
Tumor Subtyping: Making Sense of Heterogeneity with a Goal Toward Treatment
Abstract
BACKGROUND:
Bladder cancers have high total mutation burdens resulting in genomic diversity and intra- and inter-tumor heterogeneity that may impact the diversity of gene expression, biologic aggressiveness, and potentially response to therapy. To compare bladder cancers among patients, an organizational structure is necessary that describes the tumor at the histologic and molecular level. These “molecular subtypes”, or “expression subtypes” of bladder cancer were originally described in 2010 and continue to evolve secondary to next generation sequencing (NGS) and an increasing public repository of well-annotated cohorts.
OBJECTIVE:
To review the history and methodology of expression-based subtyping of non-muscle invasive (NMIBC) and muscle invasive bladder cancer (MIBC).
METHODS:
A literature review was performed of primary papers from PubMed that described subtyping methods and their descriptive feature including search terms of “subtype”, and “bladder cancer”.
RESULTS:
21 papers were identified for review. Tumor subtyping developed from N = 2 to N = 6 subtyping schemes with most subtypes comprised of at least luminal and basal tumors. Most NMIBCs are luminal cancers and luminal MIBCs may be associated with less aggressive features, while one study of basal tumors identified a better clinical outcome with systemic chemotherapy. Tumors with a P53-like may have intrinsic resistance to chemotherapy. The heterogeneity of tumors, which is likely derived from stromal components and immune cell infiltration, affect subtype calls.
CONCLUSION:
Subtyping, while still evolving, is ready for testing in clinical trials. Improved patient selection with tumor subtyping may help with tumor classification and potentially match patient or tumor to therapy.
INTRODUCTION
Bladder cancer is the ninth most common cancer worldwide [1]. Despite improvements in local and systemic therapy, risk-stratification, and smoking cessation, there have been no improvements in survival for patients with bladder cancer since 2000 [2]. The high mortality rate among patients with muscle invasive bladder cancer (MIBC) is likely secondary to molecular heterogeneity and the lack of durable responses to broadly applied systemic therapies [3]. Patients with metastatic bladder cancer have very poor survival estimated at less than 5% at 5 years [4]. With widespread adoption of next-generation sequencing (NGS), solid tumor oncology has shifted from classic histopathologic classification towards molecular-based approaches [5]. Breast and lung cancers have been at the forefront of this movement with molecular testing to stratify prognosis and select therapeutics [5]. Despite one of the highest frequencies of mutations among cancers, bladder cancer has few subtype-defining oncogene alterations that can be targeted with systemic therapy, such as ALK, BRAF, EGFR, Estrogen receptor (ER) and HER-2 [6]. As an alternative to classifying tumors by actionable DNA alterations, RNA-based tumor subtyping was developed to articulate the molecular features of the tumor, tumor microenvironment, and immune infiltrate (when present). While genomic alterations may be passed to daughter cells during mitosis and are the “blueprints” for tumors, gene signatures from the transcriptome summarize the expression landscape and are more dynamic, reflecting intracellular signaling, biologic processes, and cellular composition of the tumor epithelium and microenvironment. These gene signatures collectively can be summarized by tumor expression subtype [7]. Therefore, the potential benefit of tumor subtyping is that the subtype may be a more a more informative description of the tumor biology that may translate into improved risk stratification and clinical decision-making compared to grade and stage. Herein, we will look back on the advances made in the molecular characterization of bladder cancer, review the clinical utility of RNA-based subtyping for decision making, and look to the future for clinical application.
HOW ARE SUBTYPES CREATED?
In this review we describe the results of gene expression-based subtyping. But how is this performed? Most subtyping begins with an unsupervised clustering [8] approach in which a statistical model is applied to group samples by differential gene expression. Often, 2 to 7 subtypes can be generated depending on the number of patients in the cohort and the quality of RNA. What is the “right” number of subtypes? This topic is controversial [3, 9]. While we anticipate that most investigators would develop the same or a similar number of subtypes if the sample sizes were sufficiently large, there may be subjectivity in determining tumor subtypes. Validation of the stability of the subtypes can require repeat analysis, silhouette plots and evaluation of gene-ontology pathways that can help support the unique features of each subtype.
Molecular subtypes are usually generated using mRNA expression (“transcriptome”) data, although the Cancer Genome Atlas (TCGA) and other groups have also used DNA alterations (mutations and copy number variations), microRNA or lncRNA expression patterns, and even combinations of genomic data (“clusters of clusters”). The mRNA expression datasets are often “filtered” to select what the investigator thinks will be the most informative subset of genes (i.e., the top 10% most variable genes across the cohort), although this filtering is not necessary. The dataset is then subjected to an unsupervised analysis using an algorithm that groups tumors together based on shared gene expression. Mathematical formulas exist to define the number of clusters that fit best with the dataset. Importantly, the number of clusters that result is dependent on both the size of the dataset and its composition (i.e., its biological heterogeneity), and the results may also be sensitive to the type of tissue used to generate the transcriptome data (fresh, flash-frozen, or FFPE, for example) and the platform employed (i.e., RNA-Seq versus direct hybridization arrays). Therefore, the number of molecular subtypes observed in any discovery study will be dictated by all of these variables; however, once molecular subtypes are settled upon, tumors can be “fit” into any subtyping scheme using a supervised approach. Finally, molecular subtype assignments are typically made using only molecular data with no consideration of clinical covariates. The rationale for doing this is that clinical outcomes are probably influenced by a variety of different variables that are not directly related to underlying tumor biology. A summary of papers published on bladder cancer subtyping is included in Table 1.
