
Loss of Cxcr2 in Myeloid Cells Promotes Tumour Progression and T Cell Infiltration in Invasive Bladder Cancer
Abstract
BACKGROUND:
CXCR2 is a chemokine receptor expressed in myeloid cells, including neutrophils and macrophages. Pharmacological inhibition of CXCR2 has been shown to sensitize tumours to immune checkpoint inhibitor immunotherapies in some cancer types.
OBJECTIVE:
To investigate the effects of CXCR2 loss in regulation of tumour-infiltrating myeloid cells and their relationship to lymphocytes during bladder tumorigenesis.
METHODS:
Urothelial pathogenesis and immune contexture was investigated in an OH-BBN model of invasive bladder cancer with Cxcr2 deleted in myeloid cells (LysMCre Cxcr2flox/flox). CXCR2 gene alterations and expression in human muscle invasive bladder cancer were analysed in The Cancer Genome Atlas.
RESULTS:
Urothelial tumour pathogenesis was significantly increased upon Cxcr2 deletion compared to wildtype mice. This was associated with a suppression of myeloid cell infiltration in Cxcr2-deleted bladders shortly after the carcinogen induction. Interestingly, following a transient increase of macrophages at the outset of tumour formation, an increase in T cell infiltration was observed in Cxcr2-deleted tumours. The increased tumour burden in the Cxcr2-deleted bladder was largely independent of T cells and the status of immune suppression. The Cxcr2-deleted mouse model reflected the low CXCR2 mRNA range in human bladder cancer, which showed poor overall survival.
CONCLUSIONS:
In contrast to previous reports of increased CXCR2 signalling associated with disease progression and poor prognosis, CXCR2 was protective against bladder cancer during tumour initiation. This is likely due to a suppression of acute inflammation. The strategy for sensitizing checkpoint immunotherapy by CXCR2 inhibition in bladder cancer may benefit from an examination of immune suppressive status.
INTRODUCTION
According to GLOBOCAN data, 550,000 newly diagnosed cases and 200,000 cases of mortality were reported for bladder cancer in 2018, which equates to 3.0% and 2.1% of all cancers, respectively [1, 2]. Bladder cancer is more frequent in males than females and 90% of newly diagnosed cases are in people over 55-years old. More than 80% of the cases are attributed to preventable causes, such as smoking and occupational exposure to carcinogens [2]. Bladder cancer is classified as non-muscle invasive (NMIBC), muscle invasive (MIBC) and metastatic bladder cancer [3]. NMIBC recurs frequently, and the risk of disease progression cannot be undermined [4]. MIBC is treated by neoadjuvant chemotherapy and radical cystectomy, however few management options are available for the metastatic disease, which has dismal prognosis [5]. The burden of bladder cancer is high due to the needs of surveillance and the treatment cost of NMIBC [1]. With the recent uptake of immune checkpoint inhibitor (ICI) immunotherapy in advanced disease, the overall management cost of bladder cancer may further increase by the population growth and ageing [1].
The ICI immunotherapy has been successfully used in the treatment of advanced BC [6–8]. The ICIs restore cytotoxic and proliferative T cell functions by blocking T cell receptor-mediated immune suppressive signalling [6, 7]. The main challenges are relatively low response rates (13–48%), immune-related adverse effects and pseudo-progression [6]. The response is regulated by multiple mechanisms, including the levels of tumour antigens, PD-L1 expression in tumour and immune cells, the level and location of tumour infiltrating lymphocytes (TILs), and the factors that influence TILs, such as cytokine and chemokine production [6–8]. Approaches to increase tumours’ sensitivity to ICI immunotherapy is being intensively studied, including modulation of myeloid cells, such as neutrophils and tumour-associated macrophages (TAMs) [3].
Neutrophils are myeloid cells that account for 50–70% of the total circulating leukocytes, and normally act as the first responder to damaged and inflamed tissues, but are also involved in all stages of cancer progression, including metastasis [9–13]. Neutrophils could play both anti- and pro-tumour roles. A high level of both CD66b neutrophils and CD3 T cells was associated with better overall survival and progression-free survival [14]. Conversely, a high level of tumour infiltrating neutrophils was associated with poor prognosis in both NMIBC and MIBC [15, 16]. A high neutrophil-to-lymphocyte ratio (NLR), a parameter based on the blood level of neutrophils and lymphocytes, is a poor prognosis in many cancers, including bladder cancer [17–20]. Similarly, macrophages could bear an anti- and pro-tumour role, but in the tumour, they are mostly considered as pro-tumour “M2” macrophages and associated with poor prognosis [21–23]. Macrophages play a role in tumour initiation and progression by creating a paracrine and autocrine loop with tumour cells, causing inflammation, angiogenesis, and immunosuppression [22, 23]. In the bladder, macrophages are mostly resident in the submucosa and shown to have detrimental effects on adaptive immunity in both non-malignant and malignant diseases [21, 24].
CXCR2 is a G-protein coupled receptor that binds to chemokines known as glutamic acid-leucine-arginine (ELR)-positive chemokines (CXCL1-3, CXCL5-8), and transmigrates cells to the sites of inflammation [25, 26]. CXCR2 is reported to be expressed on the cell surface of myeloid cells, including neutrophils, monocytes, macrophage, and mast cells [25, 26]. In cancer, its role was mostly reported as pro-tumorigenic [25, 26]. Importantly, CXCR2 inhibition was shown to counteract immunosuppressive myeloid cell populations in the tumour, promoting tumour infiltration of T cells and sensitising tumours to ICI immunotherapy [27, 28]. In the bladder, increased expression of CXCR2 ligands, CXCL5 and CXCL2, was associated with higher tumour grade and worse prognosis [29, 30]. Expressed in bladder cancer cell lines, CXCL5 upregulated metalloproteinase (MMP) 2/9 and induced cell migration and invasion in a CXCR2-dependent manner [29]. CXCR2 and its ligands were also shown to be expressed in and myeloid derived suppressor cells (MDSCs) and activated their migration [30, 31].
The environmental signals coming from the host and the tumour immune microenvironment can regulate the heterogeneous nature of the myeloid cell population. Therefore, the role and the effects of tissue-infiltrating myeloid cells require an investigation in an organ and context-specific manner. Here we have characterised the role of Cxcr2 during bladder pathogenesis using genetic ablation of Cxcr2 in myeloid cells in a model of invasive bladder cancer induced by the bladder-specific carcinogen, N-butyl-N-(4- hydroxybutyl) nitrosamine (OH-BBN). CXCR2 gene alteration and transcript levels in human bladder cancer were further analysed in The Cancer Genome Atlas (TCGA) Cell 2017 dataset [32].
MATERIALS AND METHODS
Mice and carcinogen treatment
Animal experiments were carried out at the CRUK Beatson Institute, Glasgow, in accordance with the Home Office Animal (Scientific Procedures) Act 1986 in the UK (Project Licence 70/9028), adhered to ARRIVE guidelines, and approved by the University of Glasgow Animal Welfare and Ethical Review Board. Genotyping was performed by Transnetyx (Cordoba, TN, USA). The number of mice used, including gender, is presented (Table 1) and specified in each figure. LysMCre Cxcr2flox/flox (Cxcr2 flox) mice were generated by crossing LysMCre and Cxcr2flox/flox mice [28] in C57Bl/6J background. The C57Bl/6J mice (wildtype) purchased from Charles River (Tranent, UK) were used as controls. Mice aged 8-10 weeks were administered with 0.05% v/v N-butyl-N-(4-hydroxybutyl) nitrosamine (OH-BBN, TCI, Oxford, UK) in drinking water for 10 weeks (Fig. 1A). The bladders of wildtype mice aged beyond 20 weeks were monitored using the Vevo3100 ultrasound system with 25–55 MHz transducer (Fujifilm VisualSonics, Toronto, Canada) and culled when they showed clinical signs of bladder tumours. Blood was sampled in EDTA-containing tubes and white blood cell populations were analysed using ProCyte Dx Hematology Analyzer (IDEXX, Westbrook, Maine, USA).
