
Role of Chromatin Modifying Complexes and Therapeutic Opportunities in Bladder Cancer
Abstract
BACKGROUND:
Chromatin modifying enzymes, mainly through post translational modifications, regulate chromatin architecture and by extension the underlying transcriptional kinetics in normal and malignant cells. Muscle invasive bladder cancer (MIBC) has a high frequency of alterations in chromatin modifiers, with 76% of tumors exhibiting mutation in at least one chromatin modifying enzyme [1]. Additionally, clonal expansion of cells with inactivating mutations in chromatin modifiers has been identified in the normal urothelium, pointing to a currently unknown role of these proteins in normal bladder homeostasis.
OBJECTIVE:
To review current knowledge of chromatin modifications and enzymes regulating these processes in Bladder cancer (BCa).
METHODS:
By reviewing current literature, we summarize our present knowledge of external stimuli that trigger loss of equilibrium in the chromatin accessibility landscape and emerging therapeutic interventions for targeting these processes.
RESULTS:
Genetic lesions in BCa lead to altered function of chromatin modifying enzymes, resulting in coordinated dysregulation of epigenetic processes with disease progression.
CONCLUSION:
Mutations in chromatin modifying enzymes are wide-spread in BCa and several promising therapeutic targets for modulating activity of these genes are currently in clinical trials. Further research into understanding how the epigenetic landscape evolves as the disease progresses, could help identify patients who might benefit the most from these targeted therapies.
INTRODUCTION
Bladder cancer (BCa) is the fourth most common cancer in men and the sixth most common cancer overall, with an average age of diagnosis around 73 years [2, 3]. Most BCa (∼90%) develops from the uninterrupted proliferation of cells in the stratified urothelium, a specialized epithelial layer that lines the urinary tract [4–7]. Next generation sequencing of BCas has identified a heterogeneous somatic mutation landscape, with one of the highest mutation burdens, estimated at 8.2 mutations per MB (median) [8–12].
In the 2017 Cancer Genome Atlas (TCGA) MIBC cohort, after the TP53/RB tumor suppressor pathway, chromatin modifier genes are the second most frequently mutated class of genes [11]. Recent studies investigating early events in bladder tumorigenesis have identified the clonal expansion of cells with widespread somatic mutations in chromatin regulatory genes within normal urothelium [13, 14]. Identification of a high frequency of mutations in chromatin modifying enzymes in independent cohorts has heightened the need to understand the function of these gene alterations in influencing disease aggressiveness and predicting treatment outcomes in BCa patients [11, 15–22].
REGULATION OF CHROMATIN STRUCTURE
The core nucleosome structure consists of 146 bp of DNA wrapped around a histone octamer containing two molecules of H2A, H2B, H3 and H4 (Fig. 1) [23–25]. Tail-like structures protruding from the core-histones are prime sites for post-translational modifications (PTMs) that regulate chromatin structure. Chromatin packaging and accessibility is a determining factor for all cellular events from cell division during organism development to uncontrolled cell division during neoplastic transformation [26–28].
Fig. 1
Overview of chromatin organization in a nucleus.
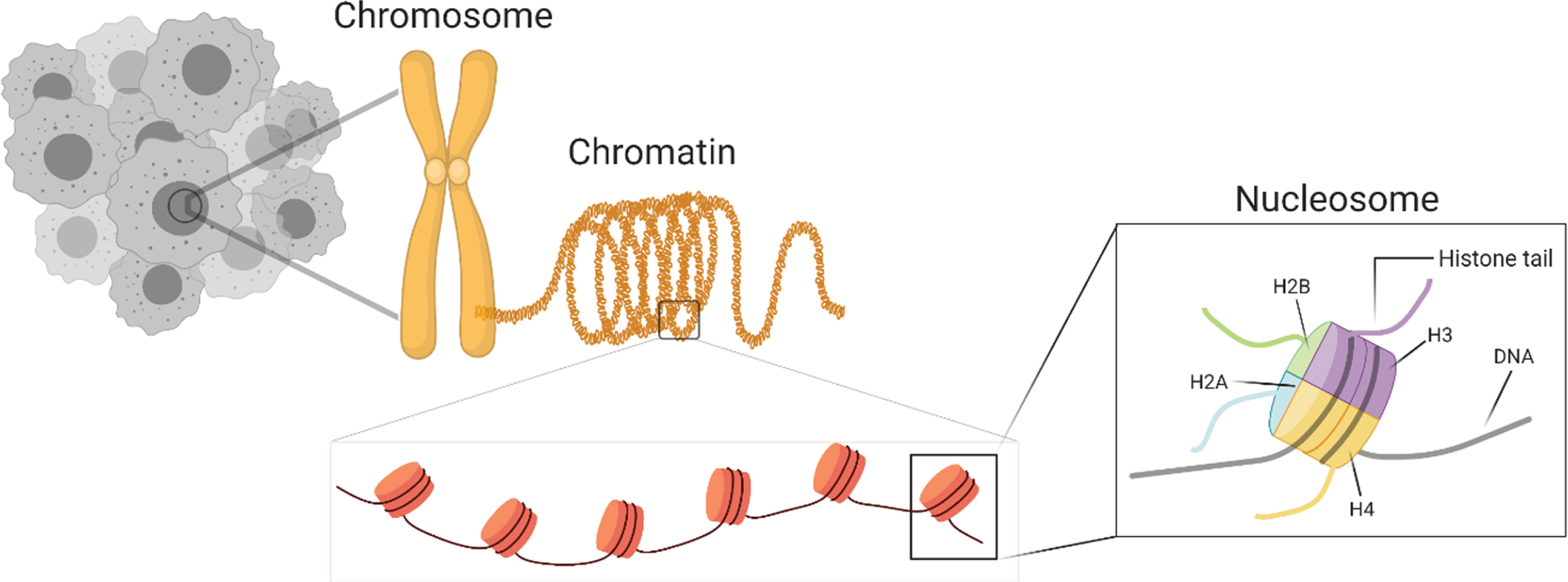
Enzymatic processes that regulate chromatin organization are targets of dysregulation in cancer. Unlike tumor suppressors or oncogenes, loss of function mutations in chromatin regulatory genes may have unique effects on the transcriptional landscape depending on the cell type. More importantly, in a manner unique to cell-of-origin, epigenetic status impacts chromatin packaging which regulates the genomic mutational landscape [29, 30].
The distorted genetic and epigenetic landscape of cancer cells supports tumor evolution by amplifying cellular signals that allow for uninhibited tumor growth [31–33]. During carcinogenesis, tumor cells create their own micro-environmental niche, that is distinctive in its metabolic requirements and signaling pathways [31]. Each of these adaptations helps the tumor gain a survival advantage while evading the immune system. The fast pace of modifying chromatin compaction is frequently exploited by cancer cells to achieve an altered transcriptional landscape [34]. Over the decades, pathologists have observed hyper-chromatic nuclei in malignant cells, likely a reflection of the massive number of organizational changes that take place during neoplastic transformation [35, 36]. With development of high sensitivity assays, the effect of chromatin packing on transcriptional plasticity and its influence on tumor heterogeneity and therapeutic outcome is better appreciated [37, 38].
