
High-throughput DNA Sequencing Identifies Novel CtIP (RBBP8) Variants in Muscle-invasive Bladder Cancer Patients
Abstract
Background:
Germline mutations in DNA damage signalling and repair genes predispose individuals to cancer. Rare germline variants may also increase cancer risk and be predictive of outcomes following cancer treatments, but require high-throughput sequencing (HTS) for detection in large cohorts.
Objective:
To use a dual indexing system on a HTS platform to detect novel variants in CtIP (RBBP8) which may be associated with clinical outcomes following radiotherapy treatment for bladder cancer.
Methods:
All exons and flanking introns of CtIP were amplified from germline DNA from bladder cancer patients using seven primer pairs by automated long-range PCR. Amplicons were pooled, fragmented and ligated to adaptor sequences. One of 96 tag sequences was introduced at each end by PCR. Sequencing was performed on a single flow cell of an Illumina MiSeq. Reads were mapped by Stampy and variants called by Platypus. For phasing experiments, target regions were amplified and cloned for Sanger sequencing.
Results:
Of 201 samples, 160 were successfully amplified. Eleven CtIP variants were called, within the exons and 15 bp adjacent intronic DNA, including eight known variants from the 1000 Genomes project, plus three previously unreported variants now confirmed by Sanger sequencing. In two individuals, phasing experiments showed two variants of interest to be on separate alleles, likely to result in stronger impairment of gene function.
Conclusions:
We have demonstrated proof of principle for dual indexing on 160 samples on one MiSeq flow cell sequencing surface, and show that for the CtIP gene multiplexing of up to 720 samples would provide sufficient coverage to achieve >98% detection power for rare germline variation, reducing HTS costs substantially.
INTRODUCTION
Genomic instability is one of the ten hallmarks of cancer [1]. Cells require functional DNA damage signalling and repair pathways to maintain genomic stability and thus prevent cancer development. Indeed, early in tumorigenesis, before the onset of genomic instability and malignant conversion, human cells activate an ATR/ATM-regulated DNA damage response network that delays or prevents the onset of cancer [2, 3].
Rare germline mutations in the DNA repair genes BRCA1 and BRCA2 predispose women to breast cancer (cancer relative risk of 5), as do more frequent, moderately-penetrant mutations (relative risk of 1.5–5) in ATM, the cell cycle kinase CHEK2 and the DNA damage signalling MRN complex genes MRE11, RAD50 and NBS1 [4]. Although more common than mutations, single nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) >1% , can also be associated with increased cancer risk. We found an MRE11 3’UTR SNP to be associated with bladder cancer risk [5], and the International Consortium of Bladder Cancer found a non-synonymous NBS1 SNP to be associated with bladder cancer risk in a large meta-analysis [6]. DNA damage signalling and repair are also important processes in the response to radiotherapy treatment, as double-strand breaks (DSB) are the lethal lesions caused by ionising radiation. In a relatively small study [7], it was found that DNA repair gene SNPs were associated with response to chemoradiation in bladder cancer.
With the advent of multiplexing allowing genotyping of known variants in large numbers of samples, SNP studies have been extended to hypothesis-free genome-wide association studies (GWAS). However, not all GWAS hits have been replicated, the GWAS approach cannot discover new variants, and only small effect sizes are seen with odds ratios of 1.2–1.3 [8]. Rarer genetic variants, with MAFs of 0.1–1% , i.e. lower than SNPs but higher than deleterious mutations, are thought to be important contributors to cancer risk, with odds ratios of >2, and with the summation of the effects of several rare variants making a significant contribution to population attributable risk [9]. However, to detect novel rare variants requires expensive Sanger sequencing or, alternatively, high-throughput sequencing (HTS).
We recently used high-throughput sequencing technology to study the DNA damage signalling gene, MRE11, in germline DNA from 186 muscle-invasive bladder cancer (MIBC) patients treated with radical radiotherapy, and identified two germline single nuclear polymorphisms (SNPs) and six new rare variants which were associated with survival following radiotherapy and late treatment toxicity [10]. Although successful, the MRE11 project required nine lanes of sequencing mainly due to the limited level of multiplexing available. If more samples could be run per lane, this would reduce costs, thus increasing the numbers of samples which could be analysed and hence increasing the power of any study.
