patients with a pathogenic gene alteration in this study was very low (only 6 randomised patients). Taken together, the available data suggest that further development of PARP inhibition in this disease should be through clinical trials that incorporate a prospectively planned approach to biomarker selection.
The optimal predictive biomarker strategy for selecting patients with MUC to receive a PARP inhibitor remains to be determined. The studies included here made pragmatic prospective decisions for the composite biomarker rules to be included within each study. This resulted in gene lists that, whilst similar, were not identical. This parallels circumstances in other cancers where optimal biomarker composition remains to be fully defined, such as in prostate cancer [11]. One consequence, that seems likely to be replicated in MUC, is that the individual relevance for each incorporated gene, at least beyond BRCA1 and BRCA2, may be challenging, if not impossible to determine. It is likely to be impossible to adequately power a prospective trial to test individual genes within a composite biomarker. Realistically, this probably requires some pragmatism in gene list choices for future studies. In addition, ATLANTIS also incorporated high genome wide LOH as a surrogate for a DNA repair deficient phenotype. This was based on prior data from ovarian cancer, however exploratory analysis from ATLANTIS suggested that presence of somatic gene alterations, rather than inclusion solely on the basis of LOH status, may have driven the clinical benefit seen in this trial [22].
The biomarker positive rate across these three trials was in the range of 13 to 30%. This reduced to an upper limit of 20% if only somatic gene alterations are considered [22, 24, 25]. This has implications for the design and feasibility of future studies of PARP inhibitors in MUC where biomarker selection is to be implemented. It is likely that phase 3 assessment of PARP inhibition in a MUC biomarker defined subset might require a population of thousands to be pre-screened for biomarker status in a similar manner to the recent experience with FGFR inhibition [27]. Getting this aspect of design correct will likely be important for future success or failure. Unlike, for example, in prostate cancer, we probably do not have the luxury of undertaking multiple definitive studies of PARP inhibition and so the design of a phase 3 study needs to be carefully considered in terms of its biomarker strategy.
Another consideration for biomarker development and operating characteristics is the recently presented data for enfortumab vedotin in combination with pembrolizumab that has redefined the optimal initial treatment for MUC patients [10]. This may have implications for both biomarker selection and practical questions relating to positioning of PARP inhibition within the disease pathway. The logical approach of linkage of PARP inhibition to patients with platinum sensitive disease, that underpinned the ATLANTIS and Meet-URO12 studies, might potentially be impacted if platinum based chemotherapy becomes a second line (or later) treatment option for some patients. An alternative, of combining PARP inhibition with enfortumab and pembrolizumab, has not been evaluated in any form to date.
There are some limitations of this systematic review, and its findings, which should be noted. Perhaps most significant is the limitation in statistical power, across each trial and in combination, based on the sample sizes included. This is of particular relevance in respect to ATLANTIS, which opted for a 90% power and 20% one-sided level of statistical significance for its prospective sample size calculation. As a result, it reported 80% confidence intervals for its primary endpoint which reduces the level of confidence in its reported outcome. The biomarker subset analyses for the BAYOU and Meet-URO12 studies are also limited by small sample size and were not primary endpoints of the respective studies. Other limitations include our inability to proceed to meta-analysis because of heterogeneity in trial design and our search restriction to English language articles.
CONCLUSION
PARP inhibitors should not yet enter routine clinical practice for MUC based on currently available data. They lack efficacy in unselected patients. However, despite limitations in statistical power, phase 2 studies do provide support for further clinical investigation of PARP inhibition within biomarker-selected patient subsets. The optimal components of a predictive biomarker for patient selection is not yet determined.
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
The authors report no funding specific to this project.
AUTHOR CONTRIBUTIONS
All authors were involved in the conception of the project, collection of data, interpretation of data and the writing of the article. All authors had access to the data.
CONFLICT OF INTEREST
SJC has received fees for either speaking or advisory work from AstraZeneca, Roche, Janssen, MSD, Astellas, Amphista Therapeutics, Pfizer, Merck and Bayer; provided expert testimony for MSD, Merck and Pfizer; travel and meeting assistance from Janssen, Bayer, BMS, and Merck; and research funding support from AstraZeneca, Roche, Astex Pharmaceuticals and Clovis Oncology.
SJC was the lead investigator for the rucaparib randomisation within the ATLANTIS clinical trial which was funded by Cancer Research UK (A18386). The rucaparib randomised comparison received funding, and provision of rucaparib and placebo, from Clovis Oncology. Clovis Oncology also provided advice in defining the components of the DRD biomarker and funding to Foundation Medicine to undertake biomarker analysis.