Table 1
Summary of the mouse cohorts
| Genotype | Time from the start of OH-BBN treatment (weeks) | Cohort size (n) | Haematuria (n) | Tumour incidents observed by histopathology (n) | ||||
| Total | Male | Female | Male | Female | Male | Female | ||
| LysMCre Cxcr2flox/flox (Cxcr2 flox) | 2 | 41 | 29 | 12 | ||||
| 16 | 5 | 5 | 0 | |||||
| 20 | 43 | 27 | 16 | 8/27 (29.6%) | 0/16 (0%) | 15/27 (55.6%) | 6/16 (37.5%) | |
| Wildtype | 2 | 37 | 28 | 9 | ||||
| 16 | 5 | 5 | 0 | |||||
| 20 | 72 | 45 | 27 | 0/45 (0%) | 0/27 (0%) | 16/45 (35.6%) | 1/27 (3.7%) | |
| > 20w | 10 | 10 | 2/10 (20%) | 10/10 (100%) | ||||
Mice were monitored for general health and clinical signs of bladder cancer, including haematuria, difficulties in micturition and weight loss. Haematuria was notable in Cxcr2 flox males at 20 weeks. None of the mice presented haematuria or tumours at 2 and 16 weeks. Wildtype mice aged beyond 20 weeks (21 –43 weeks) were monitored by ultrasound imaging for bladder tumours and culled when mice showed clinical signs of bladder tumours. Data of wildtype mice at 2 and 20 weeks from our previous study [35] were included.
Fig. 1
Bladder tumour phenotype of LysMCre Cxcr2flox/flox (Cxcr2 flox) mice. (A) Mice were given tobacco carcinogen 0.05% (v/v) OH-BBN in drinking water for 10 weeks, and bladder tissues were examined at 2, 16 and 20 weeks from the start of the carcinogen treatment. (B) An example of a bladder tumour developed at 20 weeks from the start of the carcinogen treatment in a Cxcr2 flox animal, with an invasion to the muscle (arrow) and keratinization (Kr). Representative images of wildtype (C, E) and Cxcr2 flox (D, F) bladders at 16 weeks (C, D), and 2 weeks (E, F). Enhanced tumour pathogenesis of the urothelium, such as carcinoma in situ-like lesions (arrowheads) was observed in Cxcr2 flox bladders at 16 weeks (D). At 2 weeks, the differences between wildtype and Cxcr2 flox bladders were not evident. IHC staining showed infiltrating Cxcr2+ myeloid cells in tumours at 20 weeks (G), the urothelium (J) and around the blood vessels in the bladder stroma (L) at 2 weeks in wildtype mice (arrows in G, J, L). In contrast, the staining was absent in Cxcr2 flox tumours (H, I) and in the bladder at 2 weeks’ time point (K, M). IHC staining was performed in at least n = 3 samples in each genotype. Annotations in B-M are; CIS, carcinoma in situ; Kr, keratinization; M, muscle, St, stroma; T, tumour; U, urothelium; V, blood vessel. The scale bar represents 500μm in B, 250μm in C-F, 30μm in G-M. For evaluation of the bladder and tumour phenotype, the scoring criteria from our previous study [35] were refined to optimally capture the phenotype of OH-BBN treated mice under our protocol. The “bladder phenotype” defines the overall state of the urothelial tumorigenesis and progression. The “invasiveness” describes the state of the basement membrane and invasion of tumour cells, while the “squamous transformation” describes the squamous appearance of cells and keratinization in the urothelium or in the tumour. The criterium “minimal changes” of the bladder phenotype was established to indicate the baseline appearance of the urothelium treated with OH-BBN observed [35]. One representative H&E section was scored per animal. The bladders usually showed a mixed phenotype of all criteria. The worst score was used. Bladder phenotype (N, R), invasiveness (O, S) and squamous transformation (P) are expressed as a frequency of observations (%) comparing wildtype (wt, n = 45) and Cxcr2 flox (flox, n = 27) male mice at 20 weeks (N-P) and at 16 weeks (n = 5 per cohort) (R, S). Incidents of tumour in N were presented by numbers in Table 1. The tumour area was measured by QuPath in a representative section per mouse at 20 weeks (n = 15 per cohort) (Q) and carcinoma in situ (CIS) and tumour areas at 16 weeks (n = 5 per cohort) (T). The means with standard deviation are shown in bars (Q, T). Observations in male mice are presented (N-T). The p-values (Mann-Whitney test) are indicated where significant (p < 0.05).
![Bladder tumour phenotype of LysMCre Cxcr2flox/flox (Cxcr2 flox) mice. (A) Mice were given tobacco carcinogen 0.05% (v/v) OH-BBN in drinking water for 10 weeks, and bladder tissues were examined at 2, 16 and 20 weeks from the start of the carcinogen treatment. (B) An example of a bladder tumour developed at 20 weeks from the start of the carcinogen treatment in a Cxcr2 flox animal, with an invasion to the muscle (arrow) and keratinization (Kr). Representative images of wildtype (C, E) and Cxcr2 flox (D, F) bladders at 16 weeks (C, D), and 2 weeks (E, F). Enhanced tumour pathogenesis of the urothelium, such as carcinoma in situ-like lesions (arrowheads) was observed in Cxcr2 flox bladders at 16 weeks (D). At 2 weeks, the differences between wildtype and Cxcr2 flox bladders were not evident. IHC staining showed infiltrating Cxcr2+ myeloid cells in tumours at 20 weeks (G), the urothelium (J) and around the blood vessels in the bladder stroma (L) at 2 weeks in wildtype mice (arrows in G, J, L). In contrast, the staining was absent in Cxcr2 flox tumours (H, I) and in the bladder at 2 weeks’ time point (K, M). IHC staining was performed in at least n = 3 samples in each genotype. Annotations in B-M are; CIS, carcinoma in situ; Kr, keratinization; M, muscle, St, stroma; T, tumour; U, urothelium; V, blood vessel. The scale bar represents 500μm in B, 250μm in C-F, 30μm in G-M. For evaluation of the bladder and tumour phenotype, the scoring criteria from our previous study [35] were refined to optimally capture the phenotype of OH-BBN treated mice under our protocol. The “bladder phenotype” defines the overall state of the urothelial tumorigenesis and progression. The “invasiveness” describes the state of the basement membrane and invasion of tumour cells, while the “squamous transformation” describes the squamous appearance of cells and keratinization in the urothelium or in the tumour. The criterium “minimal changes” of the bladder phenotype was established to indicate the baseline appearance of the urothelium treated with OH-BBN observed [35]. One representative H&E section was scored per animal. The bladders usually showed a mixed phenotype of all criteria. The worst score was used. Bladder phenotype (N, R), invasiveness (O, S) and squamous transformation (P) are expressed as a frequency of observations (%) comparing wildtype (wt, n = 45) and Cxcr2 flox (flox, n = 27) male mice at 20 weeks (N-P) and at 16 weeks (n = 5 per cohort) (R, S). Incidents of tumour in N were presented by numbers in Table 1. The tumour area was measured by QuPath in a representative section per mouse at 20 weeks (n = 15 per cohort) (Q) and carcinoma in situ (CIS) and tumour areas at 16 weeks (n = 5 per cohort) (T). The means with standard deviation are shown in bars (Q, T). Observations in male mice are presented (N-T). The p-values (Mann-Whitney test) are indicated where significant (p < 0.05).](https://content.iospress.com:443/media/blc/2022/8-3/blc-8-3-blc211645/blc-8-blc211645-g001.jpg)
Histology, scanning and histopathological scoring
The bladders were dissected and gently emptied of urine by pressing the tissue with forceps. The bladders were fixed in 10% neutral-buffered formalin overnight and embedded in paraffin. Four-μm sections were used for haematoxylin and eosin (H&E) staining and immunohistochemistry (IHC). Stained slides were scanned by NanoZoomer S60 and analysed by NDP View 2 (Hamamatsu, Shizuoka, Japan) or QuPath version 0.2.3 [34]. H&E images were used for histopathological analysis of bladder and tumour phenotype. The scoring criteria are described in Fig. 1 legend and Table S1. Scoring was performed by multiple observers (NFBI, NC, MF, TI) in discussion with the pathologists (JMS, JDGL).