The CtIP gene is also involved in DNA damage signalling and repair. This 93.16 kb gene with a 3288 bp transcript encodes an 897 amino acid endonuclease which, in complex with BRCA1, repairs lethal DNA double strand breaks (DSBs) caused by ionising radiation [11, 12]. CtIP interacts functionally with the MRN complex and acts downstream of ATM to promote DSB resection, ATR signalling and homologous recombination [12, 13]. Although CtIP is mainly involved in DNA DSB repair via homologous recombination (HR), recent reports also implicate the protein in an alternative end-joining pathway, called microhomology-mediated end-joining (MMEJ) [14–16], and extracts from MIBC samples preferentially use MMEJ rather than conventional non-homologous end-joining to repair DSBs in contrast to non-MIBC samples [17]. Germline variants of CtIP are likely to impact on these processes, and the most affordable and rapid way of detecting these novel variants is through the use of HTS technology [18, 19].
The tremendous increase in sequencing capacity of current HTS platforms allows a large number of samples to be analysed in each run. In this study we took advantage of the dual indexing system developed by Illumina. The strategy involves sequential reading of the indices present on adapter sequences positioned at both ends of every DNA fragment sequenced on Illumina platforms. We designed a set of custom oligos, using 96 single tag sequences developed at the Wellcome Trust Centre for Human Genetics [20], which are compatible with Illumina’s chemistry and machine recipe. This customisation allowed us to successfully sequence 160 germline DNA samples [10] on a single flow cell of an Illumina MiSeq, thereby demonstrating the proof-of-principle that this system could be used to multiplex up to 96 by 96 samples. We are not aware of others having used this technology for such a purpose in human samples to date.
We hypothesised that this dual indexing system would allow us to detect novel variants in CtIP, which may be associated with clinical outcomes following radiotherapy.
MATERIALS AND METHODS
Study population
Muscle-invasive bladder cancer patients (N = 201) were recruited prospectively having given written informed consent with local ethical approval (Leeds (East) Research Ethics Committee project 04/Q1206/62), as previously described [21, 22]. Patients were treated with three-dimensional conformal external beam radiotherapy (52.5–55 Gy in 20 fractions over 4 weeks) between August 2002 and October 2009 in Leeds, UK. DNA was extracted from blood samples collected prior to radiotherapy using standard salting-out protocols. Follow-up data were acquired prospectively on standard proformas from clinic visits and from the electronic patient record (Table 1).
Work schematic
The work schematic is outlined in Supplementary Figure S1.
Long-range PCR
The CtIP (RBBP8) reference genomic sequence (GRCh37.chr18:20,513,275-20,606,464) was used to design a total of 7 primers pairs that amplified all exons plus a minimum of 200 bases of flanking intron in 7 amplicons (total 44.88kb) (Supplementary Table S1). Primers were designed using Primer Quest (IDT) software (http://www.idtdna.com/primerquest/Home/Index). Primer lengths were between 19 and 25 base pairs, melting temperatures (Tm) were between 58.7°C and 62°C with less than 1.1°C difference between corresponding pairs. The GC content of the primers ranged from 42.9% to 52.6% with no more than a 6% difference between primer pairs. No known variants, according to dbSNP database, were contained within the primer sequences. Automated long-range PCR was performed using PrimeSTAR GXL DNA polymerase (TaKaRa). PCR reactions were performed in 384-well PCR plates in a 20 μl final volume with 100 ng of genomic DNA and PrimeSTAR GXL Taq under the following conditions: reactions were denatured at 98°C for 2 min and then cycled (98°C 10 sec, 68°C 10 min) for 30 cycles, with a final extension of 68°C for 5 min, using the primers listed in Supplementary Table S1. Optimal PCR conditions differed for amplicon 7, therefore reactions were denatured at 94°C for 30 sec and then cycled (94°C 15 sec, 55°C 15 sec, 68°C 11 min) for 30 cycles, with a final extension of 68°C for 7 min using primer set 7.
In order to identify the minimum number of cycles that would generate a single amplicon, an aliquot of each reaction was collected after 20, 25, 28 and 30 cycles. A Biomek FX was used to automate the following steps: a) genomic DNA and PCR reagents to be transferred to a 384-well PCR plate, b) PCR cleanup with 0.4 volumes of AMPure XP magnetic beads, c) Quant-iTtrademark PicoGreen dsDNA assay to assess the concentration of the amplicons, d) equimolar pooling of amplicons per patient.
Library preparation, dual indexing and sequencing
Samples were quantified using the Quant-iT PicoGreen dsDNA Kits (Invitrogen) and a GENios plate scanner (Tecan) according to manufacturer specifications. Sample integrity was assessed using 1% agarose gels (Supplementary Figure S2). For some of the samples that did not show a band on the gel, additional material was used for the amplicon generation. However, in many cases, apparently undetectable DNA successfully produced amplicons. No further quantitation or loading of DNA to rule out gel artefacts was performed to limit material wastage. For each sample 5–50 ng of pooled amplicons were fragmented using an Episonic 1000 (Epigentek) using the following settings: amplitude 40, process time 3 mins 20 secs, pulse on for 20 secs, pulse off for 20 secs.