TK, LW, GF and JS declare that they have no known conflicts of interest.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/BLC-230071.
REFERENCES
[1] | Jubber I , Ong S , Bukavina L , Black PC , Comperat E , Kamat AM , et al. Epidemiology of Bladder Cancer in A Systematic Review of Risk Factors, Eur Urol (2023) ;84: :176–90. |
[2] | Bellmunt J , de Wit R , Vaughn DJ , Fradet Y , Lee JL , Fong L , et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma, N Engl J Med (2017) ;376: :1015–26. |
[3] | Giles M , Crabb SJ Systemic Treatment-Decision Algorithms in Muscle-Invasive Bladder Cancer: Clinical Complexities and Navigating for Improved Outcomes, Res Rep Urol (2023) ;15: :321–31. |
[4] | De Santis M , Bellmunt J , Mead G , Kerst JM , Leahy M , Maroto P , et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 6, J Clin Oncol (2012) ;30: :191–9. |
[5] | Powles T , Park SH , Voog E , Caserta C , Valderrama BP , Gurney H , et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma, N Engl J Med (2020) ;383: :1218–30. |
[6] | von der Maase H , Hansen SW , Roberts JT , Dogliotti L , Oliver T , Moore MJ , et al. Gemcitabine and cisplatin versus methotrexate,vinblastine, doxorubicin, and cisplatin in advanced or metastaticbladder cancer: results of a large,randomized, multinational, multicenter, phase III study, J ClinOncol (2000) ;18: :3068–77. |
[7] | Galsky MD , Hahn NM , Rosenberg J , Sonpavde G , Hutson T , Oh WK , et al. Treatment of patients with metastatic urothelial cancer “unfit” for Cisplatin-based chemotherapy, J Clin Oncol (2011) ;29: :2432–8. |
[8] | Gupta S , Bellmunt J , Plimack ER , Sonpavde GP , Grivas P , Apolo AB , et al. Defining “platinum-ineligible” patients with metastatic urothelial cancer (mUC), Journal of Clinical Oncology (2022) ;40: :4577. |
[9] | Powles T , Rosenberg JE , Sonpavde GP , Loriot Y , Duran I , Lee JL , et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma, N Engl J Med (2021) ;384: :1125–35. |
[10] | Powles TB , Perez Valderrama B , Gupta S , Bedke J , Kikuchi E , Hoffman-Censits J , et al. LBA6 EV-302/KEYNOTE-A Open-label, randomized phase III study of enfortumab vedotin in combination with pembrolizumab (EV+P) vs chemotherapy (Chemo) in previously untreated locally advanced metastatic urothelial carcinoma (la/mUC), Annals of Oncology (2023) ;34: :S1340. |
[11] | Inderjeeth AJ , Topp M , Sanij E , Castro E , Sandhu S Clinical Application of Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Prostate Cancer. Cancers (Basel). 2022;14. |
[12] | Nambiar DK , Mishra D , Singh RP Targeting DNA repair for cancer treatment: Lessons from PARP inhibitor trials, Oncol Res (2023) ;31: :405–21. |
[13] | Wang SSY , Jie YE , Cheng SW , Ling GL , Ming HVY PARP Inhibitors in Breast and Ovarian Cancer. Cancers (Basel). 2023;15. |
[14] | Jian W , Xu HG , Chen J , Xu ZX , Levitt JM , Stanley JA , et al. Activity of CEP-a poly (ADP-ribose) polymerase inhibitor, in urothelial carcinoma correlates inversely with homologous recombination repair response to DNA damage, Anticancer Drugs (2014) ;25: :878–86. |
[15] | Miron B , Hoffman-Censits JH , Anari F , O’Neill J , Geynisman DM , Zibelman MR , et al. Defects in DNA Repair Genes Confer Improved Long-term Survival after Cisplatin-based Neoadjuvant Chemotherapy for Muscle-invasive Bladder Cancer, Eur Urol Oncol (2020) ;3: :544–7. |
[16] | Mullane SA , Werner L , Guancial EA , Lis RT , Stack EC , Loda M , et al. Expression Levels of DNA Damage Repair Proteins Are Associated With Overall Survival in Platinum-Treated Advanced Urothelial Carcinoma, Clin Genitourin Cancer (2016) ;14: :352–9. |
[17] | Nickerson ML , Dancik GM , Im KM , Edwards MG , Turan S , Brown J , et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer, Clin Cancer Res (2014) ;20: :4935–48. |