2.3Immunohistochemistry
Antigen retrieval (AR) was performed in heat-induced epitope retrieval (HIER) buffer (ThermoFisher Scientific, Waltham, Massachusetts, USA) by microwave to boiling for 1 min at full power, followed by 10 min at 20% power. Sections were incubated with 3% H2O2 for 15 minutes and blocked with 2.5% normal goat or horse serum (Vector Laboratories, Peterborough, UK) for 30 minutes. Primary antibody was applied for 1 hour at room temperature. After washing with TBST (VWR, Lutterworth, UK), secondary antibodies (ImmPRESS HRP Polymer Detection Kit, Vector Laboratories) were applied for 30 minutes. Immunoreactivity was detected by 3,3′-diaminobenzidine (DAB) substrate (Vector Laboratories) and slides were counterstained with haematoxylin. Details of the antibodies used are provided in Table S2.
Image analysis
Manual annotation in QuPath [34] was used to measure the area of the carcinoma in situ (CIS) and tumour. The immune cells stained positively by IHC was quantified in 10 fields of 100×100-μm, randomly placed in the urothelium, stroma, muscle layer, and tumour. The mean value was used to represent each sample. QuPath was also used for quantification of Ki67+ and Caspase3+ cells. The stain vector was first adjusted by selecting a representative area indicating haematoxylin and DAB. Region of interest (ROI) was drawn manually for area representing tumour. The mean size of the ROIs was 1.42 mm2, ranging from 0.18–10.00 mm2. Caspase 3+ cells were quantified manually using QuPath. For Ki67, positive cell detection command was used. The threshold 1+ on the score compartment “Nucleus: DAB OD mean” was used to categorize positive or negative cells by adjusting the value in each image. For quality control, all images were also manually scored in the test ROIs. Scoring was performed by at least two independent observers (NFBI, AREY, NXC and TI).
Total RNA extraction and cDNA synthesis
At dissection, the bladder tissue was halved at the midline. One half was processed in paraffin for histology, the other half was collected in RNAlater (Qiagen, Manchester, UK) for RNA extraction and stored at –20°C. The total RNA was extracted using the RNeasy Mini Kit (Qiagen) and was quantified using NanoDrop Microvolume Spectrophotometer (ThermoFisher Scientific). Samples with 260/280 absorbance ratio between 1.8 and 2.2 were used. The integrity of the RNA was also evaluated by agarose gel electrophoresis. RNA samples with two sharp bands, corresponding to 28S and 18S ribosomal RNA, and with the intensity ratio of the 28S:18S = 2:1, were considered intact and used for the further analysis. The RNA samples were stored at -20°C or –80°C. For the cDNA synthesis, 0.5μg of total RNA was subjected to the removal of genomic DNA elimination and reverse transcription using the RT2 First Strand Kit (Qiagen). Genomic DNA was removed by incubating 0.5μg of the total RNA in Buffer GE with a final volume of 10μl at 42°C for 5 minutes. The reverse-transcription was performed with Buffer BC3, Control P2, RE3 Reverse Transcriptase Mix and RNAse free water added to a final volume of 20μl and at 42°C for 15 minutes. The reaction was terminated by incubation at 95°C for 5 minutes and added with 91μl of RNAse-free water, to achieve the final volume of 111μl. The resultant cDNA was stored at –20°C or used immediately for RNA array.
RNA array
RNA array experiment was performed using RT2 Profiler Mouse Cancer Inflammation & Immunity Crosstalk PCR Array (Qiagen). The PCR component mix was prepared with 1350μl of 2X RT2 SYBR Green Mastermix, 102μl of cDNA synthesis reaction and RNase-free water added to the final volume of 2700μl. For each well of the RT2 Profiler PCR array, 25μl of the PCR components mix was added. PCR reaction was performed using the StepOnePlus Real-Time PCR System (ThermoFisher Scientific). Thermal cycle conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Fold changes between samples were determined using the 2–ΔΔCT method. Total RNA from wildtype (n = 3) and Cxcr2 flox (n = 2) bladders at 20 weeks were used. The ΔCT values of house-keeping gene was subtracted from those of each gene, then the mean value for wildtype and Cxcr2 flox was determined. The ΔΔCT value was determined by ΔCT (Cxcr2 flox mean) - ΔCT (wildtype mean), then converted to log2 fold by 2–ΔΔCT.
TCGA data analysis
The clinical information, genomic alternations, mRNA expression, and MIBC molecular Subtype (mRNA Cluster) of human bladder cancer (TCGA, Cell 2017) (n = 412) were downloaded from cBioPortal (https://www.cbioportal.org/public-portal/index.do) and from the reference [32].
Statistics
GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) was used. Gaussian distribution of the data was first evaluated. The differences of the means between two data groups were evaluated using the Mann-Whitney test for non-parametric distributions. Kruskal-Wallis test was used to evaluate the differences of more than three data groups. Pearson (parametric) or Spearman (non-parametric) tests were used for evaluation of correlation. Overall survival was estimated using the Kaplan–Meier method and analysed using the log-rank test. A p value of < 0.05 was considered significant.
RESULTS
Cxcr2 deletion enhanced urothelial tumorigenesis
Using the previously established 10-weeks on, 10-weeks off protocol of OH-BBN induction of invasive urothelial carcinoma [35], we have compared the bladder pathogenesis in mice deleted with Cxcr2 in the myeloid cell lineage, LysMCre Cxcr2flox/flox (Cxcr2 flox) and wildtype mice at the timepoints of 2, 16 and 20 weeks from the start of carcinogen treatment (Table 1, Fig. 1A). Human Protein Atlas data showed that CXCR2 expression can be detected in many immune cells and is enriched particularly in granulocyte lineage and neutrophils (Protein Atlas version 21.0, https://www.proteinatlas.org/ENSG00000180871-CXCR2) [36]. LysMCre mice were previously shown to drive Cre-lox recombination in the majority of neutrophils and macrophages, as well as dendritic cells to some extent [33]. The current model is therefore designed to allow evaluation of the effects of Cxcr2 loss in recruitment of myeloid cells to the bladder tissue, where inflammation was induced by OH-BBN.
Evaluating the tissue histopathology, Cxcr2 flox mice showed more frequent occurrence of bladder tumours (p = 0.005), higher invasiveness of tumour cells (p = 0.025) and squamous transformation (p = 0.001) compared to wildtype mice at 20 weeks from the start of OH-BBN treatment (Fig. 1B, N-P). The tumour area was similar (mean; wildtype 9.2 mm2, Cxcr2 flox 10.4 mm2, n = 15 per genotype) (Fig. 1Q). Cxcr2 flox bladders showed formation of carcinoma in situ (CIS) and increased invasiveness at 16 weeks, while wildtype bladders remained unchanged (Fig. 1C, D, R-T). At 2 weeks, the urothelium of Cxcr2 flox and wildtype was morphologically indistinguishable (Fig. 1E, F). Absence of Cxcr2+ myeloid cells were evident in tumours, the urothelium and around the blood vessels in Cxcr2 flox mice (Fig. 1G-M). Female Cxcr2 flox mice also showed more advanced bladder phenotype compared to wildtype females (Fig. S1). The phenotype was overall more advanced in males than female mice (Fig. 1, S1). Male mice were used in further investigations.