Table 1
| Original RNA-subtyping study | Publication year | n (tumors) | k (subtypes) | MIBC/NMIBC | Nomenclature | Study focus | Reference |
| Sjödahl et al. | 2012 | 308 | 5 | Mix | UroA, UroB, GU, Infiltrated, SCC-like | Biological charactrization | 14 |
| Choi et al. | 2014 | 73 | 3 | MIBC | Lum, p53-like, Basal | Role of PPARG/TP63. Chemotherapy response | 18 |
| Damrauer et al. | 2014 | 262 | 2 | MIBC | Lum, Basal | Comparison to breast cancer subtypes | 17 |
| TCGA | 2014 | 129 | 4 | MIBC | Cluster I-IV | Associations to genomic data | 19 |
| Rebouissou et al. | 2014 | 85 | 2 | MIBC | Basal and Non-Basal categories (MC1-MC7) | Role of EGFR in basal subtype | 38 |
| Hedegaard et al. | 2016 | 460 | 3 | NMIBC | Class 1-3 | NMIBC biology, Mutational signatures | 39 |
| Sjödahl et al. | 2017 | 307 | 6 | MIBC | Uro, GU, Epi-Inf, SCCL/Mes, SCCL/UroB, Sc/NE | Association to IHC-based subtyping | 21 |
| Robertson et al. | 2017 | 408 | 5 | MIBC | Lum-P, Lum-Inf, Lum, BASQ, Neuronal | Patient outcomes. Association to lncRNA and miRNA levels | 23 |
| Seiler et al. | 2017 | 269 | 4 | MIBC | Lum, Lum-Inf, Basal, Claudin-low | Outcome after chemotherapy treatment | 30 |
| Tan et al. | 2019 | 2411 | 6 | NMIBC/MIBC | Pap, Lum, HER2L, SCC, MES, NEURAL | Associations to pathological evaluation and signaling pathways | 25 |
| Kamoun et al. | 2019 | 1750 | 6 | MIBC | LumP, LumNS, LumU, Stroma-rich, BASQ, NE-like | Identify conensus subtypes. Clinical and biological associations | 27 |
EARLY SUBTYPING SIGNATURES
Prior to the large-scale NGS transcriptome profiling performed by TCGA, there were multiple series that compared RNA profiling of invasive, locally advanced or metastatic tumors to identify occult signatures of aggressive cancers. These experiments applied hybridization technology to classify bladder cancers by cDNA microarrays [10– 12]. These studies included 75 tumors on average, most of which were formalin-fixed and paraffin-embedded (FFPE). To the credit of these authors, the results of the arrays were made publicly available and are still incorporated in meta-cohorts investigated today (see below). Yet, no reproducible gene lists were consistent between studies and gene profiles that differentiated MIBC from NMIBC were no more helpful than pathologic staging. Thus, the clinical utility of each genomic classifier remained dependent on the specific cohort from which it was created. This may have been secondary to the methods used for RNA extraction, batch-effects or the bioinformatic analysis applied to analyze each cohort.
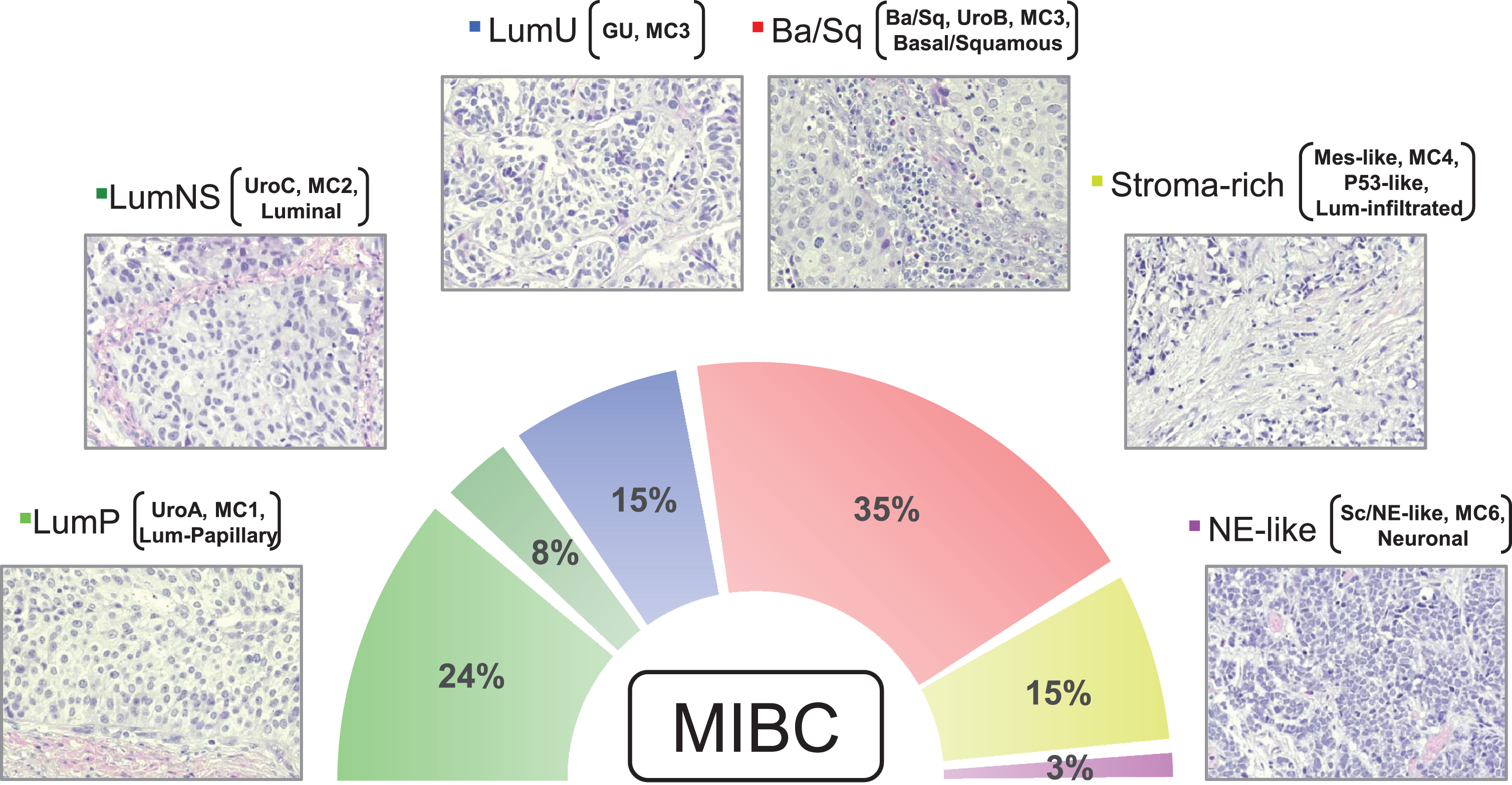
Fig. 1
Micrographs of one representative hematoxylin & eosin stained tumor for each consensus subtype is shown (200×). The figure exemplifies the typical histomorphological patterns for each consensus subtype: Luminal-papillary, relatively organized urothelial histology; LumNS, less organized urothelial histology; LumU, severely disorganized urothelial histology; Ba/Sq, squamous differentiation; Stroma rich, Infiltrative growth pattern with stromal reaction; NE-like, Neuroendocrine differentiation. While the neuroendocrine tumors have neuroendocrine molecular features, they may not have neuroendocrine histology. Percentages show the proportion of MIBC tumors belonging to each subtype based on data in Kamoun et al. (2019). In addition to the consensus subtype nomenclature, subtypes from other classification systems (Lund, CIT-Curie, MDA, and TCGA) enriched in each of the consensus subtypes are shown in brackets.
