
CDK4/6 Inhibitors in Cancer Therapy: A Novel Treatement Strategy for Bladder Cancer
Abstract
Patients with metastatic bladder cancer (mBC) treated with cisplatin-based chemotherapy have a limited median survival of only around 14 months [1]. Despite over 30 years of basic and clinical research, until recently no therapeutic options beyond cisplatin-based therapy had entered clinical routine and, at least in the US, none of the tested agents had been approved for second-line treatment. This has changed with the advent of immune checkpoint blockade, including especially PD-1/PD-L1 inhibitors. The high response rates of 24% over a 14.4 month follow up led to the first US Food and Drug Administration (FDA) approval for a second line therapy for these patients, and it is likely that this marks the beginning of a new era in the systemic treatment of muscle-invasive bladder cancer [2–4].
The strong clinical need to improve the medical management of this disease for those patients, not responding to current therapy has led to an increased molecular understanding of bladder cancer and has forstered the development of many potential molecular manipulations and targeted strategies beyond the new immune-oncologic approaches. Among the molecular alterations indentified in bladder cancer, cell cycle deregulation appears to be a key driver of disease progression. Target-directed therapy against CDK4/6 is an emerging strategy to regain control of cell cycle deregulation. Here, we provide an overview of the current status of CDK4/6 inhibitors in cancer therapy, their potential use in mBC and the challenges for their clinical use.
REGULATION OF THE CELL CYCLE PROGRESSION BY CDK4/6
The cell cycle progresses sequentially through the gap 1 (G1), synthesis (S), gap 2 (G2) and mitosis (M) stages. This progression is regulated via a complex signaling network. The CDK proteins, CDK1, CDK2, CDK4 and CDK6, are major regulators of cell cycle progression when complexed with specific cyclin proteins [5, 6]. Constitutive expression of CDKs and temporal control of various cyclins enables the regulation of specific cell cycle phases by distinct cyclin-CDK complexes [7]. CDK activity is negatively regulated by several inhibitory proteins. The various aspects of CDK biology and function have been previously reviewed comprehensively [5, 6].
CDK4 and CDK6, which show structural and functional homology, regulate the transition of quiescent cells in the G1 phase into the S phase when complexed with cyclin D proteins (Fig. 1). Cyclin D proteins have three subtypes, cyclin D1-3, and accumulate in the presence of mitogenic stimuli [6–8]. Negative regulators of CDK4/6 include the inhibitor of CDK4 (INK4) proteins, p16INK4A, p15INK4B, p18INK4C and p19INK4D, which inhibit CDK4/6 activity either by reducing their binding with cyclin D1 or by directly occupying their catalytic domains [9].
Fig.1
Schematic representation of the CDK4/6-Rb pathway: CDK4/6 phosphorylate Rb upon complexing with cyclin D proteins. This leads to the dissociation of Rb from the E2F family of transcription factors and allows transcription of E2F target genes that enable cell cycle progression from the G1 to the S phase. Small molecule inhibitors of CDK4/6 inhibit Rb phosphorylation, S phase progression and cell proliferation. CDK4/6 activity is also influenced by upstream mitogenic signaling pathways, inhibitor of CDK4 (INK4) proteins, as well as checkpoint mechanims including the p53 pathway.
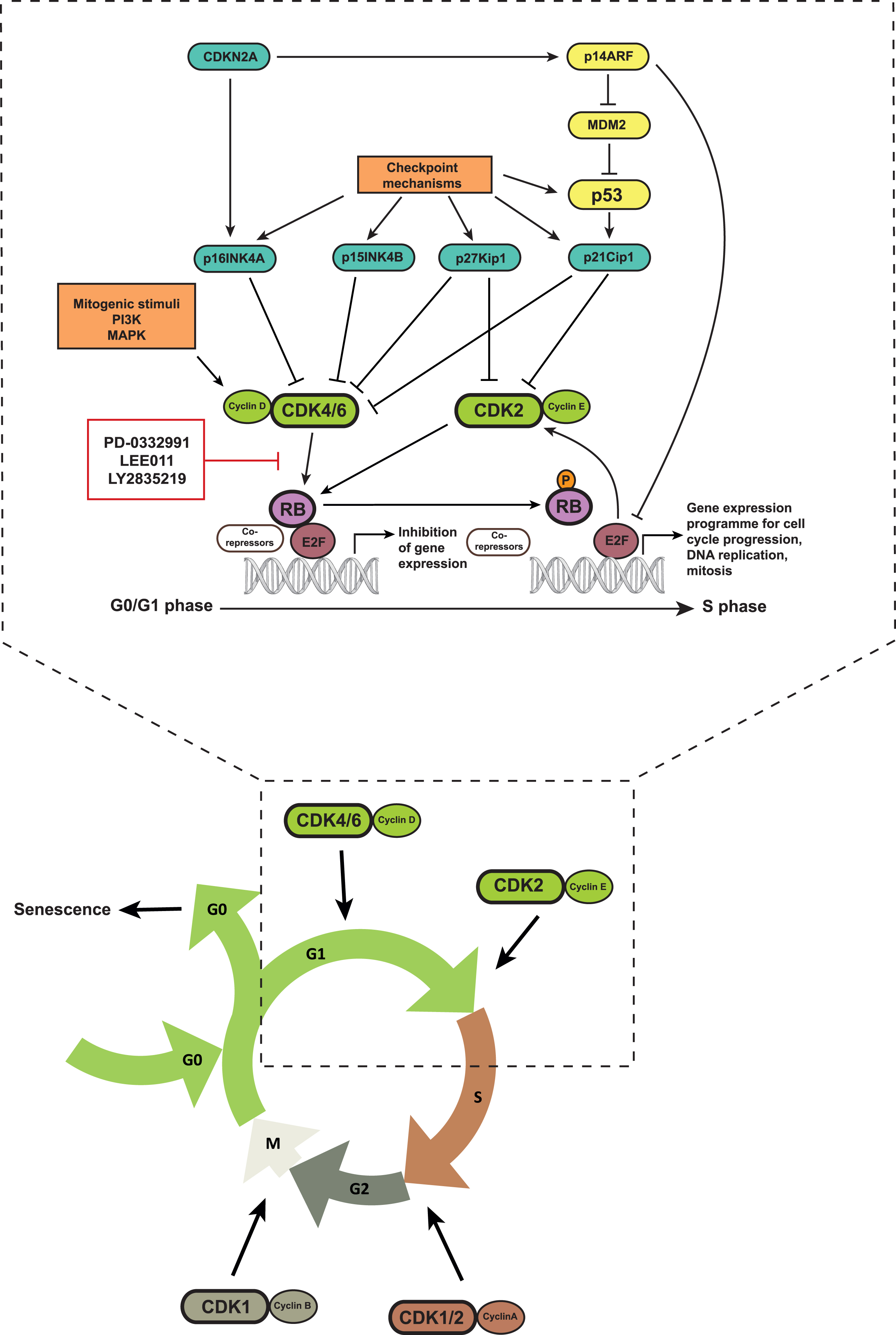
