Bladder Cancer Molecular Taxonomy: Summary from a Consensus Meeting
Abstract
The advent of Omics technologies has been key to the molecular subclassification of urothelial bladder cancer. Several groups have used different strategies to this aim, with partially overlapping findings. The meeting at the Spanish National Cancer Research Center-CNIO was held to discuss such classifications and reach consensus where appropriate. After updated presentations on the work performed by the teams attending the meeting, a consensus was reached regarding the existence of a group of Basal-Squamous-like tumors – designated BASQ – charaterized the high expression of KRT5/6 and KRT14 and low/undetectable expression of FOXA1 and GATA3. An additional tumor subgroup with urothelial differentiation features was recognized whose optimal molecular definition is required. For other subtypes described, more work is needed to determine how robust they are and how to best define them at the molecular level.
ABBREVIATIONS
BASQ | Basal/Squamous-Like |
CNV | copy number variation |
EMT | epithelial-mesenchymal transition |
GU | genomically unstable |
ICA | independent component analysis |
ICGC | International Cancer Genome Consortium |
IHC | immunohistochemistry |
MDA | MD Anderson Cancer Center |
NMI | non muscle-invasive |
RPPA | reverse phase protein arrays |
RTK | receptor tyrosine kinase |
SCCL | squamous cell carcinoma-like |
SMG | significantly mutated genes |
TCGA | The Cancer Genome Atlas |
UBC | urothelial bladder cancer |
UNC | University of North Carolina |
INTRODUCTION
The wealth of information resulting from the genomics revolution has provided the opportunity to deconvolute histology-based tumor phenotypes into a huge number of individual molecular trait-basedphenotypes. A major challenge is to summarize such large datasets into tumor subtypes through a new molecular taxonomy of cancer. This effort, pioneered in the early 2000’s in the area of breast cancer, has expanded to include essentially all tumor types in recent years through the massive genome sequencing efforts carried out under the auspices of the The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) projects. Urothelial bladder cancer (UBC) has been a relative latecomer to these studies. In the last few years, the efforts of both individual research groups and the TCGA consortium have led to several proposals for molecular subclassification that herald promise for clinical application in multiple aspects of patient management.
In UBC, the Lund group was first to use expressionprofiling of tumors covering a wide spectrum of the disease to propose of a molecular taxonomy classification. Subsequent studies - either biology-based or multidata platform-based - rapidly showed remarkably consistent evidence supporting the existence of UBC subphenotypes. Most of this research has been carried out in muscle-invasive bladder cancer. At the same time, this work unveiled unmet needs in this area, such as more detailed genomic analyses of non muscle-invasive (NMI) tumors. The TCGA Pan-Cancer integrated multidata platform analysis of 12 different cancer types also allowed placement of the UBC molecular subphenotypes in a broader context of cell and tissue-specific differentiation. Issues related to differences in genome technologies, bioinformatics strategies/tools for genome analysis, nomenclature, biology-based vs. clinical problem-targeted classification - among others - arose from the independent molecular taxonomy proposals.
In this scenario, several among us deemed that - at this relatively early stage of development of UBC molecular taxonomy - a Consensus meeting would provide a useful setting to facilitate progress in the field and stimulate collaboration. The meeting held at the Spanish National Cancer Research Center-CNIO (Madrid, Spain) on March 24, 2015 brought together all of the groups having proposed genome-based molecular subtyping of UBC in the recent years. The main aims were to:
1) present the sample selection and methodological strategies used to achieve the different molecular taxonomies;
2) discuss the overlap existing among the different molecular taxonomies;
3) develop a cooperative strategy to optimize the reported classifications; and
4) join efforts to validate the optimized classification in prospective studies.
What follows is a summary of the presentations, discussions, and consensus agreement including some suggestions to the investigators working in thisfield.
THE TCGA PROJECT
S. Lerner and K. Hoadley presented the work of the TCGA bladder cancer project. The initial TCGA report included the interim integrated genomic analysis of the first 131 patients [1]. Chemotherapy-naive, invasive urothelial cancers were analyzed for somatic mutations, DNA copy number variants (CNVs), mRNA and microRNA expression, protein and phosphoproteins (reverse phase protein arrays, RPPA), and DNA methylation. Other histologies were excluded from the study. The genetic information was integrated with comprehensive clinical and pathologic data. This invasive tumor cohort has one of the highest somatic mutation rates with a mean and median somatic mutation rate of 7.7 and 5.5 per megabase, similar to adenocarcinoma and squamous cell carcinoma (SCC) of the lung and melanoma. There were 32 significantly mutated genes (SMGs) involved in multiple pathways including cell cycle regulation (93% of tumors), chromatin remodeling (76%), DNA damage response, transcription factors, and receptor tyrosine kinase (RTK)/RAS/PI3K (72%) signaling pathways. Four SMGs involved in epigenetic regulation (ARID1A, MLL2, KDM6A, and EP300) were mutated in up to one fourth of the tumors. One-third of the tumors were characterized by cancer–specific DNA hypermethylation [1]. An additional 281 invasive tumors are in the pipeline and the TCGA team is currently analyzing the complete data set with the expectation of publishing an updated comprehensive integrated analysis. Mutation data are available on all 412 samples (including the first 131) and 54 SMGs have been identified, including 26 that were reported in the first paper. As observed in the first 131 tumors, many of these SMGs have not previously been described in bladder cancer. Combining CNV and somatic mutations, 69% of tumors harbor one or more potentially actionable targets. Three mutation/CNV groups were identified and characterized as: 1) “Focally amplified” - enriched in focal CNVs (e.g. 3p loss/PPARG) and MLL2 mutations; 2) enriched for TP53 and RB1 mutations, and E2F3 amplifications; and 3) papillary histology, FGFR3 mutant, and CDKN2A-deficient.