In 2010, the group of Mattias Höglund of Lund University undertook the first effort to subtype bladder cancer that would evolve into the current molecular characterization of bladder cancer. Analyzing 99 NMIBC and 45 MIBC tumors, the group identified two intrinsic molecular subtypes of bladder cancer, MS1 (mainly NMIBC) and MS2 (mainly MIBC) [13] (Figure). In 2012, the Höglund group validated molecular subtypes with 215 NMIBC and 93 MIBC tumors [14]. The authors suggested a framework of classifying tumors based on their epithelial differentiation. They further expanded on the MS1 and MS2 subtypes resulting in a total of 7 tumor (MS1a, MS1b, MS2a1, MS2a2, MS2b1, MS2b2.1, MS2b2.2). Biological characterization of these clusters resulted in the definition of 5 major, reproducible molecular subtypes: UroA (MS1a, MS1b), genomically unstable (MS2a1, MS2a2), infiltrated (MS2b1), UroB (MS2b2.1), and SCC-like (MS2b2.2). Importantly, each subtype showed unique gene expression profiles, were enriched for different tumor stage categories, and showed differences in survival patterns. UroA tumors were primarily low grade, papillary NMIBCs and were associated with the best prognosis of the 5 subtypes. Biologically, these tumors were characterized by elevated levels of FGFR3 and frequent mutations in FGFR3, suggesting that FGFR3 may play an important role in the biological properties of these malignancies. SCC-like and UroB tumors were predominantly MIBC, and patients with these tumors had the worst prognosis of the 5 subtypes. Both UroB and SCC-like subtypes shared similar expression of basal keratins not normally expressed in the urothelium. UroB tumors were notable for frequent co-existence of TP53 and FGFR3 mutations. Infiltrated tumors were found to be significantly enriched in several genes for collagens, proteoglycans, and basal laminal components. This would indicate that the gene expression profile of these tumors is heavily comprised of tumor-infiltrating stromal cells and endothelial cells. Genomically unstable tumors were found to have significantly higher frequency of TP53 mutations but no FGFR3 mutations and demonstrated grossly rearranged genomes.
AT LEAST TWO MIBC SUBTYPES: BASAL AND LUMINAL
Subtyping of breast cancers identified 5 molecular subtypes associated with distinct histologic and clinical outcomes including incidence, risk factors, prognosis, and treatment efficacy [15]. A similar stratification of bladder cancers was suggested by early molecular descriptions of basal and luminal differentiation states [16]. The first subtyping study to apply these breast cancer-related subtypes to bladder cancer used a two-subtype system that classified tumors in either “luminal” or “basal” cancers [17]. At a minimum, these groupings parse tumors by similarity to either more stem-like cells that originate from the base of stratified epithelium (basal tumors) of the bladder or to more differentiated epithelial cell (luminal tumors). The Kim Lab combined four publicly available datasets to generate a meta-dataset of 262 MIBCs [17]. The classification system, named BASE47, that applied a minimal set of 47 genes, was able to accurately group tumors by RNA expression profile and differentiate luminal and basal tumors. Like the Lund UroB and SCC-like subtypes, the basal-like cluster was enriched in KRT5, KRT14, KRT6B, but also the stem cell marker CD44 normally found in basal urothelial cells, suggesting a more poorly differentiated origin of these tumors [17, 18] with similarities in gene expression to basal breast tumors [17– 19]. In addition to basal cellular markers, basal tumors had increased EMT signatures such as higher level of TWIST1/2, SNAI2, ZEB and VIM and epidermal growth factor receptor (EGFR) signaling pathways. Basal tumors tended to be more biologically aggressive, with alterations identified in pathways involved in tumorigenesis, cell survival, and cellular movement [17– 19]. Clinically, basal tumors were associated with worse outcomes (overall survival reduction by 14.9 months, p = 0.098). Patients with basal tumors had an increased risk of metastasis at presentation and sarcomatoid differentiation on pathology [17– 19]. “Luminal” tumors were named due to their similarities in gene expression to luminal breast tumors and positivity for markers of differentiated urothelium [17– 19]. In contrast to basal tumors, luminal subtype cancers were enriched in low molecular weight keratins (KRT20), uroplakins (UPK1, UPK2, UPK3A), mutations in FGFR3 and TSC1, and HER2 upregulation. Clinically, luminal tumors did significantly better than their basal tumor counterparts, with improved overall survival and disease-specific survival.