The kinase activity of CDK4/6 leads to the phosphorylation of members of the retinoblastoma (Rb) protein family including Rb, p107 and p130, which results in their functional inactivation [5, 8, 10]. In quiescent cells, active hypophosphorylated Rb binds to members of the E2F transcription factor family together with other co-repressors, and suppresses E2F function. Upon phosphorylation, Rb dissociates from this complex and allows the transcription of E2F target genes including cyclin A, cyclin E and DHFR, among others, which are required for the transition of the cell cycle into the S phase [10]. Hence, inhibition of CDK4/6 activity leads to Rb dephosphorylation and repression of E2F activity, which promotes a G0/G1 arrest. This has fueled the development of CDK4/6 inhibitors as target therapy in cancer cells.
Beside the CDK4/6-RB pathway, the p53 mediated cell cycle checkpoint is also responsible for arresting cells in G1/S. Cell cycle checkpoints are surveillance mechanisms that detect DNA damage in cells, arrest their cell cycle to allow time for repair and thus ensure genome integrity in the next generation of daughter cells [11]. The kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR) function as DNA damage sensors and directly or indirectly phosphorylate p53. This phosphorylation interrupts the interaction between MDM2, a ubiquitin ligase, and p53, thereby preventing proteasomal degradation of p53 [11, 12]. Accumulation of p53 increases the transcription of p21kip1, a CDK2 inhibitor, and thereby blocks cell cycle progression into S phase [9, 13]. p53 stabilization also occurs via p14ARF that can induce MDM2 degradation [14]. p14ARF is encoded by the CDKN2A gene that also encodes for p16INK4A. Hence, the CDKN2A gene locus can negatively regulate the cell cycle via both p53- and Rb-dependent mechanisms.
THERAPEUTIC POTENTIAL OF CDK4/6 INHIBITION IN BLADDER CANCER
Cell cycle deregulation is a common feature of mBC and includes molecular alterations in the CDK4/6-Rb pathway. CDKN2A and CDKN2B are candidate tumor suppressors to be affected by chromosome 9 deletions, which occur in over 50% of mBC tumors [15, 16]. Inactivating mutations of RB1 are found in 11–20% of tumor specimens, which indicates that up to 80% of bladder cancer patients that still maintain functional RB1 might be eligible for this therapy since the presence of RB1 is necessary for the response to CDK4/6 inhibitors. Alterations in CDKN2A and RB1 are also mutually exclusive [17].
These molecular alterations or changes in expression also correlate with the molecular subtypes of mBC that have been described recently by multiple groups. FGFR3 mutations together with CDKN2A loss were observed in the UroB Lund subtype and the TCGA cluster 2 subtype [17, 18]. Meanwhile, the Lund GU subtype, basal tumors from the UNC classification and TCGA cluster 3 were characterized by RB1 mutations/deletions, and CCND1, E2F3 or cyclin E1 (CCNE1) amplifications. These basal tumors had a poorer prognosis as compared to luminal tumors [17–19]. The Lund group also identified two distinct transcriptional circuits operating in the various molecular subtypes. These were the FGFR3/CCDN1 transcriptional circuit in UroA and UroB tumors, partly characterized by high FGFR3 and CCDN1 expression together with CDKN2A deletions, and the E2F3/RB1 circuit with deletions of RB1, high CDKN2A and reduced FGFR3 in GU tumors. Patients with tumors enriched for the FGFR3/CCDN1 transcriptional circuit had a better prognosis as compared to those with the E2F3/RB1 circuit [20]. Based upon the current knowledge on response to CDK4/6 inhibitors, it is thus likely that the subtype featuring FGFR3 mutations together with CDKN2A loss is probably a responsive subtype.