Unsupervised clustering of mRNA, miRNA and RPPA data suggested the existence of four expression clusters, 3 of which were fully characterized as follows. Cluster I was enriched with papillary morphology and FGFR3 dysregulation through mutations, amplifications, gene fusions, overexpression, and miRNA regulation. Clusters I and II expressed high HER2 (ERBB2) and estrogen receptor beta signaling signatures, sharing features with Luminal A breast cancers. Cluster III shows similarities to other TCGA tumor types, including basal-like breast and SCC of the head and neck and the lung [1]. Tumors in this cluster show high expression of KRT14 and CD44, suggestive of stem cell markers, similar to the basal subtype of bladder cancer originally reported by the Baylor College of Medicine group [2, 3] and to the subtype described by Sjödahl et al. which was associated with the worst prognosis compared to the other subtypes they described [4]. This was confirmed with a recent TCGA pan-cancer analysis of 12 tumor types and 6 data types, which stratified the bladder cancer cohort into 3 groups. A small subset was similar to lung adenocarcinoma, a second subset - with most of the cluster III and squamous samples - clustered with the head and neck and lung SCC, and the third - which contained the majority of tumors - was unique to bladder cancer and had the best prognosis compared to the two other subtypes [5]. Expression cluster IV comprised the smallest number of patients and shared features similar to cluster III but also contained features of the surrounding stroma and muscle. Whole exome sequencing and copy number variation have been incorporated into the expression profiling in the expanded cohort of 412 tumors. The basal cluster had a higher proportion of females and displayed high expression of immune response genes and squamous epithelial markers, suggesting that this particular phenotype may respond to therapy targeting immune checkpoint inhibition.
The complete data set will increase the power to detect additional low frequency events (6), validate the cluster analyses, test hypotheses regarding chemotherapy resistance, and provide a host of translational opportunities for functional validation and targeted therapy trials. Outcome analyses were deliberately not included in the analysis of the first 131 tumors as the follow up data were not mature. It is expected that all this information will be included in the final analysis of the full cohort.
THE LUND MOLECULAR TAXONOMY STUDIES
M. Höglund and his group presented a summary of the work carried out in Lund. A first attempt to classify UBC in terms of genome wide gene expression was made by Lindgren et al. and included both invasive and NMI tumors [7]. This analysis of 144 samples identified two major molecular subtypes named MS1 and MS2. The split between MS1 and MS2 clearly divided the samples into grade 1 or 2 (MS1) and grade 3 (MS2) (WHO1999), and between Ta (MS1) and ≥T2 (MS2) tumors. T1 cases were distributed almost equally between the two subtypes. The MS1 and MS2 categories differed significantly with respect to the number of genomic alterations, FGFR3 and TP53 mutations, and survival. To deepen the analysis, Sjödahl et al. [8] extended the study to include 100 Ta, 100 T1, and 100 ≥ T2 tumors that made it possible to subdivide the MS1 cases into two groups (MS1a and MS1b) and the MS2 subtype into five groups (MS2a1, MS2a2, MS2b1, and MS2b2.1 and MS2b2.2). An extensive biological interpretation of gene expression data identified biological themes including immune, late cell cycle, keratin, and RTK signatures that determined the data structure. In addition, a signature associated with FGFR3 expression was derived. Based on histology, gene signature biology, and FGFR3, PIK3CA, and TP53 mutations, three major subtypes or UBC were defined: urobasal (Uro) (MS1a, MS1b, and MS2b2.1), genomically unstable (GU) (MS2a1 and MS2a2), and SCC-like (SCCL) (MS2b2.2). In addition, an “infiltrated” group was recognized in which non-tumoral inflammatory transcripts dominated expression patterns. A subset of the urobasal tumors (MS2b2.1) showed a “progressed phenotype” with increased cell cycle activity and expression of basal cell related keratins in suprabasal cell layers. This group corresponded largely to invasive tumors and was named urobasal Bto distinguish it from urobasal A that were NMI in almost all cases. An important finding was that molecular subtypes transcended pathological staging. For instance, all four subtypes (UroA, UroB, GU, and SCCL) were detected among T1 tumors and no fundamental differences were observed between invasive and NMI GU cases [8].
Lindgren et al. extended these analyses by combining gene expression data with array-CGH data for 146 cases [9]. This revealed that Uro tumors show low numbers of genomic alterations, typically loss of chromosome 9 and gains of 1q, whereas GU and SCCL tumors showed complex changes with frequent focal genomic alterations, i.e. 6p22 (E2F3/SOX4) amplifications. By an integrated approach, two major genomic circuits were found to participate in UBC: a FGFR3/CCND1 circuit operating in Uro tumors and an E2F3/RB1 circuit in GU tumors. For the SCCL subtype, no specific circuit could be established. In addition, homozygous deletions of CDKN2A (9p21) were found to represent a progression event among Uro tumors.
To validate the gene expression results using immunohistochemistry (IHC), a panel of antibodies detecting 20 proteins was used providing further support to the UroA, UroB, GU, and SCCL subtypes [10]. Urobasal tumors commonly exhibited a basal membrane with KRT5 + and P-CAD + basal cells and TP63 + transitional cells; GU tumors were KRT5 - , P-CAD - , and TP63 - but E-CAD + and ERBB2 +; and the SCCL tumors were KRT5 + and P-CAD + throughout the tumor. Using IHC, the “infiltrated” group was shown to be composed of either GU or SCCL tumor cells, infiltrated with immune cells. The previously described genomic circuits could be assessed at the IHC level to identify Uro and GU cases. More recently, the IHC-based classification system described by Sjödahl et al. [9] was applied to 165 T1 tumors, showing that molecular subtype (Uro vs. GU and SCCL) had a major impact on progression rates, supporting the clinical value of the taxonomy [11].
DNA-methylation was assessed by Lauss et al. [12] in 149 UBC and showed that the MS1 and MS2 subtypes differed significantly with respect to methylation patterns. Epigenetic structure was further analyzed in Aine et al. [13], showing that UroA tumors exhibited a methylation pattern distinct from the remaining subtypes and that the majority of GU cases revealed a Polycomb (PCR1/2)-related methylation pattern. The compiled genomic (array-CGH) and DNA-methylation data supports the relevance of the urobasal, GU, and SCCL bladder cancer subtypes.
The subtype-specific gene expression profiles described above were assigned to transcription factor regulatory modules by use of publically available Chip-Seq data and IHC [14]. Tumors of the SCCL subtype overexpressed genes enriched in STAT3 binding, showed phospho-STAT3 up-regulation, and displayed down-regulation of PPARG, RXRA, FOXA1, and GATA3 transcription factors as well as their target genes. The urobasal subtype showed up-regulation of PPARG, RXRA, FOXA1, and GATA3 - and their target genes - as well as expression of anterior HOXA and HOXB genes, and up-regulation of HOXA2 targets. The major regulatory axis operating in the GU subtype was PLK1-FOXM1, inducing a strong proliferative activity. Hence, the three major subtypes, Uro, GU, and SCCL may be explained by the activity of specific gene regulatory networks, either involved in urothelial differentiation or shared with other tumor types [14].