MOVING ON FROM TWO SUBTYPES
In 2014, two publications expanded on a two-subtype system and suggested at least three distinct MIBC subtypes were reasonable. The TCGA identified two luminal subtypes (I and II) and two basal subtypes (III and IV) [11]. Within the luminal subtypes (subtype I and II by TCGA), the MD Anderson (MDA) group identified a subset of tumors characterized by a p53 gene expression signature that was associated with an “activated” P53 pathway (although not associated with TP53 alterations [18, 19]). So-called “P53-like” tumors were luminal architecture, had some similar features to basal tumors, with low levels of luminal differentiation markers, yet were unstable on silhouette analysis suggesting these tumors could be misclassified on repeated testing (like TCGA subtype II). An important clinical feature of p53-like tumors was the poor response to cisplatin-based neoadjuvant chemotherapy (0/7 in the discovery cohort and 1/9 in the validation cohort responded to chemotherapy). This subset corresponded well to Cluster II identified by the 2014 TCGA [19].
While initially described in their 2014 BASE47 manuscript, the University of North Carolina (UNC) group further investigated the claudin-low subtype in a follow-up study in 2015 aided by additional tumors from the 2014 TCGA [17]. “Claudin-low” tumors were named after the lower expression of the tight-junction proteins claudins 3, 4, and 7 but with a corresponding increase in pro-invasive pathways such as EMT and stem cell genes [20]. Claudin low tumors represented 16% of the original UNC cohort, and clustered within the basal subtype with no difference in disease-specific or overall survival from basal tumors. After the first TCGA publication in 2014, a follow up description of claudin low tumors suggested this subtype corresponded to tumors classified within Clusters III and IV identified by the TCGA [19] with poor clinical outcomes similar to basal tumors. While broadly clustering with basal tumors, the smaller set of claudin low tumors (10% overall of the TCGA) had increased frequency of TP53 (62%), RB1 (46%), NCOR1(20%) and EP300 (31%) mutations compared to both luminal and basal subtypes, with fewer mutations in KDM6A (11%) and FGFR3 (2%). These tumors were enriched in genes involved in epithelial-to-mesenchymal transition (EMT), claudins and immune cell signatures. Claudin-low tumors had a mixed immunologic phenotype with enrichment of T-cells (CD8, CD3, CD4), B-cells, and macrophages, but also enhanced expression of exhaustion markers PDL1 (CD274), CTLA4, TIM3, LAG3 and PD1. Thus, Kim suggested that claudin low tumors were primed tumors that could potentially respond to checkpoint therapy.
The initial Lund classification included both NMIBC and MIBC samples and was not directly comparable to the other MIBC-based classifiers. In 2017, the Lund group updated their classification to the MIBC context by analysis of 307 tumors with matched RNA and immunohistochemistry data [21]. The analysis identified their previously described subtypes Uro (A/B), genomically unstable, and Basal/SCC-like as well as a minor VIM+ and ZEB2 + mesenchymal-like subtype and a minor TUBB2B+ small-cell/neuroendocrine subtype. In addition to describing these novel minor subtypes the Lund group used the immunostainings to dissect how tumor-cell intrinsic properties, versus tumor environment, drives samples to segregate or converge into gene expression clusters. The conclusion is that non-tumor cell molecular signals play a large role in determining the structure of gene expression clusters detected in MIBC. This work led to an updated Lund taxonomy that was later applied to the larger TCGA cohort where mutation and copy-number alterations were differentially distributed among the subtypes [22].
With the second comprehensive analysis of the TCGA that included 408 MIBCs, unbiased non-negative matrix factorization (NMF) consensus clustering of RNA-seq profiling identified 5 unique MIBC subtypes: luminal, luminal-papillary, luminal-infiltrated, basal-squamous, and neuronal [23]. The luminal-papillary (35% of tumors) corresponded with the luminal (UNC, MDA), Lund UroA and TCGA 2014 Cluster I group enriched in papillary tumors with high expression of FGFR3. The LP tumors had the best prognosis of the 5 subtypes. Luminal-infiltrated tumors (19%) were defined by the presence of smooth muscle, myofibroblasts, and lymphocytes and demonstrated enrichment of the wildtype p53-gene signature, similar to the MDA p53-like, Lund infiltrated and TCGA 2014 cluster II group. The luminal subtype (6%) had the highest expression of uroplakins (UPK1A and UPK2) and other markers for terminal urothelial differentiation suggestive of differentiation from a luminal umbrella cell. The basal-squamous cluster (35%) was enriched for squamous histology with high levels of basal and stem cell markers and low levels of Sonic hedgehog signaling.
A notable finding from the TCGA 2017 was the addition of a neuronal subtype of MIBC (5%). In their 2015 paper, the Lund group identified a subset of poorly differentiated tumors defined by increased expression of pluripotency-associated genes (SOX2 and SOX21) as well as an enrichment in alterations to RB1. According to the Lund classification, this cluster is termed small-cell/neuroendocrine-like [24]. The TCGA confirmed a neuronal signature identifying a subset of tumors with high expression of genes involved in neuronal differentiation and development, yet only 3 of the 20 tumors had histologic features suggestive of neuroendocrine histology. Because they lack expression of luminal urothelial differentiation genes, the neuronal tumors cluster closer to the basal squamous subtype than to luminal-like subtypes. The genetic hallmark of neuronal tumors, consisting of mutation of both TP53 and RB1, was found in 85% of tumors. Notably, the neuronal subtype had the worst prognosis of all the 5 subtypes.
