
Toward Personalised Liquid Biopsies for Urothelial Carcinoma: Characterisation of ddPCR and Urinary cfDNA for the Detection of the TERT 228 G>A/T Mutation
Abstract
Background:
TERT promotor mutations are present in >75% of bladder tumours; these mutations are also detectable in urine. Previous studies have used urinary pellet DNA, and semi-quantitative methods unsuitable for detecting very low mutant allele frequencies.
Objective:
In this proof-of-principle study we use ddPCR to count the DNA molecules with wt and mutant TERT sequences in urinary cfDNA from patients whose bladder cancers harbour TERT mutations.
Methods:
Urinary cfDNA prepared from the urine from 104 bladder cancer patients was analysed. We determined the mutant allele frequency across stages and grades of disease, analysed concordance between cfDNA and tumour DNA, compared cfDNA with pellet DNA, and analysed the quantity and size distribution of cfDNA.
Results:
In 71 of 77 patients with a 228 G>A/T mutant tumour, the mutation was also detected in urinary cfDNA by ddPCR; all 6 “false negatives” were low grade pTa tumours. Overall concordance between tissue and cfDNA mutation status was 92%, and 100% was achieved for high grade disease. Median mutant allele frequencies in urinary cfDNA were 3.4, 13.4 and 32.1% in grade 1, 2 and 3 disease. The 228 G>A/T mutation was not detected in urinary cfDNA in 26 out of 27 mutation-negative patients (96% specificity).
Conclusions:
Concordance between tumour DNA and urinary cfDNA is high, and TERT 228 G>A/T ddPCR may prove useful for monitoring patients that harbour this mutation. Mutant allele frequencies in cfDNA are often high, but assays capable of detecting very low mutant allele frequencies will be required to achieve high sensitivity in low grade disease.
INTRODUCTION
The current gold standard method for urothelial bladder cancer (UBC) detection in both the incident and surveillance settings is cystoscopy. Much research has focused on reducing dependence upon this burdensome and expensive investigation by searching for urinary biomarkers which might enable non-invasive detection. Such biomarkers might even permit surveillance via urine samples collected in patients’ own homes.
Historically, urinary biomarker research has focussed on proteins and whilst work continues on developing panels of proteins with sufficient diagnostic accuracy, most proteins lack the sensitivity and specificity required for clinical utility [1]. Other classes of biomarkers such as mRNA and miRNA profiles and volatile organic compounds show promise; however, recently DNA-based biomarkers have come to the fore driven by technological developments and an increased understanding of the genomic and epigenetic mechanisms underpinning UBC (reviewed in [2]). Bladder cancer is a heterogeneous disease so it is likely that a panel of biomarkers will be required to achieve high sensitivity [3, 4]. Detection of copy number changes, somatic mutations and DNA methylation in urine DNA have provided encouraging results [5–9]. Nonetheless, significant challenges remain to achieve clinically acceptable sensitivity and specificity: although urine should be an ideal source of biomarkers due to its direct contact with the tumour, extracting sufficient usable DNA from urine samples can be difficult, and the proportion of that DNA which originates from the tumour is variable. The optimal analytical platform must therefore be able to cope with both low nanogram quantities of input DNA and low mutant allele frequencies. In addition, all the components of a multimarker test would ideally be measurable using a single robust, rapid and inexpensive assay platform.
In 2013, mutations in the TERT promotor at position –124 and –146 bp relative to the TERT transcription start site were reported to be common events in UBC [10–13]. The mutations are at positions chr5:1,295,228 and chr5:1,295,250 (hg19), hereby referred to as “228” and “250”. The 228 G>A is a particularly strong biomarker candidate as this single base substitution has been reported to be present in 53–66% of UBCs, and across all stages and grades of disease [12, 14, 15]. Several studies have reported the detection of TERT promotor mutations in DNA from urine cell pellets [7, 10, 16, 17]. However, there is evidence that urinary cfDNA may more faithfully recapitulate whole tumour DNA than the more commonly used urinary cell pellet DNA [9, 18, 19]. The high frequency of TERT mutations in UBC make them key constituents of any somatic mutation panel for detecting primary UBC; a large mutation panel including TERT mutations may be required for detecting primary disease but following index tumour sequencing a smaller personalised mutation panel could suffice for surveillance and TERT mutations would be useful for the majority of patients. We hypothesise that because of the number of mutations required, a large panel for initial detection is likely to be next generation sequencing (NGS) based [7], whereas smaller personalised panels for surveillance could be based on digital droplet PCR (ddPCR) with its ability to detect very low mutant allele frequencies but limited ability to detect multiple mutations [19, 20].
In this study, we investigate the use of digital droplet PCR for the TERT 228 G>A mutation to determine mutant allele frequency and hence “tumour burden” in urinary cfDNA and compare with cell pellet DNA and tumour DNA next generation sequencing data. We also characterise the size and quantity of DNA across different stages and grades of bladder cancer. We conclude that the application of TERT mutation ddPCR to urinary cfDNA warrants further examination as a personalised assay for the surveillance of UBC patients whose tumours harbour this mutation.
MATERIALS AND METHODS
Patients and specimens
Urine and tissue specimens were prospectively collected for biomarker research between 2006 and 2011 as part of the Bladder Cancer Prognosis Programme (BCPP, ethics approval 06/MRE04/65) [21]. All UBC patients were newly-diagnosed and had not received treatment prior to urine or tissue collection. Inclusion and exclusion criteria are detailed elsewhere [21]. Urine specimens were placed on ice, centrifuged at 2000 g for 10 minutes within 8 hours of collection, and supernatants and pellets stored at –80°C. Tissue specimens were collected at the time of transurethral resection, snap-frozen in liquid nitrogen and stored at –80°C.
