
FGFR3 – a Central Player in Bladder Cancer Pathogenesis?
Abstract
The identification of mutations in FGFR3 in bladder tumors in 1999 led to major interest in this receptor and during the subsequent 20 years much has been learnt about the mutational profiles found in bladder cancer, the phenotypes associated with these and the potential of this mutated protein as a target for therapy. Based on mutational and expression data, it is estimated that >80% of non-muscle-invasive bladder cancers (NMIBC) and ∼40% of muscle-invasive bladder cancers (MIBC) have upregulated FGFR3 signalling, and these frequencies are likely to be even higher if alternative splicing of the receptor, expression of ligands and changes in regulatory mechanisms are taken into account. Major efforts by the pharmaceutical industry have led to development of a range of agents targeting FGFR3 and other FGF receptors. Several of these have entered clinical trials, and some have presented very encouraging early results in advanced bladder cancer. Recent reviews have summarised the drugs and related clinical trials in this area. This review will summarise what is known about the effects of FGFR3 and its mutant forms in normal urothelium and bladder tumors, will suggest when and how this protein contributes to urothelial cancer pathogenesis and will highlight areas that may benefit from further study.
FGFR3 STRUCTURE AND ACTIVATION
FGF receptors are transmembrane receptor tyrosine kinases consisting of three extracellular immunoglobulin (Ig) domains, a single-pass transmembrane domain and an intracellular split kinase domain (Fig. 1A). There are four full-length receptors that conform to this structure (FGFRs 1–4). A fifth receptor (FGFRL1) lacks the intracellular domain and is thought to antagonise FGFR signalling by acting as a decoy receptor [6]. There are 22 known human fibroblast growth factors (FGFs), 18 of which are secreted and interact with FGFRs [7]. These bind to the extracellular domain of the receptor, with differential ligand-binding specificity determined by alternative splicing of FGFRs in Ig domain III. Although there are only 4 FGFRs, alternative splicing and the large number of FGFs provide highly variable signalling potential. In FGFR3, alternative inclusion of exons 8 or 9 generates isoforms 3b and 3c respectively (Fig. 1A). FGFs in the extracellular matrix interact with heparan sulphate proteoglycans (HSPGs) and binding of HSPGs by FGFRs helps to stabilise the ligand-receptor interaction. FGFR3-3b, expression of which is confined to normal epithelial tissues, binds FGF1 strongly and FGFs 9, 16 and 20 with weaker affinity, whereas FGFR3-3c can bind FGF2 and many more FGFs [8]. Binding of an FGF to a monomeric FGFR induces receptor dimerization, autophosphorylation of multiple tyrosine residues in the kinase domain and activation of downstream signalling pathways [9]. The activated receptor binds and phosphorylates FRS2 which recruits GRB2 and SOS to activate the RAS/MAPK pathway [10]. Phosphorylation of GAB1 leads to recruitment and activation of phosphatidylinositol 3-kinase (PI3K) [11], and PLCγ binding to FGFR3 independently of FRS2 leads to activation of protein kinase C [12]. Activation of STAT [13, 14] and RSK2 [15] can also occur. Signalling output, which is highly dependent on cell context, can lead to a range of changes including proliferation, migration and differentiation [7].
Fig. 1
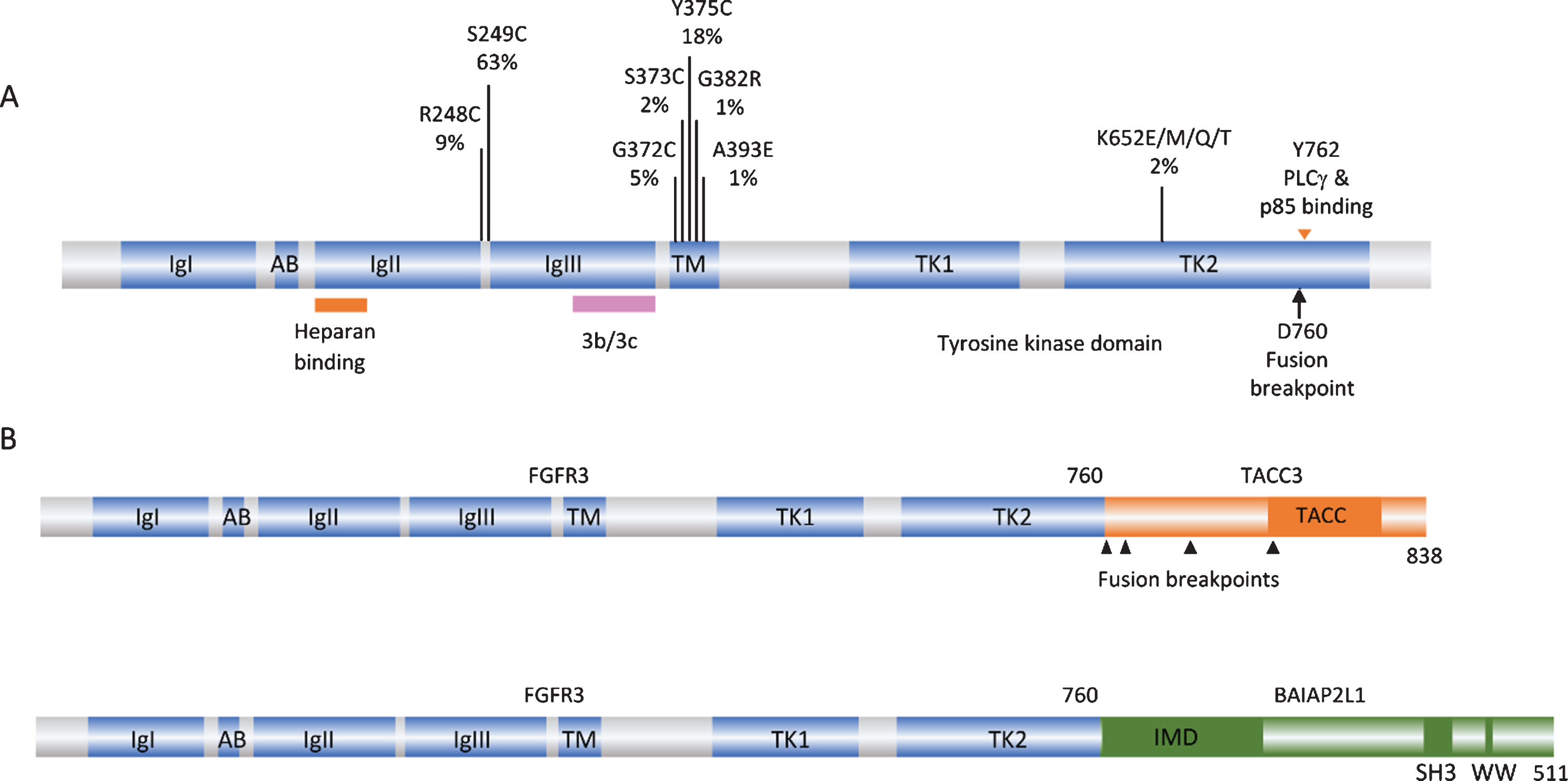
FGFR3 point mutations and translocations in bladder cancer. A. Structure of FGFR3 isoform 3b protein showing position of point mutations. Mutation data taken from tumors of all grades and stages (COSMIC, June 2020). IgI, IgII, IgIII: immunoglobulin-like domains. AB: acid box. TK1 and TK2: split tyrosine kinase domain. 3b/3c: region of exons 8 and 9 where alternative splicing generates isoforms 3b and 3c. B. Examples of FGFR3 fusion proteins identified in bladder tumors. TACC: transforming acid coiled-coil. IMD: IRDp53/MIM homology. SH3: src homology.
FGFR3 EXPRESSION IN THE NORMAL UROTHELIUM AND CULTURED NORMAL UROTHELIAL CELLS
In human urothelium, FGFR3 is the most abundantly expressed FGF receptor at both mRNA and protein levels [16, 17]. Normal human urothelial cells are highly proliferative in culture and can be maintained for multiple passages before senescence [18]. During this finite lifespan, FGFR3 mRNA and protein levels increase when cells are proliferatively quiescent at confluence and there is a major increase in expression as cells approach senescence [16]. These findings in normal urothelial cells provide clues that regulation of FGFR3 may be linked to urothelial contact inhibition and/or senescence.
The 3b isoform is the predominant full length mRNA FGFR3 isoform in normal urothelial cells. However, an isoform lacking exons 8–10 (Δ8–10) was found to dominate in cultured normal cells and, like the full length form, is upregulated at confluence. This isoform, which lacks the region encoding the second part of the third Ig-like loop and the transmembrane domain, is translated, glycosylated and secreted. It can bind FGF1 and dimerise, and was able to block the response to FGF1 in cells expressing full-length FGFR3. As Δ8–10 is expressed at lower levels in tumor cell lines, this implies that by binding and sequestering FGFs, or by binding and inhibiting signalling by the full-length receptor, this isoform may perform an important negative regulatory role in the normal urothelium [16].
ABERRANT ACTIVATION OF FGFR3 IN BLADDER CANCER
Alterations affecting FGFR3 signalling are found more frequently in bladder than in any other cancer type. Aberrant activation of the receptor can occur via several mechanisms. These are related to tumor stage and grade, and have different effects on downstream signalling and phenotypic consequences. Such aberrant activation may occur at different stages during the pathogenesis of NMIBC and MIBC and may cooperate with other events in a context-dependent manner.
FGFR3 point mutation
The first and most common mechanism of activation is missense point mutation [1, 19–24]. Mutations are located in several hotspots in the protein (Fig. 1A). By far the most common (63% of reported mutations) is S249C, followed by Y375C, these two mutations accounting for >80% of all mutations detected. These point mutations show a strong relationship to low tumor grade and stage [25]. Up to 85% of papillary urothelial neoplasms of low malignant potential (PUNLMP) and stage Ta tumors have a mutation, stage T1 tumors have lower frequency (∼20–40%) and MIBC lower still (12–15%) [17, 19, 20, 26–31].
When present in the germline, the common mutations found in bladder cancer cause severe, autosomal dominant and lethal forms of skeletal dysplasia. R248C, S249C, G370/372C and Y373/375C (3c/3b isoform numbering) cause thanatophoric dysplasia type I (TDI), and the kinase domain mutation K650/652E causes thanatophoric dysplasia type II (TDII) [32]. In this context, FGFR3 activation leads to premature cessation of proliferation of chondrocytes in the long bone growth plates [33].
Mutations resulting in replacement with a cysteine residue were assumed to cause formation of a disulphide bond leading to constitutive receptor activation. These mutant forms do indeed lead to constitutive receptor phosphorylation and ligand-independence [34], though when the strength of the dimers formed was measured by Förster resonance energy transfer (FRET) it was found to be relatively modest, suggesting that other structural perturbations in the dimer may contribute to the strong downstream signalling induced by these mutant forms [35]. In contrast, the K650/652E mutant form exists as a ligand-independent constitutively phosphorylated monomer [34, 36], and G380/382R and A391/393E remain predominantly ligand-dependent for activation [37, 38].
Although S249C is the most common mutation found in bladder cancer, R248C is much more common in the germline. These mutations are also found in the benign skin lesions seborrheic keratosis and epidermal nevi where, as in the germline, R248C is the more common mutation [39]. What determines the difference in prevalence in these situations? Several factors might contribute; there may be differential tolerance of certain mutations in the entire organism versus specific somatic tissues, somatic selection pressures may select for one rather than the other depending on context-dependent function, or the mutational processes at work may differ. An elegant study recently examined the relationship of S249C mutations and APOBEC-mediated mutagenesis in bladder cancer [40]. A large proportion of both NMIBC and MIBC have elevated expression of APOBEC enzymes and their mutational profiles contain a high load of predicted APOBEC-mediated mutational events [30, 41]. Of all FGFR3 point mutations found in bladder cancer, only S249C (TCC > TGC) shows similarity to an APOBEC-type mutation (TCN > T[G/T]N) where N is usually A or T. When the distribution of S249C mutations was related to APOBEC mutational load, a clear relationship was found in both NMIBC and MIBC [40]. APOBEC mutagenesis preferentially targets lagging strand ssDNA templates and it was shown that the coding strand of FGFR3 is mainly replicated as a lagging strand. The sequence context of S249 is predicted to allow a hairpin structure to form, also favoured by APOBEC enzymes. This study also demonstrated that the S249 codon sequence can be deaminated by APOBEC3A, providing a highly persuasive explanation for the excess of S249C mutations in bladder tumors. The differential mutation frequency in tumors of different stages, despite higher APOBEC activity in MIBC, likely reflects distinct pathogenesis pathways of these tumor groups.
Whether there are any differences in the selective advantage of the two most common mutations is not entirely clear. Although the most common mutations, S249C and Y375C, have similar oncogenic potency in immortal mesenchymal cells [40, 42], it is not certain that both confer the same advantage in urothelial cells. Knockdown of FGFR3 in bladder tumor cell lines bearing these mutations was shown to have similar transcriptional effects [40]. However, in cultured telomerase-immortalized normal urothelial cells (TERT-NHUC), a difference in the potency of these mutations has been found. Both mutations drive cells to a higher cell density at confluence, but S249C has the more potent effect [42]. Y375C is less strongly dimerised and retains some ligand dependence, suggesting that it may rely on stromal or autocrine ligand for maximal effect. Whether ligands are available in vivo at the time of receptor mutation is currently unknown. Thus S249C predominance can be explained as both APOBEC target and possibly in provision of a more potent selective advantage.