EXPANDING THE COHORTS
As the number of tumors in each cohort increases, the ability to identify subtle, but potentially important biologic variations that distinguish each subtype is possible. Most of the aforementioned studies were smaller cohorts, with the 2017 TCGA cohort the largest at 408 tumors [23]. With increased size of the cohort, the detection of low frequency alterations can be evaluated that can impact tumor subtype; more than 3,000 samples would be needed to reliably detect mutations in 2% of the samples that could identify an independent subtype [25]. While all subtyping systems share agreement that at least basal and luminal tumors are the anchor point of a subtype, the addition of more tumors could further distinguish the main subtypes based on non tumor-cell expression features such as immune cell infiltrate and stromal cells [7]. Compiling multiple retrospective cohorts to generate a large “meta-cohort” was a logical next step to identify rare subtypes of MIBC. This “meta-cohort” included 2411 unique bladder tumors (both NMIBC and MIBC) from a total of 36 publicly available bladder cancer gene expression datasets [25]. One major challenge of this effort was the inter- and intra-study variation between cohorts and platforms (Illumina and Affymetrix) that required significant batch-correction (by application of ComBat) twice (once to combine studies on each platform and once to combine platforms). The meta-cohort identified six molecular subtypes (termed BOLD subtypes) which demonstrated good concordance with prior subtypes generated by the Lund, MDA, UNC, and TCGA groups. This study identified 3 distinct luminal subtypes (luminal, papillary, and HER2-like), a distinct Basal/SCC-like subtype, a neuronal subtype, and a stem-like mesenchymal (MES) subtype that resembled claudin-low tumors. Similar to prior studies, the subtypes demonstrated significantly different clinical prognosis (p < 0.0001). In the entire meta-cohort of NMIBC and MIBC, papillary tumors had the best overall median survival (>135 months) while SCC-like tumors demonstrated the poorest survival (20.6 months) as expected given the stage differences.
One strength of BOLD was the inclusion of NMIBC tumors (20%) suggesting a common subtyping classification that could potentially be applied across urothelial carcinoma, regardless of stage. Contributing studies ranged from stage-Ta NMIBC, pelleted urine samples, formalin fixed, fresh frozen samples, metastatic tumors, and even normal samples. Merging data from such diverse sources leads to batch effects that cannot be overcome by in-silico adjustments. Indeed, the identified BOLD classes were significantly enriched for technical variables beyond what could be explained by inclusion criteria of the contributing studies, and thus we could not compare BOLD to the other subtype classification systems without a high risk for bias. The most significant drawbacks of the meta-cohort were the lack of annotation of treatment response (including intravesical and systemic therapy), and the strong heterogeneity of the included studies leading to a persisting association between subtypes and variability even after batch adjustments (see commentary [26]). To date, BOLD has not been applied prospectively in randomized trials. These consensus studies represent an important step in the right direction towards a consolidation of the multiple subtyping schemas and utilization of this data towards clinical decision making, but careful examination of the patients (stage, tissue type and preparation) could add further granularity to the analysis.
CONSENSUS MIBC SUBTYPING: ONE METHOD TO UNIFY ALL SUBTYPING
As more subtyping schemes emerged, it became apparent that achieving a standard subtyping platform could improve tumor clustering across clinical trials and move subtyping from a descriptor to a biomarker. Therefore, the Bladder Cancer Molecular Taxonomy Group developed a consensus molecular classification schema in 2019 [27] that combined 18 publicly available MIBC mRNA datasets to assemble a cohort of 1,750 tumors. Each tumor was MIBC, and aggregated from 29 subtypes that was processed into six consensus subtypes by a Markov clustering algorithm. Six distinct biologically relevant consensus molecular subtypes were identified using consensus based clustering: luminal papillary (LumP), luminal non-specified (LumNS), luminal unstable (LumU), Stroma-rich, Basal/Squamous (Ba/Sq), and Neuroendocrine-like (NE-like) (Figure). Consistent with prior investigations, the three luminal subtypes (LumP, LumNS, and LumU) overexpressed urothelial differentiation genes while the Ba/Sq and NE-like tumors were found to have high levels of genes associated with basal and neuroendocrine differentiation, respectively. LumP had the highest frequency of mutations in FGFR3 (40%) and KDM6A (38%), as well as papillary morphology and the best median survival (4 years). LumNS occurred more in elderly patients, was enriched in ELF3 mutations (35%) and fibroblast stromal infiltration, and 36% of tumors demonstrated micropapillary variant histology with a 1.8 year median survival. LumU tumors had a high cell cycle enrichment, frequent TP53 (76%) and ERCC2 mutations (22%), a high mutation burden and APOBEC signature with a median survival of 2.9 years. Stroma-rich tumors had significant smooth muscle, fibroblast and B cell infiltrate with a median survival of 3.8 years. Basal tumors had increased mutations in TP53 (61%) and RB1 (25%) with EGFR activation, CD8+ and NK-cell infiltration. Basal tumors were more common in women and presented with advanced stage and a median survival of 1.2 years. Finally, neuroendocrine tumors had both mutations in TP53 (94%) and RB1 (39%) with neuroendocrine differentiation on histology and the lowest median survival at 1 year.
ROLE OF MOLECULAR SUBTYPES ON THERAPEUTIC RESPONSE
The first seminal papers on tumor subtyping were descriptive, assigning each tumor to a subtype and attempting to identify functional or gene ontology pathways that were distinct among subtypes. Most of the clinical outcomes differentiating the subtypes were based on recurrence and/or survival, with possible systemic therapy limited to adjuvant chemotherapy. Ideally, subtyping could be applied before surgery to help select tumors for decisions involving systemic therapy. For example, in breast cancer, the molecular subtype can help determine the benefit of neoadjuvant chemotherapy as patients with basal-like and HER2-positive tumors benefit most from neoadjuvant systemic therapy [28]. While the exact subtypes of MIBC remain debated, limited studies have investigated the subtype-specific responses to surgery, chemotherapy and immunotherapy.