Using the bladder cancer TCGA data (234 invasive tumors) as a reference set, the Lund group evaluated four published gene expression-based bladder cancer classification systems: the two-tiered University of North Carolina (UNC), the three-tiered MD Anderson (MDA), the four-tiered TCGA, and the five-tiered Lund [15]. This analysis showed that a hierarchical relationship among the UNC, MDA, and TCGA classifications systems exists and that substantial biological subgroup heterogeneity remains at the highest resolution. Thus, a six-tiered classification system was reached including: a SCCL/UroB group, a GU group, an urobasal group, two groups with slightly different profiles emerging from the “infiltrated” category, and a new variant tentatively classified as “small cell/neuroendocrine-like”. Subtype assignments were validated by genomic alterations (chromosomal aberrations) and gene mutations [15].
Because the Lund group based their classification on both invasive and NMI cases and some of the subtypes cluster close together in pure invasive settings, they conclude that gene expression phenotypes converge upon progression. However, subtype-specific gene expression signatures are still present in invasive tumors.
THE UNIVERSITY OF NORTH CAROLINA CLASSIFICATION
B. Kim’s strategy focused on the analysis of invasive tumors. Using K2 consensus clustering, a robust classification of tumors enriched in basal (KRT5/6 and CD44) vs. luminal (PPARG, GATA3, KRT20, and UPK2) subtypes was reached [16]. A signature including 47 genes (BASE47) was generated by prediction analysis of microarrays (PAM) that was associated with outcome in 3 independent patient series. The basal subtype revealed similarities with the basal subtype of breast cancers, as demonstrated by applying the PAM50 signature to the UBC datasets. This group contained a claudin-low subgroup, as defined in breast cancer, that was enriched in epithelial-mesenchymal transition (EMT) and tumor initiating cell markers. Patients whose tumors were of the claudin-low subgroup had an outcome similar to that of patients in the “basal” category. Using pathway analysis and GSEA, a significant enrichment in genes related to inflammatory cell infiltration and immune checkpoint was observed in the basal subgroup and, more specifically, among the claudin-low tumors. The RNA-defined basal subgroup was also selectively enriched in RB pathway gene alterations while the luminal subtype was enriched in FGFR3 and TSC1 mutations. By contrast, there was no enrichment of TP53 pathway alterations. The basal group was significantly more common among women [16].
THE BAYLOR COLLEGE OF MEDICINE (TUMOR DIFFERENTIATION) CLASSIFICATION
K. Chan presented an update of his group’s recent work on bladder cancer subtyping based on urothelial cell differentiation [2, 3, 17]. A KRT14/Thy-1/CD44-positive self-renewing stem cell population was proposed to give rise to a partially differentiated KRT5/KRT17/CD44-positive progeny that is thought, in turn, to acquire KRT8/18 expression and terminally differentiate into luminal cells expressing uroplakins and KRT20. Knowledge on the normal urothelium differentiation program was thus used to classify tumors. The KRT14 + group (basal subtype) was characterized as having poor prognosis in multiple patient cohorts [2] and these KRT14 + basal tumors are resistant to neoadjuvant cisplatin-based chemotherapy in a small patient cohort [18]. An 18-gene classifier containing differentiation markers was applied to the TCGA series, showing that basal tumors were enriched in stem/progenitor urothelial cell markers such as KRT14/5/17, CD44 and CD49f, as well as signaling molecules including EGFR, JAK2, and STAT3. These findings echo an independent report from the CIT group demonstrating that EGFR signaling is functionally important in their basal-like tumors (see below) [19] and further support studiesfrom Lund and MDA groups that STAT3 signaling is enriched in their basal tumors [20] (see above and below). Patients with tumors classified as “basal” had a significantly worse survival than those that were “differentiated” and the P-values generated from this 18-gene classifier were more significant than those resulting from clustering methodologies utilized by TCGA or other groups to correlate with clinical outcome [17]. Baylor College of Medicine’s defined “basal” tumors were exclusively in cluster III/IV of the TCGA and overlapped with “basal” tumors as defined by the MDA group (see below). Similarly, the “differentiated” tumors overlapped well with clusters I and II of the TCGA classifier and with the “luminal” group of MDA [1, 20]. By contrast, the p53-like group of MDA was similarly split between “basal” and “differentiated” tumors (see below). A comparison was also performed with the classification of Lund: the “basal” group was modestly enriched in a subset of the Urobasal A tumors (Ms1a) and in the “infiltrated” group, and strongly enriched among the Urobasal B and SCCL tumors. By contrast, the “differentiated” group was enriched among the Ms1b subgroup of Urobasal A tumors and the Ms2a.2 subgroup of GU tumors. Interestingly, all Ms2a.1 GU tumors were “differentiated” Classification based on urothelial cell differentiation demonstrated a significant association with survival, and the basal subtype correlated well with clustering methodologies by TCGA (Cluster III/IV) and CIT [1, 19]. Nonetheless, it remains unresolved why the urothelial differentiation clustering method showed that basal tumors are more, or at least equally, resistant to chemotherapy while the MDA group claims otherwise.
THE MD ANDERSON SUBTYPES
To identify intrinsic subtypes, D. McConkey and the group at MDA modeled their approach after the pioneering breast cancer subtyping studies of Perou et al. [21, 22]. They generated genome wide mRNA expression profiling data from a cohort of 142 flash-frozen invasive and NMI UBC using Illumina chips [20]. The results revealed the presence of three distinct clusters; further analyses of the significantly differentially expressed genes defining each cluster revealed that they were enriched with biomarkers that had previously been implicated by Perou’s group and others as characteristic of the basal-like (KRT5, KRT14, CDH3, CD44) and luminal (KRT20, CD24, FOXA1, GATA3, ERBB2, ERBB3) intrinsic subtypes of breast cancer [20, 23, 24]. One of the subtypes was defined by a gene expression signature characteristic of active wild-type p53, therefore being designated “p53-like” [20]. Functional studies demonstrated that active DNp63a and STAT3 were involved in the control of basal gene expression, whereas PPARG was responsible for luminal gene expression, and DNp63a and PPARG antagonized each other. Tumors within the p53-like subtype were mostly resistant to neoadjuvant cisplatin-based combination chemotherapy [20].