The strength of the phenotype of increased proliferation and cell viability at confluence in TERT-NHUC expressing point mutant forms of FGFR3 is in the order of mutation frequency found in tumors (S249C>Y375C>K652E) despite the fact that K652E is the most highly phosphorylated form in these cells [42]. This lack of contact inhibition was reflected in changes in cell-cell and cell-substrate adhesion, with the appearance of looser cell-cell junctions in cells expressing S249C- and Y375C-FGFR3 compared to controls and cells expressing high levels of wildtype FGFR3 [43]. The cells detached more readily from culture vessels coated with collagen IV, collagen I and fibronectin, and changes in expression of genes involved in mediating cell-cell and cell-substrate adhesion and extracellular matrix remodelling were found. This included downregulation of EPCAM and genes encoding structural components of desmosomes (DSC2, DSC3, DSG1, PKP1, PKP3), adherens junctions (CDH1, CDH16) and focal adhesions (PXN, ZYX) and upregulation of several integrins (ITGA2, ITGAV, ITGB5, ITGB6) and genes involved in extracellular matrix remodelling (HAS3, PLAT, PLAU, PLAUR, MMP10). Several of these genes were conversely altered when S249C-FGFR3 was knocked down in a mutant tumor cell line [43]. MMP10 has previously been identified as an easily-accessible pharmacodynamic biomarker for FGFR-targeted therapy in bladder cancer. It was also downregulated when FGFR3 was silenced, and both MMP1 and MMP10 levels were reduced in the urine of bladder cancer patients in a phase I trial of anti-FGFR3 monoclonal antibody [44]. Taken together, these data indicate that mutant FGFR3 has effects on regulation of the cell cycle in response to cell-cell contact, elicits changes in cell junctions and cell adherence to proteins found in the urothelial basement membrane and adjacent connective tissue [45], and induces changes in expression of potent modulators of the extracellular matrix, all functions predicted to provide a selective advantage to cells early in the process of tumor development.
FGFR3 fusion proteins
FGFR3 can also be activated by the generation of fusion proteins. These were initially identified when aberrant high molecular weight forms of FGFR3 were detected in cell lines and tumors [46]. Such fusions have subsequently been found in 2–6% of MIBC [30, 31, 47–49]. FGFR3-TACC3 and FGFR3-BAIAP2L1 chimeric proteins have been identified, with FGFR3-TACC3 fusions appearing most common. Activating point mutations have not been found in these fusions. NMIBC have not been examined, though the very high frequency of point mutations in Ta tumors suggests that any fusions are likely to be found in T1 cases.
All of the fusions described to date contain the entire sequence of FGFR3 apart from the final exon (amino acids 1–760) fused to C-terminal regions of the fusion partner. Examples are shown in Fig. 1B. TACC3 lies 48 kb telomeric to FGFR3 on chromosome arm 4p and the common mechanism of fusion is tandem duplication and insertion so that the coiled-coil region of TACC3 is fused in-frame to FGFR3 exon 18. As a result of this mechanism, low-level gain of the region is detected in cases with translocation.
These fusion proteins show some constitutive dimerization and are constitutively phosphorylated, though they do retain some ligand-dependence [46, 50], and they are potently transforming in immortal rodent mesenchymal cells [46, 48]. The fusion partners are predicted to induce dimerization, as the coiled-coil domain of TACC3 is retained almost intact in all FGFR3-TACC3 fusions and the IMD domain, related to the BAR (Bin-Amphiphysin-Rvs) domain which can also promote dimerization, is retained in FGFR3-BAIAP2L1. As BAR domains are predicted to interact with membranes [51], FGFR3-BAIAP2L1 may show altered cellular localisation. Early experiments in glioblastoma, where similar FGFR3-TACC3 fusions are found, indicated increased aneuploidy when FGFR3-TACC3 was expressed [52]. TACC3 normally provides stability to the mitotic spindle and altered levels of expression are associated with changes in mitotic progression and chromosome segregation [53]. As FGFR3-TACC3 could be detected at the spindle poles, it was proposed that this directly caused defects in chromosome segregation [52]. However, the majority of FGFR3-TACC3 fusions lack TACC3 Ser558, phosphorylation of which is required for formation of the TACC3-clathrin-ch-TOG complex at the spindle [54]. More recently, it has been shown that the fusion protein recruits wildtype TACC3 away from the spindle and this was demonstrated in bladder tumor cell lines RT112 and RT4, which contain FGFR3-TACC3 fusions with different TACC3 components. The effect could be induced by the TACC3 part of the fusion protein alone and could be rescued by low-level overexpression of wildtype TACC3. Inhibition of the kinase activity of the fusion protein with the FGFR inhibitor PD173074 did not abrogate the mitotic defects, clearly indicating a role for the fusion partner component alone [55]. Thus the contribution of the FGFR3-TACC3 fusion proteins to the neoplastic phenotype comprises not only FGFR3 kinase-regulated functions but also a contribution to genomic instability via the TACC3 component.
Upregulated expression and isoform switching
Precise regulation of expression of appropriate FGFR3 isoform and ligands is required for normal physiological processes. Studies of protein expression by immunohistochemistry in stage Ta and T1 bladder tumors show upregulated expression of FGFR3 in 70–80% of Ta and 40–70% of T1 tumors [17, 56–59]. In Ta tumors this directly relates to mutation frequency but in T1 tumors exceeds the known mutation frequency and indicates likely upregulation of wildtype protein. Although activating point mutations in FGFR3 are relatively uncommon in MIBC, upregulated expression of the protein is found in 30–50% of cases [17, 59–62], though high-level amplification has not been reported. Whilst a few of the tumors expressing high levels might contain fusion genes, the majority are likely to be wildtype. Figure 2 shows the relationship of expression to mutation status in relation to tumor stage in a one-year cohort of tumors diagnosed at a single Institution [17]. Studies that have examined expression in matched tumor and lymph node metastases [60–62] have found good concordance, suggesting that upregulated FGFR3, particularly if related to FGFR3 mutation, could be a valid therapeutic target in such cases. However, some evidence suggests that upregulated expression in the absence of mutation may not denote FGFR3-dependence. Thus it is notable that upregulated expression in FGFR3 wildtype tumors in a series of patients treated by cystectomy was not associated with prognosis in the same way as mutation [63] and in a trial of Dovitinib (a multi-kinase inhibitor that inhibits FGFRs 1–3) in BCG-unresponsive NMIBC, complete responses at 6 months were only observed in patients with mutant and not over-expressing wildtype tumors [64].
In bladder tumor cell lines, expression of FGFR3 was found to be restricted to those that also expressed E-cadherin and TP63, both of which are “epithelial” characteristics, whereas cell lines expressing ZEB1 and vimentin, features of epithelial-mesenchymal transition (EMT), expressed FGFR1 rather than FGFR3. Compatible with the relationship of FGFR1 to the EMT phenotype, inhibition of FGFR1 led to decreased invasion in these cell lines [65]. In bladder tumor cell lines with epithelial phenotype, ectopic expression of FGFR1 and treatment with FGF2 can elicit an EMT [66], suggesting that there are highly distinct roles for these two receptors in bladder cancer. This is supported by studies of bladder tumors, where a non-overlapping pattern of mRNA expression of FGFR3 and FGF2, a potent ligand for FGFR1, is found, with FGF2-expressing tumors showing EMT characteristics [67]. As expected, at the protein level FGFR1 is upregulated in tumors of higher grade and stage, in which FGFR3 mutations are less common [68, 69]. However, the relative expression of FGFR1 and FGFR3 has not been examined systematically in a large tumor series.
In addition to changes in expression level, isoform switching to the 3c isoform, which binds a wide range of FGFs, is also predicted to play a role, potentially facilitating autocrine or paracrine signalling. This has been identified in MIBC-derived tumor cell lines [16] but has not been examined in tumor tissues. However, upregulated expression of FGF2 is common in advanced bladder tumors [67, 70, 71] and increased levels of FGF1 and FGF2 can be detected in the urine of bladder cancer patients [72–75]. Increased levels of FGFs 19, 21 and 23, all of which bind FGFR3-3c, have been reported in the serum of patients with bladder cancer [76], though the full repertoire of FGFs that bind FGFR3-3c remains to be examined in tumor tissues.
REGULATION OF FGFR3 EXPRESSION
As indicated above, although expression of FGFR3 mRNA and protein is strongly related to mutation status in NMIBC [17, 19, 22, 29, 77, 78], this is not the case in MIBC (Fig. 2). Understanding the mechanism for this upregulation, and of any differential significance of mutant versus upregulated wildtype FGFR3, is of clinical interest, particularly in MIBC where FGFR3 inhibitor therapy may be used. As amplification of the FGFR3 region on 4p has not been identified and low-level gains are uncommon, other mechanisms of upregulation must exist.
Fig. 2
Mutation and expression of FGFR3 in relation to tumor stage in a one-year cohort of tumors from a single Institution. A. Distribution of activating point mutations according to tumor stage. B. Expression of FGFR3 protein according to tumor stage. “High” denotes expression above that found in normal urothelium and “Low” absence of expression or levels lower than in normal urothelium. Data from [17].
![Mutation and expression of FGFR3 in relation to tumor stage in a one-year cohort of tumors from a single Institution. A. Distribution of activating point mutations according to tumor stage. B. Expression of FGFR3 protein according to tumor stage. “High” denotes expression above that found in normal urothelium and “Low” absence of expression or levels lower than in normal urothelium. Data from [17].](https://content.iospress.com:443/media/blc/2020/6-4/blc-6-4-blc200373/blc-6-blc200373-g002.jpg)
Several transcriptional regulators have been implicated, though their relative importance in the normal urothelium and in specific tumor settings has not yet been explored in detail. The p53 family members p63 and p73 have been shown to regulate FGFR3 [79]. p63 is expressed from two promoters to give rise to two major isoforms, Transcriptionally Active p63 (TAp63) with a transactivation domain at the N terminus and ΔNp63 which has an activation domain at the C terminus. p63 is expressed in basal and intermediate layers of the normal urothelium and is required for urothelial differentiation [80, 81]. ΔNp63 has been shown to inhibit EMT [82]. Levels of ΔNp63 are upregulated in low-grade tumors [83] and mirror FGFR3 mutation in distribution. Overall, loss of expression is a poor prognostic feature [80, 81, 84–87], with most invasive tumors apart from a subset of aggressive tumors with squamous differentiation showing low expression [88, 89].
Analysis of cell cycle regulators in a panel of FGFR-addicted cell lines including urothelial lines, identified MYC as a critical determinant of FGFR inhibitor sensitivity and response, and FGFR inhibition in sensitive cell lines led to phosphorylation and subsequent degradation of MYC by the proteasome. The importance of MYC in sustaining the effects of FGFRs was confirmed by inhibition of MYC expression with the BET inhibitor JQ1 which recapitulated the effect of FGFR inhibition [90]. A second study of the relationship of FGFR3 and MYC in FGFR3-driven bladder tumor cells [91], identified an FGFR3/MYC positive feedback loop whereby activated FGFR3 regulates MYC mRNA and protein levels via p38 and AKT and in turn MYC directly upregulates FGFR3 by binding to upstream enhancers. Disruption of this feedback loop though FGFR, p38, AKT or MYC inhibition was demonstrated. Importantly, whilst inhibition of FGFRs and the related phosphorylation of the adaptor protein FRS2 was independent of response outcome, downregulation of MYC in response to FGFR inhibition predicted response [90]. These findings present not only potential for therapy but a biomarker for FGFR3 dependence that may be applied in early assessment of treatment response and/or development of resistance.
FGFR3 expression is also regulated at the mRNA level by micro RNAs miR-99a/100, both of which are downregulated in low-grade NMIBC compared to normal urothelium. These miRs were shown to downregulate FGFR3 expression in cultured normal urothelial cells [92] and also to regulate the urothelial differentiation factor FOXA1 [93]. Loss of the 3’ end of FGFR3 containing the target sequence for these miRs in the FGFR3-TACC3 fusion transcript, is compatible with the high-level expression of these fusion transcripts [50]. As loss of expression of miRs-99a/100 was almost ubiquitous, and more frequent than FGFR3 mutation in low-grade tumors, and the presence of the S249C mutation was not associated with differential miR expression, it was suggested that altered miR expression may lead to FGFR3 upregulation before the acquisition of mutation in NMIBC [92]. It has been reported that under hypoxic conditions FGFR3 mRNA and protein is upregulated in a HIF1α-dependent manner in bladder tumor cell lines, whilst miR-100 is downregulated [94]. As developing papillary bladder tumors have a strongly hypoxic periphery, this may indicate that hypoxia could be an early mechanism by which FGFR3 expression is upregulated via changes in miR expression prior to acquisition of mutation.
In MIBC, expression of miRs 99a/100 is lowest in the luminal-papillary mRNA expression subtype (see below), where FGFR3 mutations are most common [30]. Upregulation of wildtype FGFR3 protein in other MIBC subtypes is unlikely to be regulated in the same way. Here, levels may be more strongly regulated by MYC, possibly related to common gain of 8q in MIBC. Although the predicted relationship of MYC and FGFR3 mRNA levels could be found in FGFR3-mutant tumor samples, this did not apply to FGFR3 wildtype tumors [91]. Indeed, overall levels of MYC expression in MIBC are highest in the basal/squamous subtype and not in groups with high frequency of FGFR3 mutation [95].
It has been shown that the position of mutations in FGFR3 has differential effects on phosphorylation, retention at the plasma membrane and ubiquitylation of the receptor, with presumed effects on duration of signalling. Thus, mutations such as R248C and Y373/375C, which form dimers at the cell surface, show reduced internalisation compared to the wildtype receptor [96] and G380/382R, which is ligand-dependent, is also retained at the membrane and escapes ligand-mediated internalisation even at saturating ligand concentrations [37].