The first group to investigate treatment response associated with each subtype was the MDA group in 2014. In their initial discovery cohort, Choi and colleagues noted that the p53-like tumors were mostly resistant to neoadjuvant chemotherapy [18]. The group was able to replicate this chemoresistance in vitro in eight bladder cancer cell lines with the p53-like gene signature that were resistant to cisplatin-induced apoptosis. While luminal tumors did not have a signature that affected response to chemotherapy, the authors found that two cohorts of chemotherapy-treated basal tumors had pathologic response only when they co-expressed immune infiltrated markers. In a follow-up study, the MDA group investigated the response of 60 patients in a neoadjuvant trial of dose-dense MVAC (DDMVAC) and bevacizumab [29]. Using a three-subtype panel, (basal, luminal and p53-like) 38 pre-treatment TUR specimens were profiled. Tumors with a p53-like signature again had the worst survival following treatment with a 5-year survival of 36%, consistent with prior data that p53-like tumors have intrinsic chemoresistance, compared to luminal tumors (73%) and a surprisingly high survival for basal tumors (91% at 5 years). Bone metastasis, with a median overall survival of only 15 months, occurred only in p53-like tumors (9/16 p53-like patients). A confirmatory cohort treated with MVAC NAC again demonstrated improved 5-year survival for basal subtype tumors (77%) compared to luminal (56%) or p53-like (56%), p < 0.021). While p53-like tumors did not have worse survival in this confirmatory cohort, the validation cohort confirmed improved survival in basal tumors treated with NAC. While the basal subtype was relatively small (n = 11), an improvement in survival may have been associated with bevacizumab targeting the HIF-like hypoxia signature identified in basal tumors. Thus, luminal tumors seemed to have the best clinical outcome regardless of treatment, basal tumors had the worst in absence of NAC, but were potentially responsive to NAC, and the p53-like tumors were unresponsive to cisplatin-chemotherapy.
A major limitation to subtyping is that subtyping strategies compare relative gene expression patterns among tumors in a cohort of patients. To apply subtyping to individual patients, a single-patient classifier would need to be developed in which individual patients could be evaluated and assigned to a subtype. Seiler et al. developed a single-patient classifier in a retrospective cohort of 269 consecutive MIBC patients from 5 institutions treated with at least three cycles of NAC. The outcomes by subtype were compared to 397 MIBC patients from other cohorts who did not receive NAC [30]. The single-patient genomic subtyping classifier (GSC) was trained to classify tumors into one of four subtypes: claudin-low, basal, luminal-infiltrated, and luminal. Luminal tumors had the best overall survival across subtyping methods regardless of NAC. Overall survival of patients with basal tumors was worst in the non-NAC cohort, but similar to the luminal tumors in the NAC cohort, even though there was no difference in pathologic response to chemotherapy. A notable limitation in this study was the lack of correlation of pathologic response with overall survival, which contrasts with most of the established literature. Consistent with prior studies, patients with luminal-infiltrated tumors (TCGA 2014 Cluster II, p53-like) fared poorly compared to other luminal subtypes. A major limitation of this study was the comparison of patient outcomes by subtype across multiple different and separate patient cohorts, in addition to the retrospective nature of the study.
Some studies have not been able to demonstrate a difference in survival across subtypes treated with chemotherapy. A 2018 Phase II trial of dose-dense gemcitabine and cisplatin (DDGC) (n = 28) and accelerated MVAC (AMVAC) (n = 43) compared survival based on the three MDA subtypes [31]. While p53-like tumors had a trend to worse survival in the DDGC cohort, a combined analysis found no significant differences in survival or response by subtype. The Bladder Cancer Molecular Taxonomy Group evaluated the association between their novel classification schema and were unable to identify any significant association of consensus class with pathologic response to NAC. [27].
Three clinical trials of immunotherapy have investigated if subtype has an impact on the clinical response of both metastatic and localized bladder cancer. IMvigor 210 was a phase II trial of metastatic platinum refractory or cisplatin-ineligible patients treated with the PD-L1 inhibitor, atezolizumab [32]. The study team sought to determine TCGA subtypes (K = 4) using a curated gene list of 8 genes and suggested that cluster II tumors, had a significantly higher clinical response compared to other subtypes. The investigators found enrichment of PD-L1 expression and immune cell infiltration in the basal subtype versus the luminal subtype, although the increased PD-L1 expression did not correlate with the objective response rate.
An integrated biomarker analysis was performed that included immunohistochemistry, tumor mutation profiling and transcriptome analysis in a more extensive evaluation of the 298 patients from the IMvigor 210 cohort. While the investigators did not identify an association with tumor subtyping using a reduced TCGA subtyping method, tumors classified as genomically unstable (GU) by the Lund Taxonomy had improved response to atezolizumab. The first version of the Lund subtyping applied here did not include a Sc/NE subtype and this subset of tumors was likely classified as GU, the closest subtyping neighbor. Their initial TCGA subtyping was likely affected by the classification methodology applied to each tumor using only a limited gene set. The TCGA single-patient classifier by Kim and colleagues using the TCGA 2017 classification identified a significantly improved response in neuronal tumors [33]. While associated with the worst survival in the TCGA 2017, the 11 neuronal tumors identified in IMVigor210 were found to have the best survival among the 5 subtypes (2 complete response, 6 partial response, and 3 unknown). The neuronal subtype was small in IMVigor210, but the results were confirmed by The Bladder Cancer Molecular Taxonomy Group with consensus MIBC subtyping that identified an enhanced response in NE-like, LumNS and LumU tumors [27]. Further investigation of improved response of neuronal tumors should be investigated in clinical trials.