DNA was extracted from 135 paired frozen tumours and urine supernatants (cfDNA); DNA was also extracted from 17 accompanying urine pellets. The 135 patients were selected from the larger BCPP study on the basis that they were within the 50% of BCPP tumours subjected to targeted NGS and hence with known TERT mutation status (manuscript in preparation) and had >10 ml of urine supernatant available for cfDNA preparation. No attempt was made to select on the basis of stage or grade.
Tumour next generation sequencing
DNA was extracted from 25 mg frozen tissue using the DNeasy Blood and Tissue kit (Qiagen, #69506). The TERT promotor mutation status of every tumour was determined by targeted next generation sequencing as described previously [7].
Urine ddPCR
Approximately 50 ml of urine was processed per patient, with 10 ml of urinary supernatant or the entire pellet utilised for DNA extraction using the Quick-DNA Urine kit (Zymo, D3061). DNA concentrations were determined using the Qubit high sensitivity DNA kit (Thermo, #Q32854) and cfDNA size determined by Bioanalyzer using high sensitivity chips (Agilent, # 5067-4626). cfDNA preparations from 104 patients yielded >10 ng DNA and were included for analysis. The overall study design and patient demographics are shown in Supplemental Figure S1 and Table S1. The number of wt and TERT 228 G>A/T DNA molecules was determined in all urinary DNAs and 21 of the tumour DNAs by digital droplet PCR using the TERT_C228T liquid biopsy assay (Thermo, #A44177), supermix for probes (Biorad, #1863010) and the Biorad QX200 droplet formation/reader system (Biorad). The PCR program consisted of 10 minutes at 96°C followed by 54 cycles of 98°C×30 sec and 55°C×2 min. Log rank tests were used to compare data across patient groups.
RESULTS
Assay validation
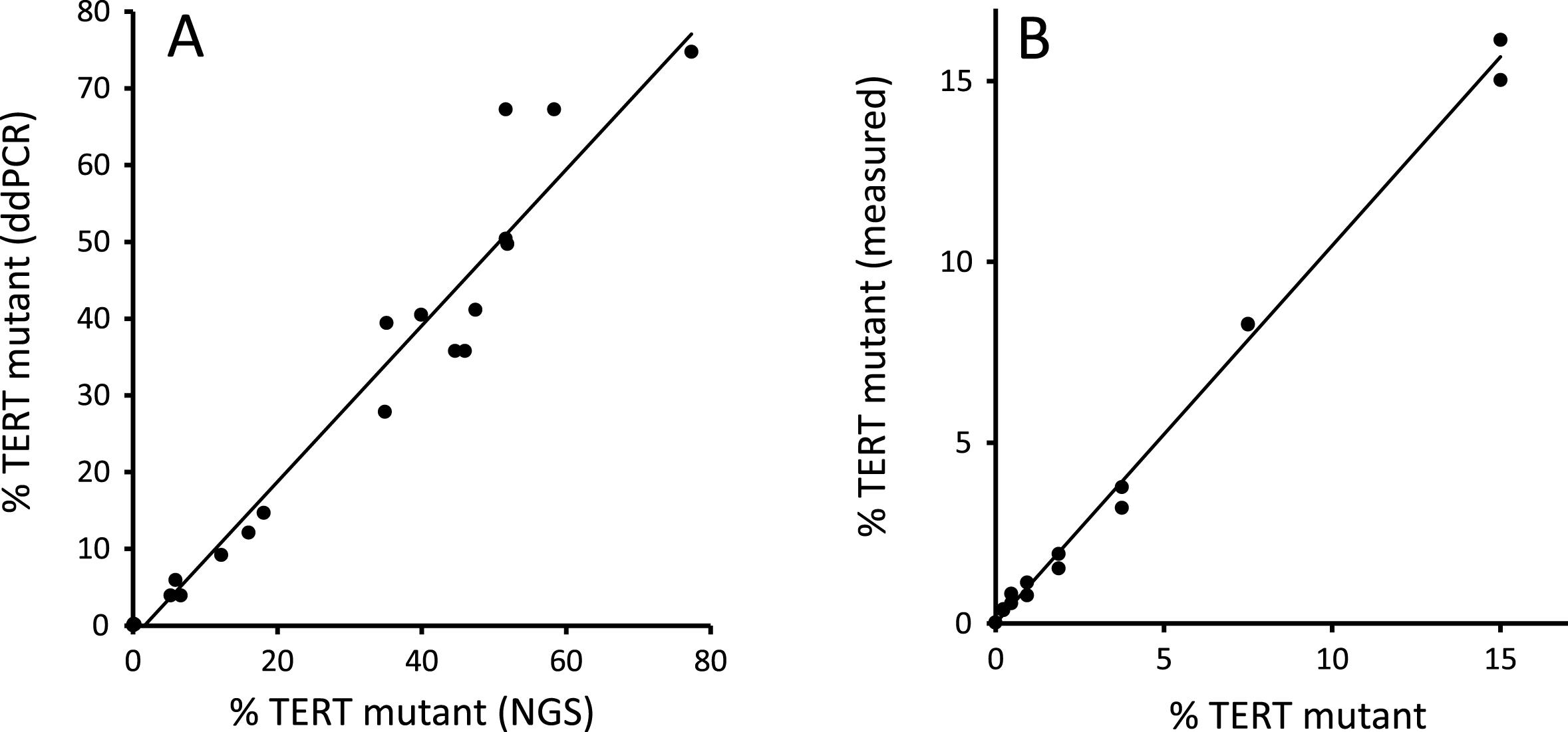
The TERT promotor sequence is both repetitive and GC rich; in our experience, many polymerases fail to amplify this region. We therefore commenced by comparing the levels of TERT mutation found by ddPCR versus an established NGS workflow [7] in DNA extracted from 21 fresh-frozen tumours. As shown in Fig. 1A, good agreement between the 2 methods was observed. We found that the ddPCR assay yields a positive result with the less common 228 G>T mutation as well as the 228 G>A mutation. The ddPCR assay also performed well in a serial dilution of mutant DNA (Fig. 1B).