Chromatin modifying proteins include chromatin remodeling complexes and DNA and histone modifying proteins. Chromatin remodeling complexes, using the energy from ATP hydrolysis, reposition nucleosomes to increase accessibility of DNA binding proteins to DNA whereas histone modifying proteins edit post-translational modifications (PTMs) on histone tails [39, 40]. Histone PTMs are a minimalistic way to control transcription at specific loci. DNA can also be directly epigenetically modified with repercussions on the transcriptional output at any specific locus (Fig. 2). PTMs and DNA modifications can weaken or strengthen non-covalent interactions between histones and DNA and local chromatin composition, thereby allowing or restricting access to selected sections of DNA. Densely packed heterochromatin is largely inaccessible to transcription machinery, while euchromatin, which is more loosely packed, contains accessible sections of the genome with a high transcriptional output. Analogous to other signaling pathways, chromatin organization is coordinated by chromatin modifying enzymes in response to external cues such as developmental stimuli [28, 41, 42].
Fig. 2
Overview of chromatin regulatory processes and the enzymes that regulate these in mammalian cells.
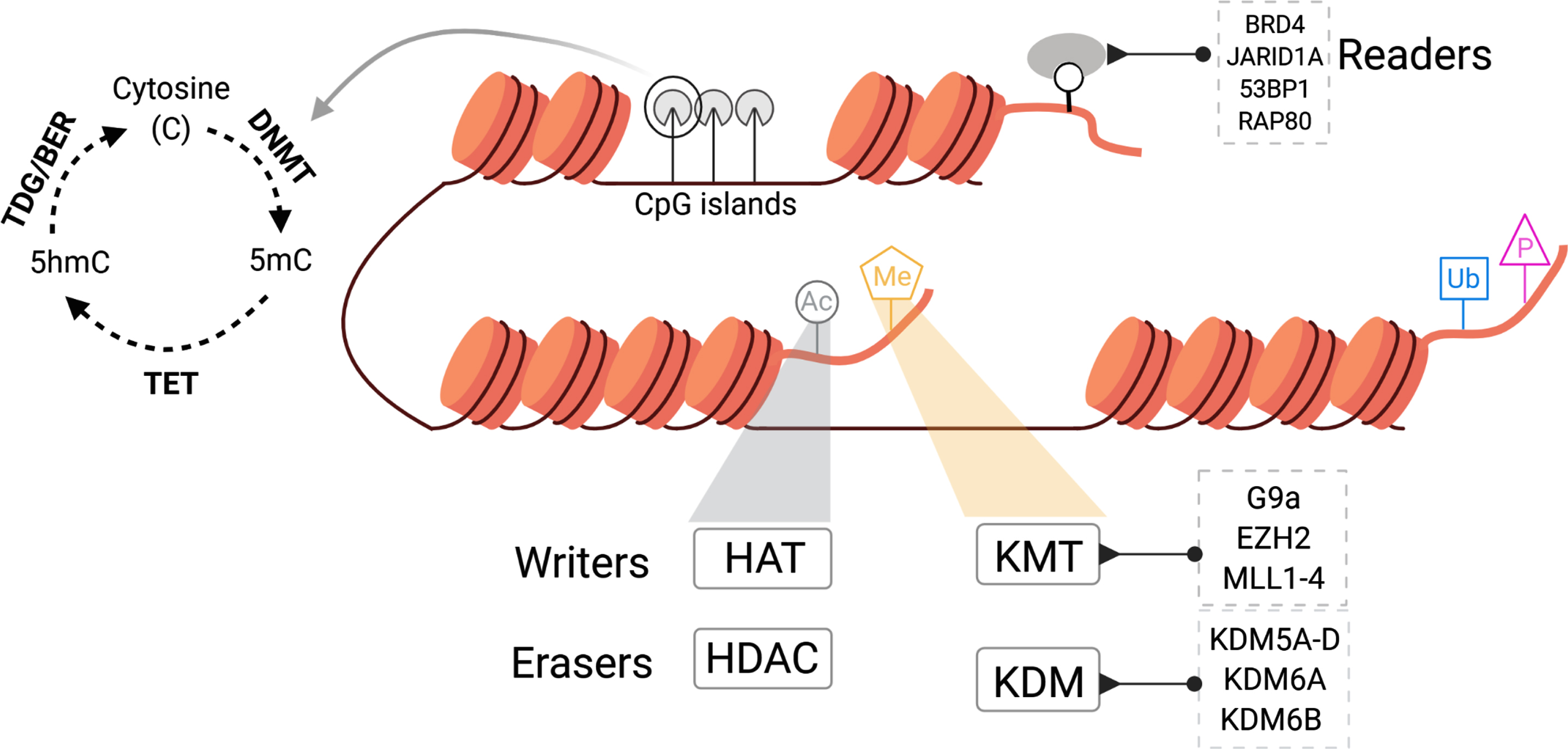
Chromatin modifying enzymes can be readers, writers, or erasers, aptly classified by their unique actions. Readers interpret the covalent modifications present on chromatin, writers add chromatin marks and erasers remove post translational modifications from the chromatin. Each of these enzymes lay down unique post-translational modifications such as methylation (Me), acetylation (Ac), phosphorylation (P), and ubiquitination (Ub) that inimitably modulate chromatin structure (Fig. 2). In this review, we highlight the state of epigenetic dysregulation in BCa focusing on current knowledge and therapeutic opportunities available to target chromatin modifiers.
DNA METHYLATION
Overview
Methylation is a dynamic, reversible, and non-sequence altering means of DNA modification that determines accessibility and transcription of DNA [43, 44]. Commonly found near gene promoters, DNA methylation is associated with transcriptional silencing. Cytosines found in palindromic CpG dinucleotide repeats are prime sites for DNA methylation. Promoters containing CpG islands, generally unmethylated, can be targeted by aberrant DNA methylation during cellular transformation, which is associated with transcriptional silencing of tumor suppressor genes. In a metastable phenomenon, DNA methylation patterns established at the time of tissue patterning and differentiation are passed on as epigenetic traits across cell generations [45]. Studies have identified silencing of genes by DNA methylation to be as common as loss-of-function mutations, if not a more prevalent mechanism of transcriptional repression in cancer [46, 47].
Regulators of DNA methylation
DNA methylation landscape is the output of DNA methyltransferases (DNMT1, DNMT3A, DNMT3B) and demethylase (TET1, TET2, TET3) activity [48, 49]. DNA methyltransferases add methyl groups at carbon-5 position of cytosine (5mC) while TET enzymes oxidize 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fc) and 5-carboxylcytosine (5caC) [50]. Mutations in DNA methyltransferases are found in 6% of MIBC tumors and 10% of MIBC cases harbor mutations in DNA demethylases. While several studies have explored DNA methylation status as a prognostic marker for BCa staging, subtyping, diagnosis or therapy outcome, the role of genes most affected by aberrant DNA methylation in triggering carcinogenesis remains unclear.
Causes of derailed DNA methylation in BCa
Epidemiological studies have identified a correlation between smoking and the risk of developing Bca [51–53]. Studies analyzing DNA methylation patterns in urothelial carcinomas have identified a CpG island hyper methylator phenotype to be associated with the intensity of cigarette smoking (Fig. 3).