Distribution of fragments after shearing was determined with a Tapestation 2200, D1200 system (Agilent). Libraries were constructed using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB- E7370L) with minor modifications and a custom automated protocol on a Biomek FX (Beckman). Ligation of adapters to both ends of the DNA (Supplementary Figure S3) was performed using adapters prepared at the Wellcome Trust Centre for Human Genetics according to the Illumina design (Multiplexing Sample Preparation Oligonucleotide Kit). Each library was PCR enriched with 25 μM each of the following custom primers for 18 cycles:
Multiplex PCR primer 1.0: 5′AATGATACGGCGACCACCGAGATCTACAC[INDEX]TCTTTCCCTACACGACGCTCTTCCGATCT3′
Index primer: 5′CAAGCAGAAGACGGCATACGAGAT[INDEX]CAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT3′
Indices used were 8 bp long, as previously published [20]. Final clean up of the libraries was performed using 0.85 volumes of AMPure XP and a Biomek NXp. For equimolar pooling of the libraries, size distribution using a Tapestation 2220 and relative concentrations were determined by PicoGreen as described above. To estimate the volume to use for sequencing, a real time PCR was performed to measure the relative concentration of the pool compared to a previously sequenced library. Sequencing was performed as 150 bp paired end on a MiSeq according to Illumina specifications.
Mapping and variant calling
Reads were mapped using Stampy [23] version 1.0.20, and variants were called by running Platypus [24] version 0.4.0 on each sample independently. Default parameters were used in both steps. Variant calling was restricted to regions covered by the union of exons in RefSeq RBBP8 transcripts, with a 15 bp shoulder to include any splicing variants (20 regions covering 4,175 bp). SIFT [25] and PolyPhen-2 [26] were used to predict damaging effects of variants.
Sanger sequencing and PCR product cloning
Sanger sequencing was performed to validate three novel variants detected in three patient samples. For phasing experiments, the region containing variants 20513374 and 20514119 was amplified in one individual and that containing variants 20513374 and 20513432 in a second individual. PCR was performed using High Fidelity Platinum Taq (Life Technologies) with 94°C 2 min, (94°C 15 sec, 55°C 30 sec, 68°C 2 min) for 30 cycles, 4°C 5 min. Products were run on 1% agarose gels before bands were excised and gel purified using a QIAquick gel extraction kit (Qiagen), according to manufacturer’s instructions. PCR products were cloned into pcDNA 4 TOPO vector (Life Technologies) as per protocol. Plasmids were transformed into One Shot TOP10 Chemically Competent E. coli (Life Technologies). DNA was isolated from three positive colonies from each patient using a QIAprep spin miniprep kit (Qiagen). DNA was sequenced by capillary sequencing with both forward and reverse primers flanking the variants.
Statistical analysis
The associations of having a rare variant with disease-specific and overall survival were examined using a Cox proportional hazards model. The model was adjusted for carrying at least one common variant, age at treatment, sex, concurrent chemotherapy, neoadjuvant chemotherapy, histology, grade, hydronephrosis, previous superficial bladder cancer and radiotherapy dose. Also an unadjusted model was fitted for each variant. The proportional hazards assumption was checked graphically using scaled Schoenfeld residuals and the corresponding chi-squared test. Statistical analysis was performed using R [27].
RESULTS
Patient demographics
Table 1 shows the demographics for 160 MIBC patients treated with radical radiotherapy where PCR amplification was successful. No patient had distant metastatic disease. Sixty-two patients died of disease. Median follow-up for patients who did not die was 61.4 months. The median follow-up time for all 160 patients with sequencing information is 28.7 months with an interquartile range of 45.2 months (Table 1).
Long range PCR (LR-PCR) optimisation
The primer design for the generation of amplicons required to satisfy two major points, namely, minimise the number of independent reactions needed to amplify the CtIP region and provide a robust assay even for DNA of relatively low quality. The primers were designed in intronic regions to encompass all the exons in the locus (Fig. 1). Optimisation of primer and PCR conditions were performed to consistently obtain a single band per amplicon of the expected molecular weight. The conditions used in this study allowed amplification of all exonic CtIP regions in seven amplicons using control human genomic DNA as a template. The amplicon sizes ranged from 3.9 kb to 10.1 kb and covered a total of 44.88 kb of the 93.16Kb of the locus. Sequencing results showed that the reads were 79.5% on target, equivalent to an enrichment of >50,000×, demonstrating that the PCR products had a high specificity for the CtIP locus.