Alterations in either p53, Rb, p16INK4A, cyclin D1 or p21 expression have been identified as markers of disease progression, decreased disease-specific survival or poorer 5-year overall survival in various studies. A worse prognosis was also observed when patients had a combination of these molecular alterations [21–23]. While inactivation of RB1 alone does not result in tumor formation in vivo, loss of all Rb family members results in the development of bladder tumors. Moreover, invasive disease has been observed in mice deficient in both p53 and Rb upon exposure to carcinogens [24, 25]. These studies demonstrate that the CDK4/6-Rb pathway is frequently altered in mBC and can influence both pathogenesis and prognosis. It is thus likely that targeting the G1-S cell cycle transition has therapeutic potential in mBC.
TARGETING THE CDK4/6-RB PATHWAY IN CANCER
The disruption of the CDK4/6-Rb signaling pathway and an uncontrolled G1-S transition of the cell cycle is a common feature of cancer cells. This can occur due to various molecular alterations including loss of function mutations or deletions of the RB1 gene (encoding for Rb), CDKN2A (encoding for p16INK4A and p14ARF) or CDKN2B (encoding for p15INK4B). Such deregulation can also result from amplification or activating mutations in CCND1 (encoding for cyclin D1), E2F1-3, CDK4, CDK6 or components of various mitogenic signaling pathways such as the PI3K or MAPK pathways [26].
Several ATP-competitive small molecule CDK inhibitors have been developed. However, first generation inhibitors such as flavopiridol are non-selective and can inhibit multiple CDKs which might result in limited efficacy and high toxicity [8]. Next generation CDK4/6 inhibitors display high selectivity and include palbociclib (PD-0332991 from Pfizer), abemaciclib (LY-2835219 from Eli Lilly) and ribociclib (LEE011 from Novartis) [27–29] (Table 1). These CDK4/6 inhibitors have been tested pre-clinically in in vitro and in vivo models of several cancer entities including leukemia, breast cancer, melanoma, glioma, pancreatic cancer, hepatocellular carcinoma, lung adenocarcinoma, sarcoma, ovarian cancer, renal cancer, prostate cancer and mBC [8, 30]. In most studies they have demonstrated a consistent molecular and functional phenotype with a dose-dependent reduction in Rb phosphorylation, protein expression and transcription of E2F target genes, which correlates with a G0/G1 arrest and inhibition of cell proliferation. Additionally, all these reports demonstrate that Rb expression is a pre-requisite for sensitivity to these inhibitors.
Table 1
Three clinically potential CDK4/6 inhibitors
| Compound | FDA approved | Tumor type | Citation |
| palbociclib (PD-0332991) | Yes | Breast cancer | 31,33 |
| ribociclib (LEE011) | Yes | Breast cancer | 34 |
| abemaciclib (LY-2835219) | Not yet |
Our group has recently investigated the effects of CDK4/6 inhibitors in mBC. In our study, palbociclib (PD-0332991) was tested on a panel of 10 mBC cell lines [30]. In Rb positive cell lines, we observed a dose dependent reduction in total Rb protein that correlated with a decrease in phosphorylated Rb. This decrease in total Rb correlated partially with a reduction in RB1 transcript levels and was associated with a reduction in the transcription of E2F target genes CCNA2 and CCNE2. A significant G0/G1 cell cycle arrest and reduction in tumor proliferation and growth was observed only in Rb positive cells, which was also confirmed in a three-dimensional tumor xenograft model using the chicken chorioallantoic membrane (CAM).
CDK4/6 inhibitors are presently being tested in over 80 clinical trials in oncology among them one in bladder cancer (https://clinicaltrials.gov, clinical trial identifier NCT02334527). Results from trials in advanced hormone positive, HER2-negative breast cancer have demonstrated a significant improvement in progression free survival (PFS) using a combination of CDK4/6 inhibitors and hormone therapy as compared to hormone therapy alone [31–34]. Based on these data, palbociclib and ribociclib have been approved or designated as breakthrough therapy, respectively, by the US FDA in this clinical setting [32, 34] The efficiency and toxicity between palbociclib and ribociclib are very similar. The principal grade 3/4 adverse events are neutropenia and leucopenia. Early results from trials in other solid or hematological tumors have also demonstrated acceptable safety and preliminary clinical benefit of CDK4/6 inhibitors [35–37].
CHALLENGES TO THE CLINICAL USE OF CDK4/6 INHIBITORS
Results from our group suggest that CDK4/6 inhibition has the potential to be an effective therapeutic strategy in mBC. However, several aspects of understanding the molecular mechanisms of treatment response need further investigation. Here, we discuss the challenges to enable an effective transition from experimental models to clinical implementation of CDK4/6 inhibitors in mBC based on a critical review of published literature to date.