The MDA group compared their subtype calls with those of the groups at the University of Lund [8], TCGA [1], and the UNC [16], using shared datasets. Overall, there was close concordance among theclassifications. The MDA “basal” tumors corresponded well with the Lund “SCC-like” subtype, TCGA’s Clusters III (“squamous”) and IV, and UNC’s “basal-like” tumors. The MDA “luminal” tumors corresponded with the Lund “genomically unstable”, TCGA cluster I and UNC’s “Luminal”, while the MDA “p53-like” tumors matched the Lund “infiltrated”, TCGA Cluster II, and merged in the “luminal” UNC subtype [12, 13, 20, 23]. Because each group used very distinct approaches to identify their subtypes, the fact that all reached similar conclusions strongly suggests that the subtypes are highly reproducible and biologically relevant, regardless of how they are named.
Many investigators now prefer consensus clustering to unsupervised hierarchical clustering to identify subtypes of cancer because it assesses the stability of clusters based on multiple re-samplings. Therefore, the MDA group investigated how their calls would have changed had they applied consensus clustering, as opposed to other clustering methods, using the TCGA RNAseq dataset (n = 128, log2-transformed) as a reference. They identified the top 5000 most variable genes as measured by median absolute deviation and median centered the data and used this subset for consensus clustering (specifically, agglomerative hierarchical clustering). Area under the curve analyses identified four clusters as the optimum but one of the clusters only contained one tumor, leading them to conclude that three groups would have been more biologically reasonable. When the analyses were performed using k = 5, TCGA’s four clusters were reproduced almost exactly, and comparison of the calls again revealed excellent concordance with the MD Anderson subtypes.
THE CIT “BASAL-LIKE” TUMOR CLASSIFICATION
F. Radvanyi and his colleagues from the CIT consortium (Institut Curie, Henri Mondor and Foch hospitals, Institut Gustave Roussy, CEPH, La Ligue Contre le Cancer) focused on the “basal-like” tumor subgroup because it was the largest homogeneous group identified by various unsupervised methods to classify invasive bladder cancer based on transcriptome data [19]. In a joint analysis of 7 datasets of invasive tumors, more advanced cases were over-represented among the “basal-like” tumors. Survival analyses showed worse outcome for this group of patients, independently of stage and grade, lymph node, and metastatic status. The survival curves of patients with basal-like tumors were very different from other bladder cancer patients, with most of the death events occurring within one year after diagnosis. At the level of DNA alterations, this group was characterized by significantly more EGFR gains or amplifications, FHIT deletions, and TP53 mutations; at the transcriptomic level, an enrichment of EGFR pathway activation was identified. In agreement with this observation, EGFR phosphorylation was higher in the basal-like subgroup. This led to testing in vitro and in vivo the sensitivity of bladder cancer cells to the EGFR inhibitor erlotinib. For this purpose, a particular effort was made to identify relevant preclinical models recapitulating human primary basal-like bladder tumors. Using a transcriptomic signature derived from human invasive bladder cancer datasets, they identified 11 human bladder cancer cell lines - out of 22 - dysplaying a basal-like profile [19]. In addition, they showed that tumors induced by N-butyl-N-(4-hydroxybutyl) nitrosamine in mice, a commonly used model of chemical bladder carcinogenesis, recapitulated keymolecular features of the human basal-like tumors including the autocrine activation of the EGFR pathway. In these models, erlotinib treatment showed anti-tumor efficacy and the basal-like phenotype was predictive of erlotinib response. This study provided, in various preclinical models, the first proof-of-concept that anti-EGFR therapies could be effective to treat human basal-like bladder cancers [19].
In another study, they went on to use Independent Component Analysis (ICA), an approach allowing a gene to be placed in multiple pathways, a feature that relates better to biological functions [25]. Using this strategy, they could characterize cancer subtypes and identify candidate genes to be involved in different pathways. The “basal” subgroup was found to correspond to the TCGA cluster III and it was mainly enriched in the interferon response pathway, had a high content in myofibroblasts and low T and B lymphocyte content. Smooth muscle and T and B lymphocytes were mainly enriched in cluster IV. Invasive cancers could be divided into two groups according to the urothelial differentiation component. Both clusters I and II presented an urothelial differentiation program in contrast to clusters III and IV. By combining ICA of transcriptome and copy number data, they could predict that PPARG was an oncogene for molecularly differentiated tumors. This was confirmed by functional analyses [25]. When they applied ICA to a Pan-Cancer cohort, some components were found in all cancer types (i.e. lymphocytes, cell cycle). By contrast, others were found only in UBC (i.e. urothelial differentiation and the carcinoma in situ pathway).
Radvanyi also discussed different strategies for identifying the basal-like subgroup comparing a 40 gene-based transcriptomic signature and IHC based on two antibodies recognizing KRT5 and KRT6 as a positive marker of the subgroup and FOXA1 as a negative marker [19] (see below). In their studies, an 85% agreement was found between both techniques.
MECHANISTIC INSIGHTS INTO THE CONTROL OF “BASALITY”
As indicated above, all the classifications included a set of tumors expressing basal or SCCL markers and F. X. Real reviewed the evidence from various classifications in search of putative transcription factors/networks that might be involved in the activation of the various molecular sub-phenotypes. There has been extensive work pointing to genes involved in urothelial differentiation, including FOXA, GATA, PPARG, ELF3, and IRF1 [26–28]. FOXA1 and GATA3 were enriched among tumors in TCGA clusters I and II and depleted from clusters III and IV. STAT3 and DNp63a have been proposed as regulating the basal phenotype, though less is known regarding the direct transcriptional regulation of the “basal” program. Work was presented pointing to the participation of FOXA and GATA proteins in the repression of the EMT and of a basal phenotype, and to the emergence of these programs upon silencing or inactivation of the GATA/FOXA proteins. Chromatin immunoprecipitation followed by massive parallel sequencing showed GATA binding to 24/47 genes from the BASE47 signature in different cell types, including urothelial cells (P. Martinelli and F. X. Real, unpublished). GATA and FOXA transcription factors also appeared to be involved in cross-regulatory networks linking epithelial differentiation to the transcriptional regulation of EGFR, its phosphorylation, and the activation of downstream ERK and PI3K pathways [29]. The evidence presented suggests that individual members of these families (GATA1-6, FOXA1-3) play important roles in different tissue types, further supported by the fact that several of these genes are selectively mutated in UBC [1, 30, 31].