Despite our efforts to design primers that would amplify reliably the available samples, 41 out of the 201 patient samples failed to produce one or more of the 7 amplicons and therefore could not be sequenced. The quality check of the failed samples showed that either quantity or integrity of the DNA available was suboptimal (Supplementary Figure S2). For the five failed samples where the DNA did not appear to be degraded or of particularly low concentration, we cannot exclude the possibility that variants at the priming sites could have prevented efficient amplification in some cases.
Dual indexing
One of the major limitations of current HTS technologies lies with the number of samples that can simultaneously be sequenced in parallel. In order to scale up the level of multiplex libraries that could be analysed on a single MiSeq run, we designed a set of oligos that allow the incorporation of two distinct indices on each library. The end product of this tagging process generates fragments that are compatible with dual indexing chemistry on Illumina platforms and therefore does not require software customisation of recipes or pipelines (Supplementary Figure S3). For this study we used the same set of 96 tags previously published [20] which implies that the theoretical potential of libraries that could be pooled together is over 9000 (96×96). This enhanced multiplex capacity allowed 160 samples to be sequenced simultaneously, with a mean and median coverage of 400× and 314× respectively. The depth of coverage for 99.8% of sites was at least 10× and 99.3% of sites were covered at least 30×, but with variation in coverage across samples (median per-sample coverage 120×–804×).
Variant discovery
In 160 patients sequencing was successful, and eleven variants were called within the exons and 15 bp intronic shoulder regions of the CtIP gene, eight of these were known variants from the 1000 Genomes Phase 1 release 3 (Fig. 2 and Table 2). Sixty-eight patients carried at least one of the 11 variants. All other 1000 Genomes known variants in the region had low (1% or less) estimated population allele frequency and were not detected.
Validation of new variants by Sanger sequencing
Three variants identified from our Illumina data which had not been previously reported in the 1000 Genomes Phase 1 release 3, namely, GRCh37.chr18:20548848 C->T, GRCh37.chr18:20573562 G->C, and GRCh37.chr18:20581662 G->A, were confirmed using Sanger sequencing (Fig. 2). We have since found one of the three variants to have been reported in http://evs.gs.washington.edu/EVS/(rs372643826, GRCh37.chr18:20548848 C->T).
Correlation with patient outcome