Identification of the molecular mechanism of CDK4/6 inhibitors
The mechanism by which CDK4/6 inhibitors are proposed to act is the inhibition of kinase activity of CDK4/6. Preclinical studies have consistently demonstrated that CDK4/6 inhibitors reduce the level of phosphorylated Rb. However, with very few exceptions, this is accompanied by a synchronous decrease in total Rb protein level as demonstrated in leukemia, breast cancer, myeloma, glioma, pancreatic cancer, ovarian cancer and mBC models [30, 38–48]. Despite being a central aspect for the biochemical response to CDK4/6 inhibition, this observation and the possible underlying mechanisms have not been adressed. Since CDK4/6 inhibitors display anti-tumor activity only in Rb positive cells, the understanding of the contribution of Rb protein level and/or its phosphorylated form to this effect can improve our understanding of the molecular determinants of therapy response.
We have demonstrated that the transcript level of Rb is down regulated upon CDK4/6 inhibition. Moreover, levels of exogenous Rb under the control of a CMV promoter were not affected upon treatment indicating that CDK4/6 inhibitors influence the RB1 promoter activity to result in transcription reduction [30]. Molecules such as members of the Rb family (Rb, p107 and 130) or E2F proteins, which can bind to the RB1 promoter, could potentially be involved in this mechanism [49–51].
We also tested if this effect depends on the proteasomal degradation of Rb. The addition of MG-132, a proteasomal inhibitor, did not rescue Rb from degradation after CDK4/6 inhibition, indicating that the decrease in total Rb protein level might not be predominantly regulated by the proteasome. Most proteasome inhibitors exhibit very high cytotoxic activity. We used MG-132 at a concentration of 100 nM where it did not exhibit cytotoxic activity in cell viability assays but should specifically inactivate proteasome activity according to the literature [52]. However, a comprehensive examination of the proteasome as part of the biochemical response to CDK4/6 inhibitors needs to be performed.
Functional effects of CDK4/6 inhibition on cell cycle arrest and senescence
All published reports have demonstrated that CDK4/6 inhibitors are capable of arresting cells in the G0/G1 phase. However, it is important to investigate whether these agents are capable of inducing senescence, which represents a state of permanent and irreversible cell cycle arrest [53]. It has been reported that CDK4/6 inhibition for 3 days or longer, can initiate senescence in vitro and in vivo in a subpopulation of examined tumor cells [54–60]. However, since the induction of senescence is incomplete, the senescence-resistant subclones are able to re-enter the cell cycle upon the removal of CDK4/6 inhibition. In vivo, chronic CDK4/6 inhibition failed to confer a permanent cell cycle arrest of tumor cells even after initial stable disease control [58, 61].
Early adaption to CDK4/6 inhibition was also observed in leukemia, breast cancer and liposarcoma cells [61–63]. Upon acute CDK4/6 inhibition representative cell lines of these tumor entities (MCF-7, U937, THP1) showed 24 hours after treatment a better inhibition of Rb phosphorylation and cell cycle arrest than at later time points where a partial recovery could be observed. This has been demonstrated to result from a compensatory activation of the CDK2 pathway, probably due to increased adaptive PI3K signaling or down regulation of p27Kip1 [61, 63]. Cells can also acquire Rb mutations during long term treatment which correlates with therapy resistance, indicating that an appropriate combination or sequential therapy design is necessary to overcome such resistance mechanims [61].
Some studies have also reported functional effects of CDK4/6 inhibition on inducing apoptosis and epithelial-mesenchymal transition (EMT) [41, 42, 44, 46, 64]. Abemaciclib (LY2835219) could induce massive apoptosis in a tumor-derived vemurafenib-resistant melanoma cell line, while no apoptosis was observed in its parental cell line [64]. Prolonged treatment with PD-0332991 for 5 days induced apoptosis in a proportion of ovarian cancer cells [44]. CDK4/6 inhibition could induce EMT and enhanced the invasion of SMAD-4 wild-type pancreatic cancer cells through activation of the TGF-beta pathway [42]. Additional investigations are warranted into these phenotypes to understand how they impact the overall anti-tumor effect of these agents.
Potential predictive and stratifying biomarkers for personalizing CDK4/6 target therapy
It is likely that various molecular mechanisms regulate the sensitivity or resistance to CDK4/6 inhibitors. Their identification can enable the development of stratifying molecular biomarkers according to tumor genetics. Preclinical studies have unanimously demonstrated that only Rb expressing cells respond to CDK4/6 inhibitors based mainly on correlation studies in Rb positive or negative cell lines [29, 30, 39, 44, 57, 65, 66]. Our group demonstrated that reconstitution of recombinant Rb protein in Rb negative cells fails to sensitize them to CDK4/6 inhibition [30]. RNAi-mediated silencing of Rb in Rb-expressing cells induces resistance to CDK4/6 inhibition [62]. One study also demonstrated by using a retrovirus that stably expresses microRNAs against Rb that CDK4/6 inhibitors mediate their effects independent of Rb expression [45, 67]. These seemingly contradictory findings might be explained by differences in cell cycle regulation in Rb positive and negative cells. For instance, Rb deficient cells can regulate the G1/S transition independent of the CDK4/6-Rb pathway and these inhibitors might be ineffective despite Rb reconstitution [26, 68, 69]. Conversely, acute Rb loss using RNAi might not mirror the molecular pathways that are active in intrinsically Rb negative cells and could also be compensated by p107 or p130 Rb family members [45, 67]. Hence, the direct assessment of the contribution of Rb expression to the response to CDK4/6 inhibitors remains challenging.