DEFINING “INTRINSIC” BLADDER CANCER SUBTYPES
The term “intrinsic” was first applied to the molecular classification of breast cancers. The work by Perou’s group demonstrated that tumor subtypeusually remained stable regardless of where or when a given tumor was sampled [21], leading to the conclusion that subtype membership was an “intrinsic” property of a given breast cancer. The original definition of “intrinsic” referred to tumor cell properties. Although the MDA group consistently observed their three subtypes in multiple independent datasets, this did not necessarily indicate that all of the subtypes were “intrinsic”. Direct measurements of cluster stability using silhouette score analyses indicated that many of the “p53-like” tumors in the MDA discovery cohort were unstable, unlike the “basal” and “luminal” tumors [20]. In addition, analyses of matched tumors before and after neoadjuvant chemotherapy demonstrated that many of the “luminal” tumors acquired “p53-like” features after therapy [20]. This observation has been confirmed in every neoadjuvant chemotherapy cohort analyzed to date (n = 5) (W. Choi, unpublished observations). Analyses of matched primary tumors and lymph node metastases (n = 33) indicated that basal tumors almost never switched subtypes whereas luminal and p53-like tumors display much more subtype “switching” (W. Choi, unpublished observations). Therefore, basal or luminal subtype membership appears to be an intrinsic tumor property but p53-like tumors may enter or exit the subtype as a consequence of environmental stimuli (“plasticity”). Finally, a working definition of “intrinsic bladder cancer subtypes” should be agreed upon in a future meeting.
THE CONSENSUS
The need to reach consensus about the UBC subtypes and how they can be best defined is important in, at least, two different areas: first, to achieve an improved understanding of the underlying biology; second, using this information to stratify patients with the aim of improving management, based either on differences in outcome or in response to therapy. The latter aspect concerns both standard and novel, targeted, therapies towards precision medicine.
The above subtype descriptions clearly indicate that all groups involved in the definition of invasive bladder cancer subtypes identified a tumor subset characterized by the expression of markers typical of basal cells in stratified epithelia, most notably KRT5/6 and KRT14. The low expression levels of FOXA1 and GATA3 at the RNA and protein levels also characterize this tumor subtype. Furthermore, several groups working on UBC, as well as in other tumor types, have shown that there are good reagents to detect the proteins encoded by these four genes in formalin-fixed paraffin-embedded tissue sections. Therefore, the group reached the consensus conclusion that a subgroup of invasive bladder cancers can be identified as being KRT5/6 + KRT14 + FOXA1 - GATA3 - (Fig. 1). In these tumors, KRT5/6 and KRT14 are expressed broadly in cancer cells, without epithelial compartmentalization (Fig. 2). This group appears to be consistently associated with a poor prognosis. Future studies should refine this molecular definition, determine the optimal techniques that can be applied for tumor classification, as well as clinical-pathological, and etiological associations. The Lund data suggest that this tumor subtype can be recognized among T1 tumors [8, 11]. Independent studies should be useful to confirm these findings.
All attendees agreed that there is robust and concordant evidence supporting the existence of an invasive tumor subgroup with more differentiated, urothelial features. This group is enriched in FGFR3 alterations and expression of GATA3, FOXA1, KRT20, and miR99/miR100, all of them features that are common among the Urobasal A tumors from the Lund classification. Considering the data from NMI tumors will likely enhance the definition of this category. However, the group felt that there was not sufficient data to reach a consensus on the optimal molecular definition of such subgroup and that future work should address this question in the context of both NMI and invasive bladder tumors. This remains an important task for futurestudies.
As of the other subtypes described (i.e. the GU from Lund and the p53-like from MDA) more work is required to determine how robust they are, how to best define them across transcriptomic datasets, and how to best identify them using RNA-based or IHC-based assays.
NOMENCLATURE
The group concurred that names are important as they deliver implicit information that should both be helpful and clear. The designations that are proposed here are operational, do not strictly correspond to pathological classifications, and do not imply cell-of-origin relationships. The group makes the following recommendations:
1. To use Basal/Squamous-like (proposed acronym, BASQ) to designate the tumors displaying the KRT5/6 + KRT14 + FOXA1 - GATA3 - phenotype. This designation responds to the fact that there is expression of basal keratins and an enrichment of tumors displaying histological squamous differentiation in this subgroup [10, 19, 20] and that the molecular pan-cancer analysis assigned this group together with squamous tumors of the lung and head and neck [5]. However, it should be emphasized that this relationship is not unequivocal as not all tumors in this group will present with histological squamousfeatures and some tumors with squamous features may not fall within the group.
2. The Lund group has suggested that the previously “Urobasal” subtype defined by them change name to “Urothelial-like” abbreviated as Uro, and that “UroA” and “UroB” be retained as for the previously described subtypes which, in full, should be referred to as “Urothelial-like A” and “Urothelial-like B” subgroups (therefore, for the sake of clarity, it is not recommended the use of the term “urobasal” for these subgroups).
The group agreed to reconvene in the future to update the consensus evidence and to develop strategies for future collaboration.
Meeting attendees: M. Aine, Y. Allory, E. Carrillo-de Santa Pau, D. G. Pisano, K. Chan, L. Dyrskjot, A. Hartmann, K. Hoadley, M. Höglund, W. Kim, S. P. Lerner, N. Malats, M. Marqués, D. J. McConkey, B. Mellado, T. C. McKee, F. Radvanyi, F. X. Real, A. de Reyniès, G. Sjödahl.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
ACKNOWLEDGMENTS
We would like to thank all the meeting attendants for their participation and discussion, and N. Malats and E. Carrillo-de Santa Pau for contributions to a previous version of the paper. This manuscript and the Bladder Cancer Classification Consensus Meeting that led to its development were supported by a grant from Astellas Pharma Europe Ltd given to Universitat Pompeu Fabra, UPF. Astellas Pharma Europe Ltd holds no responsibility for the origination, development and content of either the manuscript or the meeting, including the agenda, choice of participants, manuscript author, editorial control ordistribution.