FGFR3 SIGNALLING IN NORMAL AND TUMOR-DERIVED UROTHELIA
Whilst inhibition or knockdown of FGFR3 in cells containing point mutant or fusion proteins inhibits cell proliferation e.g.[97–102], cell cycle arrest rather than apoptosis is induced [100], and escape from inhibition can occur relatively rapidly [103, 104]. Thus, there is much interest in understanding the downstream effects of FGFR3 in order to identify combinatorial or second line approaches that can enhance the effects of FGFR inhibition and/or prevent the development of resistance.
Key to understanding how FGFR3 contributes to bladder cancer development is the critical role of cellular context in determining its signalling output. This is exemplified by the differential effect of point mutant forms on chondrocyte proliferation and in epithelial cancers [42, 105–107]. Signalling downstream of FGFR3 via RAS/MAPK, PI3K-AKT, PLCγ and STATs has been reported in chondrocytes and other non-epithelial cell types and malignancies such as multiple myeloma where FGFR3 is upregulated following a translocation event involving the immunoglobin heavy chain [15, 108–110]. Many studies using chondrocytes or immortalised mesenchymal cells have focussed on the K650/652E mutant form. An early study of the tyrosine phosphorylation sites in the receptor, and their relative importance for subsequent phenotypic and signalling effects, used the 3c isoform, its K650E derivative and assays in NIH-3T3 cells [13]. In this system STAT1/3 activation was shown to be a major consequence of FGFR3 activation [14]. Whether phosphorylation of these sites has similar consequences in urothelial cells is unknown.
In TERT-NHUC, RAS/MAPK and PLCγ signalling are activated following FGF1 stimulation of ectopically expressed wildtype FGFR3-3b. This contrasts with the situation in NIH-3T3 where there is strong additional activation of the PI3K pathway and src [42]. In these normal cells, MAPK and PLCγ signalling are also activated following ectopic expression of mutant forms of FGFR3, with no changes in the levels of activated p38, JNK, SRC, STAT1 and AKT [42]. In tumor cells, the situation is less clear. It cannot be assumed that the same pathways are activated as in normal urothelial cells and it is likely that responses depend on the availability of docking and effector proteins and the overall mutational and expression landscape. In FGFR3-altered bladder tumor cell lines, the MAPK pathway is active downstream of FGFR3 as demonstrated by major inhibition of p-ERK but not p-AKT following FGFR inhibitor treatment [103, 111]. This preferential activation of the MAPK pathway in urothelial cells provides a rationale for the finding that FGFR3 and RAS gene mutations are mutually exclusive in bladder tumors [112]. However, dominance of the MAPK pathway has not been confirmed by assessment of pathway activation status in tumor tissues. Indeed, p-AKT rather than p-ERK detected by immunohistochemistry was reported to be associated with FGFR3 mutation [113]. Possibly both pathways are activated in the majority of tumors. Plasticity in signalling via these pathways is now well-documented and is exemplified in cell lines by resistance to or escape from FGFR3 inhibition in RT112 via PI3K/AKT signalling [111, 114]. Maintenance of MAPK pathway activity via activation of EGFR or ERBB2/3 signalling have also been reported as mechanisms of escape from or resistance to FGFR inhibition in bladder tumor cell lines [103, 115].
FGFR3 can interact with both p85α, the negative regulatory subunit of PI3K, via the PLCγ interaction site Y762 [116] and TGFα-activated kinase 1 (TAK1) via amino acids 589–806 [117]. The consequences of p85α interaction in urothelial cells have not been studied but in the bladder cancer cell line MGH-U3, which contains a Y375C mutation, FGFR3 signalling via TAK1 was shown to positively regulate NF-κB activity [117]. The juxtamembrane domain of FGFR3 has also been shown to interact with the N-terminal region of the kinase domain of EphA4 leading to mutual transactivation and ephrin-A1 potentiation of the FGF response of wildtype FGFRs [118]. It has also been shown that wildtype FGFRs 1–3 can form all possible stable heterodimers in the absence of ligand. Interestingly, largest effects were found for heterodimers of mutant FGFR3 (G380R and A391E) with wildtype FGFR3 [119]. These experiments were carried out using ectopically-expressed proteins in HEK293 and CHO cells and thus confirmation in urothelial models is required. It will be of particularly interest to examine the effects of common mutant forms of FGFR3 found in bladder cancer in this context.
Although S249C, Y375C and K652E mutant forms are all able to activate both MAPK and PLCγ during active proliferation of normal urothelial cells, only S249C and Y375C forms show strong activation of PLCγ at confluence. S249C with the PLCγ binding site (amino acid Y762)[120] mutated to phenylalanine shows a major reduction in this response and in continued proliferation and viability at confluence. K652E fails to activate PLCγ and is unable to elicit this phenotype [42]. This may indicate that there is positive selection for mutant forms with this capability and explain the relatively infrequent finding of K652E mutations in bladder tumors.
In urothelial and other cell types, FGFR3 fusions activate MAPK signalling [46, 52, 121], but other aspects of downstream signalling are likely to be altered. The region of FGFR3 that is lost in these fusions (amino acids 761–806) contains Y762, which is implicated in PLCγ activation and binding of p85α [116], and part of the region implicated in interaction with TAK1 [117]. As the fusions fail to activate PLCγ, unlike point mutant forms, they are unable to elicit the overgrowth of normal urothelial cells at confluence [42, 46]. The consequences of altered interaction of the fusion proteins with p85α and TAK1 have not been examined.
ETV5, a member of the PEA subfamily of ETS transcription factors [122] is an FGF effector during embryonic development [123–125]. It is upregulated and has been implicated in regulation of several aspects of the malignant phenotype in other cancers [126–130] and was identified as a pharmacodynamic biomarker for inhibition of FGFRs 1–3 in a variety of FGFR-driven tumor cell lines [131]. In normal urothelial cells, ETV5 is upregulated downstream of MAPK activation by ligand-stimulated wildtype or point mutant forms of FGFR3, with S249C inducing the highest level of upregulation. In these cells, ETV5 had an effect on confluent cell density independent of FGFR3 expression, implicating it as an effector of this FGFR3-stimulated phenotype. In bladder tumor cell lines, it conferred a proliferative advantage and interestingly, in some MIBC-derived lines, genes previously implicated in ETV5-induced EMT in other tumour types were upregulated, implying a context-dependent effect of FGFR3/ETV5 in these cases [132].
In the FGFR3-driven tumor cell line 97-7, knockdown of ETV5 was found to modulate expression of WWTR1 (TAZ), and TAZ and its transcriptional targets were also upregulated in TERT-NHUC expressing mutant FGFR3 [132]. TAZ is a co-transcriptional regulator that together with the related protein YAP1 mediates transcription of pro-proliferative and anti-apoptotic genes. YAP/TAZ are negatively regulated through the Hippo pathway in response to cell-cell contact and cell density. Thus upregulation of TAZ as a consequence of FGFR3 signalling via ETV5 provides a rational explanation for FGFR3-induced loss of contact inhibition.
Downstream effects of signalling by ectopically-expressed wildtype FGFR3 and an FGFR3-TACC3 fusion have been examined using phosphoproteomics and network analysis in TERT-NHUC in the presence and absence of FGF1 stimulation. Pathways uniquely implicated by the fusion protein included chaperone activation pathways and stress response. Interestingly, two pathways related to TP53 expression and degradation were implicated and in both TERT-NHUC and RT112 tumor cells it was shown that TP53 is downregulated when FGFR3-TACC3 fusion and not wildtype FGFR3 is stimulated by FGF1 [133].
Further information on downstream effects of FGFR3-TACC3 has also come from examination of expression changes elicited in astrocytes by the active fusion protein or a kinase-dead version in the presence and absence of the FGFR inhibitor PD173074 [134]. This revealed that FGFR3-TACC3 activates genes involved in oxidative phosphorylation and mitochondrial biogenesis. Cells expressing the fusion showed increased oxygen consumption rate and mitochondrial inhibitors were shown to be inhibitory. PIN4 was identified as a phosphorylation substrate of FGFR3-TACC3 that triggered the biogenesis of peroxisomes and the production of intracellular reactive oxygen species (ROS) that in turn activated the transcriptional coactivator PGCIα, a regulator of mitochondrial biogenesis [134]. Dependence of tumors with FGFR3-TACC3 fusions on mitochondrial metabolism suggests that inhibitors of oxidative phosphorylation may be beneficial for this subset of patients. As this effect is driven by the kinase activity of the fusion protein, it is possible that other fusions and point mutant forms may have the same effect. Whether these effects are relevant in the urothelial context remains to be examined.
The ultimate effect of FGFR3 activation also depends on a range of feedback regulatory mechanisms [135]. These include the sprouty proteins (SPRY1-4) that are upregulated in response to FGFR signalling and bind to GRB2 and SOS1 to provide negative feedback [136] and SEF and DUSP proteins. Changes in other key proteins such as FRS2, or regulatory phosphatases [137, 138] may also have major effects on signalling. Ultimately all of these determine the context in which FGFR3 signals and may require consideration in interpreting the results of clinical trials of FGFR inhibitors.
FGFR3 ALTERATIONS AND BLADDER CANCER SUBTYPES
Several classification systems based on mRNA expression have been reported for bladder cancer. Most have focussed on MIBC [30, 88, 95, 139–141], one included two thirds NMIBC cases [142] and one has focussed on NMIBC [143, 144]. Broad classification of MIBC into two subtypes termed “luminal” and “basal-like”, showed that FGFR3 mutation was confined to the luminal type [139]. Further sub-classification into three [88], four [140] or more [30, 141] sub-groups has confirmed that mutation and upregulated expression of FGFR3 are largely focussed in subgroups of luminal tumors many of which have high levels of expression of markers of urothelial differentiation such as uroplakins, transcription factors (TFs) involved in urothelial differentiation (PPARG, FOXA1, GATA3, ELF3), ERBB2 and ERBB3. Recent evaluation of both mRNA and protein expression by immunohistochemistry (IHC) has allowed tumor cell phenotype to be determined independent of the overall mRNA features of the entire cellular population, and this defined 5 phenotypic classes of MIBC: urothelial-like, genomically unstable, basal/SCC-like, mesenchymal-like, and small-cell/neuroendocrine-like, with high FGFR3 expression within the urothelial-like group. This large group could be further subdivided into UroA, UroB and UroC, the latter two of which showed some features in common with basal/SCC-like and GU groups respectively [141]. An mRNA expression classifier (LundTax), that captures these IHC and RNA features was subsequently developed [95] and used to examine the large TCGA MIBC dataset [30]. The UroA subtype, which is hypothesised to represent UroA NMIBC that have progressed, has high frequency of FGFR3 mutations (44%), high expression of urothelial differentiation-associated TFs and high expression of uroplakins. UroB also has a high frequency of mutations (50%) but lacks expression of these TFs and markers of differentiation and has upregulated expression of the basal-type keratins KRT5 and KRT14 which are associated with squamous differentiation. In contrast, Uro C tumors contain few FGFR3 mutations (4%) and lack a previously-derived FGFR3 signature [142], but retain urothelial TFs and differentiation markers. This analysis reveals considerable heterogeneity in the “luminal” class of MIBC that is reflected in differential survival, with Uro A and UroC showing significantly better outcome than UroB. Given the high frequency of FGFR3 mutations in both UroA and UroB, it will be of great interest to compare the responses of these groups to FGFR inhibitors.
Three (1–3) [143] and more recently four (1, 2a, 2b and 3) transcriptional classes of NMIBC have recently been described by the UROMOL group [144]. Classes 1 and 3 contain mostly stage Ta tumors, with relatively stable genomes measured by SNP array analysis. These express high levels of FGFR3 and have a high frequency of FGFR3 mutation. Fewest mutations were detected in class 2a, which contained the largest proportion of T1 tumors and showed significantly reduced recurrence-free survival.
Overall, the luminal/urothelial-like subtypes of MIBC have relatively low levels of immune cell infiltration and stromal markers. T-cell infiltration is very low in FGFR3-mutant tumors [145] and this led to speculation that FGFR3 may be causally related to poor infiltration and that such tumors may show reduced response to immune checkpoint inhibitors. Whether FGFR3 status directly influences the associated non-infiltrated phenotype and response to checkpoint inhibitors is under debate. During induction of tumors by BBN in a genetically engineered mouse model expressing S249C in the urothelium, no difference in T-cell infiltration compared to that in wildtype mice was reported, though an early decrease in neutrophil infiltration prior to the development of tumors in S249C mice was evident [146]. Although some initial studies reported that luminal phenotype tumors responded less well to immune checkpoint inhibitors [147, 148], analysis of response in direct relationship to FGFR3 status in two large trials has recently found no statistically significant relationship. Data from this study suggests that stromal TGF-β signals, which are known to adversely influence response [149], are lower in FGFR3-altered cases, potentially balancing out the effect of low T-cell infiltration [150].
TIMING OF FGFR3 MUTATION AND MUTATIONAL LANDSCAPE OF FGFR3-ALTERED TUMORS
When does FGFR3 mutation occur during bladder cancer pathogenesis? Although there is considerable information for NMIBC, the overall picture is not completely clear and there are several caveats that preclude straightforward interpretation of current data. Common mutational events in NMIBC that could represent initiating or very early events are mutations in the TERT promoter [151, 152], FGFR3 mutation and loss of heterozygosity (LOH) of 9q [153, 154], with mutation frequency in the order TERT > FGFR3 >9q (∼80% >70% >50%). Does this imply an order of events? One major caveat is that as discussed above it is now clear that several molecular subtypes of both NMIBC and MIBC exist and mutational profiles and potentially the preferred order of molecular events are also expected to differ.