Altogether, Cxcr2 deletion in myeloid cells led to a significant increase in bladder tumorigenesis, with an earlier and more frequent occurrence of tumours with morphological appearances indicative of enhanced progression.
Effects of Cxcr2 deletion in tissue infiltration of immune cells
We next investigated how Cxcr2 deletion influenced the tissue infiltration of immune cells. At 2 weeks timepoint, acute inflammation is normally observed in response to OH-BBN treatment [35]. As expected, infiltration of neutrophils, assessed by the canonical IHC markers, including Ly6G, NIMP and S100 calcium-binding protein A9 (S100A9), was suppressed in the Cxcr2 flox bladder stroma compared to those in the wildtype (p = 0.004) (Fig. 2A, B). Similarly, infiltration of F4/80+ macrophages were also downregulated as expected (p = 0.004) (Fig. 2D). Furthermore, TILs, including CD3+, Granzyme B+ (GrB+), CD4+, and FoxP3+ cells, was significantly reduced in the Cxcr2 flox bladder stroma (Fig. 2F). Similar changes were observed throughout the bladder tissue, including the urothelium and the bladder muscle (Fig. S2).
Fig. 2
Suppression of Ly6G+ neutrophil infiltration in Cxcr2 flox mouse bladders, with upregulation of macrophages at 16 weeks and CD3+ T cells at 20 weeks. Populations of neutrophils (A-C), macrophages (D, E) and T cells (F, G) were characterised in the bladder stroma at 2 and 16 weeks, and in the tumour of wildtype (wt) and Cxcr2 flox (flox) mice at 20 weeks from the start of OH-BBN treatment. IHC markers used were Ly6G, NIMP, S100A9 and MPO for neutrophils, F4/80 for macrophages, CD3, CD8, Granzyme B (GrB), CD4, and FoxP3, for general, cytotoxic, activated, helper, and regulatory T cells, respectively. (A, D, F) Density of each population was quantified. The number of samples used were; wildtype, n = 5-7, Cxcr2 flox n = 6-7 at 2 weeks; wildtype, n = 3-5, Cxcr2 flox n = 4-5 at 16 weeks; wildtype, n = 5-8, Cxcr2 flox n = 8–11 at 20 weeks. Male mice were analysed. The bars represent the means with standard deviation. The p-values are indicated where significant (p < 0.05). Kruskal-Wallis test were used to evaluate differences in immune infiltrations during time course (indicated at the top of each graph). The differences between Cxcr2 flox and wildtype at the same timepoint were evaluated by Mann-Whitney test (indicated above each value). Images are representative of Ly6G+ neutrophils in tumours at 2 weeks (B) and 20 weeks (C), F4/80+ macrophages in the bladder tissue stroma at 16 weeks (E), and CD3+ T cells in tumours at 20 weeks (G), with arrowheads indicating the positively stained cells. (H) A similar pattern of Ki67+ cells (arrowheads) were found in CIS and tumours in wildtype and Cxcr2 flox mice at 20 weeks. (I) Density of Ki67+ cells were quantified in wildtype (n = 5) and Cxcr2 flox tumours (n = 4). (J) A representative image of Caspase 3 IHC staining showing the positive cell (black arrowhead) and a necrotic area (red arrowhead) in wildtype mice at 20 weeks. (K) Density of Caspase 3+ cells were quantified in wildtype (n = 5) and Cxcr2 flox tumours (n = 4). Scale bars indicate 50 μm in B, I, K, 100μm in C, E, and G. 100μm and 50μm in images of CIS and tumours, respectively (I)
At 16 weeks, reduced infiltration of neutrophils and T cells was observed (Fig. 2A, F, S2), likely to be in reflection of the cessation of carcinogen at 10-week time point. Interestingly, however, a significant increase in macrophages was observed in the Cxcr2 flox bladder stroma compared to wildtype (p = 0.029) (Fig. 2D, E). At 20 weeks, infiltration of Ly6G+ and S100A9+ neutrophils was continued to be suppressed in Cxcr2 flox tumours compared to wildtype (p = 0.039, 0.048, respectively, Fig. 2A, C), while levels of macrophages remained low (Fig. 2D). Unexpectedly, overall levels of TILs were higher in Cxcr2 flox mice compared to wildtype (CD3+, p = 0.049. Fig. 2F). TILs were mostly observed in the tumour stroma, as shown in the example of CD3+ cells (Fig. 2G).
We then investigated how genotype and tumour phenotype are correlated with immune cell levels. Cross-examining the relationship between genotype, phenotype and immune cell infiltrations observed in each sample (Fig. S3), genotype was associated with infiltrating immune cells at 2 weeks as expected. At 20 weeks, association between genotype remained with Ly6G and CD3 levels, with little association between tumour phenotype and TILs, indicating that Cxcr2 deletion is more likely to have influenced the changes in the level of CD3+ T cells at 20 weeks rather than the tumour phenotype.
Finally, to see how changes in the immune cell levels have influenced the tumour burden, we analysed the status of cell proliferation and apoptosis in tumour tissues at 20 weeks (Fig. 2H-K). Ki67+ proliferating cells were observed mostly in the basal edge of CIS and in tumours (Fig. 2H). Levels of Ki67+ cells were similar in wildtype and Cxcr2 flox tumours (Fig. 2I), indicating that the increased tumour burden in Cxcr2 flox may have originated at an earlier stage. Caspase 3+ cells were relatively infrequent in tumours of both genotypes (Fig. 2J). No significant difference was observed in the density of Caspase 3+ cells in wildtype and Cxcr2 flox tumours (Fig. 2K). No obvious similarity was found in the location of Caspase3+ cells with those of Ly6G+ neutrophils or TILs (Fig. 2C, G, J).
Taken together, following a transient increase of macrophages at the outset of tumour formation, an increase in T cell infiltration was observed in Cxcr2 flox tumours. However, the elevated level of TILs was not associated with changes in cell proliferation or cell death in the absence of Cxcr2. Therefore, the increased tumour burden upon Cxcr2 deletion may have resulted from a suppression of acute inflammation in the bladder at the time of tumour initiation.
Levels of blood immune cells did not always correlate with those in the bladder tissue
The effects of Cxcr2 deletion on blood leukocyte populations were compared to those in the tissues (Fig. S4). At 2 weeks from the start of OH-BBN treatment, the level of neutrophils in the blood appeared lower in Cxcr2 flox mice compared to wildtype, as expected, while basophils appeared to have increased (Fig. S4A, B, not statistically significant). At 16 weeks, the blood levels of monocytes were higher in Cxcr2 flox mice (p = 0.016) (Fig. S4B), similar to the observations in the tissue (Fig. 2B). In addition, the lower population of neutrophils within the whole blood (p = 0.024, Fig. S4A), and higher blood concentration of lymphocytes were observed (p = 0.032, Fig. S4B), leading to the lower NLR value (p = 0.032, Fig. S4A, B). While this was not detected in the tissue level at 16 weeks, it resembled the observations in the tumour tissue at 20 weeks (Fig. 2A, F).
In contrast, the lower densities of tissue-infiltrating neutrophil and the higher TILs observed in Cxcr2 flox compared to wildtype (Fig. 2A, F) were not detected in the blood in tumour-bearing mice at 20 weeks (Fig. S4A, B). Instead, the higher monocyte and lower lymphocyte concentrations were observed in the blood in non-tumour bearing Cxcr2 flox mice compared to wildtype (Fig. S4B).