Apart from Rb expression, correlation studies suggest that loss of p16INK4A, overexpression of cyclin D1, amplification of CDK4 and down regulation of MDM2 are associated with sensitivity to CDK4/6 inhibitors in different cancer models [39, 41, 44, 47, 54, 56, 65, 70]. Increased expression of E2F1, compensatory activation of the CDK2 pathway via up regulation of cyclin D1, E or A, as well as increased PI3K and MAPK signaling have all been associated with resistance to CDK4/6 inhibitors [5, 40, 44, 45, 63, 67, 71–74].
Preliminary genetic analyses from tumors undergoing treatment in clinical trials also provide information about potential biomarkers. CDK4/6 inhibitors showed significant clinical benefit in trials of advanced estrogen receptor (ER) positive breast cancer, which is commonly Rb positive, and well differentiated or de-differentiated liposarcoma, which is commonly Rb positive and CDK4 amplified [31–35, 75]. However, since some patients did not show clinical benefit, it is likely that other molecular alterations also play a role in determining response to treatment. While ER expression was significantly associated with response to palbociclib in combination with hormone therapy in breast cancer, additional biomarkers such as CCND1 amplification or CDKN2A loss were not predictive of response [32, 75]. No obvious association was seen between alterations in cell cycle genes such as CCND1, CDKN2A, CDKN2B or CCNE1 and treatment response in early exploratory analyses in phase 1 studies in various solid tumors [36, 37]. These data also indicate correlations between alterations in TP53, KRAS, EGFR and treatment response, which also vary in different tumor entities [37]. The benefit of these alterations as stratifying biomarkers needs to be explored in larger patient cohorts.
Hence, the genetic determinants of response to CDK4/6 inhibitors are likely to be highly complex and not restricted to obvious molecules that regulate Rb function, namely CDKN2A, CDKN2B, CCND1. Future investigations applying advanced technologies such as functional genomics in preclinical studies and genomic analyses of clinical samples can potentially lead to the development of a personalized medicine strategy for their clinical use. For mBC, it will also be interesting to analyze if specific molecular subtypes influence the response to CDK4/6 inhibition given their distinct association with cell cycle deregulation.
Combination therapies that might improve the response to CDK4/6 inhibitors
Stable disease characterized a large proportion of clinical benefit seen in trials with different CDK4/6 inhibitors, possibly reflecting its cytostatic effect. Combination therapy with other agents thus has the potential to improve efficacy by promoting tumor eradication. Such strategies can also help to overcome resistance to CDK4/6 inhibitors. Moreover, synergistic combinations can allow for a dose reduction of individual drugs and have the potential to reduce toxicity.
This strategy can be used to improve responses seen with standard chemotherapy regimens. However, the efficacy of various chemotherapy agents is dependenet on growth arrest in specific cell cycle stages. This can lead to either antagonistic, additive or synergistic effects in combination with CDK4/6 inhibitors, which may also depend on the relative timing of combination or sequential therapy. We have observed that the combination of PD-0332991 and cisplatin have a synergistic effect in mBC cell lines [30]. Interestingly, this combination has antagonistic effects on pancreatic ductal adenocarcinoma cell growth [40]. Combination of CDK4/6 inhibition with carboplatin or paclitaxel chemotherapy in ovarian cancer cells showed additive or synergistic interactions when used concomitantly, while treating cells with CDK4/6 inhibitors 24 hours before chemotherapy caused an antagonistic effect [44].
CDK4/6 inhibitors have also been combined with different targeted therapies. Their combination with hormone therapy has clearly demonstrated benefit in preclinical studies and clinical trials in hormone positive breast cancer. This benefit might stem from the association between cyclin D1 and Rb phosphorylation in the pathogenesis and treatment response of hormone positive breast cancer [76]. Multiple studies have demonstrated that increased signaling in the PI3K-mTOR pathway contributes to resistance to CDK4/6 inhibitors. Combinations of CDK4/6 and PI3K pathway inhibitors have shown synergistic effects in reducing proliferation and increasing senescence [55, 58]. Early data from clinical trials combining CDK4/6 inhibitors, endocrine therapy and PI3K or mTOR inhibitors has also demonstrated an acceptable safety profile and preliminary clinical activity [77, 78]. Table 2 shows an overview of current clinical trials that are ongoing to explore the effects of combination therapy with CDK4/6 inhibitors in a variety of different tumor entities.