One approach to investigate the timing of events has been to examine frequency of events in cohorts of samples that are predicted to represent “stages” in disease pathogenesis. Several studies have been made of morphologically “normal” urothelium from patients with bladder cancer and potential precursor lesions. Studies of histologically normal urothelium from NMIBC-bearing bladders have found LOH of 9q or deletion detected by FISH [155–158], suggesting that 9q deletion can precede the appearance of an overt tumor. To date only a single study has examined FGFR3 in such samples [159]. This study examined normal samples from 38 patients with FGFR3-mutant bladder tumors and found no mutations. 9q was not examined, though it seems likely that a significant number of the tumors would have had 9q LOH and that, as in other studies, the surrounding “normal” urothelium in such samples would contain some 9q deletions. Studies of flat and papillary hyperplasia, now defined as “urothelial proliferation of uncertain malignant potential” [160], which may represent precursors of NMIBC, also report 9q LOH [161–163] and in a single study that examined both FGFR3 and 9q, 9q LOH was found to be the more common event [164]. These studies suggest that 9q LOH precedes FGFR3 mutation, at least in a subset of NMIBC. However, there may be different requirements and timing of events in different NMIBC subtypes. Thus, it is noteworthy that two studies aimed at defining molecular subtypes of NMIBC have described one group of low-grade tumors with FGFR3 mutation but retention of 9q and a second with both FGFR3 mutation and 9q LOH [77, 165]. This indicates that the ordering of these so-called “early” events is not uniform, and points to two distinct subsets of tumors, only one with 9q loss as an early event but both with common FGFR3 mutation.
As indicated above, downregulation of miRs 99/100 and upregulation of FGFR3 may precede acquisition of mutations [92]. Compatible with this suggestion is the finding that a germline sequence variant close to FGFR3 is associated with low-grade and low-stage bladder cancer. This variant, at least in adipose tissue, is associated with increased FGFR3 expression and importantly, the frequency of the variant is higher in FGFR3-mutant tumors [166].
Urothelial papilloma and inverted urothelial papilloma are low-grade tumors with low frequency of recurrence, often occurring in young patients, and with debated relationship to urothelial carcinoma. In many of these cases, FGFR3 mutation is absent, with a high frequency of HRAS or KRAS mutations [167–169], indicating a separate molecular profile from both PUNLMP and low-grade non-invasive urothelial carcinoma for this benign lesion.
In flat dysplasia and carcinoma in situ (CIS) that are predicted precursors of non-papillary MIBC, TP53 mutation/17p LOH and 9q LOH are common, but FGFR3 mutations are absent [170, 171], indicating that FGFR3 has no early role in the development of these tumors. TP53 mutations and/or upregulated expression are detected frequently in flat dysplasia and CIS [171–173] and appear to precede deletion of chromosome 9 [171, 174].
The distinct mutational profiles of NMIBC and MIBC and their precursor lesions provided evidence for the early concept that bladder tumor development may follow two distinct pathways, the non-invasive/papillary pathway with FGFR3 alterations as a major feature and the pathway initiated in CIS, containing many TP53 mutations [171–173]. This concept remains, and in general is supported by more profound current understanding of tumors of both types. However, it now seems clear that papillary/luminal/urothelial-like MIBC with FGFR3 alteration as a major feature represent NMIBC that have progressed. Several studies indicate that loss of the 9p21 locus CDKN2A, which encodes the RB1 and TP53 regulators p16INK4A and p14ARF, may allow progression of FGFR3-mutant NMIBC. p16INK4A is a regulator of cell cycle arrest and senescence via binding to CDK4/6, which retains the retinoblastoma protein RB1 in a hypophosphorylated and active state. It is upregulated in response to various oncogenic stimuli, inducing a state termed “oncogene-induced senescence” [175], and in the setting of an FGFR3 mutation may be partially induced and restrain tumor growth. CDKN2A homozygous deletion is found more frequently in FGFR3-mutant tumors [30, 95, 176] and strikingly, this was found in 79% of FGFR3-mutant MIBC [176]. The role of CKDN2A/RB1 inactivation in FGFR3-mutant tumor pathogenesis has also been indicated in a genetically-engineered mouse model with inducible expression of Fgfr-S243C (mouse equivalent of human S249C) in the urothelium. Although increased levels of p-AKT and p-MAPK were induced, there was no increase in urothelial proliferation and p16, p19 and p53 protein levels increased. However, when crossed with mice with urothelial expression of SV40T, which inactivates both p53 and Rb1, high-grade papillary tumors developed [177]. Thus, loss of CDKN2A/RB1-regulated cell cycle arrest and/or the oncogene-induced senescence function appears essential in allowing tumor formation in the Fgfr3-driven mouse model and for tumor progression in human NMIBC. This has important implications for follow-up of FGFR3-mutant NMIBC with deletion of CDKN2A, as these may represent a high-risk group.
Examination of the TCGA data supports this concept. Separation of MIBC based on CDKN2A deletion status reveals that FGFR3 mutation is more common in samples with deletion, compatible with this predominantly luminal-papillary subtype tumors representing progressed NMIBC. These tumors also contain a higher frequency of mutations that are common in NMIBC such as PIK3CA and STAG2. TP53 mutation is more common in tumors with retention of CDKN2A that lack FGFR3 mutation and is skewed towards the basal-squamous and other subtypes (Fig. 3A).
Fig. 3
Oncoplots showing distribution of common mutations according to chromosome 9 status. A. Oncoplot for selected genes in muscle-invasive bladder tumours with and without deletion of the CDKN2A locus. Data from [30]. B. Oncoplot for selected genes in stage Ta tumors with and without 9q loss. Data from [165].
![Oncoplots showing distribution of common mutations according to chromosome 9 status. A. Oncoplot for selected genes in muscle-invasive bladder tumours with and without deletion of the CDKN2A locus. Data from [30]. B. Oncoplot for selected genes in stage Ta tumors with and without 9q loss. Data from [165].](https://content.iospress.com:443/media/blc/2020/6-4/blc-6-4-blc200373/blc-6-blc200373-g003.jpg)
An alternative mechanism of progression for FGFR3-mutant tumors is suggested by studies that have shown the presence of mutation in a non-invasive/superficial part of a tumor and lack of mutation in a deeper or invasive component [17, 62]. It is possible that cases with discordant mutation status contain more than one tumor clone but alternatively, loss of FGFR3 mutation may have occurred, potentially alleviating oncogene-induced upregulation of p16 and related cell cycle checkpoint genes and allowing tumor progression.
Where does mutation of the TERT promoter fit within these pathogenesis pathways? Such mutations are more common than FGFR3 mutation in NMIBC and have been reported in the “normal” urothelium of tumor-bearing bladders [178] and in urine samples up to 10 years before diagnosis of bladder cancer [179]. As mutations are equally common in NMIBC and MIBC (70–80%), these mutations may precede all other molecular events identified to date and represent an essential and potentially initiating event. Telomerase is known to allow cells to overcome oncogene-induced senescence [180] and thus early mutation may allow cellular tolerance of later FGFR3 mutations.
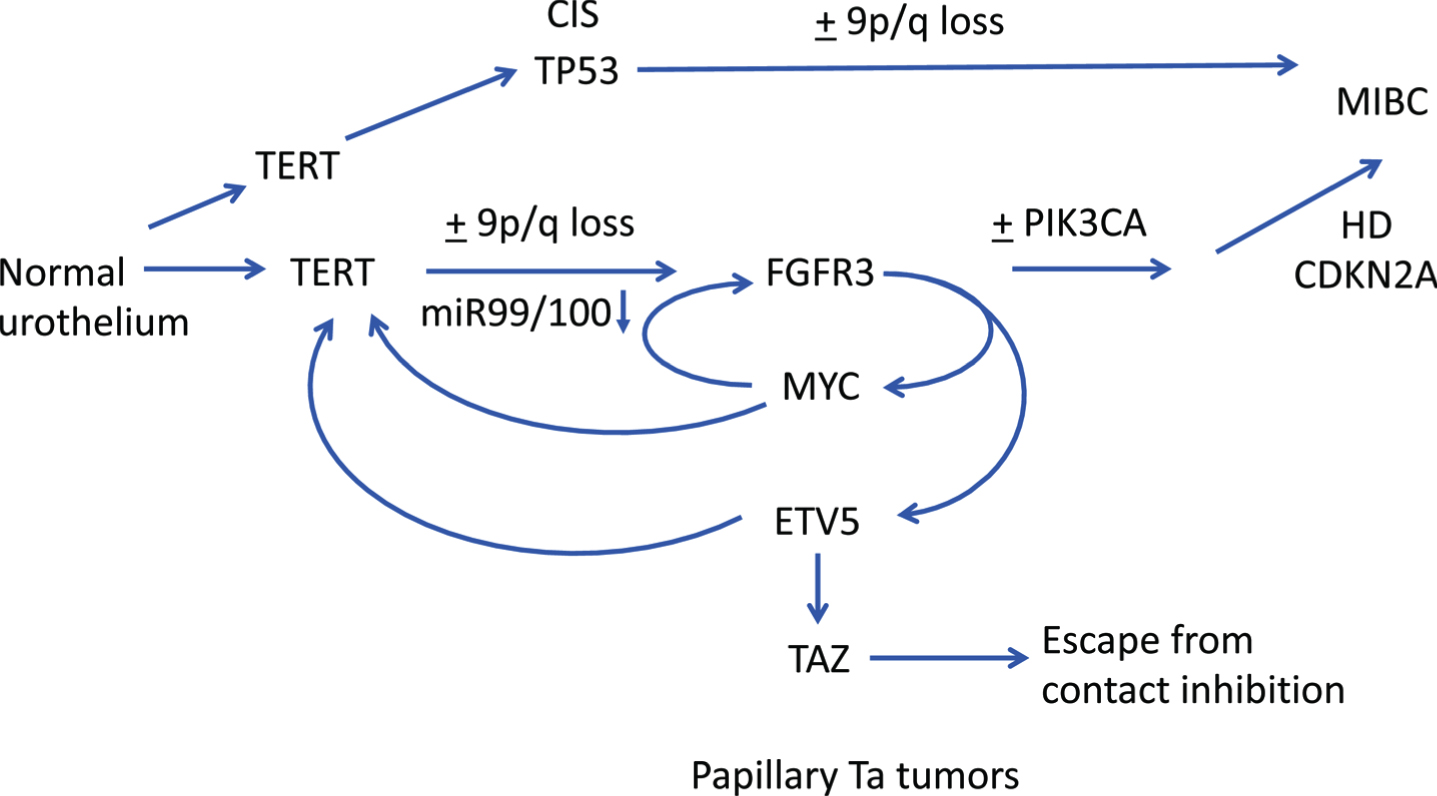
Recent findings suggest potential complex interplay between FGFR3 and several other genes early in NMIBC pathogenesis. In thyroid cancer, ETV5 is upregulated downstream of mutant BRAF and binds to the TERT promoter [181]. Interestingly, binding is preferentially to the –124 bp(T) TERT promoter mutation that is the most common mutation found in both thyroid and bladder tumors [182]. It is also known that MYC can activate TERT [183] and recent data show that ETV5 and MYC can cooperate in de-repressing the TERT promoter [184]. Taken together, this could indicate a perfect storm between TERT, FGFR3, ETV5, MYC and TAZ early in bladder cancer pathogenesis with FGFR3 mutation playing a central role (Fig. 4).
Fig. 4
Hypothetical pathways of pathogenesis of non-invasive and invasive bladder cancer. Straight arrows show potential timing of selected events during development of NMIBC and MIBC based on data from dysplasia, CIS and urothelial hyperplasia and subtype analysis of NMIBC and MIBC. FGFR3, PIK3CA: activating point mutation. TERT: promoter point mutation. TP53: inactivating mutation. HD: homozygous deletion. Curved arrows indicate interrelated regulation of expression of FGFR3, TERT, MYC, ETV5 and TAZ.
What is the final mutational context of FGFR3-altered tumors? FGFR3 alterations are generally found in genomically stable tumors with low mutational burden. When examined across the entire bladder cancer spectrum, mutations in FGFR3 and TP53 mutation are almost mutually exclusive [27, 185–188]. Similarly, few FGFR3-altered tumors have mutations in RB1, PTEN or high-level amplification of E2F3 [30], all features of non-luminal MIBC. As indicated above, FGFR3 and RAS gene mutations are mutually exclusive [112] and FGFR3 and ERBB2 mutations are reported to be mutually exclusive in high-grade NMIBC [29].
Common features accompanying FGFR3 mutation in stage Ta tumors are mutations in PIK3CA, KDM6A and STAG2 [165]. When the mutational profiles of Ta tumors with and without loss of 9q are examined, both groups have high frequency of FGFR3 mutation (Fig. 3B), but interestingly, those with no loss of 9q have a greater number of RAS gene mutations and a higher frequency of STAG2 mutation [165]. It is assumed that, as tumors with 9q LOH usually have hemizygous deletion of the entire chromosome, this group contains those at risk of progression to MIBC via loss of the second allele of CDKN2A. How these different mutational constellations contribute to phenotype and whether this affects clinical outcome remains to be evaluated. Larger studies of NMIBC including methylation and non-coding RNA profiles may elucidate exact requirements and interrelationships more fully in the future. However, it is clear that in Ta tumors mutations in PIK3CA almost invariably occur in tumors with FGFR3 or RAS mutation [113, 165, 189–192] (Fig. 3B). Why PIK3CA mutation in the absence of FGFR3 mutation is common in MIBC (Fig. 3A) but not found in NMIBC remains an unanswered question.