Finally, tumour-bearing mice showed higher levels of neutrophils, NLR, and eosinophils, compared to non-tumour bearing mice in wildtype (p = 0.028, 0.049, 0.025, respectively) (Fig. S4B). This indicated that, in contrast to the findings in the tissue, the presence of tumours had more effects on granulocyte and lymphocyte levels in circulation, and that it confirms the association of higher NLR values with worse prognosis in terms of tumour presence.
Taken together, the levels of immune cells in the blood did not always correspond with those in the bladder tissue. Interestingly, changes may have occurred in a specific population of granulocytes depending on the stage of tumour pathogenesis.
Both low and high CXCR2 expression ranges are associated with a poor prognosis in human bladder cancer
To elucidate the role of CXCR2 in human bladder cancer, we analysed the data available from TCGA Bladder Cancer (Cell 2017) cohort (n = 412) [32]. The OncoPrinter function in the cBioPortal revealed that CXCR2 deletions occurred in 8 samples (n = 8/404, 2.0%) together with deletions of CXCR1 (n = 8/8) and those of chemokine receptor CCR2 (n = 2/8) (Fig. S5A, B). Interestingly, deletions of CXCR2 occurred together with those of T cell inhibitors, PD-1 (n = 6) and CTLA4 (n = 3) (Fig. S5C), however mostly mutually exclusive with genomic alterations common in bladder cancer, such as FGFR3 and TP53 (Fig. S5D). By Spearman correlation analysis, we found that the level of CXCR2 mRNA was inversely correlated with age, disease stage, metastatic stage, histological grade and mutation count (Fig. S6A). CXCR2 was not associated with specific MIBC molecular subtype, unlike the example of FGFR3 that is associated with luminal papillary subtype [32, 37] (Fig. S6B).
Kaplan-Meier analysis showed a worse overall survival associated with a lower level of CXCR2 mRNA expression, with a median survival of 32.0 months, in contrast to 92.9 months in patients with higher expression (log-rank, p = 0.044) (Fig. 3A). Previous studies, however, mostly reported the pro-tumour role associated with an increase of CXCR2 signalling [25, 26, 28, 38, 39], including those in bladder cancer [29–31]. Investigating the highest level of CXCR2 mRNA (≥80 log2 expression), we found that the median survival was 46.8 months, while 97.0 months in the medium range of CXCR2 mRNA (≥40, < 80 log2 expression) (Fig. S7A). CXCR2 expression was positively associated with the levels of CXC ligands, including CXCL1, CXCL6 and CXCL8, in both CXCR2low (< 40 log2 expression) and CXCR2high (≥80 log2 expression) ranges (Figs. 3B, S7B). Interestingly, in the CXCR2low group, PD-L1 expression level was inversely correlated with CXCR2 expression (Spearman r -0.124, p = 0.041). In contrast, in the CXCR2high group, the correlation was positive (Spearman r 0.281, p = 0.023) (Figs. 3C, S7), which is in line with the previous studies where increased CXCR2 expression was associated with an immune suppressive, pro-tumour role.
Fig. 3
Low CXCR2 mRNA expression is associated with poor overall survival in human bladder cancer. (A) Kaplan-Meier survival analysis was performed using a bladder cancer dataset (TCGA, Cell 2017) with information on CXCR2 mRNA expression and overall survival (n = 400), categorised according to low (< 40 log2 mRNA expression, n = 269), and high (≥40 log2 mRNA expression, n = 131) CXCR2 mRNA expression (log2). (B, C) Using the MIBC cases of the Bladder Cancer, TCGA, Cell 2017 cohort (> T2, n = 398), correlation between CXCR2 and CXC ligands, and that of PD-1/PD-L1 was analysed. The mRNA expression of CXCR2 and CXCL1, CXCL6 and CXCL8 were positively correlated (B). The mRNA expression of CXCR2 and PD-L1 were inversely correlated in the CXCR2 “low” range (< 40 log2 mRNA expression, n = 269), while positively correlated in the CXCR2 “high” range (≥80 log2 mRNA expression, n = 65) (C). (D) Mice aged beyond 20 weeks (21–43 weeks) were monitored by ultrasound imaging for the presence of bladder tumours and culled when mice showed clinical signs of bladder tumour, such as haematuria and weight loss. (E) Relationship between the timepoint of tumour development and tumour area size, as well as immune cell density was evaluated by Spearman correlation in wildtype mice that developed tumour at 20 weeks (n = 12) and beyond 20 weeks (n = 10) from the start of OH-BBN treatment. wildtype mice were treated with 0.05% OH-BBN for 10 weeks. Mice aged beyond 20 weeks (21–43 weeks) were monitored by ultrasound imaging for the presence of bladder tumours and culled when mice showed clinical signs of bladder tumour, such as haematuria and weight loss. (F) The status of immune cell infiltration was evaluated using Spearman correlation between Ly6G+ neutrophil density and T cell populations positive with CD3+, CD8+, Granzyme B+, and FoxP3+ in tumours as in E.
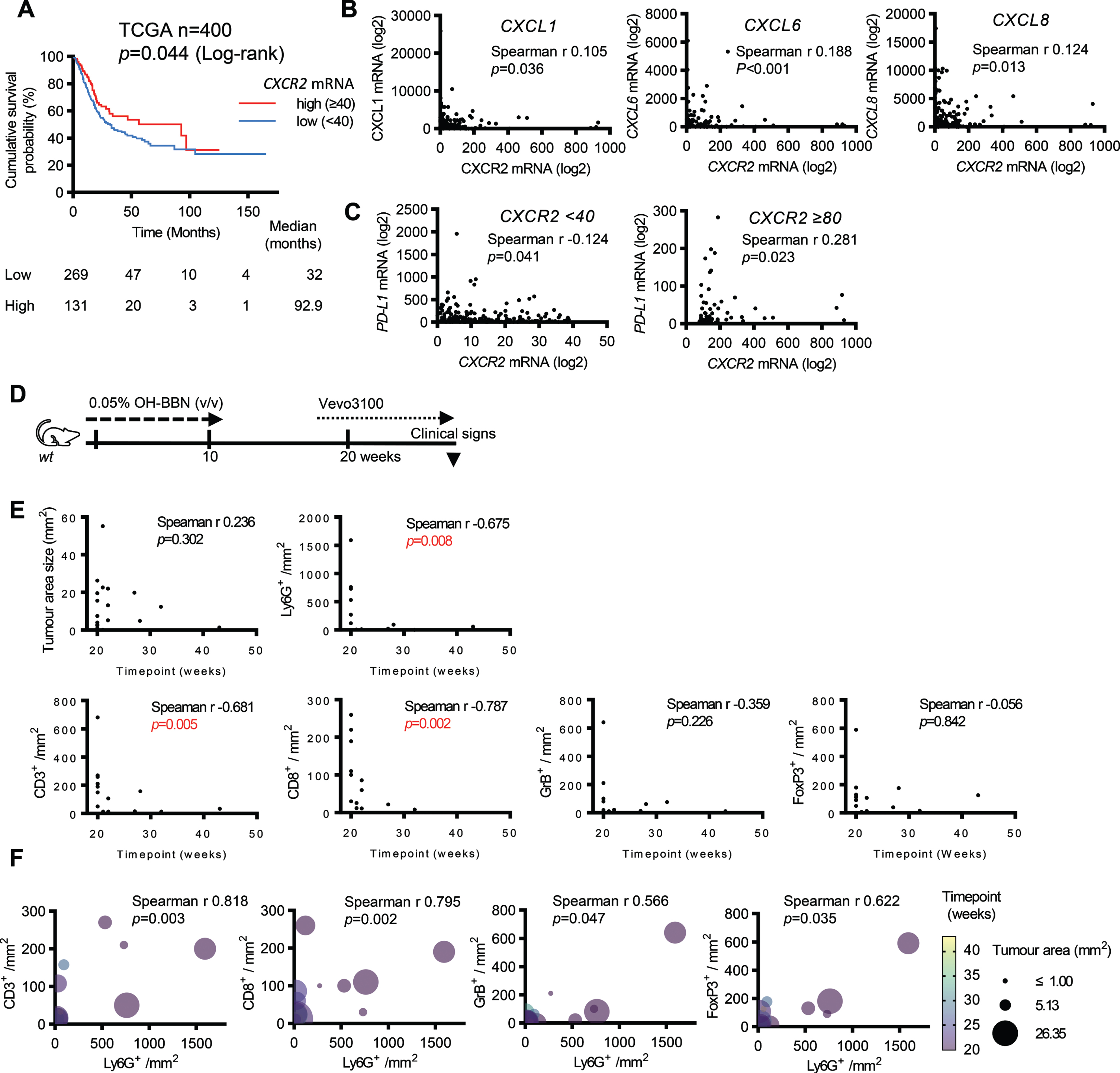
Status of immune suppression in the OH-BBN treated mice
To better understand how the above observations in human bladder cancer reflect in our mouse model, we examined the expression of regulatory genes in inflammation and immunity using an RNA array approach. Upregulation was observed in both immunostimulatory and immunosuppressive factors in Cxcr2 flox tumours comparing to wildtype tumours at 20 weeks (n = 3 wildtype, n = 2 Cxcr2 flox) (Table S3), including that of a pro-inflammatory factor, interferon γ (Ifng), as well as immunosuppressive factors produced by regulatory T cells, Transforming growth factor β (Tgf β) and interleukin 10 (Il10). Pcdc-1/Pd-1 and Cd274/Pd-L1 were also upregulated (4.55 and 2.84 log2 fold, respectively) in the absence of Cxcr2. Interestingly, Ccr2, a chemokine receptor that plays a major role in monocyte chemotaxis [40], and its ligand Ccl2, were highly upregulated in Cxcr2 flox tumours (6.68 and 3.16 log2 fold, respectively).