Table 2
Ongoing clinical trials combining CDK4/6 inhibitors with drugs apart from endocrine therapy
| Combined agents with CDK4/6 inhibitors | Tumor types | ClinicalTrials | Phase |
| Identifier | |||
| EGFR inhibitor, cetuximab | Locally Advanced Squamous Cell Carcinoma | NCT03024489 | Phase 1/2 |
| ALK inhibitor, ceritinib | ALK-positive non-small cell lung cancer | NCT02292550 | Phase 1/2 |
| PI3K/mTOR Inhibitor Gedatolisib | Advanced Squamous Cell Lung, Pancreatic, Head &Neck and Other Solid Tumors | NCT03065062 | Phase 1 |
| PI3K inhibitors, taselisib or pictilisib | Advanced Solid Tumours, Breast Cancer | NCT02389842 | Phase 1 |
| Chemotherapy, paclitaxel | Metastatic Pancreatic Ductal Adenocarcinoma | NCT02501902 | Phase 1 |
| Proteasome inhibitor, bortezomib | Mantle Cell Lymphoma | NCT01111188 | Phase 1 |
| BRAF inhibitor, encorafenib | Locally Advanced Metastatic BRAF Mutant Melanoma | NCT01777776 | Phase 1/2 |
CONCLUSIONS
CDK4/6 inhibitors have recently been approved for clinical use in breast cancer and have an established safety profile. Frequent cell cycle deregulation makes CDK4/6 a compelling target in mBC. We have demonstrated that CDK4/6 inhibition is a promising strategy in mBC to limit cellular proliferation and tumor growth. Palbociclib is also being investigated in a clinical trial for patients with mBC (clinical trial identifier NCT02334527). Although CDK4/6 is a promising target in mBC, further research is required to improve its adoption in the clinic. We anticipate that a thorough understanding of the molecular mechanisms and functional consequences of CDK4/6 inhibition can guide the identification of genetic determinants of response to these inhibitors. This can also aid in the development of a personalized medicine strategy by using stratifying biomarkers. It should also provide the rationale for the development of effective combination therapy strategies. Since alterations in the CDK4/6-Rb pathway correlate with distinct subtypes of mBC, an intriguing hypothesis that deserves additional investigation is that CDK4/6 inhibitors might be beneficial in particular molecular subtypes.
REFERENCES
[1] | Witjes JA , Comperat E , Cowan NC , De Santis M , Gakis G , et al. EAU guidelines on muscle-invasive and metastatic bladder cancer: Summary of the 2013 guidelines. Eur Urol (2014) ;65: :778–92. |
[2] | Powles T , Eder JP , Fine GD , Braiteh FS , Loriot Y , et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) ;515: :558–62. |
[3] | Rosenberg JE , Hoffman-Censits J , Powles T , van der Heijden MS , Balar AV , et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet (2016) ;387: :1909–20. |
[4] | Sharma P , Callahan MK , Bono P , Kim J , Spiliopoulou P , et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): A multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol (2016) ;17: :1590–8. |
[5] | Peyressatre M , Prevel C , Pellerano M , Morris MC . Targeting cyclin-dependent kinases in human cancers: From small molecules to Peptide inhibitors. Cancers (Basel) (2015) ;7: :179–237. |
[6] | Lim S , Kaldis P . Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development (2013) ;140: :3079–93. |
[7] | Satyanarayana A , Kaldis P . Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene (2009) ;28: :2925–39. |
[8] | Asghar U , Witkiewicz AK , Turner NC , Knudsen ES . The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov (2015) ;14: :130–46. |
[9] | Sherr CJ , Roberts JM . CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev (1999) ;13: :1501–12. |
[10] | Giacinti C , Giordano A . RB and cell cycle progression. Oncogene (2006) ;25: :5220–7. |
[11] | Abraham RT . Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev (2001) ;15: :2177–96. |
[12] | Moll UM , Petrenko O . The MDM2-p53 interaction. Mol Cancer Res (2003) ;1: :1001–8. |
[13] | el-Deiry WS , Tokino T , Velculescu VE , Levy DB , Parsons R , et al. WAF1, a potential mediator of p53 tumor suppression. Cell (1993) ;75: :817–25. |
[14] | Pomerantz J , Schreiber-Agus N , Liegeois NJ , Silverman A , Alland L , et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell (1998) ;92: :713–23. |
[15] | Knowles MA , Hurst CD . Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat Rev Cancer (2015) ;15: :25–41. |
[16] | Goebell PJ , Knowles MA . Bladder cancer or bladder cancers? Genetically distinct malignant conditions of the urothelium. Urol Oncol (2010) ;28: :409–28. |
[17] | Cancer Genome Atlas Research Group, et al., Comprehensive molecular characterization of urothelial bladder carcinoma. Nature (2014) ;507: (7492):315–22. |
[18] | Sjodahl G , Lovgren K , Lauss M , Patschan O , Gudjonsson S , et al. Toward a molecular pathologic classification of urothelial carcinoma. Am J Pathol (2013) ;183: :681–91. |
[19] | Damrauer JS , Hoadley KA , Chism DD , Fan C , Tiganelli CJ , et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A (2014) ;111: :3110–5. |
[20] | Lindgren D , Sjodahl G , Lauss M , Staaf J , Chebil G , et al. Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma. PLoS One (2012) ;7: :e38863. |