[18] | Plimack ER , Dunbrack RL , Brennan TA , Andrake MD , Zhou Y , Serebriiskii IG , et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer, Eur Urol (2015) ;68: :959–67. |
[19] | Yap KL , Kiyotani K , Tamura K , Antic T , Jang M , Montoya M , et al. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival, Clin Cancer Res (2014) ;20: :6605–17. |
[20] | Yin M , Grivas P , Emamekhoo H , Mendiratta P , Ali S , Hsu J , et al. ATM/RB1 mutations predict shorter overallsurvival in urothelial cancer, Oncotarget (2018) ;9: :16891–8. |
[21] | Page MJ , McKenzie JE , Bossuyt PM , Boutron I , Hoffmann TC , Mulrow CD , et al. The PRISMA statement: an updated guideline for reporting systematic reviews, BMJ (2021) ;372: :n71. |
[22] | Crabb SJ , Hussain S , Soulis E , Hinsley S , Dempsey L , Trevethan A , et al. A Randomized, Double-Blind, Biomarker-Selected, Phase II Clinical Trial of Maintenance Poly ADP-Ribose Polymerase Inhibition With Rucaparib Following Chemotherapy for Metastatic Urothelial Carcinoma, J Clin Oncol (2023) ;41: :54–64. |
[23] | Sterne JAC , Savovic J , Page MJ , Elbers RG , Blencowe NS , Boutron I , et al. RoB a revised tool for assessing risk of bias in randomised trials, BMJ.l (2019) ;366: :4898. |
[24] | Rosenberg JE , Park SH , Kozlov V , Dao TV , Castellano D , Li JR , et al. Durvalumab Plus Olaparib in Previously Untreated, Platinum-Ineligible Patients With Metastatic Urothelial Carcinoma: A Multicenter, Randomized, Phase II Trial (BAYOU), J Clin Oncol (2023) ;41: :43–53. |
[25] | Vignani F , Tambaro R , De Giorgi U , Giannatempo P , Bimbatti D , Carella C , et al. Addition of Niraparib to Best Supportive Care as Maintenance Treatment in Patients with Advanced Urothelial Carcinoma Whose Disease Did Not Progress After First-line Platinum-based Chemotherapy: The Meet-URO12 Randomized Phase 2 Trial, Eur Urol (2023) ;83: :82–9. |
[26] | Fulton B , Jones R , Powles T , Crabb S , Paul J , Birtle A , et al. ATLANTIS: a randomised multi-arm phase II biomarker-directed umbrella screening trial of maintenance targeted therapy after chemotherapy in patients with advanced or metastatic urothelial cancer, Trials (2020) ;21: :344. |
[27] | Loriot Y , Matsubara N , Park SH , Huddart RA , Burgess EF , Houede N , et al. Erdafitinib or Chemotherapy in Advanced or Metastatic Urothelial Carcinoma, N Engl J Med (2023) ;389: :1961–1971. |
[28] | Grivas P , Loriot Y , Morales-Barrera R , Teo MY , Zakharia Y , Feyerabend S , et al. Efficacy and safety of rucaparib in previously treated, locally advanced or metastatic urothelial carcinoma from a phase 2, open-label trial (ATLAS), BMC Cancer (2021) ;21: :593. |
[29] | Powles T , Carroll D , Chowdhury S , Gravis G , Joly F , Carles J , et al. An adaptive, biomarker-directed platform study of durvalumab in combination with targeted therapies in advanced urothelial cancer, Nat Med (2021) ;27: :793–801. |
[30] | Necchi A , Raggi D , Giannatempo P , Alessi A , Serafini G , Colecchia M , et al. Exceptional response to olaparib in BRCA2-altered urothelial carcinoma after PD-L1 inhibitor and chemotherapy failure, Eur J Cancer (2018) ;96: :128–30. |
[31] | Martin Romano P , Roubaud G , Lavaud P , Cabart M , Pages A , Vasseur D , et al. P Phase II study of rucaparib and atezolizumab (ARIANES): Results in patients (pts) with platinum-sensitive metastatic urothelial cancer (mUC) and metastatic castration-resistant prostate cancer (mCRPC), Annals of Oncology (2022) ;33: :S1334. |
[32] | Rodriguez-Moreno JF , de Velasco G , Alvarez-Fernandez C , Collado R , Fernandez-Rodriguez R , Estevez SV , et al. 761P Impact of the combination of durvalumab (MEDIplus olaparib (AZDadministered prior to surgery in the molecular profile of resectable urothelial bladder cancer, NEODURVARIB trial. Annals of Oncology (2020) ;31: :S589. |