Fig.1
ddPCR validation. Figure 1(A) shows the TERT 228 G>A/T mutant allele frequency in DNA extracted from 21 fresh-frozen bladder tumours measured by ddPCR and NGS. Figure 1(B) shows ddPCR analysis of a 2-fold serial dilution of pooled TERT mutant tumour DNA (15% mutant by NGS) diluted in pooled TERT wt tumour DNA.
cfDNA analysis of urinary DNA
ddPCR was used to analyse urinary cfDNAs from 104 UBC patients. All of these patients had the TERT promotor sequenced in their tumour DNA by NGS (12×wt, 15×250A, 75×228A and 2×228T). The mutant allele frequency as determined by ddPCR is shown for every cfDNA analysed in Fig. 2. cfDNA from the 27 patients with tumours negative for a 228 mutation gave very low levels of mutant positive droplets and allowed us to define a threshold of 1% mutant allele frequency (MAF) for a “positive result” (mean + 2SD). The cfDNA result from 26 of the 27 wt or 250 G>A tumours were below this threshold. Analysing the cfDNA from patients with a TERT 228 G>A/T positive tumour by NGS gave a positive result in 71 out of 77 cases (92%): 24 of 30 mutant positive pTa tumours (80%) gave a positive result, and all pT1 (n = 19) and MIBC (n = 27) cases gave a positive result. All 6 “false-negative” results were from patients with grade 1 pTa disease, and there were no false-negatives amongst higher grades and stages. The cfDNA MAF data is summarised in Fig. 3. These data show that MAF is strongly influenced by tumour grade and size, but less so by stage of disease, cfDNA concentration or the number of tumours.
Fig.2
TERT 228 G>A/T mutant allele frequencies in the cfDNA of 104 UBC patients. The mutant allele frequency for each patient in the study is represented by a single bar. Patients are sorted within each group by mutant allele frequency. The groups are: UBCs with wt TERT promotor, UBCs with TERT 250 G>A mutant promotor and UBCs with TERT G > 228A/T separated into pTa, pT1 and pT2 + disease.
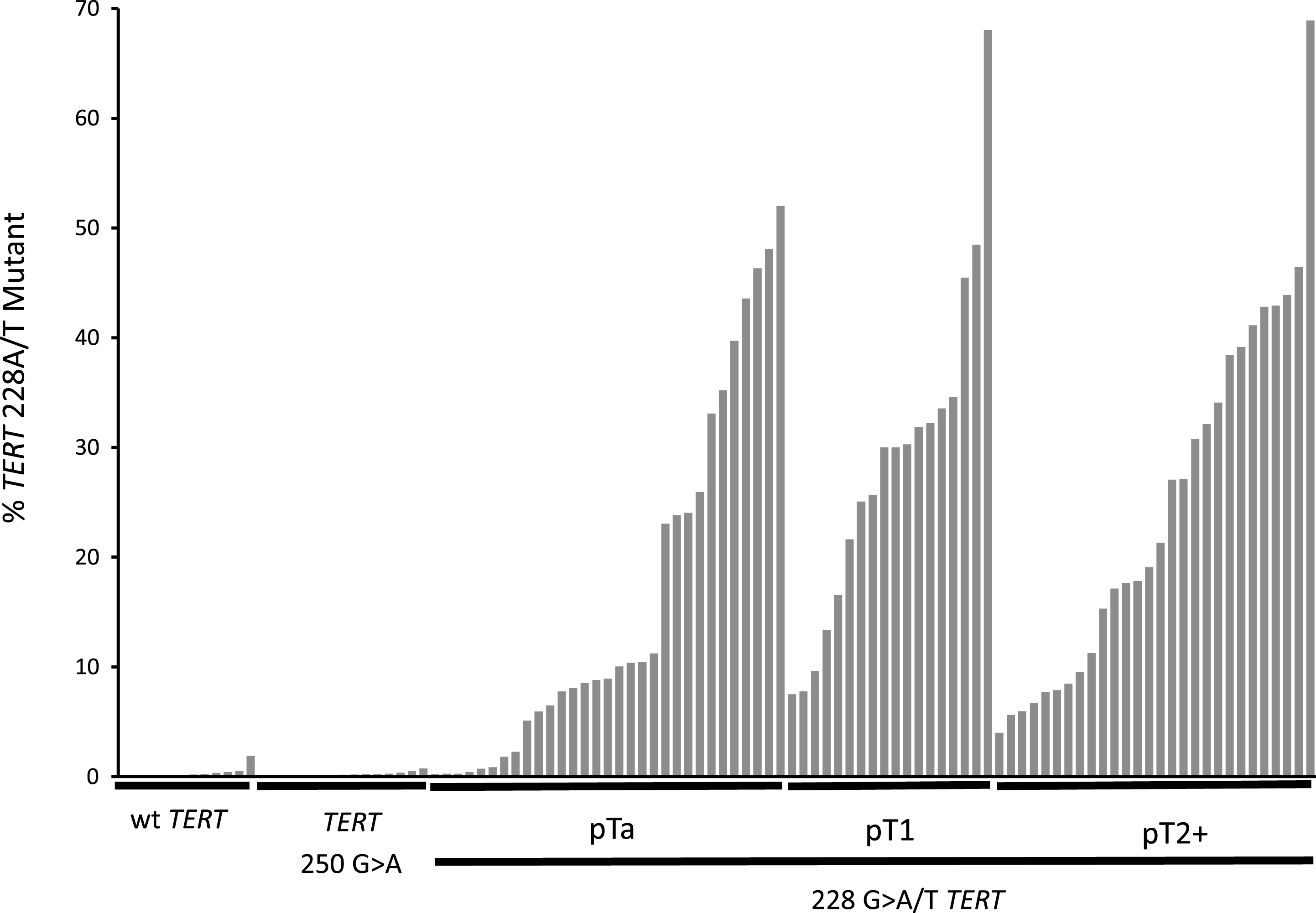
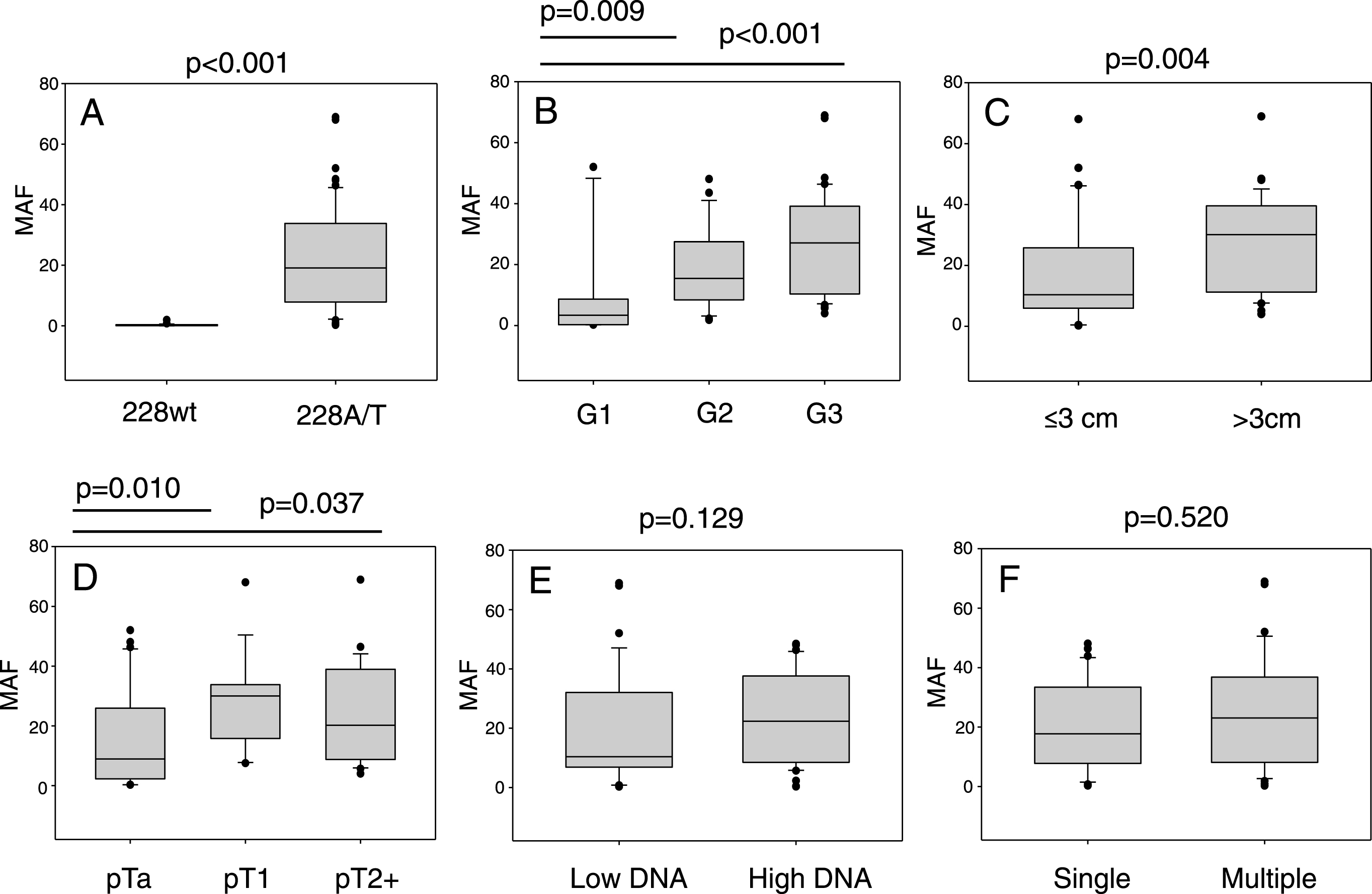
Fig.3
TERT mutant allele frequencies in cfDNA. The 6 panels show the influence of, A: tumour mutation status, B: tumour grade, C: tumour size, D: disease stage, E: cfDNA concentration (< />median level) and F: number of tumours on cfDNA TERT mutant allele frequency determined by ddPCR.