REFERENCES
[1] | The Cancer Genome Atlas Research network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature (2014) ;507: (7492):315–322. |
[2] | Volkmer JP , Sahoo D , Chin RK , Ho PL , Tang C , Kurtova AV , Willingham SB , Pazhanisamy SK , Contreras-Trujillo H , Storm TA , Lotan Y , Beck AH , Chung BI , Alizadeh AA , Godoy G , Lerner SP , van de Rijn M , Shortliffe LD , Weissman IL , Chan KS . Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc Natl Acad Sci USA (2012) ;109: (6):2078–2083. |
[3] | Ho PL , Kurtova A , Chan KS . Normal and neoplastic urothelial stem cells: Getting to the root of the problem. Nat Rev Urol (2012) ;9: (10):583–594. |
[4] | Sjödahl G , Lauss M , Lovgren K , Chebil G , Gudjonsson S , Veerla S , Patschan O , Aine M , Fernö M , Ringnér M , Månsson W , Liedberg F , Lindgren D , Höglund M . A molecular taxonomy for urothelial carcinoma. Clin Cancer Res (2012) ;18: (12):3377–3386. |
[5] | Hoadley KA , Yau C , Wolf DM , Cherniack AD , Tamborero D , Ng S , Leiserson MD , Niu B , McLellan MD , Uzunangelov V , Zhang J , Kandoth C , Akbani R , Shen H , Omberg L , Chu A , Margolin AA , Van’t Veer LJ , Lopez-Bigas N , Laird PW , Raphael BJ , Ding L , Robertson AG , Byers LA , Mills GB , Weinstein JN , Van Waes C , Chen Z , Collisson EA ; Cancer Genome Atlas Research Network, Benz CC , Perou CM , Stuart JM . Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell (2014) ;158: (4):929–944. |
[6] | Lawrence MS , Stojanov P , Mermel CH , Robinson JT , Garraway LA , Golub TR , Meyerson M , Gabriel SB , Lander ES , Getz G . Discovery and saturation analysis of cancer genes across 21 tumour types. Nature (2014) ;505: (7484):495–501. |
[7] | Lindgren D , Frigyesi A , Gudjonsson S , Sjödahl G , Hallden C , Chebil G , Veerla S , Ryden T , Månsson W , Liedberg F , Höglund M . Combined gene expression and genomic profiling define two intrinsic molecular subtypes of urothelial carcinoma and gene signatures for molecular grading and outcome. Cancer Res (2010) ;70: (9):3463–3472. |
[8] | Sjödahl G , Lauss M , Lövgren K , Chebil G , Gudjonsson S , Veerla S , Patschan O , Aine M , Fernö M , Ringnér M , Månsson W , Liedberg F , Lindgren D , Höglund M . A molecular taxonomy for urothelial carcinoma. Clin Cancer Res (2012) ;18: (12):3377–3386. |
[9] | Lindgren D , Sjödahl G , Lauss M , Staaf J , Chebil G , Lövgren K , Gudjonsson S , Liedberg F , Patschan O , Månsson W , Fernö M , Höglund M . Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma. PLoS One (2012) ;7: (6):e38863. |
[10] | Sjödahl G , Lövgren K , Lauss M , Patschan O , Gudjonsson S , Chebil G , Aine M , Eriksson P , Månsson W , Lindgren D , Fernö M , Liedberg F , Höglund M . Toward a molecular pathologic classification of urothelial carcinoma. Am J Pathol (2013) ;183: (3):681–691. |
[11] | Patschan O , Sjödahl G , Chebil G , Lövgren K , Lauss M , Gudjonsson S , Kollberg P , Eriksson P , Aine M , Månsson W , Fernö M , Liedberg F , Höglund M . A Molecular Pathologic Framework for Risk Stratification of Stage T1 Urothelial Carcinoma. Eur Urol (2015) ; In press PMID 25770486. |