Fig. 3
Carcinogen exposure leads to promoter hypermethylation in BCa.
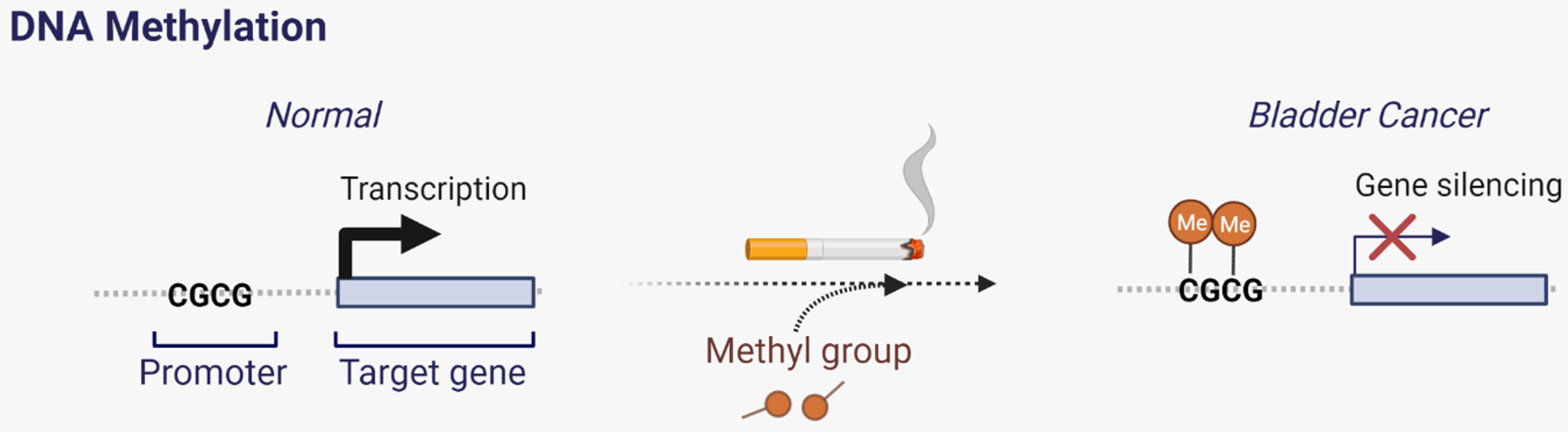
Furthermore, this increase in methylation was associated with silencing of tumor suppressor genes in high grade invasive bladder tumors [1, 54–57].
Additionally, an increase in methylated metabolites is observed in high grade tumors from smokers. Overall, there is strong evidence to suggest that external stimuli (such as smoking) can alter DNA methylation landscape in BCa [49, 55, 58]. In-depth analysis of variation in local DNA methylation levels between normal and disease state in larger cohorts can help establish prognostic biomarkers for early diagnosis of BCa.
Insights from clinical trials with DNA methylation inhibitors
Given the multidimensional effects of DNA methylation, inhibition of this process has wide ranging outcomes from inhibition of proliferation to induction of an anti-viral state manifested by de-repression of genes involved in antigen presentation and inflammatory responses [59]. Inhibition of DNA methylation has been explored as a therapeutic strategy in blood cancers and solid tumors. Cytidine analogues, azacytidine and decitabine have been approved for patient use in acute myeloid leukemia and myelodysplastic syndrome [60].
With the goal of targeting the altered DNA methylation landscape, S110, a derivative of decitabine showed tumor regression in mouse xenograft models [61]. DNMT1 inhibitors are currently in clinical trials for advanced BCa (NCT00030615, NCT02030067, NCT01478685) [62–64]. Results from a phase I trial designed to test the efficacy of 5-azacytidine combination with sodium phenylbutyrate failed to identify any clinical benefit associated with the combination in a cohort of patients with refractory solid tumors [65]. In-vitro studies in BCa cells exposed to decitabine point to enhanced expression of tumor antigens such as cancer testis antigens (CTA) leading to activation of immune-regulatory pathways. This opens up the avenue for utilizing immunotherapy to enhance the efficacy of DNMT inhibitors [66, 67].
REGULATION OF TRANSCRIPTIONAL STATE BY HISTONE MODIFICATIONS
Overview
Charged histone tails extending from nucleosome cores are prime sites for post-translational modifications. Several histone modifications such as deamination, citrullination, ADP-ribosylation, and O-GlcNAcylation play critical roles in regulation of chromatin structure during diverse cellular processes. Among these the most widely studied histone modifications are acetylation and methylation which are reviewed here [68].
Histone acetylation and deacetylation
Altered state of histone acetylation has been identified as a distinguishing feature of human tumors [69]. Relaxation of chromatin allows transcription machinery to access DNA thereby increasing transcriptional output from the locus. Acetyl marks added to positively charged lysine residues on histone tails neutralize electrostatic interactions, thereby allowing for localized unwrapping of chromatin. Acetylation is often associated with open chromatin, occurring mostly at promoter and enhancer elements and is regulated by activity of writers: histone acetyltransferases (also referred to as lysine acetyltransferases, HAT) and erasers: histone deacetylases (HDAC).
HATs belong to two major subgroups classified based on their functions: nuclear type-A HATs and cytoplasmic type-B HATs. Nuclear type-A HATs are comprised of three protein families: GNAT (KAT2A, KAT2B), MYST (KAT5, KAT6A, KAT6B, KAT7, KAT8) and p300/CBP (KAT3A, KAT3B) each with unique substrate specificity that regulates acetylation events leading to gene expression [70]. Cytoplasmic type-B HATs coordinate acetylation activities on newly formed histones prior to nucleosome assembly. Approximately 36% of MIBC tumors in TCGA cohort have a mutation in one or more HAT enzymes [11].
To date, 18 enzymes with histone deacetylase activity have been identified. Depending on structural homology and function, these enzymes are broadly classified into four classes: Class I (HDAC 1-3 and HDAC8), Class II (HDAC 4-7, HDAC9 and HDAC10), Class III (SIRT 1-7) and Class IV (HDAC11) [71].
Elevated expression levels of HDAC-1, HDAC-2 and HDAC-3 are noted in high grade urothelial tumors, however a definitive function of these proteins in bladder carcinogenesis remains unclear [72]. Silencing of histone deacetylases has been shown to inhibit cell growth in-vitro [73]. Additionally, studies investigating the usefulness of HDAC inhibitors in in-vitro models and animal studies have observed a decrease in cellular proliferation and a reduction in tumor volumes [74]. However, despite the encouraging pre-clinical results, HDAC inhibitors have failed to show efficacy in BCa patients (NCT02236195, NCT00363883) [75–78].