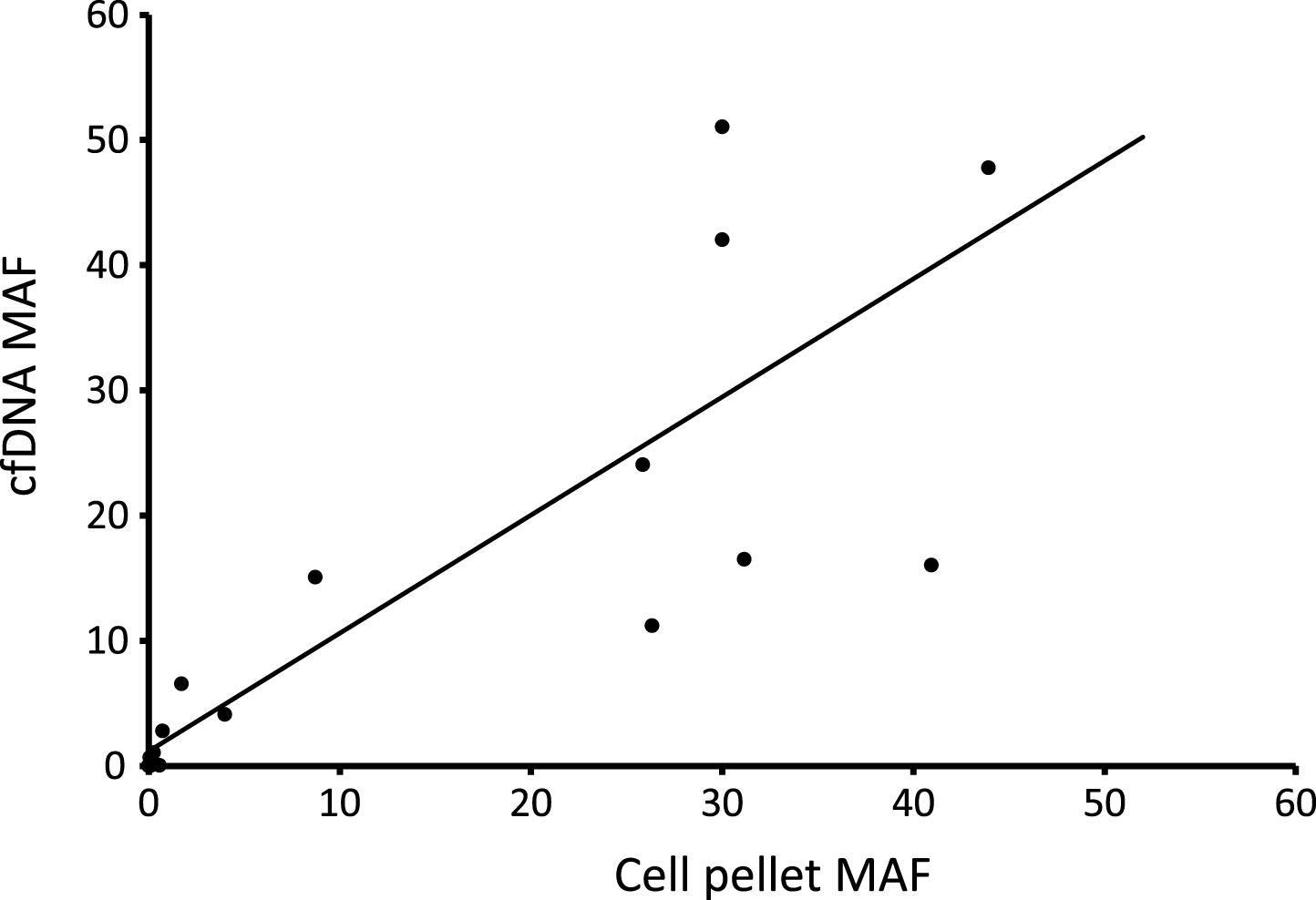
ddPCR analysis of urinary cfDNA and cell pellet DNA
Figure 4 plots the TERT MAF detected by ddPCR for 17 paired cfDNAs and cell pellet DNAs (both types of DNA extracted from the same urine void). There is a positive correlation between tumour content in the two types of DNA (r2 = 0.73), although there is also scatter in the data indicating that the MAF in the two types of DNA may differ in any individual patient. The average MAF was not significantly different between cfDNA and pellet DNA.
Fig.4
Comparison of mutant allele frequencies in urinary cfDNA and cell pellet DNA. 17 paired samples were analysed by ddPCR.
Quantification and size analysis of urinary cfDNA in UBC patients
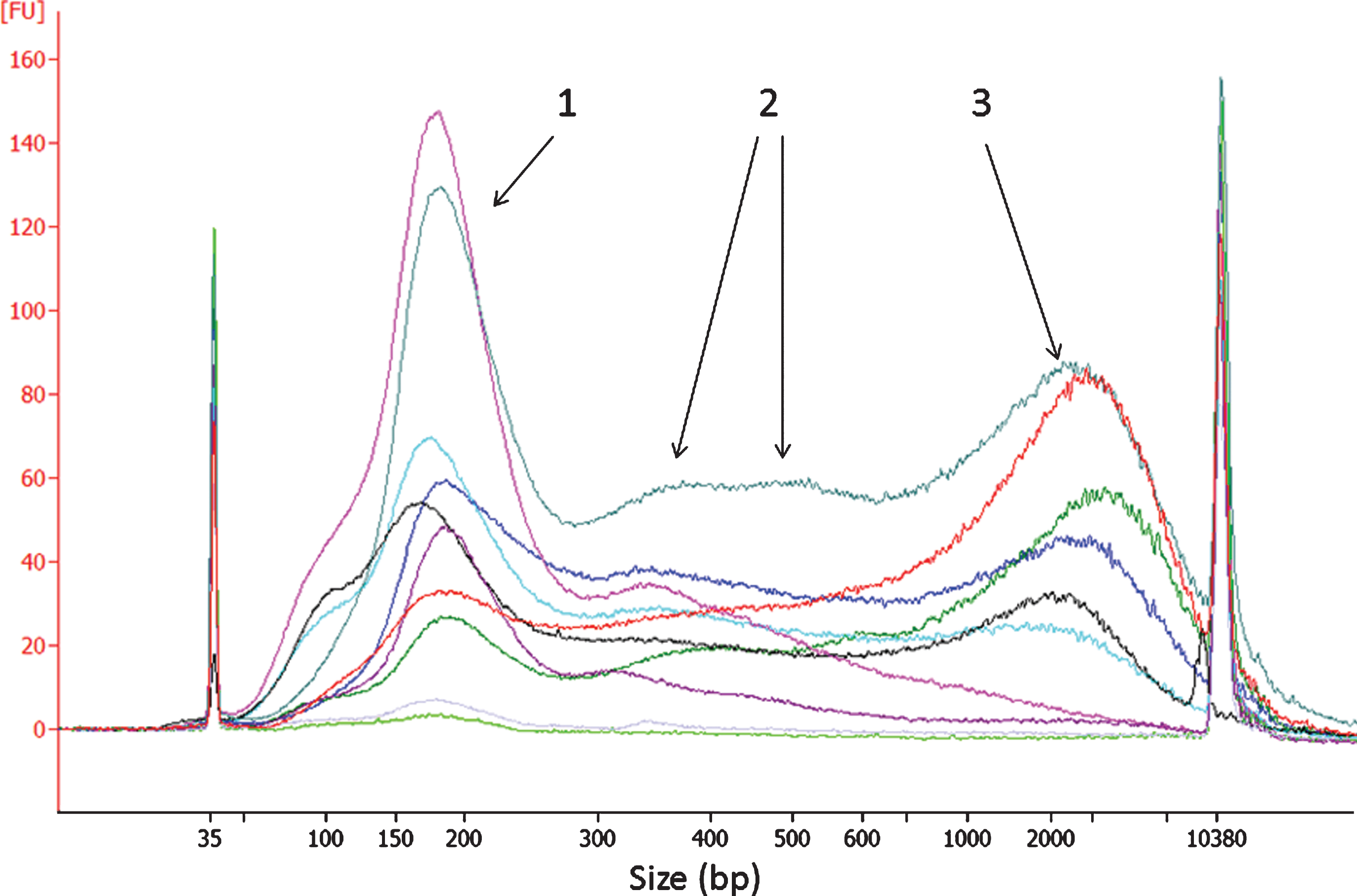
cfDNA was prepared from 10 ml of each urine supernatant. The median yield of cfDNA was 39 ng (3.9 ng/ml urine); however, yield was highly variable and 10 samples yielded less than 1 ng of cfDNA. Yield was dependent on the stage, grade and size but not the number of tumours (Fig. 5). The Bioanalyzer gel electrophoresis system was used to analyse the size distribution of urinary cfDNA in UBC patients. The vast majority of samples presented with a peak of approximately 178 bp consistent with mononucleosomal cfDNA (Fig. 6). Di- and trinucleosomal DNA was also evident in most samples along with