[21] | Chatterjee SJ , Datar R , Youssefzadeh D , George B , Goebell PJ , et al. Combined effects of p53, p21, and pRb expression in the progression of bladder transitional cell carcinoma. J Clin Oncol (2004) ;22: :1007–13. |
[22] | Shariat SF , Tokunaga H , Zhou J , Kim J , Ayala GE , et al. p53, p21, pRB, and p16 expression predict clinical outcome in cystectomy with bladder cancer. J Clin Oncol (2004) ;22: :1014–24. |
[23] | Lee K , Jung ES , Choi YJ , Lee KY , Lee A . Expression of pRb, p53, p16 and cyclin D1 and their clinical implications in urothelial carcinoma. J Korean Med Sci (2010) ;25: :1449–55. |
[24] | Santos M , Martinez-Fernandez M , Duenas M , Garcia-Escudero R , Alfaya B , et al. In vivo disruption of an Rb-E2F-Ezh2 signaling loop causes bladder cancer. Cancer Res (2014) ;74: :6565–77. |
[25] | He F , Mo L , Zheng XY , Hu C , Lepor H , et al. Deficiency of pRb family proteins and p53 in invasive urothelial tumorigenesis. Cancer Res (2009) ;69: :9413–21. |
[26] | Knudsen ES , Knudsen KE . Tailoring to RB: Tumour suppressor status and therapeutic response. Nat Rev Cancer (2008) ;8: :714–24. |
[27] | Tate SC , Cai S , Ajamie RT , Burke T , Beckmann RP , et al. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin Cancer Res (2014) ;20: :3763–74. |
[28] | Rader J , Russell MR , Hart LS , Nakazawa MS , Belcastro LT , et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res (2013) ;19: :6173–82. |
[29] | Fry DW , Harvey PJ , Keller PR , Elliott WL , Meade M , et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther (2004) ;3: :1427–38. |
[30] | Sathe A , Koshy N , Schmid SC , Thalgott M , Schwarzenbock SM , et al. CDK4/6-inhibition controls proliferation of bladder cancer and transcription of RB1. J Urol (2016) ;195: (3):771–9. |
[31] | Finn RS , Martin M , Rugo HS , Jones S , Im SA , et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med (2016) ;375: :1925–36. |
[32] | Finn RS , Crown JP , Lang I , Boer K , Bondarenko IM , et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol (2015) ;16: :25–35. |
[33] | Cristofanilli M , Turner NC , Bondarenko I , Ro J , Im SA , et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol (2016) ;17: :425–39. |
[34] | Hortobagyi GN , Stemmer SM , Burris HA , Yap YS , Sonke GS , et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med (2016) ;375: :1738–48. |
[35] | Dickson MA , Schwartz GK , Keohan ML , D’Angelo SP , Gounder MM , et al. Progression-Free Survival Among Patients With Well-Differentiated or Dedifferentiated Liposarcoma Treated With CDK4 Inhibitor Palbociclib: A Phase 2 Clinical Trial. JAMA Oncol (2016) ;2: :937–40. |
[36] | Infante JR , Cassier PA , Gerecitano JF , Witteveen PO , Chugh R , et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin Cancer Res (2016) ;22: :5696–705. |
[37] | Patnaik A , Rosen LS , Tolaney SM , Tolcher AW , Goldman JW , et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, Non-Small cell lung cancer, and other solid tumors. Cancer Discov (2016) ;6: :740–53. |
[38] | Witkiewicz AK , Borja NA , Franco J , Brody JR , Yeo CJ , et al. Selective impact of CDK4/6 suppression on patient-derived models of pancreatic cancer. Oncotarget (2015) ;6: :15788–801. |
[39] | von Witzleben A , Goerttler LT , Marienfeld R , Barth H , Lechel A , et al. Preclinical characterization of novel chordoma cell systems and their targeting by pharmocological Inhibitors of the CDK4/6 cell-cycle pathway. Cancer Res (2015) ;75: :3823–31. |
[40] | Franco J , Witkiewicz AK , Knudsen ES . CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget (2014) ;5: :6512–25. |
[41] | Barton KL , Misuraca K , Cordero F , Dobrikova E , Min HD , et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One (2013) ;8: :e77639. |
[42] | Liu F , Korc M . Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther (2012) ;11: :2138–48. |
[43] | Miller TW , Balko JM , Fox EM , Ghazoui Z , Dunbier A , et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov (2011) ;1: :338–51. |
[44] | Konecny GE , Winterhoff B , Kolarova T , Qi J , Manivong K , et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res (2011) ;17: :1591–602. |
[45] | Dean JL , Thangavel C , McClendon AK , Reed CA , Knudsen ES . Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene (2010) ;29: :4018–32. |
[46] | Sawai CM , Freund J , Oh P , Ndiaye-Lobry D , Bretz JC , et al. Therapeutic targeting of the cyclin DCDK4/6 complex in T cell leukemia. Cancer Cell (2012) ;22: :452–65. |
[47] | Finn RS , Dering J , Conklin D , Kalous O , Cohen DJ , et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res (2009) ;11: :R77. |
[48] | Menu E , Garcia J , Huang X , Di Liberto M , Toogood PL , et al. A novel therapeutic combination using PD 0332991 and bortezomib: Study in the 5T33MM myeloma model. Cancer Res (2008) ;68: :5519–23. |
[49] | Burkhart DL , Ngai LK , Roake CM , Viatour P , Thangavel C , et al. Regulation of RB transcription in vivo by RB family members. Mol Cell Biol (2010) ;30: :1729–45. |