Histone methylation and demethylation
Histone methylation mainly occurs on the positively charged tails of histones H3 and H4 at sites of lysine and arginine residues [79]. The location of the methylated residue in the sidechain and the number of methyl-groups present on the amino-acid residue determine the effect of the modification on chromatin conformation and hence transcriptional state of the locus. Methylation at H3K4, H3K36 and H3K79 is associated with transcriptional activation whereas H3K9 and H3K27 are transcriptional repressor marks. H3K4me1 is highly enriched at enhancers while H3K4me3 is predominantly present at active promoters [80]. Histone methylation is coordinated by activity of histone methyltransferases and demethylases. More than 30 genes function as writers of histone methylation and approximately 18 genes belong to the KDM family of lysine demethylases [81].
Key players in histone methylation
The most well-studied proteins for their role in regulating transcription through histone methylation are the Polycomb (PcG) and Trithorax (TrxG) group of proteins [82, 83]. PcG and TrxG group of proteins play contrasting roles in regulating transcription. PcG proteins which encompass two major complexes, PRC1 and PRC2, repress transcription through histone methylation at H3K27 (PRC2) and ubiquitination (PRC1). TrxG proteins made up of COMPASS and COMPASS-like protein complexes behave as histone methyltransferases. SET1A (KMT2F), SET1B (KMT2G) and MLL1 (KMT2A), MLL2 (KMT2D), MLL3 (KMT2C), MLL4 (KMT2B) make up the catalytic subunits of distinct multi-subunit COMPASS complexes, each with discrete functions. Furthermore, KDM6A, a histone H3K27 demethylase, has also been identified as a member of the COMPASS complex [84]. Proteins within these structures regulate assembly, stability, and activity of the enzymatic complex [85]. Each of these enzymatic complexes regulates H3K4 methyltransferase in a unique manner impacting different functions. For example, SET1 is responsible for H3K4me3 at promoters of housekeeping genes, MLL1/2 complexes deposit H3K4me2/me3 at promoters of Hox genes and are hence crucial for the gene expression pattern of developmental genes and MLL3/4 protein complexes facilitate H3K4me1 marks at transcriptional enhancers.
Consequence of dysregulated histone methylation in BCa
Analysis of 11,000 tumors from 33 different types of cancer identified alterations in methyltransferases in 49% and alterations in demethylases in 30% of MIBC tumors [86]. In BCa, alterations in one or more histone methyltransferases and demethylases were identified in 48% and 36% of MIBC cases, respectively [11].
Role of H3K4 methyltransferases in BCa
Somatic mutations in the MLL genes occur in 46% of MIBC tumors [87]. The enzymatic SET domain within the MLL (KMT) family members confers histone methyltransferase activity to this group of proteins. While a relatively small fraction of MIBC tumors have mutations in the SET domain of these genes, a significantly large fraction of tumors harbor nonsense mutations or truncating deletions resulting in a non-functional protein.
Given the crucial role of MLL gene family during development and hematopoiesis, studying the role of MLL genes in cancer using mouse models has been a difficult task [88, 89]. While deletion of Mll1 is embryonic lethal, targeted deletion of Mll1 SET domain is sufficient to rescue viability, but mice exhibit visible developmental defects [89, 90]. Dependent on the developmental stage at which Mll2 is deleted, embryos are either non-viable or develop into mice with developmental defects [91].
Mll3 deletion is embryonically lethal and like Mll1, deletion of Mll3 SET domain has been shown to rescue viability in these mice, however, ureteral epithelial tumors were observed in nearly 50% of mice carrying a homozygous deletion of Mll3 SET domain. In a p53 + /- background, this phenotype was exacerbated with 100% of mice exhibiting accelerated carcinogenesis with formation of aggressive tumors [92–94]. Downregulation of MLL3 has been demonstrated to influence expression of DNA damage response and repair genes, with higher genome instability observed in cells with low levels of MLL3 [95]. Levels of MLL2 are lower in carcinomas compared to adjacent normal tissues, in addition, loss of MLL2 has been associated with a decrease in expression of tumor suppressors PTEN, P53 [96].
It is important to note that mutations in MLL3 are found in patients with Kleefstra syndrome, an intellectual disability disorder, de novo mutations in MLL2 are found in patients with Kabuki syndrome and heterozygous loss of function mutations in MLL1 are detected in patients with Wiedemann-Steiner syndrome [97–101]. While a small frequency of patients with Kabuki syndrome have developed cancer, further research is needed to determine whether mutations in MLL2 predispose to cancer [102]. Future mechanistic studies will be crucial to determine whether loss-of-function of MLL methyltransferases is sufficient to drive bladder carcinogenesis or if epigenetic reorganization caused due to loss of MLL activity creates an accommodating environment to accelerate oncogenic transformation in cases with synchronized loss of tumor suppressor genes.
Role of H3K9 and H3K27 methyltransferases in BCa
A unique multi-regional epigenetic silencing phenotype driven by variable H3K9 and H3K27 methylation levels was found to be associated with the carcinoma in-situ expression signature in MIBC [103]. Studies have identified a correlation between global H4K20 trimethylation levels and stage of BCa, with lower levels of H4K20me1/2/3 observed with progression to higher stage cancer [104]. In an independent cohort, a decrease in H3K9 and H3K27 methylation levels was observed in cancer tissue compared to normal controls and a further decrease in these levels was observed with disease stage [105]. Studies have identified a basal and luminal sub-type specific H3K4me1 peak pattern in MIBC tumors [106, 107]. Overall, evidence from multiple studies points to a decrease in transcriptional repressive marks with an increase in stage and invasiveness of BCa.
E2F1 has been identified as a regulator of EZH2 and SUZ12 expression levels and a high expression of the E2F1-EZH2-SUZ12 pathway is associated with aggressive Bca [108]. BRD4 has also been identified as an indirect transcriptional regulator of EZH2 [109]. Several studies have shown an increase in expression of EZH2 to be associated with more aggressive disease [110–113]. Ramakrishnan et al. have noted an EZH2 dependency in SWI/SNF mutated BCa cell-lines. Furthermore, using cell-line derived xenograft models, they demonstrated combination treatment with EZH2i and cisplatin to be an effective therapy for tumors with SWI/SNF mutations, and hypothesized EZH2i cell death in animal models to be dependent on immune responses via natural killer (NK) cell signaling [114]. A recent study identified a role for the PRC2 complex in evading immune surveillance by suppression of MHC-I antigen processing genes and a consequent increase in anti-tumor immunity upon pharmacological inhibition of this complex [115].
Methylation at H3K9 is associated with transcriptional repression. G9a, a H3K9 methyltransferase is over-expressed in BCa and its inhibition has been shown to impact cell survival by inducing autophagy [116–119]. Crosstalk between G9a and DNA methyltransferase DNMT1 has been shown to induce transcriptional silencing of target genes [120]. High levels of G9a and EZH2 have been detected in tumors from BCa patients who do not respond to anti-PD-1 immunotherapy [121].