a variable amount of longer DNA (1–10 kb). The ratio of 178 bp DNA to longer fragments was highly variable although on average 50% of the intensity of the Bioanalyzer traces was >300bp. We found no relationship between the ratio of 178 bp DNA to longer fragments and MAF suggesting that the tumour:normal ratio is similar in both types of DNA and that targeting urinary cfDNA of a particular size category would not aid in biomarker detection. To corroborate this result, we applied Pronex bead fractionation (Promega) to 8 of the higher concentration cfDNAs. This method produced 2 fractions for each sample: one enriched for shorter fragments and one enriched for longer fragments (Figure S2 Supplemental Information). There was no significant difference in the TERT MAF measured by ddPCR in the 2 fractions.
Fig.5
Factors which influence urinary cfDNA concentration. All DNA concentrations were determined using the Qubit HS double-stranded DNA kit. Figure A) the effect of tumour grade on urinary cfDNA, B) effect of stage, C) effect of tumour size, D) effect of the number of tumours.
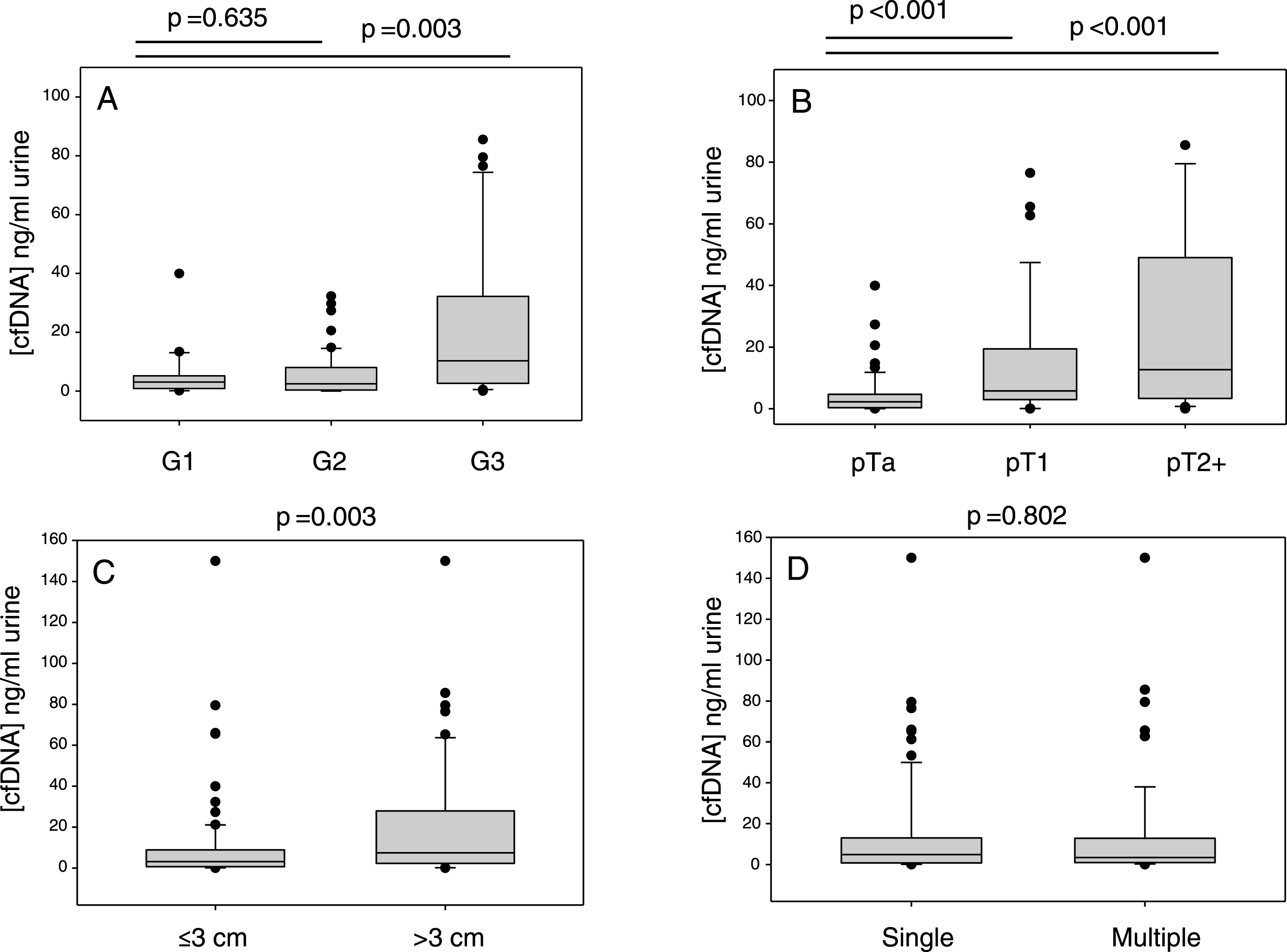
Fig.6
Bioanalyzer analysis of urinary cfDNA from UBC patients. Each trace is for a cfDNA from a different patient. The traces show the amounts of 1: mononucleosomal, 2: di- and tri-nucleosomal and 3: longer (1–10 kb) DNA fragments.