As our current OH-BBN model is optimised for the evaluation of tumour formation and initial tumour pathogenesis, it is possible that tumours had not been allowed to develop long enough to acquire a strong immunosuppressive tumour microenvironment. To assess whether immune suppression could be developed in the tumour microenvironment of our OH-BBN model, we quantified the density of Ly6G+ neutrophils, F4/80+ macrophages and TILs in tumour-bearing wildtype mice, at later timepoints, up to 43 weeks from the start of the carcinogen treatment (Fig. 3D). The majority of F4/80+ macrophages were observed in the stroma and rarely observed in tumours at 20 weeks and the timepoints beyond (Fig. 2B and data not shown). Longer timepoints were associated with fewer Ly6G+, CD3+ and CD8+ cells, but not with tumour area size (Fig. 3E). Tumour area size was not corelated with immune cell densities (Fig. S8A). In contrast, a higher density of Ly6G+ cells was associated with a higher overall populations of TILs (Fig. 3F), indicating that Ly6G+ neutrophils remained to bear an anti-tumour role, being populated together with T cells in tumours.
Taken together, our OH-BBN-induced Cxcr2 flox mouse model may reflect human bladder cancer whereby CXCR2 expression is low, in terms of worse disease prognosis, and an elevated level of immunosuppressive factors, such as Pd-1/Pd-L1. The status of immune suppression in Cxcr2 flox mice remained ambiguous, as upregulation was found in both immunostimulatory and immunosuppressive factors. However, it is unlikely that tumours in our current OH-BBN model developed an immunosuppressive tumour microenvironment, in which myeloid cells turned pro-tumour and suppressed T cell functions.
DISCUSSION
In this study, we showed that the loss of Cxcr2 in myeloid cells promoted bladder tumour progression, evidenced by a robust enhancement of earlier and frequent tumour phenotype in the Cxcr2 flox mice (Figs. 1, S1, S8B). Despite an increase in TILs (Fig. 2), the increased tumour burden in Cxcr2 flox mice resulted largely independently of their cytotoxic function (Fig. 2). Instead, this tumour phenotype may have resulted from a suppression of acute inflammation in the bladder in the absence of Cxcr2 at the time of tumour formation. Our Cxcr2 flox model reflected the worse prognosis associated with the CXCR2low expression range in TCGA MIBC cohort (Fig. 3).
CXCR2 is reported to be expressed mainly in myeloid cell lineage, including neutrophils, monocytes, macrophage, and mast cells [25, 26]. Therefore, a reduced recruitment of neutrophils and macrophages to the bladder in response to OH-BBN at 2-weeks timepoint (Fig. 2) was expected in a model of Cxcr2 loss. In addition, the level of T populations was also reduced (Fig. 2). Single Cell Type data in Human Protein Atlas, proteinatlas.org [36] indicates that CXCR2 is also present in T cells. In the LysMCre mice used in the study, efficiency of Cre-lox recombination was previously shown in the majority of neutrophils and macrophages, but little recombination was reported in B and T cells [33]. Therefore, the reduction of T cells is not caused directly by the loss of Cxcr2, but likely to be secondary to the changes in myeloid cells.
We have observed a transient increase in macrophage infiltration in Cxcr2 flox bladders at the onset of tumour formation (Fig. 2D). The increase of macrophages may be a part of the compensatory signalling in the absence of Cxcr2. Indeed, expression of Ccr2, which is mainly expressed in monocytes and macrophages, and its ligand Ccl2, was highly upregulated in Cxcr2 flox tumours comparing to wildtype (Table S3). This is consistent with the report in which Cxcr2-/- mice showed an increase in macrophage infiltration at the acutely inflamed sites with exaggerated cutaneous inflammation [41], and in a model of pancreatic adenocarcinoma, where inhibition of Cxcr2 led to an increase of TAM, while inhibition of Ccr2 and TAM infiltration lead to an increase of tumour infiltrating neutrophils [40].
The levels of immune cells in the blood circulation and those of tissue-infiltrating immune cells were not always comparable (Figs. 2, S4). Consequences of CXCR2 deletion in the bone marrow may appear in the blood more directly, rather than in the tissues where infiltrations could be regulated by the microenvironment. An increase in monocytes in Cxcr2 flox mice in the blood at 16 weeks (Fig. S4) was reflected in the tissue (Fig. 2D). However, an increase in monocytes at 20 weeks was observed in non-tumour bearing mice (Fig. S4), therefore unlikely to have been caused by tumour phenotype. Instead, the increase in blood monocyte levels may have occurred in compensation to the earlier suppression of acute inflammation by Cxcr2 deletion. The predictive value of NLR was only valid comparing tumour-bearing and non-tumour bearing mice at 20 weeks (Fig. S4). The blood analysis also indicated that basophils may have increased at 2 weeks and that there was a significant upregulation of eosinophils in tumour-bearing mice comparing to non-tumour bearing mice at 20 weeks (Fig. S5), suggesting granulocyte-specific changes depending on a disease stage.
Multiple types of myeloid cells, including neutrophils, monocytes and MDSCs, could influence the tumour microenvironment [9–13, 21–23]. Tumour-infiltrating myeloid cells were shown to account for 10-20% of cells found in bladder tumour tissues, and that 30-40% were granulocytic or polymorphonuclear (G/PMN)-MDSCs in both non-invasive and invasive tumours, with the rest being monocytic (M)-MDSCs [42]. In benign conditions, MDSCs contribute to homeostasis of T cell-mediated inflammatory response, however, in cancer, G/PMN-MDSC and M-MDSC are both immune suppressive [9, 43]. Triggered by chronic inflammation or malignancy, MDSCs induce immunosuppression, by producing, for example, inhibitory cytokines and TGFβ, and inhibiting IFNγ-mediated proinflammatory signals. High MDSC is associated with worse prognosis and poor response to BCG, chemo- and ICI therapy in bladder cancer [43]. Pro-tumour CXCR2 signalling in bladder cancer has so far been attributed to its expression in immunosuppressive MDSCs [30, 31]. A higher proportion of cells were CXCR2+ in MDSCs in patients’ tumours than those in the peripheral blood [30]. Cxcr2 was also overexpressed in MDSCs in mice with MB49 tumours as well as those with chemo-resistant MB49B [31]. Upon close examination of TCGA Bladder Cancer (Cell 2017) dataset, we have shown that both CXCR2low and CXCR2high expression ranges is associated with poor prognosis and increased PD-L1 expression (Figs. 3, S7). We speculate that the CXCR2low cases are reflected in our mouse model, and CXCR2high, the previous reports of CXCR2 overexpression [30, 31].