In an analysis of 177 second line metastatic UC patients treated with the anti-PD1 antibody Nivo-lumab in Checkmate 275, greater response was identified in basal (TCGA III) tumors compared to other subtypes (30% response). The methodology applied to perform subtyping was not discussed in the methods section of the manuscript, but the basal tumors were also enriched for chemokines CXCL9 and CXCL10 and CD8 expression, which was independently associated with response to Nivolumab [34]. Powles et al found no difference in pathologic response of tumors subtyped by the Lund Taxonomy when treated with two cycles of neoadjuvant atezolizumab in the ABACUS trial. The subtyping analysis in ABACUS may have been limited by the relatively small numbers of patients (n = 95 patients) and 5 subtype groups in the Lund Taxonomy. In contrast, an evaluation of the PURE01 neoadjuvant immunotherapy cohort that included 84 patients identified an enhanced response of basal tumors (63– 65% partial or complete response) and luminal infiltrated tumors (68%) [35].
Most analyses of treatment response stratified by subtype have focused on the poor outcomes at radical cystectomy for basal tumors. Yet, luminal tumors were described to have lower clinical stage in the TCGA 2017 (T2, 55% versus 23% for non-luminal, p < 10–8), suggesting that luminal tumors might be managed with radical cystectomy alone, thereby potentially avoiding NAC. Lotan et al evaluated 206 patients with clinical T1 and T2 tumors who underwent cystectomy and compared pathologic upstaging rates between luminal (n = 100) and non-luminal tumors (n = 106) grouped into subtype by Decipher Bioscience’s GSC platform [36]. Upstaging to pT3-4 was found in only 24% of luminal tumors compared to 47% of non-luminal tumors (p < 0.001), but there was no difference in the rates of node metastasis (21% vs 26%). The improved pathologic staging in luminal tumors may be secondary to higher inclusion of T1 tumors (87), of which 47 (54%) were luminal cancers.
Bladder preservation with chemoradiotherapy (CRT) is increasingly offered for MIBC and has become a category I treatment recommendation by the NCCN. Efstathiou et al. evaluated molecular subtypes of 136 patients treated with CRT, but did not identify significant differences in complete response, disease specific survival, and overall survival among four subtypes using the Decipher Bioscience’s GSC platform [37]. The investigators identified that T-cell infiltration and a stromal signature were both positively associated with survival after CRT, but not after NAC and radical cystectomy.
CONCLUSIONS AND FUTURE DIRECTIONS
While we have reached a point in which most contributors to subtyping feel we have sufficient subtypes to describe the biology of MIBC, future efforts to apply tumor subtyping will likely involve the application of subtyping to prospective trials of chemo and/or immunotherapy. A major improvement in the application of subtyping is the common ground provided by the consensus classification. The subtyping work of the UNC, MDA, Lund, CIT (Cartes d’Identité des Tumeurs) [38] and TCGA groups all directly relates to this common ground. Among the commonly used classifiers it is only the GSC-classifier that remains to be compared to the consensus subtypes and hopefully this will be achieved in the near future. Hopefully, this will improve the accuracy of tumor subtyping across trials and formats that will involve FFPE specimens. Trials, such as COXEN (S1314; NCT 02177695) in which RNA is available, may validate the response of different subtypes to NAC. To apply subtyping prospectively, a relatively agile platform with reproducible results will be essential. Prospective evaluation of treatment response may include tumor subtype and/or other biomarkers such as DNA damage repair gene alterations, TMB, and PDL1 status to help guide treatment decisions. While most studies have focused on MIBC, a thorough investigation of NMIBC [39] will be essential to better understand the genesis of bladder cancer.
ACKNOWLEDGMENTS
The authors have no acknowledgements.
FUNDING
JM: supported by grants from VHA BX003692-01, DoD (W81XWH-18-0257), Hope Foundation
AUTHOR CONTRIBUTIONS
Conception: JM, PB, SL, GS; Performance of work: AD, JM, PB, SL, GS; Interpretation or analysis of data: AD, JM, PB, SL, GS; Writing the article: AD, JM, PB, SL, GS
ETHICAL CONSIDERATIONS
This study, as a literature review is exempt from any requirement for Institutional Review Board approval.
Peer review and the editorial decision making process have been conducted shielded from Editor-in-Chief SL.
CONFLICT OF INTERESTS:
JM is a consultant for Ferring, AstraZeneca, Janssen and participated in advisory boards for Foundation Medicine and Nucleix, research funding from Abbvie, Tesaro, Epizyme, Prostate Cancer Foundation with trial support from SWOG, Genentech, Merck, AstraZeneca. PB is a member of an advisory board or equivalent with a commercial organization: AbbVie, Asieris, AstraZeneca, Astellas, Bayer, Biosyent, BMS, EMD-Serono, Ferring, Fergene, H3-Biomedicine, Janssen, Merck, Roche, Sanofi, Urogen, Speakers bureau for AbbVie, Biosyent, Janssen, Ferring, TerSera, Pfizer, received honoraria from: iProgen, Sanofi, Bayer, GSK and is currently participating in or have participated in a clinical trial within the past two years. Genentech, Janssen, BMS, Astellas, Sitka, MDx Health, AstraZeneca, Therelase, Pacific Edge. PB share a patent with Decipher Biosciences. SL: clinical trial grant support from Endo, FKD, JBL, Genentech, Urogen, Vaxion, Viventia; Consulting or ad board: FerGene, Genentech, Merck, Pfizer, QED, UroGen, Vaxiion, Verity; Ppatent TCGA classifier; Honoraria: UroToday, Grand Rounds Urology. DM: Grant support from Rainier Pharmaceuticals, Astra-Zeneca and consulting from Janssen, H3 Biomedicine, Rainie. JM, SL and DM have a patent for a TCGA classifier. GB: None. AD: None.