No significant association was found between carriage of at least one of the 11 variants and disease-specific survival and overall survival (Fig. 3, Table 3 and Supplementary Figure S4), nor was there a significant association between carriage of a ‘rare’ variant, defined as minor allele frequency <0.01 or novel variant, but excluding GRCh37.chr18:20548848 (rs34780140, MAF 0.005) which resulted in the synonymous change Asp548Asp (Supplementary Figure S4 and Supplementary Table S3). When we excluded the two synonymous variants, GRCh37.chr18:20573434 and GRCh37.chr18:20577669, there was no significant association between carriage of at least one of the remaining nine variants and disease-specific and overall survival (data not shown).
Phasing
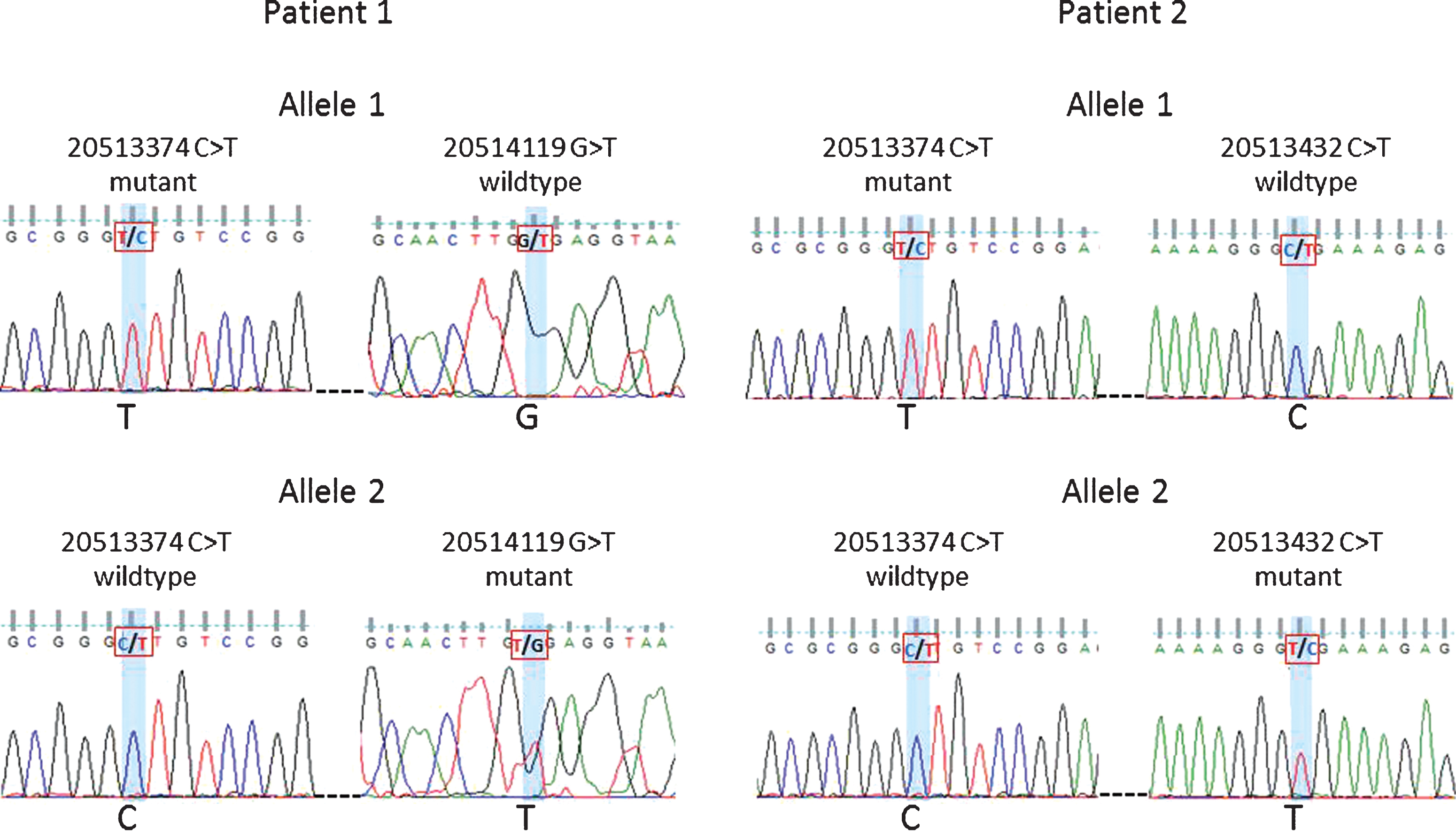
Forty-three patients carried multiple variants. Forty individuals carried two variants, namely, GRCh37.chr18:20513374 (5’UTR, uncertain significance) and GRCh37.chr18:20577669 (synonymous variant). The remaining three patients carried more than two variants, with only one variant (GRCh37.chr18:20577669) being synonymous and the others potentially deleterious (Supplementary Table S4). To establish whether individuals with two non-synonymous variants still carry a functional copy of CtIP we investigated the relationship between some of the variants. For two of the individuals, the variants at positions GRCh37.chr18:20513374 and GRCh37.chr18:20514119 or GRCh37.chr18:20513374 and GRCh37.chr18:20513432 were studied by PCR amplification of the target region and cloning. Sanger sequencing confirmed that the mutations were on separate alleles (Fig. 4). Although some of the variants were too far apart to be tested for phasing, even using novel techniques such as droplet PCR, our findings clearly indicate that the patients studied had two mutant CtIP alleles. With both copies of the gene affected it is possible that these patients have a stronger impairment of gene function.
DISCUSSION
The CtIP gene plays a key role in DSB DNA repair and signalling. Having studied MRE11, which is closely associated with CtIP, and identified novel variants which were associated with bladder cancer survival following radiotherapy, we proceeded to investigate the CtIP gene for genetic variants with the potential to be predictive of patient outcome. Germline CtIP mutations cause Seckel and Jawad syndromes [28], of which one characteristic is impaired ATR-regulated DNA damage signalling and predisposition to cancer. Rare variants might be overlooked as clinically insignificant as they confer smaller population attributable risk due to their low frequency in the population. However, the effects of a group of rare variants, each contributing moderately could increase the risk of disease development or influence response to treatment, as proposed by the ‘common disease- multiple rare variant (CDRV) hypothesis’ [9]. Most rare variants, like ones detected in this study are missense variants, with amino acid changes likely to have a functional impact on protein– protein interactions or levels of transcription. Although we found no evidence of a clinical association for the CtIP variants, our study was underpowered to find these and we had previously found associations for MRE11, so it is possible that associations may be found for rare variants of other genes.