[50] | Gill RM , Hamel PA , Zhe J , Zacksenhaus E , Gallie BL , et al. Characterization of the human RB1 promoter and of elements involved in transcriptional regulation. Cell Growth Differ (1994) ;5: :467–74. |
[51] | Hamel PA , Gill RM , Phillips RA , Gallie BL . Transcriptional repression of the E2-containing promoters EIIaE, c-myc, and RB1 by the product of the RB1 gene. Mol Cell Biol (1992) ;12: :3431–38. |
[52] | Tsubuki S , Saito Y , Tomioka M , Ito H , Kawashima S . Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem (1996) ;119: :572–76. |
[53] | Hayflick L , Koprowski H . Direct agar isolation of mycoplasmas from human leukaemic bone marrow. Nature (1965) ;205: :713–4. |
[54] | Kovatcheva M , Liu DD , Dickson MA , Klein ME , O’Connor R , et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget (2015) ;6: :8226–43. |
[55] | Heilmann AM , Perera RM , Ecker V , Nicolay BN , Bardeesy N , et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res (2014) ;74: :3947–58. |
[56] | Wiedemeyer WR , Dunn IF , Quayle SN , Zhang J , Chheda MG , et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A (2010) ;107: :11501–06. |
[57] | Michaud K , Solomon DA , Oermann E , Kim JS , Zhong WZ , et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res (2010) ;70: :3228–38. |
[58] | Yoshida A , Lee EK , Diehl JA . Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res (2016) ;76: :2990–3002. |
[59] | Acevedo M , Vernier M , Mignacca L , Lessard F , Huot G , et al. A CDK4/6-dependent epigenetic mechanism protects cancer cells from PML-induced senescence. Cancer Res (2016) ;76: (11):3252–64. |
[60] | Perez M , Munoz-Galvan S , Jimenez-Garcia MP , Marin JJ , Carnero A . Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget (2015) ;6: :40557–74. |
[61] | Herrera-Abreu MT , Palafox M , Asghar U , Rivas MA , Cutts RJ , et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res (2016) ;76: :2301–13. |
[62] | Zhang YX , Sicinska E , Czaplinski JT , Remillard SP , Moss S , et al. Antiproliferative effects of CDK4/6 inhibition in CDK4-amplified human liposarcoma in vitro and in vivo. Mol Cancer Ther (2014) ;13: :2184–93. |
[63] | Wang L , Wang J , Blaser BW , Duchemin AM , Kusewitt DF , et al. Pharmacologic inhibition of CDK4/ Mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood (2007) ;110: :2075–83. |
[64] | Yadav V , Burke TF , Huber L , Van Horn RD , Zhang Y , et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther (2014) ;13: :2253–63. |
[65] | Young RJ , Waldeck K , Martin C , Foo JH , Cameron DP , et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res (2014) ;27: :590–600. |
[66] | Comstock CE , Augello MA , Goodwin JF , de Leeuw R , Schiewer MJ , et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene (2013) ;32: :5481–91. |
[67] | Rivadeneira DB , Mayhew CN , Thangavel C , Sotillo E , Reed CA , et al. Proliferative suppression by CDK4/6 inhibition: Complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology (2010) ;138: :1920–30. |
[68] | Kaye FJ . RB and cyclin dependent kinase pathways: Defining a distinction between RB and p16 loss in lung cancer. Oncogene (2002) ;21: :6908–14. |
[69] | Liu H , Tang X , Srivastava A , Pecot T , Daniel P , et al. Redeployment of Myc and E2f1-3 drives Rb-deficient cell cycles. Nat Cell Biol (2015) ;17: :1036–48. |
[70] | Puyol M , Martin A , Dubus P , Mulero F , Pizcueta P , et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell (2010) ;18: :63–73. |
[71] | Knudsen ES , Witkiewicz AK . Defining the transcriptional and biological response to CDK4/6 inhibition in relation to ER+/HER2- breast cancer. Oncotarget (2016) ;7: (43):69111–69123. |
[72] | Santamaria D , Barriere C , Cerqueira A , Hunt S , Tardy C , et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature (2007) ;448: :811–5. |
[73] | Franco J , Balaji U , Freinkman E , Witkiewicz AK , Knudsen ES . Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep (2016) ;14: :979–90. |
[74] | Logan JE , Mostofizadeh N , Desai AJ , VONE E , Conklin D , et al. PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer Res (2013) ;33: :2997–3004. |
[75] | DeMichele A , Clark AS , Tan KS , Heitjan DF , Gramlich K , et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res (2015) ;21: :995–1001. |
[76] | Barroso-Sousa R , Shapiro GI , Tolaney SM . Clinical development of the CDK4/6 inhibitors ribociclib and abemaciclib in breast cancer. Breast Care (Basel) (2016) ;11: :167–73. |
[77] | Bardia A , Modi S , Oliveira M, Campone M, Ma B, Dirix L, Weise A, Nardi L, Zhang V, Bhansali SG, Hewes B, Chavez-MacGregor M., Triplet therapy with ribociclib, everolimus, and exemestane in women with HR+/HER2–advanced breast cancer. Cancer Res (2016) ;76: (Suppl 4):Abstract nr P6-13-01. |
[78] | Juric D , et al. Phase Ib/II study of LEE011 and BYL719 and letrozole in ER+, HER2–breast cancer: Safety, preliminary efficacy and molecular analysis. Cancer Res (2015) ;75: (Suppl 9):Abstract nr P5-19-24. |