Targeting histone methylation
Inhibition of hyperactive EZH2 has been explored as a therapeutic avenue in several different cancers [122]. EZH2 has two pockets for binding to S-adenosyl methionine (SAM), the methyl group donor for methylation reactions. Most EZH2 inhibitors currently in development target this SAM binding site within the SET domain [122]. Tazemostat, a SAM competitive inhibitor of EZH2 has recently been approved for treatment of advanced epithelioid sarcoma and refractory follicular lymphoma [123]. Several other inhibitors that function like tazemostat through competitive inhibition with SAM are currently in clinical trials [122]. Few studies have identified a role for EZH2 in epigenetic silencing of tumor immune signaling pathways [124, 125]. Pharmacological inhibition of EZH2 could enhance immunogenicity and sensitize tumors to immune checkpoint blockade approaches. Clinical trials investigating EZH2 inhibition along with anti-PD-1 immunotherapy (pembrolizumab) are currently ongoing (NCT03854474) [126]. Combinatorial inhibition of G9a and DNMT using the recently described dual G9a/DNMT inhibitor CM-272 has also been shown to effectively kill BCa cells in-vitro and in vivo in an immunocompetent transgenic mouse model of metastatic MIBC [121]. High levels of G9a and EZH2 have been detected in tumors from BCa patients who do not respond to anti-PD-1 immunotherapy. Combinatorial treatment with CM-272 and anti-PD-L1 was shown to decrease incidence of tumor as well as metastasis in a genetically engineered mouse model of MIBC.
Key players in histone demethylation
The process of demethylation in eukaryotes is carried out by two families of histone demethylases: Lysine specific demethylases (LSD) and Jumonji C domain (JMJC) containing proteins [127]. Each of these proteins differs in their substrate specificity [128]. KDM6 removes methyl groups at H3K27 site and KDM5 family of proteins remove methyl groups at H3K4 site.
Role of demethylases in BCa
The KDM5 family is altered in 9% of MIBC cases and KDM6A mutations are found in 26% of patients in TCGA MIBC cohort. A large fraction of these mutations have been identified to be loss-of-function mutations.
KDM6A (UTX) is a lysine demethylase located on the X-chromosome. Mutations in KDM6A are found at a higher frequency in low-grade NMIBC tumors (52%) in comparison to high grade tumors (24%) [129]. Additionally, a higher frequency of KDM6A mutations are detected in non-invasive bladder tumors from females compared to males [130]. In a mouse model, loss of KDM6A has been shown to increase risk of BCa incidence in female mice [131]. Ahn et al have proposed a compensatory role for KDM6A homolog UTY in KDM6A deficient cells [132]. It remains to be determined if combinatorial loss of KDM6A and UTY is a requirement for initiation of bladder carcinogenesis in males. Kobatake et al described activation of inflammatory pathways in KDM6A deficient mice and a cooperative effect between KDM6A and p53 on bladder tumorigenesis [133]. EZH2 hyper-dependence has been noted in tumors with loss-of-function alterations in KDM6A [134]. Given the high frequency of KDM6A mutations in BCa, this opens up the avenue for EZH2 inhibitors in treatment of KDM6A mutated BCa [135]. However, further research will be needed to determine if all KDM6A mutations will create epigenetic susceptibility to EZH2i.
In addition to KDM6A, several other histone demethylases have been studied in BCa. For example, JMJD1A (KDM3A) expression is elevated in BCa and has been associated with metabolic reprogramming that contributes to BCa progression [136–138]. High levels of KDM5B are detected in BCa tissues and have been shown to regulate expression of E2F1, thereby impacting cell proliferation [139]. High levels of KDM7A have been associated with increased proliferative capacity of BCa cells and cisplatin resistance in vitro [140].
Given the high frequency of loss-of-function mutations in chromatin modifying genes, the function of these proteins has been classified as oncogenic driver or tumor suppressive in BCa. However, high frequency of mutations in this group (in particular MLL2 and KDM6A) have also been identified in nonmalignant bladder urothelium. These results suggest that while clonal expansion of cells with mutations in chromatin modifiers is high in the normal urothelium, these cells are non-malignant and require additional events to trigger the process of carcinogenesis [13, 14]. For example, FGFR3 activation and KDM6A loss-of-function mutations commonly co-occur in MIBC. Recently, KDM6A loss of function mutations have been demonstrated to create a more permissive environment for FGFR3 activating mutations to drive bladder tumorigenesis [141]. While further work is needed to understand the mechanisms that drive this cooperativity, these results are encouraging and point to new roles for chromatin modifying enzymes in BCa.
Conclusions and future outlook
Dysregulation of the epigenomic landscape results in aberrant gene expression and is associated with a wide spectrum of diseases from developmental disorders to cancer [142–144]. Endeavors dedicated to sequencing tumors at different stages have identified genetic and epigenetic changes that define the evolving landscape of BCa [1, 11, 15]. While our review is focused primarily on DNA and histone methylation and histone acetylation, chromatin remodeling proteins are also frequently mutated in BCa and provide additional opportunities for therapeutic intervention [145, 146]. More mechanistic questions of epigenetic regulators require further investigation, it remains clear that chromatin modifying enzymes play a crucial role in maintaining bladder homeostasis. Even prior to malignant transformation, mutations in chromatin modifying enzymes are found at a high frequency in the urothelium, pointing to a currently unidentified role that these enzymes play in non-malignant urothelium [13, 14]. Further research dedicated to identifying dependencies specific to chromatin modifying enzymes in a BCa model may improve the range of therapeutic strategies that can be used to target this subset of tumors.
FUNDING
JJM is supported by a grant from the VHA BX003692, AP is supported by a grant from NCI: K99CA234434, LC is an American Urologic Association Research Scholar and a grant from the Society of Women in Urology.
AUTHOR CONTRIBUTIONS
KM: Conception, performance of work, interpretation of results, writing and review of article. LC: performance of work, interpretation of results, writing and review of article, AP: Conception, performance of work, interpretation of results, writing and review of article, JM: Conception, performance of work, interpretation of results, writing and review of article. All authors had access to data and manuscript.
ACKNOWLEDGMENTS
The authors have no acknowledgments
CONFLICTS
LC, KM: No conflicts of interest to report, AP: owns stock with Epizyme, JM: JJM participated in advisory boards for Ferring, AstraZeneca, BMS, Janssen, Foundation Medicine, and Merck and UroGen.