There is increasingly a demand to find efficient ways to validate hypotheses through identification of variants in candidate genes. Methods based on hybridisation often require large amounts of sample DNA and PCR-mediated techniques are, therefore, preferred when samples quantities are limited. Another advantage of PCR-based methods is that each reaction can be quality checked to determine whether to proceed with library preparation and sequencing or not. Here we opted for a long-range PCR strategy to limit the number of reactions and amount of DNA used. The complete coverage of all the exons of CtIP required seven separate reactions. Based on December 2014 pricing, we calculated a total cost (excluding labour) of £18 per sample for the final 160 samples. The single major cost was the construction of Illumina libraries (£4.47) (Supplementary Table S2). However, the ability to sequence 160 samples in parallel in a single run, as opposed to a minimum of two runs if using Illumina consumables, resulted in an approximate saving of £4 per sample. It is self-evident that cost savings for large studies can be achieved simply by pooling more samples into fewer runs.
Given the current sequencing output of Illumina platforms, there is the potential to sequence thousands of samples in parallel. In this study, our enhanced multiplex capacity allowed 160 samples to be sequenced simultaneously, with a mean and median coverage of 400× and 314× respectively, and 99.8% of sites covered at least 10×. This high coverage suggests that more samples could be multiplexed without affecting variant calling, however, variation in coverage across samples (median per-sample coverage 120×–804×) reduces the theoretical level of multiplexing significantly. Using the empirical coverage data we generated, we estimate that we can multiplex 720 samples per MiSeq lane, while still obtaining 10× coverage at 98% of sites. This depth of coverage is widely used and is thought to be sensitive enough for detecting germline variants [29]. An increase from 160 samples per run to 720 samples per run, would increase capacity 4.5 fold.
Here we described the use of custom oligos and indices to generate sequencing libraries that are compatible with Illumina chemistry and de-multiplexing technology. The dual indexing strategy described in this work could support such large numbers. However, there are two major features that still need to be addressed in order to maximise the quantity of samples that can efficiently be sequenced at adequate coverage, namely, the even representation of each amplicon and even representation of each sample. We anticipate that more streamlined methods and instrument that could reliably quantify amplicons and samples prior to pooling will help solve these problems. Similarly, the ability to amplify multiple targets in a single reaction (multiplex PCR) will help reducing the cost associated with the LR-PCR. Our methodology is also good for detecting scattered variants across a region.
In this study of 160 radiotherapy patients’ germline DNA samples, we have demonstrated proof of principle for dual indexing, which allows simultaneous sequencing of candidate genes in hundreds of samples at appropriate depth of coverage, thereby reducing high-throughput sequencing costs substantially thus permitting very large scale studies to be performed.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
ACKNOWLEDGMENTS
This project was funded by the Nuffield Department of Clinical Medicine/University of Oxford Research Fund for Proof of Principle High Throughput Sequencing Experiments, with High-Throughput Sequencing data generated by the High-Throughput Genomics groups at the Oxford Genomics Centre, Wellcome Trust Centre for Human Genetics funded by the Wellcome Trust grant reference 090532/Z/09/Z and MRC Hub grant G0900747 91070. SJ was funded by an MRC studentship awarded to the Department of Oncology, AK was funded by CRUK programme grant C5255/A15935.
REFERENCES
1 | Hanahan D, Weinberg RA(2011) Hallmarks of cancer: The next generationCell144: 5646674 |
2 | Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K(2011) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesisNature434: 864870 |
3 | Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T(2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesionsNature434: 907913 |
4 | Economopoulou P, Dimitriadis G, Psyrri A(2015) Beyond BRCA: New hereditary breast cancer susceptibility genesCancer Treatment Reviews41: 118 |
5 | Choudhury A, Elliott F, Iles MM, Churchman M, Bristow RG, Bishop DT(2008) Analysis of variants in DNA damage signalling genes in bladder cancerBMC Medical Genetics9: 69 |
6 | Stern MC, Lin J, Figueroa JD, Kelsey KT, Kiltie AE, Yuan JM(2009) Polymorphisms in DNA repair genes, smoking, and bladder cancer risk: Findings from the international consortium of bladder cancerCancer Research69: 1768576864 |