REFERENCES
[1] | Antoni S , et al. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur Urol. (2017) ;71: (1):96–108. |
[2] | Al-Husseini MJ , et al. Trends in the incidence and mortality of transitional cell carcinoma of the bladder for the last four decades in the USA: a SEER-based analysis. BMC Cancer. (2019) ;19: (1):46. |
[3] | McConkey DJ , Choi W . Molecular Subtypes of Bladder Cancer. Curr Oncol Rep. (2018) ;20: (10):77. |
[4] | Siegel RL , Miller KD , Jemal A . Cancer Statistics. 2019. CA Cancer J Clin. (2019) ;69: (1):7–34. |
[5] | Zhao L , et al. Molecular subtyping of cancer: current status and moving toward clinical applications. Brief Bioinform. (2019) ;20: (2):572–84. |
[6] | Meeks JJ , et al. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nat Rev Urol. (2020) ;17: (5):259–70. |
[7] | Lerner SP , et al. Bladder Cancer Molecular Taxonomy: Summary from a Consensus Meeting. Bladder Cancer. (2016) ;2: (1):37–47. |
[8] | Wilkerson MD , Hayes DN . ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. (2010) ;26: (12):1572–3. |
[9] | Choi W , et al. Genetic Alterations in the Molecular Subtypes of Bladder Cancer: Illustration in the Cancer Genome Atlas Dataset. Eur Urol. (2017) ;72: (3):354–65. |
[10] | Dyrskjot L , et al. Identifying distinct classes of bladder carcinoma using microarrays. Nat Genet. (2003) ;33: (1):90–6. |
[11] | Blaveri E , et al. Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res. (2005) ;11: (11):4044–55. |
[12] | Sanchez-Carbayo M , et al. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. (2006) ;24: (5):778–89. |
[13] | Lindgren D , et al. Combined gene expression and genomic profiling define two intrinsic molecular subtypes of urothelial carcinoma and gene signatures for molecular grading and outcome. Cancer Res. (2010) ;70: (9):3463–72. |
[14] | Sjodahl G , et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. (2012) ;18: (12):3377–86. |
[15] | Prat A , et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast. (2015) ;24: Suppl 2:S26–35 |
[16] | Volkmer J-P , et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proceedings of the National Academy of Sciences. (2012) ;109: :2078–83. |
[17] | Damrauer JS , et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. (2014) ;111: (8):3110–5. |
[18] | Choi W , et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. (2014) ;25: (2):152–65. |
[19] | Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. (2014) ;507: (7492):315–22. |
[20] | Prat A , et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Research. (2010) ;12: (5):R68. |
[21] | Sjödahl G , et al. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol. (2017) ;242: (1):113–25. |
[22] | Marzouka N . -a.-d, et al. A validation and extended description of the Lund taxonomy for urothelial carcinoma using the TCGA cohort. Scientific Reports. (2018) ;8: (1):3737. |
[23] | Robertson AG , et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. (2017) ;171: (3):540–56 e25. |
[24] | Aine M , et al. Biological determinants of bladder cancer gene expression subtypes. Sci Rep. (2015) ;5: :10957. |
[25] | Tan TZ , et al. Molecular Subtypes of Urothelial Bladder Cancer: Results from a Meta-cohort Analysis of 2411 Tumors. Eur Urol. (2019) ;75: (3):423–32. |
[26] | Eriksson P , Sjödahl G . Re: Tuan Zea Tan, Mathieu Rouanne, Kien Thiam Tan, Ruby Yun-Ju Huang, Jean-Paul Thiery. Molecular Subtypes of Urothelial Bladder Cancer: Results from a Meta-cohort Analysis of 2411 Tumors. Eur Urol. (2019) ;75: (4):423–32. Eur Urol. 2019;75(4):e106-e107. |
[27] | Kamoun A , et al. A consensus molecular classification of muscle-invasive bladder cancer. bioRxiv. 2019. |
[28] | Broglio KR , et al. Association of Pathologic Complete Response to Neoadjuvant Therapy in HER2-Positive Breast Cancer With Long-Term Outcomes: A Meta-Analysis. JAMA Oncol. (2016) ;2: (6):751–60. |
[29] | McConkey DJ , et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naive Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur Urol. (2016) ;69: (5):855–62. |
[30] | Seiler R , et al. , Impact of Molecular Subtypes in Muscle-invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur Urol. (2017) ;72: (4):544–54. |
[31] | Anari F , et al. Neoadjuvant Dose-dense Gemcitabine and Cisplatin in Muscle-invasive Bladder Cancer: Results of a Phase 2 Trial. Eur Urol Oncol. (2018) ;1: (1):54–60. |
[32] | Rosenberg JE , et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. (2016) ;387: (10031):1909–20. |
[33] | Kim J , et al. The Cancer Genome Atlas Expression Subtypes Stratify Response to Checkpoint Inhibition in Advanced Urothelial Cancer and Identify a Subset of Patients with High Survival Probability. Eur Urol. (2019) ;75: (6):961–4. |
[34] | Sharma P , et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2017) ;18: (3):312–22. |
[35] | Necchi A , et al. Impact of Molecular Subtyping and Immune Infiltration on Pathological Response and Outcome Following Neoadjuvant Pembrolizumab in Muscle-invasive Bladder Cancer. Eur Urol. (2020) ;77: (6):701–10. |
[36] | Lotan Y , et al. Molecular Subtyping of Clinically Localized Urothelial Carcinoma Reveals Lower Rates of Pathological Upstaging at Radical Cystectomy Among Luminal Tumors. Eur Urol. (2019) ;76: (2):200–6. |
[37] | Efstathiou JA , et al. Impact of Immune and Stromal Infiltration on Outcomes Following Bladder-Sparing Trimodality Therapy for Muscle-Invasive Bladder Cancer. Eur Urol. (2019) ;76: (1):59–68. |
[38] | Rebouissou S , et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med. (2014) ;6: (244):244–91. |
[39] | Hedegaard J , et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell. (2016) ;30: (1):27–42. |