PROGNOSTIC AND PREDICTIVE VALUE OF FGFR3 ALTERATIONS
As FGFR3 point mutations are most common in tumors of low grade and stage, there is a strong relationship of these mutations with good clinical outcome. Several studies have examined the relationship of point mutation or FGFR3 protein expression to recurrence in NMIBC and most report no significant relationship [59, 186, 193–195], though when Ta tumors were stratified by grade, an association was reported in TaG1 tumors only [24]. In univariate analysis of data from all NMIBC (Ta and T1), FGFR3 wildtype or tumors expressing low levels of FGFR3 protein have higher risk of progression, though this has not been found to be an independent prognostic factor in multivariate analyses [59, 194, 195]. In the subset of FGFR3-mutant tumors, hemizygous or homozygous deletion of the CDKN2A locus is a predictor of progression that is independent of tumor grade and stage [176]. When FGFR3 mutation is assessed in combination with MIB-1 expression as a measure of proliferative index this “molecular grade” showed independent significance with FGFR3 mutant, MIB-1 low tumors having best outcome. When used with EORTC risk score [196], this provided improved prediction of progression [195]. When primary T1 tumors alone were evaluated, a significant association of FGFR3 mutation or expression status with disease progression has been reported in multivariate analysis in some [197, 198] but not all studies [27]. Overall, these data indicate that FGFR3 mutation identifies patients with favourable NMIBC disease, though only grade and stage remain single independent predictors of outcome.
In a very large cohort of patients with MIBC or high-risk NMIBC that were treated with radical cystectomy, mutation was related to longer disease-specific survival [63]. Interestingly, although over-expression of wildtype protein was associated with lower tumor stage and grade, it was not associated with outcome, potentially indicating a functional distinction between mutation and over-expression of wildtype FGFR3, with a driver role for mutant FGFR3, and possibly a passenger role only for upregulated expression of wildtype protein. This provides additional evidence that FGFR3 mutation rather than upregulated expression may represent the better predictive biomarker for FGFR inhibitor therapies. A response rate to the FGFR inhibitor Erdafitinib of 40% has been reported in patients whose tumors contained FGFR3 point mutations or FGFR2/3 fusions [199] leading to FDA approval for this drug in locally advanced or metastatic bladder cancer. Whilst this response rate is very encouraging, it will be important to explain the lack of response in the remaining 60% of patients. Although inhibition of FGFR3 in cell lines with mutation has been widely demonstrated, as discussed above, it is clear that several mechanisms allow escape from inhibition or development of stable resistance. Detailed discussion is beyond the scope of this review but it will be important to understand these mechanisms if FGFR inhibitors are to be applied appropriately and relevant combination therapies developed in the future. Lack of response may also indicate loss of dependence on FGFR3 during tumor progression or intratumor heterogeneity with outgrowth of a non-addicted variant in these advanced tumors. It is hypothesised that heterogeneity and cellular signalling plasticity may be much less prominent in low-grade NMIBC. Thus FGFR3 as a therapeutic target may be more relevant in these patients if localised means of targeting can be developed.
The significance of FGFR3 status in predicting response to chemotherapy has not yet been examined extensively. As previous studies have indicated that better response to cisplatin-based chemotherapy is associated with basal rather than luminal expression subtype [200, 201], worse response of FGFR3 mutant tumors might be expected as mutations and upregulated expression of FGFR3 are far more common in the luminal subtype. Findings to date suggest that this is the case. One study that evaluated FGFR3 mutation status and protein expression in 72 MIBC patients, 42 of whom were treated with adjuvant chemotherapy, reported significantly shorter overall survival and borderline significance for disease-free survival related to FGFR3 overexpression. This remained a significant independent prognostic factor in multivariate analysis. The relationship to mutation was not significant, but few samples in the chemotherapy group contained mutation [202]. A recent retrospective study that examined three groups of patients treated with cisplatin-based chemotherapy supports these findings. Lower rates of pathologic response to neoadjuvant chemotherapy and reduced recurrence-free survival were recorded in patients whose tumors contained FGFR3 point mutations or fusions. In patients included in the TCGA study of MIBC who received adjuvant chemotherapy reduced RFS was also found in FGFR3 altered tumors. In a third group of metastatic patients treated with first line platinum-based chemotherapy FGFR3 alteration was associated with a different pattern of metastatic spread including higher rates of pulmonary metastases and with lower response rate. Patients with FGFR3-altered tumors that did not receive chemotherapy had superior clinical outcome [203]. Overall this suggests that despite the relationship of FGFR3 status to better outcome in MIBC, chemotherapy may be detrimental to this group and that other treatment options including FGFR inhibitor therapy may be more suitable. Interestingly, an in vitro study has also reported decreased sensitivity to cisplatin of a bladder tumor cell line (97-7) containing an S249C mutation compared to wildtype cell lines. In this case, a high level of p-AKT in the mutant line was decreased following FGFR3 inhibition and this was associated with increased cisplatin sensitivity [204]. Larger studies of patients treated with cisplatin-based neoadjuvant chemotherapy in combination with molecular profiling may allow the basis for these effects to be elucidated.
CONCLUSIONS
Whilst a great deal has been learnt about FGFR3 and its role in the pathogenesis of NMIBC and MIBC, much remains to be clarified if it is to be optimally targeted in these settings. Current data suggests that it is an early event in NMIBC, though we lack understanding of exactly how it contributes during the initial development of urothelial hyperplasia and the elicitation of a branching vasculature and papillary tumor architecture. More detailed analyses of these early stages and the development of mechanisms for localised FGFR3 targeting or targeting of the FGFR3-related phenotype have potential for major impact on the clinical management of the very large NMIBC population. In vitro and in vivo models may make a major contribution to understanding these early processes and more complex models may improve understanding of any influences of FGFR3 signalling on tumor infiltration. Unlike other epithelial tissues, the ability to culture normal urothelial cells with relative ease is a major advantage and this should allow the hypothetical interactions of TERT, FGFR3 and MYC in NMIBC pathogenesis hypothesised here to be tested. In MIBC, the excitement related to responses to FGFR inhibitors is tempered by lack of good predictive biomarkers. More detailed molecular profiling that takes into account some of the broader mechanisms of FGFR3 regulation and cross-talk should facilitate improved biomarker-driven treatment selection.
ACKNOWLEDGMENTS
I am extremely grateful to past members of my group, Darren Tomlinson, Erica di Martino and Sarah Williams for their practical and intellectual contributions over many years that have shaped my thinking about this most interesting protein, and to Julie Burns for her critical reading of this manuscript.
FUNDING
Funding of our past work on FGFR3 has been from Cancer Research UK (C6228/A5433; C6228/A12512) and Yorkshire Cancer Research (L376PA).
ETHICAL CONSIDERATIONS
As a review, this work is exempt from any requirement for Institutional Review Board approval.
CONFLICT OF INTEREST
Consulting/Advisory Role-Janssen Oncology, Bioclin Therapeutics.
REFERENCES
[1] | Cappellen D , De Oliveira C , Ricol D , de Medina S , Bourdin J , Sastre-Garau X , et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. (1999) ;23: (1):18–20. |
[2] | Casadei C , Dizman N , Schepisi G , Cursano MC , Basso U , Santini D , et al. Targeted therapies for advanced bladder cancer: new strategies with FGFR inhibitors. Ther Adv Med Oncol. (2019) ;11: , 1758835919890285. |
[3] | Morales-Barrera R , Suarez C , Gonzalez M , Valverde C , Serra E , Mateo J , et al. The future of bladder cancer therapy: Optimizing the inhibition of the fibroblast growth factor receptor. Cancer Treat Rev. (2020) ;86: :102000. |
[4] | Montazeri K , Bellmunt J . Erdafitinib for the treatment of metastatic bladder cancer. Expert Rev Clin Pharmacol. (2020) ;13: (1):1–6. |
[5] | Qin Q , Patel V , Galsky MD . Urothelial carcinoma: the development of FGFR inhibitors in combination with immune checkpoint inhibitors. Expert Rev Anticancer Ther. (2020) ;20: (6):503–12. |
[6] | Trueb B . Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell Mol Life Sci. (2011) ;68: (6):951–64. |
[7] | Ornitz DM , Itoh N . The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol. (2015) ;4: (3):215–66. |
[8] | Zhang X , Ibrahimi OA , Olsen SK , Umemori H , Mohammadi M , Ornitz DM . Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. (2006) ;281: (23):15694–700. |
[9] | Eswarakumar VP , Lax I , Schlessinger J . Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. (2005) ;16: (2):139–49. |
[10] | Tsang M , Dawid IB . Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci STKE. (2004) )228 pe17. |
[11] | Gotoh N . Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci. (2008) ;99: (7):1319–25. |
[12] | Kadamur G , Ross EM . Mammalian phospholipase C. Annu Rev Physiol. (2013) ;75: :127–54. |
[13] | Hart KC , Robertson SC , Donoghue DJ . Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol Biol Cell. (2001) ;12: (4):931–42. |
[14] | Hart KC , Robertson SC , Kanemitsu MY , Meyer AN , Tynan JA , Donoghue DJ . Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene. (2000) ;19: (29):3309–20. |
[15] | Kang S , Dong S , Gu TL , Guo A , Cohen MS , Lonial S , et al. FGFR3 activates RSK2 to mediate hematopoietic transformation through tyrosine phosphorylation of RSK2 and activation of the MEK/ERK pathway. Cancer Cell. (2007) ;12: (3):201–14. |
[16] | Tomlinson DC , L’Hote CG , Kennedy W , Pitt E , Knowles MA . Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Res. (2005) ;65: (22):10441–9. |
[17] | Tomlinson DC , Baldo O , Harnden P , Knowles MA . FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol. (2007) ;213: (1):91–8. |
[18] | Southgate J , Hutton KA , Thomas DF , Trejdosiewicz LK . Normal human urothelial cells in vitro: proliferation and induction of stratification. Lab Invest. (1994) ;71: (4):583–94. |
[19] | Billerey C , Chopin D , Aubriot-Lorton MH , Ricol D , Gil Diez de Medina S , Van Rhijn B , et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. (2001) ;158: (6):1955–9. |
[20] | Kimura T , Suzuki H , Ohashi T , Asano K , Kiyota H , Eto Y . The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. (2001) ;92: (10):2555–61. |
[21] | Sibley K , Cuthbert-Heavens D , Knowles M . Loss of heterozygosity at 4p16. 3 and mutation of FGFR3 in transitional cell carcinoma. Oncogene. (2001) ;20: :686–91. |
[22] | van Rhijn BW , Lurkin I , Radvanyi F , Kirkels WJ , van der Kwast TH , Zwarthoff EC . The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. (2001) ;61: (4):1265–8. |
[23] | van Rhijn BW , van Tilborg AA , Lurkin I , Bonaventure J , de Vries A , Thiery JP , et al. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur J Hum Genet. (2002) ;10: (12):819–24. |
[24] | Hernandez S , Lopez-Knowles E , Lloreta J , Kogevinas M , Amoros A , Tardon A , et al. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J Clin Oncol. (2006) ;24: (22):3664–71. |
[25] | Liu X , Zhang W , Geng D , He J , Zhao Y , Yu L . Clinical significance of fibroblast growth factor receptor-3 mutations in bladder cancer: a systematic review and meta-analysis. Genet Mol Res. (2014) ;13: (1):1109–20. |
[26] | van Rhijn BW , Montironi R , Zwarthoff EC , Jobsis AC , van der Kwast TH . Frequent FGFR3 mutations in urothelial papilloma. J Pathol. (2002) ;198: (2):245–51. |