DISCUSSION
We have used ddPCR to count the number of wt and TERT 228A mutant molecules in the urinary cfDNA of patients with varying stages and grades of UBC. In patients whose tumours contained the TERT 228 G>A/T mutation, we were able to detect the mutation in urinary cfDNA with a sensitivity of 92%. It is therefore likely that the combination of ddPCR and cfDNA for this common mutation in bladder cancer could play a role in disease surveillance. It should be noted that 31 patients had <10 ng of DNA extracted from the 10mls of urine, and were excluded from the analyses. Thus, the reported sensitivities and specificities relate only to those samples which did generate enough DNA to be assayed. The excluded patients comprised 4 MIBC, 5 pT1 and 22 pTa and had a mean tumour diameter of 2.7 cm (compared with 3.7 cm for the remainder of the cohort). DNA extraction from a larger volume of urine would likely resolve this issue. We found that the assay detects both G > A and G > T substitutions at position 228 (but not at the 250 position), and that 74% of the patients in this study had one or other substitution at position 228.
We envisage that index tumour sequencing is likely to be routine in the not too distant future and that this would enable selection of a small panel of somatic mutations for personalised non-invasive surveillance. Such panels could be ddPCR based and the TERT 228 G>A/T mutation would be a core component for many patients. Although limited in its ability to multiplex biomarkers, ddPCR is well suited to detecting rare mutations and is inexpensive, fast, reliable and gives absolute quantitation [22]. Tumour evolution might compromise the use of mutations for surveillance; however, in NMIBC recurrences occur as a result of incomplete resection of the primary urothelial cancer, tumour cell re-implantation, growth of microscopic tumours present at the time of the previous resection, or genuine new tumour formation [23]. Although bladder cancer is a heterogeneous disease, it is also a disease in which synchronous and metachronous tumours demonstrate “clonality” [2]. Thus, it can be reasonably assumed that mutations in the primary index tumour will remain as characteristics of new tumours and be detectable in body fluids, as others have demonstrated [19, 20, 24]. In the specific case of TERT, mutations are considered to be early events in bladder carcinogenesis and are frequent across all stages of disease suggesting that loss of TERT mutations during tumour evolution is rare [14]. Moreover, Brown et al recently reported that TERT mutations are conserved both spatially and temporally in UBCs [25]. Additional benefits of facile TERT mutation detection via urinary DNA could include treatment selection and prediction of recurrence and survival [12, 16, 26].
We found that the MAF in urinary cfDNA is surprisingly high (median = 18%), but high inter-patient variability is observed and levels are much lower in low grade disease. We also analysed the size of urinary cfDNA and found that whilst both mononucleosomal and longer DNA fragments are present in variable proportions, both appear to contain tumour DNA. Our data suggest that an analytical method with a limit of detection of 1% mutant allele frequency would detect most grade 2 and 3 tumours, but that an even higher analytical sensitivity is required to detect elusive grade 1 cases. In principle, ddPCR should allow detection of mutant alleles at much lower than 1% frequency; however, in our hands a small number of TERT mutation positive droplets were detected in DNA derived from patients with TERT wt tumours (see Figure S3) and this background “noise” limits analytical sensitivity. The positive droplets could be due to PCR errors arising from the GC-rich nature of the TERT-promotor, sample contamination, or possibly in some cases, sub-clonality, i.e. TERT mutations in areas of the tumour or urothelium that were not sequenced.
In contrast to previous studies favouring urinary cfDNA over pellet DNA for recapitulating tumour genomics [9, 18, 19], the limited comparison of urinary cfDNA and cell pellet DNA in this study (n = 17, Fig. 4) did not show a clear advantage to using cfDNA over pellet DNA for the detection of the TERT 228 G>A/T mutation. In this scenario, the higher DNA yield typically achieved from cell pellets may favour their use in diagnostic testing: larger scale studies are required for comprehensive comparison of the two sources of DNA.
In conclusion, ddPCR detection of TERT mutations in urinary cfDNA holds promise for the non-invasive detection of UBCs known to harbour these mutations (approximately three quarters of all patients). The current work has analysed urinary cfDNA from more individual patients than previous studies and adds to a body of evidence supporting urinary cfDNA as a suitable substrate for the non-invasive detection of UBC [9, 18–20].
CONFLICT OF INTEREST
RT Bryan has contributed to advisory boards for Olympus Medical Systems with regard to narrow band imaging cystoscopy. ND James has contributed to advisory boards for Merck USA and Pierre Fabre. The other authors have no conflicts of interest.
ACKNOWLEDGMENTS
IJR was supported by an Erasmus studentship. BCPP was funded by Cancer Research UK, and other costs were met by a philanthropic donation to the University of Birmingham in support of bladder cancer research.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/BLC-170152.