REFERENCES
[1] | TCGA Research Network et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. (2014) ;507: :315–22. |
[2] | Ferlay J , et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. International Journal of Cancer vol. 144 (Wiley-Liss Inc.). |
[3] | Key Statistics for Bladder Cancer. https://www.cancer.org/cancer/bladder-cancer/about/key-statistics.html. |
[4] | Sanli O , et al. Bladder cancer. Nature Reviews Disease Primers. (2017) ;3: :17022. |
[5] | Jackson AR , Ching CB , McHugh KM , Becknell B . Roles for urothelium in normal and aberrant urinary tract development. Nature Reviews Urology. (2020) ;17: :459–68. |
[6] | Amin MB . No Title. Modern Pathology. (2009) ;22: :S96–S118 (Nature Publishing Group, 2009). |
[7] | Meeks JJ , et al. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nature Reviews Urology. (2020) ;17: :259–70. |
[8] | Meeks JJ , et al. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nature Reviews Urology. (2020) ;17: . |
[9] | Glaser AP , Fantini D , Shilatifard A , Schaeffer EM , Meeks JJ . The evolving genomic landscape of urothelial carcinoma. Nature Reviews Urology. (2017) ;14: :215–29. |
[10] | Chalmers ZR , et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Medicine. (2017) ;9: :34. |
[11] | Robertson AG , et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. (2017) ;171: :540–556.e25. |
[12] | Kazanov MD , et al. APOBEC-Induced Cancer Mutations Are Uniquely Enriched in Early-Replicating, Gene-Dense, and Active Chromatin Regions. Cell Reports. (2015) ;13: :1103–9. |
[13] | Lawson ARJ , et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science (New York, N.Y.). (2020) ;370: :75–82. |
[14] | Li R , et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science (New York, N.Y.). (2020) ;370: :82–9. |
[15] | Nordentoft I , et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. (2014) ;7: :1649–63. |
[16] | Garczyk S , et al. Next-Generation Sequencing Reveals PotentialPredictive Biomarkers and Targets of Therapy for Urothelial Carcinoma in Situ of the Urinary Bladder. American Journal of Pathology. (2020) ;190: :323–32. |
[17] | Gui Y , et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature Genetics. (2011) ;43: :875–8. |
[18] | Guo G , et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nature Genetics. (2013) ;45: :1459–63. |
[19] | Kim PH , et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. European Urology. (2015) ;67: :198–201. |
[20] | Tan TZ , Rouanne M , Tan KT , Huang RYJ , Thiery JP . Molecular Subtypes of Urothelial Bladder Cancer: Results from a Meta-cohort Analysis of Tumors. European Urology. (2019) ;75: :423–32. |
[21] | Cazier JB , et al. Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nature Communications. (2014) ;5: :1–13. |
[22] | Pietzak EJ , et al. No Title. European Urology. (2017) ;72: :952–9. |
[23] | Woodcock CL , Ghosh RP . Chromatin higher-order structure and dynamics. Cold Spring Harbor Perspectives in Biology. (2010) ;2: . |
[24] | Hergeth SP , Schneider R . The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Reports. (2015) ;16: :1439–53. |
[25] | Olins DE , Olins AL . Chromatin history: Our view from the bridge. Nature Reviews Molecular Cell Biology. (2003) ;4: :809–14. |
[26] | Perino M , Jan G , Veenstra C . Developmental Cell Chromatin Control of Developmental Dynamics and Plasticity. (2016) doi: 10.1016/j.devcel.2016.08.004. |
[27] | Morgan MA , Shilatifard A . Chromatin signatures of cancer. Genes and Development. (2015) ;29: :238–49. |
[28] | Klemm SL , Shipony Z , Greenleaf WJ . Chromatin accessibility and the regulatory epigenome. Nature Reviews Genetics. (2019) ;20: :207–20. |
[29] | Polak P , et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. (2015) ;518: :360–4. |
[30] | Akdemir KC , et al. Somatic mutation distributions in cancer genomes vary with three-dimensional chromatin structure. Nature Genetics 2020;1-11. doi: 10.1038/s41588-020-0708-0. |
[31] | Hanahan D , Weinberg RA . Hallmarks of cancer: The next generation. Cell. (2011) ;144: :646–74. |
[32] | Esteller M . Cancer epigenomics: DNA methylomes and histone-modification maps. Nature Reviews Genetics. (2007) ;8: :286–98. |
[33] | Jones PA , Baylin SB . The Epigenomics of Cancer. Cell. (2007) ;128: :683–92. |
[34] | Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harbor Perspectives in Biology. (2016) ;8: . |
[35] | Amin MB . No Title. Modern Pathology. (2009) ;22: :S96–S118 (Nature Publishing Group 2009). |
[36] | Sanfrancesco J , Jones JS , Hansel DE . Diagnostically Challenging Cases. What are Atypia and Dysplasia? Urologic Clinics of North America. (2013) ;40: :281–93. |
[37] | Virk RKA , et al. Disordered chromatin packing regulates phenotypic plasticity. Science Advances. (2020) ;6: :eaax6232. |
[38] | Liu D , et al. Mutational patterns in chemotherapy resistant muscle-invasive bladder cancer. Nature Communications. (2017) ;8: :1–11. |
[39] | Kadoch C , Crabtree GR . Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Science Advancese. (2015) ;1: :1500447. |
[40] | Bowman GD , Poirier MG . Post-translational modifications of histones that influence nucleosome dynamics. Chemical Reviews. (2015) ;115: :2274–95. |
[41] | Wu C , Wong YC , Elgin SCR . The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell. (1979) ;16: :807–14. |
[42] | Schreiber SL , Bernstein BE . Signaling network model of chromatin. Cell. (2002) ;111: :771–8. |
[43] | Michalak EM , Burr ML , Bannister AJ , Dawson MA . The roles of DNA, RNA and histone methylation in ageing and cancer. Nature Reviews Molecular Cell Biology. (2019) ;20: :573–89. |
[44] | Lee JY , Lee TH . Effects of DNA methylation on the structure of nucleosomes. Journal of the American Chemical Society. (2012) ;134: :173–5. |
[45] | Horsthemke B . A critical view on transgenerational epigenetic inheritance in humans. Nature Communications. (2018) ;9: . |
[46] | Herman JG , Baylin SB . Gene Silencing in Cancer in Association with Promoter Hypermethylation. New England Journal of Medicine. (2003) ;349: :2042–54. |
[47] | Serizawa RR , et al. Integrated genetic and epigenetic analysis of bladder cancer reveals an additive diagnostic value of FGFR3 mutations and hypermethylation events. International Journal of Cancer. (2011) ;129: :78–87. |
[48] | Czamara D , et al. Integrated analysis of environmental and genetic influences on cord blood DNA methylation in new-borns. Nature Communications. (2019) ;10: :1–18. |
[49] | Marsit CJ , et al. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. (2006) ;27: :112–6. |
[50] | Wu X , Zhang Y . TET-mediated active DNA demethylation: mechanism, function and beyond. Nature Reviews Genetics. (2017) ;18: :517–34. |
[51] | Siegel RL , et al. Deaths Due to Cigarette Smoking for 12 Smoking-Related Cancers in the United States. JAMA Internal Medicine. (2015) ;175: :1574. |
[52] | Alexandrov LB , et al. Mutational signatures associated with tobacco smoking in human cancer. Science (New York, N.Y.). (2016) ;354: :618–22. |
[53] | Kim J , et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nature Genetics. (2016) ;48: :600–6. |
[54] | Wolff EM , et al. RUNX3 methylation reveals that bladder tumors are older in patients with a history of smoking. Cancer Research. (2008) ;68: :6208–14. |