[27] | Hernandez S , Lopez-Knowles E , Lloreta J , Kogevinas M , Jaramillo R , Amoros A , et al. FGFR3 and Tp53 mutations in T1G3 transitional bladder carcinomas: independent distribution and lack of association with prognosis. Clin Cancer Res. (2005) ;11: (15):5444–50. |
[28] | Rodriguez Pena MDC , Tregnago AC , Eich ML , Springer S , Wang Y , Taheri D , et al. Spectrum of genetic mutations in de novo PUNLMP of the urinary bladder. Virchows Arch. (2017) ;471: (6):761–7. |
[29] | Pietzak EJ , Bagrodia A , Cha EK , Drill EN , Iyer G , Isharwal S , et al. Next-generation Sequencing of Nonmuscle Invasive Bladder Cancer Reveals Potential Biomarkers and Rational Therapeutic Targets. Eur Urol. (2017) ;72: (6):952–59. |
[30] | Robertson AG , Kim J , Al-Ahmadie H , Bellmunt J , Guo G , Cherniack AD , et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. (2017) ;171: (3):540–56 e25. |
[31] | Kim YS , Kim K , Kwon GY , Lee SJ , Park SH . Fibroblast growth factor receptor 3 (FGFR3) aberrations in muscle-invasive urothelial carcinoma. BMC Urol. (2018) ;18: (1):68. |
[32] | Passos-Bueno MR , Wilcox WR , Jabs EW , Sertie AL , Alonso LG , Kitoh H . Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. (1999) ;14: (2):115–25. |
[33] | Ornitz DM , Marie PJ . Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. (2015) ;29: (14):1463–86. |
[34] | Naski MC , Wang Q , Xu J , Ornitz DM . Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet. (1996) ;13: (2):233–7. |
[35] | Del Piccolo N , Placone J , Hristova K . Effect of thanatophoric dysplasia type I mutations on FGFR3 dimerization. Biophys J. (2015) ;108: (2):272–8. |
[36] | Huang Z , Chen H , Blais S , Neubert TA , Li X , Mohammadi M . Structural mimicry of a-loop tyrosine phosphorylation by a pathogenic FGF receptor 3 mutation. Structure. (2013) ;21: (10):1889–96. |
[37] | Monsonego-Ornan E , Adar R , Feferman T , Segev O , Yayon A . The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol Cell Biol. (2000) ;20: (2):516–22. |
[38] | Chen F , Sarabipour S , Hristova K . Multiple consequences of a single amino acid pathogenic RTK mutation: the A391E mutation in FGFR3. PLoS One. (2013) ;8: (2):e56521. |
[39] | Hafner C , van Oers JM , Hartmann A , Landthaler M , Stoehr R , Blaszyk H , et al. High frequency of FGFR3 mutations in adenoid seborrheic keratoses. J Invest Dermatol. (2006) ;126: (11):2404–7. |
[40] | Shi MJ , Meng XY , Lamy P , Banday AR , Yang J , Moreno-Vega A , et al. APOBEC-mediated Mutagenesis as a Likely Cause of FGFR3 S249C Mutation Over-representation in Bladder Cancer. Eur Urol. (2019) ;76: (1):9–13. |
[41] | Lamy P , Nordentoft I , Birkenkamp-Demtroder K , Thomsen MB , Villesen P , Vang S , et al. Paired Exome Analysis Reveals Clonal Evolution and Potential Therapeutic Targets in Urothelial Carcinoma. Cancer Res. (2016) ;76: (19):5894–906. |
[42] | di Martino E , L’Hote CG , Kennedy W , Tomlinson DC , Knowles MA . Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type- and mutation-specific manner. Oncogene. (2009) ;28: (48):4306–16. |
[43] | di Martino E , Kelly G , Roulson JA , Knowles MA . Alteration of cell-cell and cell-matrix adhesion in urothelial cells: an oncogenic mechanism for mutant FGFR3. Mol Cancer Res. (2015) ;13: (1):138–48. |
[44] | Du X , Lin BC , Wang QR , Li H , Ingalla E , Tien J , et al. MMP-1 and Pro-MMP-10 as potential urinary pharmacodynamic biomarkers of FGFR3-targeted therapy in patients with bladder cancer. Clin Cancer Res. (2014) ;20: (24):6324–35. |
[45] | Wilson CB , Leopard J , Cheresh DA , Nakamura RM . Extracellular matrix and integrin composition of the normal bladder wall. World J Urol. (1996) ;14: (Suppl 1):S30–7. |
[46] | Williams SV , Hurst CD , Knowles MA . Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet. (2013) ;22: (4):795–803. |
[47] | Guo G , Sun X , Chen C , Wu S , Huang P , Li Z , et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. (2013) ;45: :1459–63. |
[48] | Nakanishi Y , Akiyama N , Tsukaguchi T , Fujii T , Satoh Y , Ishii N , et al. Mechanism of Oncogenic Signal Activation by the Novel Fusion Kinase FGFR3-BAIAP2L1. Mol Cancer Ther. (2015) ;14: (3):704–12. |
[49] | Ross JS , Wang K , Khaira D , Ali SM , Fisher HA , Mian B , et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer. (2016) ;122: (5):702–11. |
[50] | Parker BC , Annala MJ , Cogdell DE , Granberg KJ , Sun Y , Ji P , et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Invest. (2013) ;123: (2):855–65. |
[51] | Peter BJ , Kent HM , Mills IG , Vallis Y , Butler PJ , Evans PR , et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. (2004) ;303: (5657):495–9. |
[52] | Singh D , Chan JM , Zoppoli P , Niola F , Sullivan R , Castano A , et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. (2012) ;337: (6099):1231–5. |
[53] | Ding ZM , Huang CJ , Jiao XF , Wu D , Huo LJ . The role of TACC3 in mitotic spindle organization. Cytoskeleton (Hoboken). (2017) ;74: (10):369–78. |
[54] | Royle SJ . The role of clathrin in mitotic spindle organisation. J Cell Sci. (2012) ;125: (1):19–28. |
[55] | Sarkar S , Ryan EL , Royle SJ . FGFR3-TACC3 cancer gene fusions cause mitotic defects by removal of endogenous TACC3 from the mitotic spindle. Open Biol. (2017) ;7: (8):170080. |
[56] | Gomez-Roman JJ , Saenz P , Molina M , Cuevas Gonzalez J , Escuredo K , Santa Cruz S , et al. Fibroblast growth factor receptor 3 is overexpressed in urinary tract carcinomas and modulates the neoplastic cell growth. Clin Cancer Res. (2005) ;11: (2 Pt 1):459–65. |
[57] | Maeng YH , Eun SY , Huh JS . Expression of fibroblast growth factor receptor 3 in the recurrence of non-muscle-invasive urothelial carcinoma of the bladder. Korean J Urol. (2010) ;51: (2):94–100. |
[58] | Mhawech-Fauceglia P , Cheney RT , Fischer G , Beck A , Herrmann FR . FGFR3 and p53 protein expressions in patients with pTa and pT1 urothelial bladder cancer. Eur J Surg Oncol. (2006) ;32: (2):231–7. |
[59] | Poyet C , Hermanns T , Zhong Q , Drescher E , Eberli D , Burger M , et al. Positive fibroblast growth factor receptor 3 immunoreactivity is associated with low-grade non-invasive urothelial bladder cancer. Oncol Lett. (2015) ;10: (5):2753–60. |
[60] | Guancial EA , Werner L , Bellmunt J , Bamias A , Choueiri TK , Ross R , et al. FGFR3 expression in primary and metastatic urothelial carcinoma of the bladder. Cancer Med. (2014) ;3: (4):835–44. |
[61] | Turo R , Harnden P , Thygesen H , Fleischmann A , Thalmann GN , Seiler R , et al. FGFR3 expression in primary invasive bladder cancers and matched lymph node metastases. J Urol. (2015) ;193: (1):325–30. |
[62] | Pouessel D , Neuzillet Y , Mertens LS , van der Heijden MS , de Jong J , Sanders J , et al. Tumor heterogeneity of fibroblast growth factor receptor 3 (FGFR3) mutations in invasive bladder cancer: implications for perioperative anti-FGFR3 treatment. Ann Oncol. (2016) ;27: (7):1311–6. |
[63] | van Rhijn BWG , Mertens LS , Mayr R , Bostrom PJ , Real FX , Zwarthoff EC , et al. FGFR3 Mutation Status and FGFR3 Expression in a Large Bladder Cancer Cohort Treated by Radical Cystectomy: Implications for Anti-FGFR3 Treatment?(dagger). Eur Urol. 2020;78(5):682-7. |
[64] | Hahn NM , Bivalacqua TJ , Ross AE , Netto GJ , Baras A , Park JC , et al. A Phase II Trial of Dovitinib in BCG-Unresponsive Urothelial Carcinoma with FGFR3 Mutations or Overexpression: Hoosier Cancer Research Network Trial HCRN 12-157. Clin Cancer Res. (2017) ;23: (12):3003–11. |
[65] | Cheng T , Roth B , Choi W , Black PC , Dinney C , McConkey DJ . Fibroblast growth factor receptors-1 and -3 play distinct roles in the regulation of bladder cancer growth and metastasis: implications for therapeutic targeting. PLoS One. (2013) ;8: (2):e57284. |
[66] | Tomlinson DC , Baxter EW , Loadman PM , Hull MA , Knowles MA . FGFR1-induced epithelial to mesenchymal transition through MAPK/PLCgamma/COX-2-mediated mechanisms. PLoS One. (2012) ;7: (6):e38972. |
[67] | McNiel EA , Tsichlis PN . Analyses of publicly available genomics resources define FGF-2-expressing bladder carcinomas as EMT-prone, proliferative tumors with low mutation rates and high expression of CTLA-4, PD-1 and PD-L1. Signal Transduct Target Ther. 2017;2. |
[68] | Abdul-Maksoud RS , Shalaby SM , Elsayed WS , Elkady S . Fibroblast growth factor receptor 1 and cytokeratin 20 expressions and their relation to prognostic variables in bladder cancer. Gene. (2016) ;591: (2):320–6. |
[69] | Lim S , Koh MJ , Jeong HJ , Cho NH , Choi YD , Cho do Y , et al. Fibroblast Growth Factor Receptor 1 Overexpression Is Associated with Poor Survival in Patients with Resected Muscle Invasive Urothelial Carcinoma. Yonsei Med J. (2016) ;57: (4):831–9. |
[70] | Zaravinos A , Volanis D , Lambrou GI , Delakas D , Spandidos DA . Role of the angiogenic components, VEGFA, FGF2, OPN and RHOC, in urothelial cell carcinoma of the urinary bladder. Oncol Rep. (2012) ;28: (4):1159–66. |
[71] | Shariat SF , Youssef RF , Gupta A , Chade DC , Karakiewicz PI , Isbarn H , et al. Association of angiogenesis related markers with bladder cancer outcomes and other molecular markers. J Urol. (2010) ;183: (5):1744–50. |
[72] | Chodak GW , Hospelhorn V , Judge SM , Mayforth R , Koeppen H , Sasse J . Increased levels of fibroblast growth factor-like activity in urine from patients with bladder or kidney cancer. Cancer Res. (1988) ;48: (8):2083–8. |
[73] | Nguyen M , Watanabe H , Budson AE , Richie JP , Folkman J . Elevated levels of the angiogenic peptide basic fibroblast growth factor in urine of bladder cancer patients. J Natl Cancer Inst. (1993) ;85: (3):241–2. |
[74] | O’Brien TS , Smith K , Cranston D , Fuggle S , Bicknell R , Harris AL . Urinary basic fibroblast growth factor in patients with bladder cancer and benign prostatic hypertrophy. Br J Urol. (1995) ;76: (3):311–4. |
[75] | Chopin DK , Caruelle JP , Colombel M , Palcy S , Ravery V , Caruelle D , et al. Increased immunodetection of acidic fibroblast growth factor in bladder cancer, detectable in urine. J Urol. (1993) ;150: (4):1126–30. |
[76] | Li JR , Chiu KY , Ou YC , Wang SS , Chen CS , Yang CK , et al. Alteration in serum concentrations of FGF19, FGF21, and FGF23 in patients with urothelial carcinoma. Biofactors. (2019) ;45: (1):62–8. |
[77] | Lindgren D , Liedberg F , Andersson A , Chebil G , Gudjonsson S , Borg A , et al. Molecular characterization of early-stage bladder carcinomas by expression profiles, FGFR3 mutation status, and loss of 9q. Oncogene. (2006) ;25: (18):2685–96. |
[78] | Bernard-Pierrot I , Brams A , Dunois-Larde C , Caillault A , Diez de Medina SG , Cappellen D , et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis. (2006) ;27: (4):740–7. |
[79] | Sayan AE , D’Angelo B , Sayan BS , Tucci P , Cimini A , Ceru MP , et al. p73 and p63 regulate the expression of fibroblast growth factor receptor 3. Biochem Biophys Res Commun. (2010) ;394: (3):824–8. |
[80] | Karni-Schmidt O , Castillo-Martin M , Shen TH , Gladoun N , Domingo-Domenech J , Sanchez-Carbayo M , et al. Distinct expression profiles of p63 variants during urothelial development and bladder cancer progression. Am J Pathol. (2011) ;178: (3):1350–60. |
[81] | Urist MJ , Di Como CJ , Lu ML , Charytonowicz E , Verbel D , Crum CP , et al. Loss of p63 expression is associated with tumor progression in bladder cancer. Am J Pathol. (2002) ;161: (4):1199–206. |
[82] | Tran MN , Choi W , Wszolek MF , Navai N , Lee IL , Nitti G , et al. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem. (2013) ;288: (5):3275–88. |
[83] | Comperat E , Bieche I , Dargere D , Ferlicot S , Laurendeau I , Benoit G , et al. p63 gene expression study and early bladder carcinogenesis. Urology. (2007) ;70: (3):459–62. |
[84] | Fukushima H , Koga F , Kawakami S , Fujii Y , Yoshida S , Ratovitski E , et al. Loss of DeltaNp63alpha promotes invasion of urothelial carcinomas via N-cadherin/Src homology and collagen/extracellular signal-regulated kinase pathway. Cancer Res. (2009) ;69: (24):9263–70. |
[85] | Gaya JM , Lopez-Martinez JM , Karni-Schmidt O , Bonal DM , Algaba F , Palou J , et al. DeltaNp63 expression is a protective factor of progression in clinical high grade T1 bladder cancer. J Urol. (2015) ;193: (4):1144–50. |
[86] | Koga F , Kawakami S , Fujii Y , Saito K , Ohtsuka Y , Iwai A , et al. Impaired p63 expression associates with poor prognosis and uroplakin III expression in invasive urothelial carcinoma of the bladder. Clin Cancer Res. (2003) ;9: (15):5501–7. |