The pro-tumour role of CXCR2 was reported previously in colon cancer [44], breast cancer [45], rhabdomyosarcoma [27], and pancreatic ductal adenocarcinoma [28, 46]. The role of CXCR2 could also differ dependent on tissue types and the stages of tumour progression. In an early stage of lung cancer, a subset of tumour infiltrating neutrophils showed antigen-presenting cell (APC)-like characteristics and was able to activate T cell proliferation and IFNγ release, therefore not immunosuppressive [47, 48]. The anti-tumour role of CXCR2 could be mediated by re-enforcement of oncogene-induced senescence in tumour cells [49, 50]. A possibility that Cxcr2 deleted in myeloid cells have perturbated the autocrine chemokine signalling in tumours, influencing the senescence-associated secretory phenotype, cannot be excluded.
The limitations of the current study are as follows. Firstly, the current OH-BBN protocol is restricted to model early-stage tumours and tumour microenvironment remained immune competent, with tumour-infiltrating neutrophils maintaining a positive correlation with TIL density (Fig. 3F). Therefore, the role of CXCR2 in an immune suppressive environment and in metastasis cannot be assessed [9, 11, 13] (Fig. S8B). Secondly, the study was conducted with a genetically modified mouse model, and the frequency and size of the tumours were not sufficiently large to allow characterization of myeloid cell populations by flow cytometry and cytokine expression by ELISA. Finally, deletion or pharmacological inhibition of Cxcr2 in mice led to an enhanced T cell infiltration in the tumour in pancreatic ductal carcinoma [28]. While a similar elevated level of TILs was observed in Cxcr2 flox tumours (Fig. 2F), our current model does not provide evidence that supports such modulation to sensitize bladder tumours to ICIs. Mechanistic understanding of tumour immune microenvironment requires suitable models that reflect the advanced status of tumour progression and immune suppression. In such models, the status of CXCR2 and myeloid cell diversity could be further investigated for their influence in regulating response towards PD-1/PD-L1 inhibitors. The use of CXCR2 inhibitors requires a cautious approach and should be investigated together with a marker that indicates the status of immune suppression in invasive urothelial carcinoma.
ACKNOWLEDGMENTS
We thank Biological and Histology Services at Cancer Research UK Beatson Institute for their technical support, Dr Catherine Winchester for editing the manuscript. We are grateful to Dr Seth Coffelt and Professor Owen J. Sansom, for insights in neutrophil biology and for project conception and support, respectively. We also thank Professor Margaret A Knowles, St James’s University Hospital, for helpful discussions and encouragement.
FUNDING
This work was funded by Public Department of Malaysia (NFBI), Ministry of Higher Education of Libya (AREY), MRC MR/N021800/1 (JDGL), CRUK A17196 and A31287 core funding to the Beatson Institute (TJ, SAK, JPM), CRUK A21139 core funding to Sansom lab (TJ), and CRUK A29996 core funding to JPM lab (JPM).
AUTHOR CONTRIBUTIONS
Conception: NFBI, MF, JPM, TI; Performance of work: NFBI, MF, AREY, NC, TJ, SAK, TI; Interpretation of analysis of data: NFBI, MF, AREY, NC, JDGL, TJ, SAK, JMS, JPM, TI; Writing of the article: NFBI, JPM, TI. All authors critically reviewed and edited the manuscript drafts.
CONFLICT OF INTEREST
No conflicts of interest were declared by NFBI, MF, AREY, NC, JDGL, TJ, SAK, JMS, JPM, and TI.
SUPPLEMENTARY MATERIAL
[1] Supplementary Tables S1-S3 and Supplementary Figures S1-S8 are available in the electronic version of this article. https://dx.doi.org/10.3233/BLC-211645.
REFERENCES
[1] | Richters A , Aben KKH , Kiemeney L , The global burden of urinary bladder cancer: An update, World Journal of Urology (2020) ;38: :1895–904. |
[2] | Saginala K , Barsouk A , Aluru JS , Rawla P , Padala SA , Barsouk A , Epidemiology of bladder cancer. Med Sci (Basel). 2020;8. |
[3] | Tran L , Xiao JF , Agarwal N , Duex JE , Theodorescu D , Advances in bladder cancer biology and therapy, Nat Rev Cancer (2021) ;21: :104–21. |
[4] | Babjuk M , Bohle A , Burger M , Capoun O , Cohen D , Comperat EM , et al., EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: Update 2016. Eur Urol (2017) ;71: :447–61. |
[5] | Alfred Witjes J , Lebret T , Comperat EM , Cowan NC , De Santis M , Bruins HM , et al., Updated EAU guidelines on muscle-invasive and metastatic bladder cancer, Eur Urol (2017) ;71: :462–75. |
[6] | Siefker-Radtke A , Curti B , Immunotherapy in metastatic urothelial carcinoma: Focus on immune checkpoint inhibition, Nature Reviews Urology (2018) ;15: :112–24. |
[7] | Song D , Powles T , Shi L , Zhang L , Ingersoll MA , Lu YJ , Bladder cancer, a unique model to understand cancer immunity and develop immunotherapy approaches, The Journal of Pathology (2019) ;249: :151–65. |
[8] | Lopez-Beltran A , Cimadamore A , Blanca A , Massari F , Vau N , Scarpelli M , et al., Immune checkpoint inhibitors for the treatment of bladder cancer. Cancers (Basel). 2021;13. |
[9] | Coffelt SB , Wellenstein MD , de Visser KE , Neutrophils in cancer: Neutral no more, Nat Rev Cancer (2016) ;16: :431–46. |
[10] | Powell DR , Huttenlocher A , Neutrophils in the tumor microenvironment, Trends Immunol (2016) ;37: :41–52. |
[11] | Leach J , Morton JP , Sansom OJ , Neutrophils: Homing in on the myeloid mechanisms of metastasis, Mol Immunol (2019) ;110: :69–76. |
[12] | Mackey JBG , Coffelt SB , Carlin LM , Neutrophil maturity in cancer, Front Immunol (2019) ;10: :1912. |
[13] | Masucci MT , Minopoli M , Carriero MV , Tumor associated neutrophils, Their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol (2019) ;9: :1146. |
[14] | Mandelli GE , Missale F , Bresciani D , Gatta LB , Scapini P , Caveggion E , et al., Tumor infiltrating neutrophils are enriched in basal-type urothelial bladder cancer. Cells. 2020;9. |
[15] | Zhou L , Xu L , Chen L , Fu Q , Liu Z , Chang Y , et al., Tumor-infiltrating neutrophils predict benefit from adjuvant chemotherapy in patients with muscle invasive bladder cancer, Oncoimmunology (2017) ;6: :e1293211. |
[16] | Liu K , Zhao K , Wang L , Sun E , The prognostic values of tumor-infiltrating neutrophils, lymphocytes and neutrophil/lymphocyte rates in bladder urothelial cancer, Pathol Res Pract (2018) ;214: :1074–80. |
[17] | Kawahara T , Furuya K , Nakamura M , Sakamaki K , Osaka K , Ito H , et al., Neutrophil-to-lymphocyte ratio is a prognostic marker in bladder cancer patients after radical cystectomy, BMC Cancer (2016) ;16: :185;. |