7 | Sakano S, Wada T, Matsumoto H, Sugiyama S, Inoue R, Eguchi S(2006) Single nucleotide polymorphisms in DNA repair genes might be prognostic factors in muscle-invasive bladder cancer patients treated with chemoradiotherapyBritish Journal of Cancer95: 5561570 |
8 | Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN(2008) Sequence variant on 8q24 confers susceptibility to urinary bladder cancerNature Genetics40: 1113071312 |
9 | Bodmer W, Bonilla C(2008) Common and rare variants in multifactorial susceptibility to common diseasesNature Genetics40: 6695701 |
10 | Teo MT, Dyrskjot L, Nsengimana J, Buchwald C, Snowden H, Morgan J(2014) Next-generation sequencing identifies germline MRE11A variants as markers of radiotherapy outcomes in muscle-invasive bladder cancer Annals of Oncology: Official Journal of the European Society for Medical Oncology/ESMO25: 4877883 |
11 | Yu X, Chen J(2004) DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domainsMolecular and Cellular Biology24: 2194789486 |
12 | You Z, Shi LZ, Zhu Q, Wu P, Zhang YW, Basilio A(2009) CtIP links DNA double-strand break sensing to resectionMolecular Cell36: 6954969 |
13 | Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J(2009) Human CtIP promotes DNA end resectionNature450: 509514 |
14 | Yun MH, Hiom K(2007) CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycleNature459: 460463 |
15 | Lee K, Lee SE(2007) Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joiningGenetics176: 420032014 |
16 | Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN(2013) The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repairPLoS Genetics9: 2e1003277 |
17 | Bentley J, L’Hote C, Platt F, Hurst CD, Lowery J, Taylor C(2009) Papillary and muscle invasive bladder tumors with distinct genomic stability profiles have different DNA repair fidelity and KU DNA-binding activitiesGenes, Chromosomes & Cancer48: 4310321 |
18 | Kozarewa I, Rosa-Rosa JM, Wardell CP, Walker BA, Fenwick K, Assiotis I(2012) A modified method for whole exome resequencing from minimal amounts of starting DNAPloS One7: 3e32617 |
19 | Rohland N, Reich D(2012) Cost-effective, high-throughput DNA sequencing libraries for multiplexed target captureGenome Research22: 5939946 |
20 | Lamble S, Batty E, Attar M, Buck D, Bowden R, Lunter G(2013) Improved workflows for high throughput library preparation using the transposome-based Nextera systemBMC Biotechnol13: 104 |
21 | Kotwal S, Choudhury A, Johnston C, Paul AB, Whelan P, Kiltie AE(2008) Similar treatment outcomes for radical cystectomy and radical radiotherapy in invasive bladder cancer treated at a United Kingdom specialist treatment centerInternational Journal of Radiation Oncology, Biology, Physics70: 2456463 |
22 | Teo MT, Landi D, Taylor CF, Elliott F, Vaslin L, Cox DG(2012) The role of microRNA-binding site polymorphisms in DNA repair genes as risk factors for bladder cancer and breast cancer and their impact on radiotherapy outcomesCarcinogenesis33: 3581586 |
23 | Lunter G, Goodson M(2011) Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence readsGenome Research21: 6936939 |
24 | Rimmer A, Phan A, Mathieson I, Iqbal Z, Twigg SRWGS500 Consortium(2014) Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applicationsNature Genetics46: 8912920 |
25 | Ng PC, Henikoff S(2001) Predicting deleterious amino acid substitutionsGenome Research11: 5863874 |
26 | Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P(2010) A method and server for predicting damaging missense mutationsNature Methods7: 4248249 |
27 | R Core Team: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013; URL http://www.R-project.org/ |
28 | Qvist P, Huertas P, Jimeno S, Nyegaard M, Hassan MJ, Jackson SP(2011) CtIP mutations cause seckel and jawad syndromesPLoS Genetics7: 10e1002310 |
29 | Philips AK, Siren A, Avela K, Somer M, Peippo M, Ahvenainen M(2014) X-exome sequencing in Finnish families with intellectual disability– four novel mutations and two novel syndromic phenotypesOrphanet Journal of Rare Diseases9: 49 |
30 | Dubin MJ, Stokes PH, Sum EY, Williams RS, Valova VA, Robinson PJ(2004) Dimerization of CtIP, a BRCA1- and CtBP-interacting protein, is mediated by an N-terminal coiled-coil motifThe Journal of Biological Chemistry279: 262693226938 |
31 | Yuan J, Chen J(2009) N terminus of CtIP is critical for homologous recombination-mediated double-strand break repairThe Journal of Biological Chemistry284: 463174631752 |
32 | Li S, Ting NS, Zheng L, Chen PL, Ziv Y, Shiloh Y(2009) Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage responseNature406: 210215 |
Figures and Tables
Fig.1
Amplicon position and coverage. The relative position of the 18 exons of CtIP is shown at the top. Each exon is represented by a vertical bar. Grey boxes highlight the region encompassed by each of the 7 amplicons used in this study. The representative read depth for each amplicon is shown at the bottom as a density plot. No reads were identified outside the amplified regions.