[55] | Jin F , et al. Tobacco-Specific Carcinogens Induce Hypermethylation, DNA Adducts, and DNA Damage in Bladder Cancer. 759 (2017) doi:10.1158/1940-6207.CAPR-17-0198. |
[56] | Kitchen MO , et al. Quantitative genome-wide methylation analysis of high-grade non-muscle invasive bladder cancer. Epigenetics. (2016) ;11: :237–46. |
[57] | Bilgrami SM , Qureshi SA , Pervez S , Abbas F . Promoter hypermethylation of tumor suppressor genes correlates with tumor grade and invasiveness in patients with urothelial bladder cancer. Springer Plus. (2014) ;3: :1–9. |
[58] | Wilhelm-Benartzi CS , et al. Association of secondhand smoke exposures with DNA methylation in bladder carcinomas. Cancer Causes and Control. (2011) ;22: :1205–13. |
[59] | Tsai HC , et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell. (2012) ;21: :430–46. |
[60] | Bohl SR , Bullinger L , Rücker FG . Epigenetic therapy: azacytidine and decitabine in acute myeloid leukemia. Expert Review of Hematology. (2018) ;11: :361–71. |
[61] | Chuang JC , et al. S110, a 5-Aza-2′-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Molecular Cancer Therapeutics. (2010) ;9: :1443–50. |
[62] | Decitabine in Treating Patients With Advanced Solid Tumors - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT00030615. |
[63] | Dose-Finding and Safety Study for Oral Single-Agent to Treat Advanced Malignancies - Full Text View - ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT02030067. |
[64] | A Phase 1 Study of CC-486 as a Single Agent and in Combination With Carboplatin or ABI-007 in Subjects With Relapsed or Refractory Solid Tumors - Full Text View - ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT?term=NCT&draw=2&rank=1. |
[65] | Lin J , et al. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clinical Cancer Research. (2009) ;15: :6241–9. |
[66] | Ramakrishnan S , et al. Decitabine, a DNA-demethylating agent, promotes differentiation via NOTCH1 signaling and alters immune-related pathways in muscle-invasive bladder cancer. Cell Death and Disease. (2017) ;8: . |
[67] | Lobo J , Jerónimo C , Henrique R . Targeting the Immune system and Epigenetic Landscape of Urological Tumors. International Journal of Molecular Sciences. (2020) ;21: :829 21, 829. |
[68] | Audia JE , Campbell RM . Histone Modifications and Cancer. doi: 10.1101/cshperspect.a019521. |
[69] | Fraga MF , et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. (2005) ;37: :391–400. |
[70] | Bannister AJ , Kouzarides T . Regulation of chromatin by histone modifications. Cell Research. (2011) ;21: :381–95. |
[71] | Seto E , Yoshida M . Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harbor Perspectives in Biology. (2014) ;6: . |
[72] | Poyet C , et al. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clinical Pathology. (2014) ;14: . |
[73] | Junqueira-Neto S , et al. Phenotypic impact of deregulated expression of class I histone deacetylases in urothelial cell carcinoma of the bladder. Molecular Carcinogenesis. (2015) ;54: :523–31. |
[74] | Giannopoulou AF , et al. Revisiting histone deacetylases in human tumorigenesis: The paradigm of urothelial bladder cancer. International Journal of Molecular Sciences. (2019) ;20: . |
[75] | Grivas P , et al. Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes. Cancer. (2019) ;125: :533–40. |
[76] | Phase I Study of Vorinostat in Combination With Docetaxel in Patients With Advanced and Relapsed Solid Malignancies. - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT?term=NCT&draw=2&rank=1. |
[77] | Vorinostat in Treating Patients With Locally Recurrent or Metastatic Cancer of the Urothelium - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT?term=NCT&draw=2&rank=1. |
[78] | Quinn DI , et al. Phase II study of the histone deacetylase inhibitor vorinostat (Suberoylanilide Hydroxamic Acid; SAHA) in recurrent or metastatic transitional cell carcinoma of the urothelium - an NCI-CTEP sponsored: California Cancer Consortium trial, NCI 6879. Investigational New Drugs. (2021) ;39: :3 39, 812–820. |
[79] | Zhang Y , Reinberg D . Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes and Development. (2001) ;15: :2343–60. |
[80] | Heintzman ND , et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics. (2007) ;39: :311–8. |
[81] | Hyun K , Jeon J , Park K , Kim J . Writing, erasing and reading histone lysine methylations. Experimental and Molecular Medicine. (2017) ;49: :324. |
[82] | Andrea P , Ali S . The roles of Polycomb repressive complexes in mammalian development and cancer. Nature reviews. Molecular Cell Biology. (2021) ;22: :326–45. |
[83] | Andrea P , Ali S . Epigenetic balance of gene expression by Polycomb and COMPASS families. Science (New York, N.Y.). (2016) ;352: . |
[84] | Issaeva I , et al. Knockdown of ALR (MLL2) Reveals ALR Target Genes and Leads to Alterations in Cell Adhesion and Growth. Molecular and Cellular Biology. (2007) ;27: :1889–903. |
[85] | Shilatifard A . The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annual Review of Biochemistry. (2012) ;81: :65–95. |
[86] | Ding L , et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell. (2018) ;173: :305–320.e10. |
[87] | Rao RC , Dou Y . Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nature Reviews Cancer. (2015) ;15: :334–46. |
[88] | Yu BD , Hess JL , Horning SE , Brown GAJ , Korsmeyer SJ . Altered Hox expression and segmental identity in Mll-mutant mice. Nature. (1995) ;378: :505–8. |
[89] | Terranova R , Agherbi H , Boned A , Meresse S , Djabali M . Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proceedings of the National Academy of Sciences of the United States of America. (2006) ;103: :6629–34. |
[90] | Mishra BP , et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Reports. (2014) ;7: :1239–47. |
[91] | Glaser S , et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics and Chromatin. (2009) ;2: :5. |
[92] | Lee JWJ , et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proceedings of the National Academy of Sciences of the United States of America. (2009) ;106: :8513–8. (National Academy of Sciences, 2009). |
[93] | Lee S , et al. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferase. Proceedings of the National Academy of Sciences of the United States of America. (2006) ;103: :15392–7. |
[94] | Lee J , et al. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proceedings of the National Academy of Sciences of the United States of America. (2008) ;105: :19229–34. |
[95] | Rampias T , et al. The lysine-specific methyltransferase KMT 2C/ MLL 3 regulates DNA repair components in cancer. EMBO Reports. (2019) ;20: . |
[96] | Sun P , et al. KMT2D inhibits the growth and metastasis of bladder Cancer cells by maintaining the tumor suppressor genes. Biomedicine and Pharmacotherapy. (2019) ;115: :108924. |
[97] | Bögershausen N , et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Human Mutation. (2016) ;37: :847–64. |
[98] | Lebrun N , et al. Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. European Journal of Human Genetics. (2018) ;26: :107–16. |
[99] | Jones WD , et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. American Journal of Human Genetics. (2012) ;91: :358–64. |
[100] | Ng SB , et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature Genetics. (2010) ;42: :790–3. |
[101] | Kleefstra T , et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. American Journal of Human Genetics. (2012) ;91: :73–82. |