[18] | Kaiser J , Li H , North SA , Leibowitz-Amit R , Seah JA , Morshed N , et al., The prognostic role of the change in neutrophil-to-lymphocyte ratio during neoadjuvant chemotherapy in patients with muscle-invasive bladder cancer: A retrospective, multi-institutional study, Bladder Cancer (2018) ;4: :185–94. |
[19] | Vartolomei MD , Porav-Hodade D , Ferro M , Mathieu R , Abufaraj M , Foerster B , et al., Prognostic role of pretreatment neutrophil-to-lymphocyte ratio (NLR) in patients with non-muscle-invasive bladder cancer (NMIBC): A systematic review and meta-analysis, Urol Oncol (2018) ;36: :389–99. |
[20] | Black AJ , Zargar H , Zargar-Shoshtari K , Fairey AS , Mertens LS , Dinney CP , et al., The prognostic value of the neutrophil-to-lymphocyte ratio in patients with muscle-invasive bladder cancer treated with neoadjuvant chemotherapy and radical cystectomy, Urol Oncol (2020) ;38: :3 e17–3 e27. |
[21] | Leblond MM , Zdimerova H , Desponds E , Verdeil G , Tumor-associated macrophages in bladder cancer: Biological role, impact on therapeutic response and perspectives for immunotherapy. Cancers (Basel). 2021;13. |
[22] | Cassetta L , Pollard JW , Targeting macrophages: Therapeutic approaches in cancer, Nat Rev Drug Discov (2018) ;17: :887–904. |
[23] | Mantovani A , Marchesi F , Malesci A , Laghi L , Allavena P , Tumour-associated macrophages as treatment targets in oncology, Nat Rev Clin Oncol (2017) ;14: :399–416. |
[24] | Mora-Bau G , Platt AM , van Rooijen N , Randolph GJ , Albert ML , Ingersoll MA , Macrophages subvert adaptive immunity to urinary tract infection, PLoS Pathog (2015) ;11: :e1005044. |
[25] | Cheng Y , Ma XL , Wei YQ , Wei XW , Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer. (2019) ;1871: :289–312. |
[26] | Jaffer T , Ma DQ , The emerging role of chemokine receptor CXCR2 in cancer progression, Transl Cancer Res (2016) ;5: :S616–28. |
[27] | Highfill SL , Cui Y , Giles AJ , Smith JP , Zhang H , Morse E , et al., Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy, Sci Transl Med (2014) ;6: :237ra67. |
[28] | Steele CW , Karim SA , Leach JDG , Bailey P , Upstill-Goddard R , Rishi L , et al., CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell.(2016) ;29: :832–45. |
[29] | Gao Y , Guan Z , Chen J , Xie H , Yang Z , Fan J , et al., CXCL5/CXCR2 axis promotes bladder cancer cell migration and invasion by activating PI3K/AKT-induced upregulation of MMP2/MMP9, Int J Oncol. (2015) ;47: :690–700. |
[30] | Zhang H , Ye YL , Li MX , Ye SB , Huang WR , Cai TT , et al., CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer, Oncogene. (2017) ;36: :2095–104. |
[31] | Takeyama Y , Kato M , Tamada S , Azuma Y , Shimizu Y , Iguchi T , et al., Myeloid-derived suppressor cells are essential partners for immune checkpoint inhibitors in the treatment of cisplatin-resistant bladder cancer, Cancer Lett (2020) ;479: :89–99. |
[32] | Robertson AG , Kim J , Al-Ahmadie H , Bellmunt J , Guo G , Cherniack AD , et al., Comprehensive molecular characterization of muscle-invasive bladder cancer, Cell (2017) ;171: :540–56 e22. |
[33] | Clausen BE , Burkhardt C , Reith W , Renkawitz R , Forster I , Conditional gene targeting in macrophages and granulocytes using LysMcre mice, Transgenic Res (1999) ;8: :265–77. |
[34] | Bankhead P , Loughrey MB , Fernandez JA , Dombrowski Y , McArt DG , Dunne PD , et al., QuPath: Open source software for digital pathology image analysis, Sci Rep (2017) ;7: :16878. |
[35] | Foth M , Ismail NFB , Kung JSC , Tomlinson D , Knowles MA , Eriksson P , et al., FGFR3 mutation increases bladder tumourigenesis by suppressing acute inflammation, The Journal of Pathology (2018) ;246: :331–43. |
[36] | Uhlen M , Fagerberg L , Hallstrom BM , Lindskog C , Oksvold P , Mardinoglu A , et al., Proteomics, Tissue-based map of the human proteome. Science (2015) ;347: :1260419. |
[37] | Sweis RF , Spranger S , Bao R , Paner GP , Stadler WM , Steinberg G , et al., Molecular drivers of the non-t-cell-inflamed tumor microenvironment in urothelial bladder cancer, Cancer Immunol Res (2016) ;4: :563–8. |
[38] | Jamieson T , Clarke M , Steele CW , Samuel MS , Neumann J , Jung A , et al., Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis, J Clin Invest (2012) ;122: :3127–44. |
[39] | Steele CW , Karim SA , Foth M , Rishi L , Leach JD , Porter RJ , et al., CXCR2 inhibition suppresses acute and chronic pancreatic inflammation, The Journal of Pathology (2015) ;237: :85–97. |
[40] | Nywening TM , Belt BA , Cullinan DR , Panni RZ , Han BJ , Sanford DE , et al., Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma, Gut (2018) ;67: :1112–23. |
[41] | Dyer DP , Pallas K , Medina-Ruiz L , Schuette F , Wilson GJ , Graham GJ , CXCR2 deficient mice display macrophage-dependent exaggerated acute inflammatory responses, Sci Rep (2017) ;7: :42681. |
[42] | Eruslanov E , Neuberger M , Daurkin I , Perrin GQ , Algood C , Dahm P , et al., Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer, Int J Cancer (2012) ;130: :1109–19. |
[43] | Puttmann K , Duggan M , Mortazavi A , Diaz DA , Carson WE , Sundi D , The role of myeloid derived suppressor cells in urothelial carcinoma immunotherapy, Bladder Cancer (2019) ;5: :103–14. |
[44] | Yamamoto M , Kikuchi H , Ohta M , Kawabata T , Hiramatsu Y , Kondo K , et al., TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche, Cancer Res (2008) ;68: :9754–62. |
[45] | Acharyya S , Oskarsson T , Vanharanta S , Malladi S , Kim J , Morris PG , et al., A CXCL1 paracrine network links cancer chemoresistance and metastasis, Cell (2012) ;150: :165–78. |
[46] | Chao T , Furth EE , Vonderheide RH , CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma, Cancer Immunol Res (2016) ;4: :968–82. |
[47] | Eruslanov EB , Bhojnagarwala PS , Quatromoni JG , Stephen TL , Ranganathan A , Deshpande C , et al., Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer, J Clin Invest (2014) ;124: :5466–80. |
[48] | Singhal S , Bhojnagarwala PS , O’Brien S , Moon EK , Garfall AL , Rao AS , et al., Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer, Cancer Cell (2016) ;30: :120–35. |
[49] | Acosta JC , O’Loghlen A , Banito A , Guijarro MV , Augert A , Raguz S , et al., Chemokine signaling via the CXCR2 receptor reinforces senescence, Cell (2008) ;133: :1006–18. |
[50] | Lesina M , Wormann SM , Morton J , Diakopoulos KN , Korneeva O , Wimmer M , et al., RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis, J Clin Invest (2016) ;126: :2919–32. |