[12] | Lauss M , Aine M , Sjödahl G , Veerla S , Patschan O , Gudjonsson S , Chebil G , Lövgren K , Fernö M , Månsson W , Liedberg F , Ringnér M , Lindgren D , Höglund M . DNA methylation analyses of urothelial carcinoma reveal distinct epigenetic subtypes and an association between gene copy number and methylation status. Epigenetics (2012) ;7: (8):858–867. |
[13] | Aine M , Sjödahl G , Eriksson P , Veerla S , Lindgren D , Ringnér M , Höglund M . Integrative epigenomic analysis of differential DNA methylation in urothelial carcinoma. Genome Med (2015) ;7: (1):23. |
[14] | Eriksson P , Aine M , Veerla V , Liedberg F , Sjödahl G , Höglund M . Molecular subtypes of urothelial carcinoma are defined by specific gene regulatory systems. BMC Medical Genomics (2015) ;8: :25. doi: 10.1186/s12920-015-0101-5. |
[15] | Aine M , Eriksson P , Liedber F , Sjödahl G , Höglund M . Biological determinants of bladder cancer gene expression subtypes. Sci Rep (2015) ;5: , 10957. |
[16] | Damrauer JS , Hoadley KA , Chism DD , Fan C , Tiganelli CJ , Wobker SE , Yeh JJ , Milowsky MI , Iyer G , Parker JS , Kim WY . Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci USA (2014) ;111: (8):3110–3115. |
[17] | Chan KS , Espinosa I , Chao M , Wong D , Ailles L , Diehn M , Gill H , Presti J Jr , Chang HY , van de Rijn M , Shortliffe L , Weissman IL . Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci USA (2009) ;106: (33):14016–14021. |
[18] | Kurtova AV , Xiao J , Mo Q , Pazhanisamy S , Krasnow R , Lerner SP , Chen F , Roh TT , Lay E , Ho PL , Chan KS . Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature (2015) ;517: (7533):209–213. |
[19] | Rebouissou S , Bernard-Pierrot I , de Reynies A , Lepage ML , Krucker C , Chapeaublanc E , Herault A , Kamoun A , Caillault A , Letouze E , Elarouci N , Neuzillet Y , Denoux Y , Molinié V , Vordos D , Laplanche A , Maillé P , Soyeux P , Ofualuka K , Reyal F , Biton A , Sibony M , Paoletti X , Southgate J , Benhamou S , Lebret T , Allory Y , Radvanyi F . EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med (2014) ;6: :244ra291 |
[20] | Choi W , Porten S , Kim S , Willis D , Plimack ER , Hoffman-Censits J , Roth B , Cheng T , Tran M , Lee IL , Melquist J , Bondaruk J , Majewski T , Zhang S , Pretzsch S , Baggerly K , Siefker-Radtke A , Czerniak B , Dinney CP , McConkey DJ . Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell (2014) ;25: (2):152–165. |
[21] | Perou CM , Sorlie T , Eisen MB , van de Rijn M , Jeffrey SS , Rees CA , Pollack JR , Ross DT , Johnsen H , Akslen LA , Fluge O , Pergamenschikov A , Williams C , Zhu SX , Lønning PE , Børresen-Dale AL , Brown PO , Botstein D . Molecular portraits of human breast tumours. Nature (2000) ;406: (6797):747–752. |
[22] | Sorlie T , Perou CM , Tibshirani R , Aas T , Geisler S , Johnsen H , Hastie T , Eisen MB , van de Rijn M , Jeffrey SS , Thorsen T , Quist H , Matese JC , Brown PO , Botstein D , Lønning PE , Børresen-Dale AL . Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA (2001) ;98: (19):10869–10874. |
[23] | Choi W , Czerniak B , Ochoa A , Su X , Siefker-Radtke A , Dinney C , McConkey DJ . Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat Rev Urol (2014) ;11: (7):400–410. |