REFERENCES
[1] | D’Costa J , Goldsmith J , Wilson J , Bryan R , Ward D . A Systematic Review of the Diagnostic and Prognostic Value of Urinary Protein Biomarkers in Urothelial Bladder Cancer. Bladder Cancer (2016) ;2: (3):301–17. |
[2] | Knowles M , Hurst C . Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat Rev Cancer (2015) ;15: (1):25–41. |
[3] | Robertson A , Kim J , Al-Ahmadie H , Bellmunt J , Guo G , Cherniack A , et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell (2017) ;171: (3):540–56. |
[4] | Pietzak E , Bagrodia A , Cha E , Drill E , Iyer G , Isharwal S , et al. Next-generation Sequencing of Nonmuscle Invasive Bladder Cancer Reveals Potential Biomarkers and Rational Therapeutic Targets. European Urology (2017) :In press. |
[5] | Feber A , Dhami P , Dong L , de Winter P , Tan W , Martínez-Fernández M , et al. UroMark-a urinary biomarker assay for the detection of bladder cancer. Clin Epigenetics (2017) ;9: :8. |
[6] | van Kessel K , Beukers W , Lurkin I , Ziel-van der Made A , van der Keur K , Boormans J , et al. Validation of a DNA methylation-mutation urine assay to select patients with hematuria for cystoscopy. J Urol (2016) :in press. |
[7] | Ward D , Baxter L , Gordon N , Ott S , Savage R , Beggs A , et al. Multiplex PCR and next generation sequencing for the non-invasive detection of bladder cancer. PLoS One (2016) ;11: (2). |
[8] | Larré S , Camparo P , Comperat E , Gil Diez De Medina S , Traxer O , Roupret M , et al. Diagnostic, staging, and grading of urothelial carcinomas from urine: Performance of BCA-1, a mini-array comparative genomic hybridisation-based test. Eur Urol (2011) ;59: (2):250–7. |
[9] | Tognieri F , Ward D , Foster J , Devall A , Wojtowicz P , Alyas S , et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. European Journal of Human Genetics (2016) :1–8. |
[10] | Kinde I , Munari E , Faraj S , Hruban R , Schoenberg M , Bivalacqua T , et al. TERT promoter mutations occur early in urothelial neoplasia and are biomarkers of early disease and disease recurrence in urine. Cancer Res (2013) ;73: (24):7162–7. |
[11] | Liu X , Wu G , Shan Y , Hartmann C , von Deimling A , Xing M . Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle (2013) ;12: (10):1637–8. |
[12] | Rachakonda P , Hosen I , de Verdier P , Fallah M , Heidenreich B , Ryk C , et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A (2013) ;110: (43):17426–31. |
[13] | Vinagre J , Almeida A , Pópulo H , Batista R , Lyra J , Pinto V , et al. Frequency of TERT promoter mutations in human cancers. Nat Commun (2013) ;4: (2185). |
[14] | Allory Y , Beukers W , Sagrera A , Flández M , Marqués M , Márquez M , et al. Telomerase Reverse Transcriptase Promoter Mutations in Bladder Cancer: High Frequency Across Stages, Detection in Urine, and Lack of Association with Outcome. Eur Urol (2013) ; epub ahead of print. |
[15] | Hurst C , Platt F , Knowles M . Comprehensive mutation analysis of the TERT promoter in bladder cancer and detection of mutations in voided urine. Eur Urol (2014) ;65: (2):367–9. |
[16] | Descotes F , Kara N , Decaussin-Petrucci M , Piaton E , Geiguer F , Rodriguez-Lafrasse C , et al. Non-invasive prediction of recurrence in bladder cancer by detecting somatic TERT promoter mutations in urine. Br J Cancer (2017) ;117: (4):583–7. |
[17] | Wang K , Liu T , Liu C , Meng Y , Yuan X , Liu L , et al. TERT promoter mutations and TERT mRNA but not FGFR3 mutations are urinary biomarkers in Han Chinese patients with urothelial bladder cancer. Oncologist (2015) ;20: (3):263–9. |
[18] | Szarvas T , Kovalszky I , Bedi K , Szendroi A , Majoros A , Riesz P , et al. Deletion analysis of tumor and urinary DNA to detect bladder cancer: Urine supernatant versus urine sediment. Oncol Rep (2007) ;18: (2):405–9. |
[19] | Birkenkamp-Demtröder K , Nordentoft I , Christensen E , Høyer S , Reinert T , Vang S , et al. Genomic Alterations in Liquid Biopsies from Patients with Bladder Cancer. Eur Urol (2016) ;70: (1):75–82. |
[20] | Christensen E , Birkenkamp-Demtröder K , Nordentoft I , Høyer S , van der Keur K , van Kessel K , et al. Liquid Biopsy Analysis of FGFR3 and PIK3CA Hotspot Mutations for Disease Surveillance in Bladder Cancer. Eur Urol (2017) ; Epub ahead of print. |
[21] | Zeegers M , Bryan R , Langford C , Billingham L , Murray P , Deshmukh N , et al. The West Midlands Bladder Cancer Prognosis Programme: Rationale and design. BJU Int (2010) ;105: (6):784–8. |
[22] | Olmedillas-López S , García-Arranz M , García-Olmo D . Current and Emerging Applications of Droplet Digital PCR in Oncology. Mol Diagn Ther (2017) ;21: :493–510. |
[23] | Bryan R , Collins S , Daykin M , Zeegers M , Cheng K , Wallace D , et al. Mechanisms of recurrence of Ta/T1 bladder cancer. Ann R Coll Surg Engl (2010) ;92: (6):51–524. |
[24] | Beukers W , van der Keur K , Kandimalla R , Vergouwe Y , Steyerberg E , Boormans J , et al. FGFR3, TERT and OTX1 as a Urinary Biomarker Combination for Surveillance of Patients with Bladder Cancer in a Large Prospective Multicenter Study. J Urol (2017) ;197: (6):1410–8. |
[25] | Brown N , Lew M , Weigelin H , Weizer A , Montgomery J , Betz B , et al. Comparative study of TERT promoter mutation status within spatially, temporally and morphologically distinct components of urothelial carcinoma. Histopathology (2017) :In press. |
[26] | Isharwal S , Audenet F , Drill E , Pietzak E , Iyer G , Ostrovnaya I , et al. Prognostic Value of TERT Alterations, Mutational and Copy Number Alterations Burden in Urothelial Carcinoma. Eur Urol Focus (2017) :In press. |