[102] | Cheon CK , Ko JM . Kabuki syndrome: Clinical and molecular characteristics. Korean Journal of Pediatrics. (2015) ;58: :317–24. |
[103] | Vallot C , et al. A novel epigenetic phenotype associated with the most aggressive pathway of bladder tumor progression. Journal of the National Cancer Institute. (2011) ;103: :47–60. |
[104] | Schneider A.-C et al. , . Global histone H4K20 trimethylation predicts cancer-specific survival in patients with muscle-invasive bladder cancer. BJU International. (2011) ;108: :E290–E296. |
[105] | Ellinger J , et al. No Title. Urologia Internationalis. (2014) ;93: :113–8. |
[106] | van der Vos KE , et al. Epigenetic profiling demarcates molecular subtypes of muscle-invasive bladder cancer. Scientific Reports. (2020) ;10: :10952. |
[107] | Iyyanki T , et al. Subtype-specific epigenomic landscape and 3D genome structure in bladder cancer. doi: 10.1101/2020.02.26.966697. |
[108] | Lee SR , et al. Activation of EZH2 and SUZ12 Regulated by E2F1 Predicts the Disease Progression and Aggressive Characteristics of Bladder Cancer. Clinical Cancer Research. (2015) ;21: :5391–403. |
[109] | Wu X , et al. BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Molecular Cancer Therapeutics. (2016) ;15: :1029–42. |
[110] | Wang H , et al. Increased EZH2 protein expression is associated with invasive urothelial carcinoma of the bladder. Urologic Oncology: Seminars and Original Investigations. (2012) ;30: :428–33. |
[111] | Zhou X , et al. Increased expression of EZH2 indicates aggressive potential of urothelial carcinoma of the bladder in a Chinese population. Scientific Reports. (2018) ;8: :1–9. |
[112] | Warrick JI , et al. Enhancer of zeste homolog 2 (EZH2) expression in bladder cancer. Urologic Oncology: Seminars and Original Investigations. (2016) ;34: :258.e1–258.e6. |
[113] | Raman JD , et al. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clinical Cancer Research. (2005) ;11: :8570–6. |
[114] | Ramakrishnan S , et al. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death and Differentiation. (2019) ;26: :2100–14. |
[115] | Burr ML , et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell. (2019) ;36: :385–401.e8. |
[116] | Zhu D , Barry E , Sankin AI . Enhancing response to immunotherapy in urothelial carcinoma by targeted inhibition of the histone methyltransferase G9a pathway. Translational Andrology and Urology. (2019) ;8: :S469–S471. |
[117] | Li F , et al. G9a Inhibition Induces Autophagic Cell Death via AMPK/mTOR Pathway in Bladder Transitional Cell Carcinoma. PLOS ONE. (2015) ;10: :e0138390. |
[118] | Cao Y , peng AI et al. Inhibition of G9a by a small molecule inhibitor, UNCinduces apoptosis of human bladder cancer cells. Acta Pharmacologica Sinica. (2019) ;40: :1076–84. |
[119] | Cho HS , et al. Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia. (2011) ;13: :676–84. |
[120] | Tachibana M , Matsumura Y , Fukuda M , Kimura H , Shinkai Y . G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO Journal. (2008) ;27: :2681–90. |
[121] | Segovia C , San José-Enériz E , Munera-Maravilla E . Inhibition of a G9a/DNMT network triggers immunemediated bladder cancer regression. Nature Medicine doi: 10.1038/s41591-019-0499-y. |
[122] | Kim KH , Roberts CWM . Targeting EZH2 in cancer. Nature Medicine. (2016) ;22: :128–34. |
[123] | TAZVERIKTM (tazemetostat) tablets. https://www.tazverik.com/?gclid=EAIaIQobChMIzvOmi7Wt6wIVWgOzAB380wuQEAAYASAAEgLEEvD_BwE. |
[124] | Peng D , et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. (2015) ;527: :249–53. |
[125] | Nagarsheth N , et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Research. (2016) ;76: :275–82. |
[126] | Tazemetostat and Pembrolizumab in Treating Patients With Locally Advanced or Metastatic Urothelial Carcinoma - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03854474?term=Tazemetostat&cond=Bladder+Cancer&draw=2&rank=1. |
[127] | Kooistra SM , Helin K . Post-translational modifications: Molecular mechanisms and potential functions of histone demethylases. Nature Reviews Molecular Cell Biology. (2012) ;13: :297–311. |
[128] | D’Oto A , Tian Q.-W , Davidoff AM , Yang J . Histone demethylases and their roles in cancer epigenetics. Journal of Medical Oncology and Therapeutics. (2016) ;1: :34–40. |
[129] | Pietzak EJ , et al. Next-generation Sequencing of Nonmuscle Invasive Bladder Cancer Reveals Potential Biomarkers and Rational Therapeutic Targets. European Urology. (2017) ;72: :952–9. |
[130] | Hurst CD , et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell. (2017) ;32: :701–715.e7. |
[131] | Kaneko S , Li X . X chromosome protects against bladder cancer in females via a KDM6A-dependent epigenetic mechanism. Science Advances. (2018) ;4: :5598. |
[132] | Ahn J , et al. Target sequencing and CRISPR/Cas editing reveal simultaneous loss of UTX and UTY in urothelial bladder cancer. Oncotarget. (2016) ;7: :63252–60. |
[133] | Kohei Kobatake A , et al. Kdm6a deficiency activates inflammatory pathways, promotes M2 macrophage polarization, and causes bladder cancer in cooperation with p53 dysfunction Running title Kdm6a deficiency in bladder cancer. https://bloodcancerdiscov.aacrjournals.org. |
[134] | Wang L , et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nature Medicine. (2018) ;24: :758–69. |
[135] | Ler LD , et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Science Translational Medicine. (2017) ;9: . |
[136] | Wan W , et al. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α. Oncogene. (2017) ;36: :3868–77. |
[137] | Lan W , Zhang D , Jiang J . The roles of LSD1-mediated epigenetic modifications in maintaining the pluripotency of bladder cancer stem cells. Medical Hypotheses. (2013) ;81: :823–5. |
[138] | Hayami S , et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. International Journal of Cancer. (2011) ;128: :574–86. |
[139] | Hayami S , et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: Involvement in the proliferation of cancer cells through the E2F/RB pathway. Molecular Cancer. (2010) ;9: :59. |
[140] | Lee KH , et al. Histone Demethylase KDM7A Regulates Androgen Receptor Activity, and Its Chemical Inhibitor TC-E Overcomes Cisplatin-Resistance in Bladder Cancer Cells. International Journal of Molecular Sciences. (2020) ;21: . |
[141] | Barrows D , Feng L , Carroll TS , Allis CD . Loss of UTX/KDM6A and the activation of FGFR3 converge to regulate differentiation gene-expression programs in bladder cancer. Proceedings of the National Academy of Sciences of the United States of America. (2020) ;117: :25732–41. |
[142] | Luo C , Hajkova P , Ecker JR . Dynamic DNA methylation: In the right place at the right time. Science. (2018) ;361: :1336–40. |
[143] | Xie P , Zang LQ , Li XK , Shu Q . An epigenetic view of developmental diseases: new targets, new therapies. World Journal of Pediatrics. (2016) ;12: :291–7. |
[144] | Goldberg AD , Allis CD , Bernstein E . Epigenetics: A Landscape Takes Shape. Cell. (2007) ;128: :635–8. |
[145] | Hoffmann MJ , Schulz WA . Alterations of chromatin regulators in the pathogenesis of urinary bladder urothelial carcinoma. Cancers. (2021) ;13: :6040. |
[146] | Valencia AM , Kadoch C . Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nature Cell Biology. (2019) ;21: :152–61 (Nature Publishing Group 2019). |