[24] | McConkey DJ , Choi W , Dinney CP . New insights into subtypes of invasive bladder cancer: Considerations of the clinician. Eur Urol (2014) ;66: (4):609–610. |
[25] | Biton A , Bernard-Pierrot I , Lou Y , Krucker C , Chapeaublanc E , Rubio-Pérez C , López-Bigas N , Kamoun A , Neuzillet Y , Gestraud P , Grieco L , Rebouissou S , de Reyniès A , Benhamou S , Lebret T , Southgate J , Barillot E , Allory Y , Zinovyev A , Radvanyi F . Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep (2014) ;9: (4):1235–1245. |
[26] | Varley CL , Bacon EJ , Holder JC , Southgate J . FOXA1 and IRF-1 intermediary transcriptional regulators of PPARgamma-induced urothelial cytodifferentiation. Cell Death Differ (2009) ;16: (1):103–114. |
[27] | Böck M , Hinley J , Schmitt C , Wahlicht T , Kramer S , Southgate J . Identification of ELF3 as an early transcriptional regulator of human urothelium. Dev Biol (2014) ;386: (2):321–330. |
[28] | Adam RM , DeGraff DJ . Molecular mechanisms of squamous differentiation in urothelial cell carcinoma: A paradigm for molecular subtyping of urothelial cell carcinoma of the bladder. Urol Oncol (2015) : pii:S1078–S1439. |
[29] | Martinelli P , Madriles F , Canamero M , Carrillo-de-Santa Pau E , del Pozo N , Guerra C , Real FX . The acinar regulator GATA6 suppresses KRasG12V-driven pancreatic tumorigenesis in mice. Gut (2015) ; In press PMID 25596178. |
[30] | Guo G , Sun X , Chen C , Wu S , Huang P , Li Z , Dean M , Huang Y , Jia W , Zhou Q , Tang A , Yang Z , Li X , Song P , Zhao X , Ye R , Zhang S , Lin Z , Qi M , Wan S , Xie L , Fan F , Nickerson ML , Zou X , Hu X , Xing L , Lv Z , Mei H , Gao S , Liang C , Gao Z , Lu J , Yu Y , Liu C , Li L , Fang X , Jiang Z , Yang J , Li C , Zhao X , Chen J , Zhang F , Lai Y , Lin Z , Zhou F , Chen H , Chan HC , Tsang S , Theodorescu D , Li Y , Zhang X , Wang J , Yang H , Gui Y , Wang J , Cai Z . Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet (2013) ;45: (12):1459–1463. |
[31] | Yap KL , Kiyotani K , Tamura K , Antic T , Jang M , Montoya M , Campanile A , Yew PY , Ganshert C , Fujioka T , Steinberg GD , O’Donnell PH , Nakamura Y . Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res (2014) ;20: (24):6605–6617. |
Figures and Tables
Fig.1
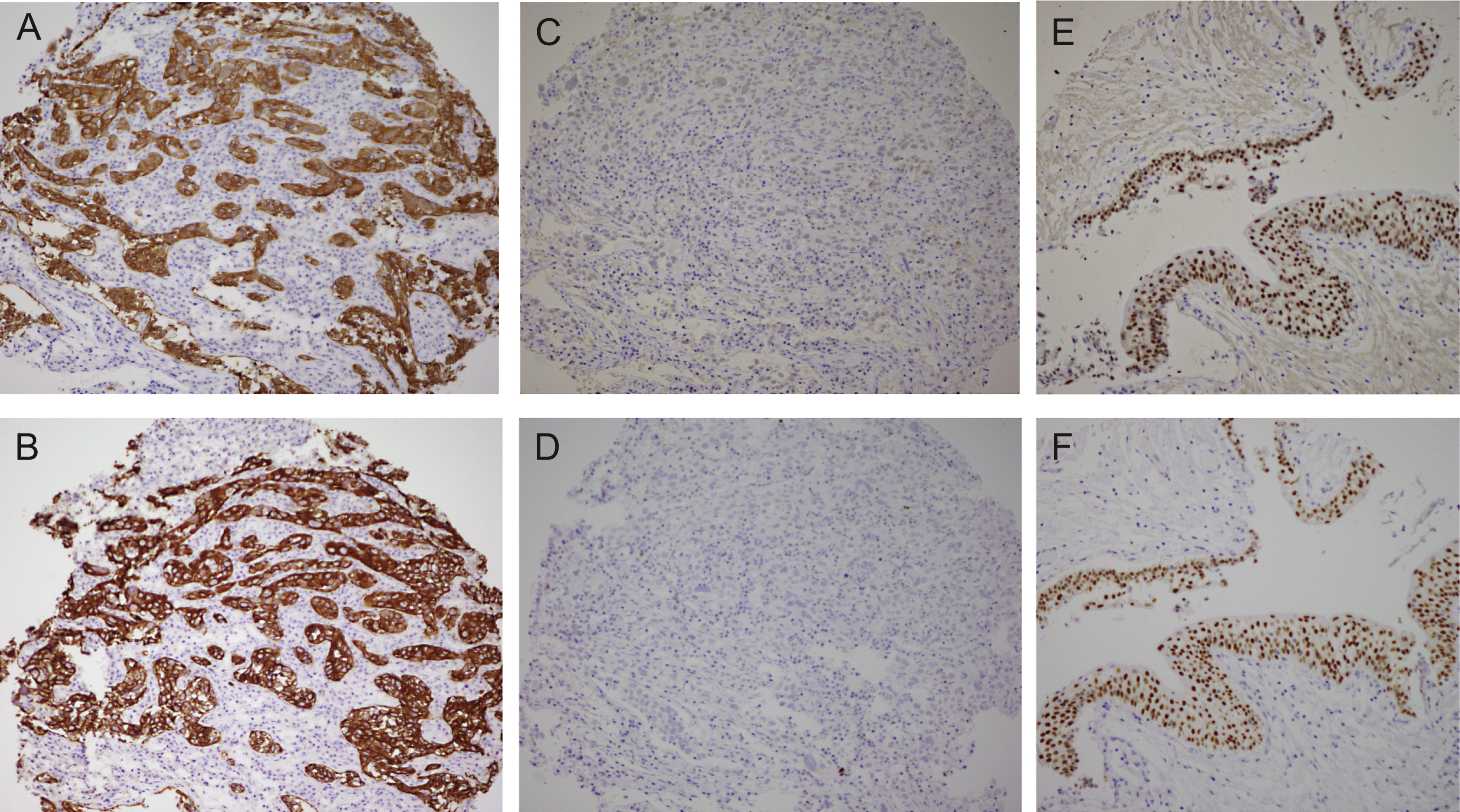
Comparison of the bladder cancer classifications as they relate to the “BASQ” (Basal-Squamous-like) consensus group. In red background, the subtypes that are enriched in this group. Tumor subclasses in other colors (p53-like, TCGA II, Infiltrated) comprise samples that would be included in the BASQ group and others that would not. Tumors in these three categories also express markers typical of urothelial differentiation to a variable extent. In red, the consensus definition of the “BASQ” subtype.
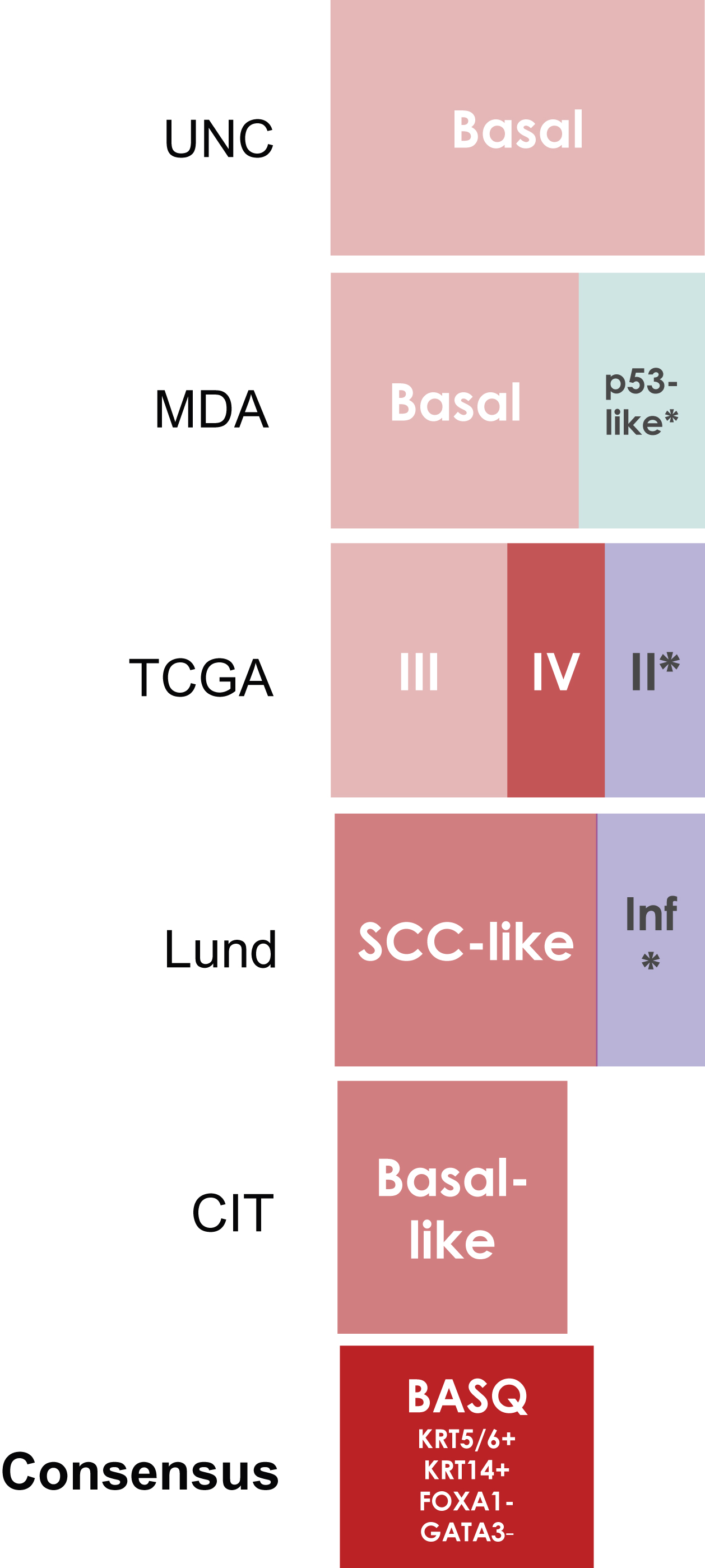
Fig.2
Basal/Squamous-like (BASQ) tumors are characterized by strong expression of KRT5/6 (A) and KRT14 (B) and low or undetectable expression of FOXA1 (C) and GATA3 (D). By contrast to them, normal urothelial cells display strong expression of FOXA1 (E) andGATA3 (F).