[87] | Koga F , Kawakami S , Kumagai J , Takizawa T , Ando N , Arai G , et al. Impaired Delta Np63 expression associates with reduced beta-catenin and aggressive phenotypes of urothelial neoplasms. Br J Cancer. (2003) ;88: (5):740–7. |
[88] | Choi W , Porten S , Kim SS , Willis D , Plimack ER , Hoffman-Censits J , et al. Identification of Distinct Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer with Different Sensitivities to Frontline Chemotherapy. Cancer Cell. (2014) ;25: :152–65. |
[89] | Choi W , Shah JB , Tran M , Svatek R , Marquis L , Lee IL , et al. p63 expression defines a lethal subset of muscle-invasive bladder cancers. PLoS One. (2012) ;7: (1):e30206. |
[90] | Liu H , Ai J , Shen A , Chen Y , Wang X , Peng X , et al. c-Myc Alteration Determines the Therapeutic Response to FGFR Inhibitors. Clin Cancer Res. (2017) ;23: (4):974–84. |
[91] | Mahe M , Dufour F , Neyret-Kahn H , Moreno-Vega A , Beraud C , Shi M , et al. An FGFR3/MYC positive feedback loop provides new opportunities for targeted therapies in bladder cancers. EMBO Mol Med. (2018) ;10: (4). |
[92] | Catto JW , Miah S , Owen HC , Bryant H , Myers K , Dudziec E , et al. Distinct microRNA alterations characterize high- and low-grade bladder cancer. Cancer Res. (2009) ;69: (21):8472–81. |
[93] | Drayton RM , Peter S , Myers K , Miah S , Dudziec E , Bryant HE , et al. MicroRNA-99a and 100 mediated upregulation of FOXA1 in bladder cancer. Oncotarget. (2014) ;5: (15):6375–86. |
[94] | Blick C , Ramachandran A , Wigfield S , McCormick R , Jubb A , Buffa FM , et al. Hypoxia regulates FGFR3 expression via HIF-1alpha and miR-100 and contributes to cell survival in non-muscle invasive bladder cancer. Br J Cancer. (2013) ;109: (1):50–9. |
[95] | Marzouka NA , Eriksson P , Rovira C , Liedberg F , Sjodahl G , Hoglund M . A validation and extended description of the Lund taxonomy for urothelial carcinoma using the TCGA cohort. Sci Rep. (2018) ;8: (1):3737. |
[96] | Bonaventure J , Horne WC , Baron R . The localization of FGFR3 mutations causing thanatophoric dysplasia type I differentially affects phosphorylation, processing and ubiquitylation of the receptor. FEBS J. (2007) ;274: (12):3078–93. |
[97] | Tomlinson DC , Hurst CD , Knowles MA . Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene. (2007) ;26: (40):5889–99. |
[98] | Qing J , Du X , Chen Y , Chan P , Li H , Wu P , et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J Clin Invest. (2009) ;119: (5):1216–29. |
[99] | Miyake M , Ishii M , Koyama N , Kawashima K , Kodama T , Anai S , et al. 1-tert-butyl-3-[6-(3,5-dimethoxy-phenyl)-2-(4-diethylamino-butylamino)-pyrido2,3 -d]pyrimidin-7-yl]-urea (PD74), a selective tyrosine kinase inhibitor of fibroblast growth factor receptor-3 (FGFR3), inhibits cell proliferation of bladder cancer carrying the FGFR3 gene mutation along with up-regulation of p27/Kip1 and G1/G0 arrest. J Pharmacol Exp Ther. (2010) ;332: (3):795–802. |
[100] | Lamont FR , Tomlinson DC , Cooper PA , Shnyder SD , Chester JD , Knowles MA . Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br J Cancer. (2011) ;104: (1):75–82. |
[101] | Guagnano V , Kauffmann A , Wohrle S , Stamm C , Ito M , Barys L , et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. (2012) ;2: (12):1118–33. |
[102] | Gust KM , McConkey DJ , Awrey S , Hegarty PK , Qing J , Bondaruk J , et al. Fibroblast growth factor receptor 3 is a rational therapeutic target in bladder cancer. Mol Cancer Ther. (2013) ;12: (7):1245–54. |
[103] | Herrera-Abreu MT , Pearson A , Campbell J , Shnyder SD , Knowles MA , Ashworth A , et al. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. (2013) ;3: (9):1058–71. |
[104] | Lima NC , Atkinson E , Bunney TD , Katan M , Huang PH . Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancers with FGFR3 Alterations. Int J Mol Sci. (2020) ;21: (9). |
[105] | Krejci P . The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutat Res Rev Mutat Res. (2014) ;759: :40–8. |
[106] | Legeai-Mallet L , Benoist-Lasselin C , Munnich A , Bonaventure J . Overexpression of FGFR3, Stat1, Stat5 and p21Cip1 correlates with phenotypic severity and defective chondrocyte differentiation in FGFR3-related chondrodysplasias. Bone. (2004) ;34: (1):26–36. |
[107] | Sahni M , Ambrosetti DC , Mansukhani A , Gertner R , Levy D , Basilico C . FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. (1999) ;13: (11):1361–6. |
[108] | Chesi M , Nardini E , Brents LA , Schrock E , Ried T , Kuehl WM , et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. (1997) ;16: (3):260–4. |
[109] | Chen J , Williams IR , Lee BH , Duclos N , Huntly BJ , Donoghue DJ , et al. Constitutively activated FGFR3 mutants signal through PLCgamma-dependent and -independent pathways for hematopoietic transformation. Blood. (2005) ;106: (1):328–37. |
[110] | Chesi M , Brents LA , Ely SA , Bais C , Robbiani DF , Mesri EA , et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. (2001) ;97: (3):729–36. |
[111] | Wang L , Sustic T , Leite de Oliveira R , Lieftink C , Halonen P , van de Ven M , et al. A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur Urol. (2017) ;71: (6):858–62. |
[112] | Jebar AH , Hurst CD , Tomlinson DC , Johnston C , Taylor CF , Knowles MA . FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. (2005) ;24: (33):5218–25. |
[113] | Juanpere N , Agell L , Lorenzo M , de Muga S , Lopez-Vilaro L , Murillo R , et al. Mutations in FGFR3 and PIK3CA, singly or combined with RAS and AKT1, are associated with AKT but not with MAPK pathway activation in urothelial bladder cancer. Hum Pathol. (2012) ;43: (10):1573–82. |
[114] | Datta J , Damodaran S , Parks H , Ocrainiciuc C , Miya J , Yu L , et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol Cancer Ther. (2017) ;16: (4):614–24. |
[115] | Wang J , Mikse O , Liao RG , Li Y , Tan L , Janne PA , et al. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene. (2015) ;34: (17):2167–77. |
[116] | Salazar L , Kashiwada T , Krejci P , Muchowski P , Donoghue D , Wilcox WR , et al. A novel interaction between fibroblast growth factor receptor 3 and the p85 subunit of phosphoinositide 3-kinase: activation-dependent regulation of ERK by p85 in multiple myeloma cells. Hum Mol Genet. (2009) ;18: (11):1951–61. |
[117] | Salazar L , Kashiwada T , Krejci P , Meyer AN , Casale M , Hallowell M , et al. Fibroblast growth factor receptor 3 interacts with and activates TGFbeta-activated kinase 1 tyrosine phosphorylation and NFkappaB signaling in multiple myeloma and bladder cancer. PLoS One. (2014) ;9: (1):e86470. |
[118] | Yokote H , Fujita K , Jing X , Sawada T , Liang S , Yao L , et al. Trans-activation of EphA4 and FGF receptors mediated by direct interactions between their cytoplasmic domains. Proc Natl Acad Sci U S A. (2005) ;102: (52):18866–71. |
[119] | Del Piccolo N , Sarabipour S , Hristova K . A New Method to Study Heterodimerization of Membrane Proteins and Its Application to Fibroblast Growth Factor Receptors. J Biol Chem. (2017) ;292: (4):1288–301. |
[120] | Mohammadi M , Honegger AM , Rotin D , Fischer R , Bellot F , Li W , et al. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol Cell Biol. (1991) ;11: (10):5068–78. |
[121] | Nelson KN , Meyer AN , Siari A , Campos AR , Motamedchaboki K , Donoghue DJ . Oncogenic Gene Fusion FGFR3-TACC3 Is Regulated by Tyrosine Phosphorylation. Mol Cancer Res. (2016) ;14: (5):458–69. |
[122] | Oh S , Shin S , Janknecht R . ETV1, 4 and an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. (2012) ;1826: (1):1–12. |
[123] | Mao J , McGlinn E , Huang P , Tabin CJ , McMahon AP . Fgf-dependent Etv4/5 activity is required for posterior restriction of Sonic Hedgehog and promoting outgrowth of the vertebrate limb. Dev Cell. (2009) ;16: (4):600–6. |
[124] | Zhang Z , Verheyden JM , Hassell JA , Sun X . FGF-regulated Etv genes are essential for repressing Shh expression in mouse limb buds. Dev Cell. (2009) ;16: (4):607–13. |
[125] | Znosko WA , Yu S , Thomas K , Molina GA , Li C , Tsang W , et al. Overlapping functions of Pea3 ETS transcription factors in FGF signaling during zebrafish development. Developmental biology. (2010) ;342: (1):11–25. |
[126] | Chotteau-Lelievre A , Revillion F , Lhotellier V , Hornez L , Desbiens X , Cabaret V , et al. Prognostic value of ERM gene expression in human primary breast cancers. Clin Cancer Res. (2004) ;10: (21):7297–303. |
[127] | Planaguma J , Abal M , Gil-Moreno A , Diaz-Fuertes M , Monge M , Garcia A , et al. Up-regulation of ERM/ETV5 correlates with the degree of myometrial infiltration in endometrioid endometrial carcinoma. J Pathol. (2005) ;207: (4):422–9. |
[128] | Monge M , Colas E , Doll A , Gonzalez M , Gil-Moreno A , Planaguma J , et al. ERM/ETV5 up-regulation plays a role during myometrial infiltration through matrix metalloproteinase-2 activation in endometrial cancer. Cancer Res. (2007) ;67: (14):6753–9. |
[129] | Firlej V , Ladam F , Brysbaert G , Dumont P , Fuks F , de Launoit Y , et al. Reduced tumorigenesis in mouse mammary cancer cells following inhibition of Pea3- or Erm-dependent transcription. J Cell Sci. (2008) ;121: (Pt 20):3393–402. |
[130] | Llaurado M , Abal M , Castellvi J , Cabrera S , Gil-Moreno A , Perez-Benavente A , et al. ETV5 transcription factor is overexpressed in ovarian cancer and regulates cell adhesion in ovarian cancer cells. Int J Cancer. (2012) ;130: (7):1532–43. |
[131] | Delpuech O , Rooney C , Mooney L , Baker D , Shaw R , Dymond M , et al. Identification of Pharmacodynamic Transcript Biomarkers in Response to FGFR Inhibition by AZD4547. Mol Cancer Ther. (2016) ;15: (11):2802–13. |
[132] | di Martino E , Alder O , Hurst CD , Knowles MA . ETV5 links the FGFR3 and Hippo signalling pathways in bladder cancer. Sci Rep. (2019) ;9: (1):5740. |
[133] | Lombardi B , Ashford P , Moya-Garcia AA , Rust A , Crawford M , Williams SV , et al. Unique signalling connectivity of FGFR3-TACC3 oncoprotein revealed by quantitative phosphoproteomics and differential network analysis. Oncotarget. (2017) ;8: (61):102898–911. |
[134] | Frattini V , Pagnotta SM , Tala , Fan JJ , Russo MV , Lee SB , et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. (2018) ;553: (7687):222–7. |
[135] | Neben CL , Lo M , Jura N , Klein OD . Feedback regulation of RTK signaling in development. Dev Biol. (2019) ;447: (1):71–89. |
[136] | Masoumi-Moghaddam S , Amini A , Morris DL . The developing story of Sprouty and cancer. Cancer Metastasis Rev. (2014) ;33: (2-3):695–720. |
[137] | Agazie YM , Movilla N , Ischenko I , Hayman MJ . The phosphotyrosine phosphatase SHP2 is a critical mediator of transformation induced by the oncogenic fibroblast growth factor receptor 3. Oncogene. (2003) ;22: (44):6909–18. |
[138] | St-Germain JR , Taylor P , Zhang W , Li Z , Ketela T , Moffat J , et al. Differential regulation of FGFR3 by PTPN1 and PTPN2. Proteomics. (2015) ;15: (2-3):419–33. |
[139] | Damrauer JS , Hoadley KA , Chism DD , Fan C , Tiganelli CJ , Wobker SE , et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci USA. (2014) ;111: (8):3110–5. |
[140] | The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. (2014) ;507: (7492):315–22. |
[141] | Sjodahl G , Eriksson P , Liedberg F , Hoglund M . Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol. (2017) ;242: (1):113–25. |
[142] | Sjodahl G , Lauss M , Lovgren K , Chebil G , Gudjonsson S , Veerla S , et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. (2012) ;18: (12):3377–86. |
[143] | Hedegaard J , Lamy P , Nordentoft I , Algaba F , Hoyer S , Ulhoi BP , et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell. (2016) ;30: (1):27–42. |
[144] | Lindskrog SV , Prip FF , Lamy P , Taber A , Groeneveld CS , Birkenkamp-Demtroder K , et al. An integrated multi-omics analysis identifies clinically relevant molecular subtypes of non-muscle-invasive bladder cancer. medRxiv. 2020; doi.org/10.1101/2020.06.19.20054809 |
[145] | Sweis RF , Spranger S , Bao R , Paner GP , Stadler WM , Steinberg G , et al. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res. (2016) ;4: (7):563–8. |
[146] | Foth M , Ismail NFB , Kung JSC , Tomlinson D , Knowles MA , Eriksson P , et al. FGFR3 mutation increases bladder tumourigenesis by suppressing acute inflammation. J Pathol. (2018) ;246: (3):331–43. |
[147] | Rosenberg JE , Hoffman-Censits J , Powles T , van der Heijden MS , Balar AV , Necchi A , et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. (2016) ;387: (10031):1909–20. |
[148] | Sharma P , Retz M , Siefker-Radtke A , Baron A , Necchi A , Bedke J , et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2017) ;18: (3):312–22. |
[149] | Mariathasan S , Turley SJ , Nickles D , Castiglioni A , Yuen K , Wang Y , et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) ;554: (7693):544–8. |
[150] | Wang L , Gong Y , Saci A , Szabo PM , Martini A , Necchi A , et al. Fibroblast Growth Factor Receptor 3 Alterations and Response to PD-1/PD-L1 Blockade in Patients with Metastatic Urothelial Cancer. Eur Urol. (2019) ;76: (5):599–603. |
[151] | Allory Y , Beukers W , Sagrera A , Flandez M , Marques M , Marquez M , et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: high frequency across stages, detection in urine, and lack of association with outcome. Eur Urol. (2014) ;65: (2):360–6. |
[152] | Hurst CD , Platt FM , Knowles MA . Comprehensive mutation analysis of the TERT promoter in bladder cancer and detection of mutations in voided urine. Eur Urol. (2014) ;65: (2):367–9. |
[153] | Tsai YC , Nichols PW , Hiti AL , Williams Z , Skinner DG , Jones PA . Allelic losses of chromosomes 9, 11, and 17 in human bladder cancer. Cancer Res. (1990) ;50: (1):44–7. |
[154] | Cairns P , Shaw ME , Knowles MA . Initiation of bladder cancer may involve deletion of a tumour-suppressor gene on chromosome 9. Oncogene. (1993) ;8: (4):1083–5. |
[155] | Czerniak B , Chaturvedi V , Li L , Hodges S , Johnston D , Roy JY , et al. Superimposed histologic and genetic mapping of chromosome 9 in progression of human urinary bladder neoplasia: implications for a genetic model of multistep urothelial carcinogenesis and early detection of urinary bladder cancer. Oncogene. (1999) ;18: (5):1185–96. |
[156] | Junker K , Boerner D , Schulze W , Utting M , Schubert J , Werner W . Analysis of genetic alterations in normal bladder urothelium. Urology. (2003) ;62: (6):1134–8. |
[157] | Obermann EC , Meyer S , Hellge D , Zaak D , Filbeck T , Stoehr R , et al. Fluorescence in situ hybridization detects frequent chromosome 9 deletions and aneuploidy in histologically normal urothelium of bladder cancer patients. Oncol Rep. (2004) ;11: (4):745–51. |
[158] | Stoehr R , Zietz S , Burger M , Filbeck T , Denzinger S , Obermann EC , et al. Deletions of chromosomes 9 and 8p in histologically normal urothelium of patients with bladder cancer. Eur Urol. (2005) ;47: (1):58–63. |
[159] | Otto W , Denzinger S , Bertz S , Gaumann A , Wild PJ , Hartmann A , et al. No mutations of FGFR3 in normal urothelium in the vicinity of urothelial carcinoma of the bladder harbouring activating FGFR3 mutations in patients with bladder cancer. Int J Cancer. (2009) ;125: (9):2205–8. |
[160] | WHO. WHO Classification of Tumours of the Urinary System and Male Genital Organs. 4th ed. Lyons: IARC Publications; (2016) . |
[161] | Hartmann A , Moser K , Kriegmair M , Hofstetter A , Hofstaedter F , Knuechel R . Frequent genetic alterations in simple urothelial hyperplasias of the bladder in patients with papillary urothelial carcinoma [In Process Citation]. Am J Pathol. (1999) ;154: (3):721–7. |
[162] | Obermann EC , Junker K , Stoehr R , Dietmaier W , Zaak D , Schubert J , et al. Frequent genetic alterations in flat urothelial hyperplasias and concomitant papillary bladder cancer as detected by CGH, LOH, and FISH analyses. J Pathol. (2003) ;199: (1):50–7. |
[163] | Chow NH , Cairns P , Eisenberger CF , Schoenberg MP , Taylor DC , Epstein JI , et al. Papillary urothelial hyperplasia is a clonal precursor to papillary transitional cell bladder cancer. Int J Cancer. (2000) ;89: (6):514–8. |
[164] | van Oers JM , Adam C , Denzinger S , Stoehr R , Bertz S , Zaak D , et al. Chromosome 9 deletions are more frequent than FGFR3 mutations in flat urothelial hyperplasias of the bladder. Int J Cancer. (2006) ;119: (5):1212–5. |
[165] | Hurst CD , Alder O , Platt FM , Droop A , Stead LF , Burns JE , et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell. (2017) ;32: (5):701–15 e7. |
[166] | Kiemeney LA , Sulem P , Besenbacher S , Vermeulen SH , Sigurdsson A , Thorleifsson G , et al. A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer. Nat Genet. (2010) ;42: (5):415–9. |
[167] | Isharwal S , Audenet F , Drill E , Pietzak EJ , Iyer G , Ostrovnaya I , et al. Prognostic Value of TERT Alterations, Mutational and Copy Number Alterations Burden in Urothelial Carcinoma. Eur Urol Focus. (2019) ;5: (2):201–4. |
[168] | McDaniel AS , Zhai Y , Cho KR , Dhanasekaran SM , Montgomery JS , Palapattu G , et al. HRAS mutations are frequent in inverted urothelial neoplasms. Hum Pathol. (2014) ;45: (9):1957–65. |
[169] | Eiber M , van Oers JM , Zwarthoff EC , van der Kwast TH , Ulrich O , Helpap B , et al. Low frequency of molecular changes and tumor recurrence in inverted papillomas of the urinary tract. Am J Surg Pathol. (2007) ;31: (6):938–46. |
[170] | Zieger K , Marcussen N , Borre M , Orntoft TF , Dyrskjot L . Consistent genomic alterations in carcinoma in situ of the urinary bladder confirm the presence of two major pathways in bladder cancer development. Int J Cancer. (2009) ;125: (9):2095–103. |
[171] | Hartmann A , Schlake G , Zaak D , Hungerhuber E , Hofstetter A , Hofstaedter F , et al. Occurrence of chromosome 9 and p53 alterations in multifocal dysplasia and carcinoma in situ of human urinary bladder. Cancer Res. (2002) ;62: (3):809–18. |
[172] | Spruck CH 3rd , Ohneseit PF , Gonzalez-Zulueta M , Esrig D , Miyao N , Tsai YC , et al. Two molecular pathways to transitional cell carcinoma of the bladder. Cancer Res. (1994) ;54: (3):784–8. |
[173] | Wagner U , Sauter G , Moch H , Novotna H , Epper R , Mihatsch MJ , et al. Patterns of p53, erbB-2 and EGF-r expression in premalignant lesions of the urinary bladder. Human Pathology. (1995) ;26: :970–8. |
[174] | Hopman AH , Kamps MA , Speel EJ , Schapers RF , Sauter G , Ramaekers FC . Identification of chromosome 9 alterations and p53 accumulation in isolated carcinoma in situ of the urinary bladder versus carcinoma in situ associated with carcinoma. Am J Pathol. (2002) ;161: (4):1119–25. |
[175] | Chandeck C , Mooi WJ . Oncogene-induced cellular senescence. Adv Anat Pathol. (2010) ;17: (1):42–8. |
[176] | Rebouissou S , Herault A , Letouze E , Neuzillet Y , Laplanche A , Ofualuka K , et al. CDKN2A homozygous deletion is associated with muscle invasion in FGFR3-mutated urothelial bladder carcinoma. J Pathol. (2012) ;227: (3):315–24. |
[177] | Zhou H , He F , Mendelsohn CL , Tang MS , Huang C , Wu XR . FGFR3b Extracellular Loop Mutation Lacks Tumorigenicity In Vivo but Collaborates with p53/pRB Deficiency to Induce High-grade Papillary Urothelial Carcinoma. Sci Rep. (2016) ;6: :25596. |
[178] | Hayashi Y , Fujita K , Nojima S , Tomiyama E , Matsushita M , Koh Y , et al. TERT C228T mutation in non-malignant bladder urothelium is associated with intravesical recurrence for patients with non-muscle invasive bladder cancer. Mol Oncol. 2020. |
[179] | Hosen MI , Sheikh M , Zvereva M , Scelo G , Forey N , Durand G , et al. Urinary TERT promoter mutations are detectable up to 10 years prior to clinical diagnosis of bladder cancer: Evidence from the Golestan Cohort Study. EBioMedicine. (2020) ;53: :102643. |
[180] | Patel PL , Suram A , Mirani N , Bischof O , Herbig U . Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc Natl Acad Sci U S A. (2016) ;113: (34):E5024–33. |
[181] | Song YS , Yoo SK , Kim HH , Jung G , Oh AR , Cha JY , et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr Relat Cancer. (2019) ;26: (6):629–41. |
[182] | Bullock M , Lim G , Zhu Y , Aberg H , Kurdyukov S , Clifton-Bligh R . ETS Factor ETV5 Activates the Mutant Telomerase Reverse Transcriptase Promoter in Thyroid Cancer. Thyroid. (2019) ;29: (11):1623–33. |
[183] | Khattar E , Tergaonkar V . Transcriptional Regulation of Telomerase Reverse Transcriptase (TERT) by MYC. Front Cell Dev Biol. 2017;5:1. |
[184] | Zhang F , Wang S , Zhu J . ETS variant transcription factor 5 and c-Myc cooperate in derepressing the human telomerase gene promoter via composite Ets/E-box motifs. J Biol Chem. 2020. |
[185] | van Rhijn BW , van der Kwast TH , Vis AN , Kirkels WJ , Boeve ER , Jobsis AC , et al. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. (2004) ;64: (6):1911–4. |
[186] | Lamy A , Gobet F , Laurent M , Blanchard F , Varin C , Moulin C , et al. Molecular profiling of bladder tumors based on the detection of FGFR3 and TP53 mutations. J Urol. (2006) ;176: (6 Pt 1):2686–9. |
[187] | Neuzillet Y , Paoletti X , Ouerhani S , Mongiat-Artus P , Soliman H , de The H , et al. A meta-analysis of the relationship between FGFR3 and TP53 mutations in bladder cancer. PLoS One. (2012) ;7: (12):e48993. |
[188] | Bakkar AA , Wallerand H , Radvanyi F , Lahaye JB , Pissard S , Lecerf L , et al. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res. (2003) ;63: (23):8108–12. |
[189] | Lopez-Knowles E , Hernandez S , Malats N , Kogevinas M , Lloreta J , Carrato A , et al. PIK3CA mutations are an early genetic alteration associated with FGFR3 mutations in superficial papillary bladder tumors. Cancer Res. (2006) ;66: (15):7401–4. |
[190] | Kompier LC , Lurkin I , van der Aa MN , van Rhijn BW , van der Kwast TH , Zwarthoff EC . FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS One. (2010) ;5: (11):e13821. |
[191] | Sjodahl G , Lauss M , Gudjonsson S , Liedberg F , Hallden C , Chebil G , et al. A systematic study of gene mutations in urothelial carcinoma; inactivating mutations in TSC2 and PIK3R1. PLoS One. (2011) ;6: (4):e18583. |
[192] | Platt FM , Hurst CD , Taylor CF , Gregory WM , Harnden P , Knowles MA . Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. (2009) ;15: (19):6008–17. |
[193] | Kompier LC , van der Aa MN , Lurkin I , Vermeij M , Kirkels WJ , Bangma CH , et al. The development of multiple bladder tumour recurrences in relation to the FGFR3 mutation status of the primary tumour. J Pathol. (2009) ;218: (1):104–12. |
[194] | Burger M , van der Aa MN , van Oers JM , Brinkmann A , van der Kwast TH , Steyerberg EC , et al. Prediction of progression of non-muscle-invasive bladder cancer by WHO 1973 and 2004 grading and by FGFR3 mutation status: a prospective study. Eur Urol. (2008) ;54: (4):835–43. |
[195] | van Rhijn BW , Zuiverloon TC , Vis AN , Radvanyi F , van Leenders GJ , Ooms BC , et al. Molecular grade (FGFR3/MIB-1) and EORTC risk scores are predictive in primary non-muscle-invasive bladder cancer. Eur Urol. (2010) ;58: (3):433–41. |
[196] | Sylvester RJ , van der Meijden AP , Oosterlinck W , Witjes JA , Bouffioux C , Denis L , et al. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: a combined analysis of 2596 patients from seven EORTC trials. Eur Urol. (2006) ;49: (3):466–77. |
[197] | van Rhijn BW , van der Kwast TH , Liu L , Fleshner NE , Bostrom PJ , Vis AN , et al. The FGFR3 mutation is related to favorable pT1 bladder cancer. J Urol. (2012) ;187: (1):310–4. |
[198] | Kang HW , Kim YH , Jeong P , Park C , Kim WT , Ryu DH , et al. Expression levels of FGFR3 as a prognostic marker for the progression of primary pT1 bladder cancer and its association with mutation status. Oncol Lett. (2017) ;14: (3):3817–24. |
[199] | Loriot Y , Necchi A , Park SH , Garcia-Donas J , Huddart R , Burgess E , et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. (2019) ;381: (4):338–48. |
[200] | McConkey DJ , Choi W , Shen Y , Lee IL , Porten S , Matin SF , et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naive Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur Urol. (2016) ;69: (5):855–62. |
[201] | Seiler R , Ashab HAD , Erho N , van Rhijn BWG , Winters B , Douglas J , et al. Impact of Molecular Subtypes in Muscle-invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur Urol. (2017) ;72: (4):544–54. |
[202] | Sung JY , Sun JM , Chang Jeong B , Il Seo S , Soo Jeon S , Moo Lee H , et al. FGFR3 overexpression is prognostic of adverse outcome for muscle-invasive bladder carcinoma treated with adjuvant chemotherapy. Urol Oncol. (2014) ;32: (1), 49 e23–31. |
[203] | Teo MY , Mota JM , Whiting KA , Li HA , Funt SA , Lee C-H , et al. Fibroblast Growth Factor Receptor 3 Alteration Status is Associated with Differential Sensitivity to Platinum-based Chemotherapy in Locally Advanced and Metastatic Urothelial Carcinoma. European Urology. 2020;In Press. https://doi.org/10.1016/j.eururo.2020.07.018 |
[204] | Xie X , Lin J , Zhong Y , Fu M , Tang A . FGFR(3S249C) mutation promotes chemoresistance by activating Akt signaling in bladder cancer cells. Exp Ther Med. (2019) ;18: (2):1226–34. |