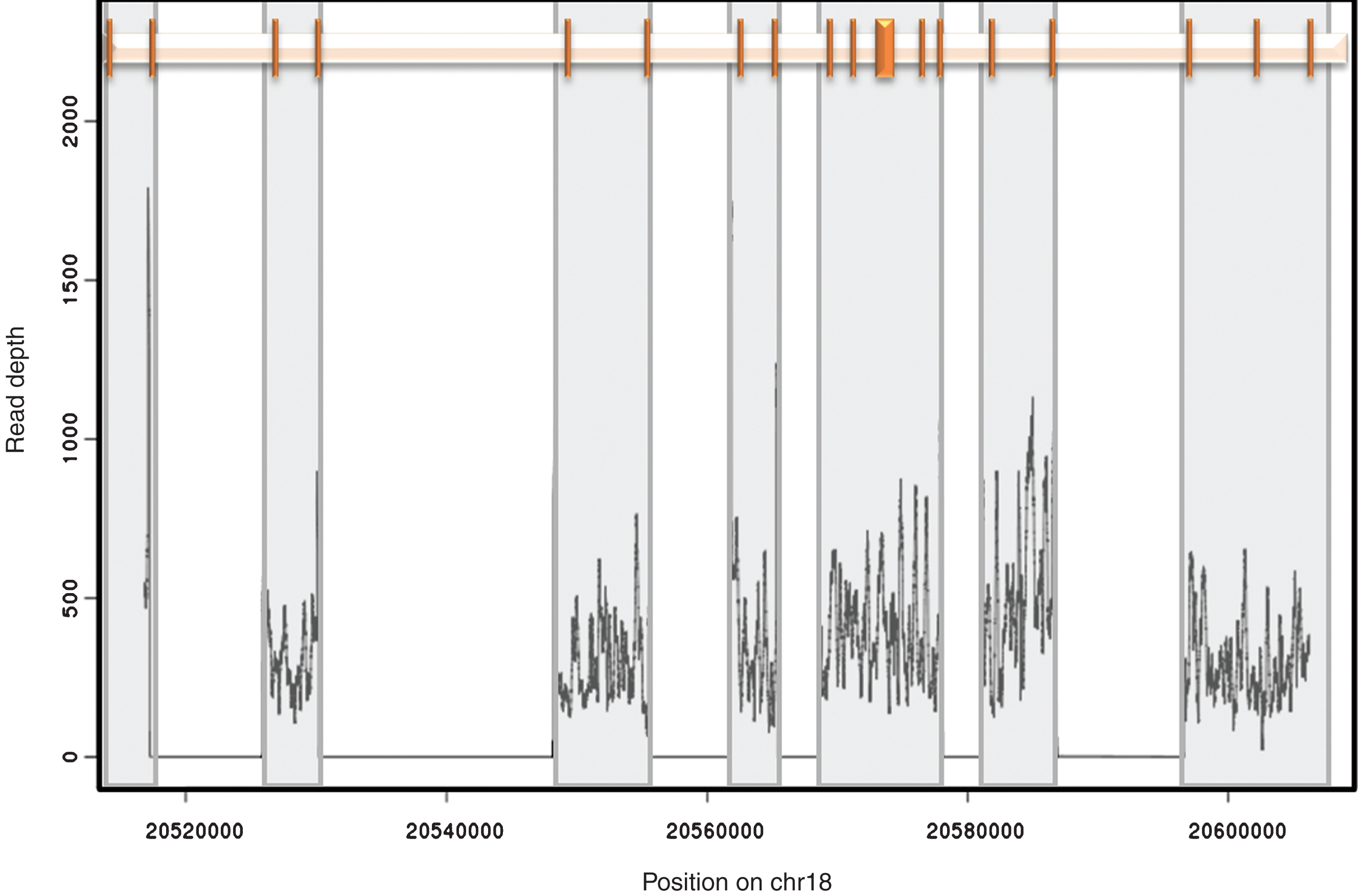
Fig.2
Top panel: Protein domains of human CtIP. The N-terminus of human CtIP contains the dimerization domain (amino acids 40–165) [30]. In vitro CtIP shows DNA-binding activity, which could play a role in recruitment to DSBs (amino acids 509–557) [12]. Interaction with the MRN complex has been shown to occur at both the N-terminal region (amino acids 22–45) and the C- terminal region (amino acids 650–897) of CtIP [13, 31]. Signalling via two ATM phosphorylation sites (S664 and S745) are conserved in vertebrates [32]. The location of non-synonymous variants are shown. Known variants 1–4, 6 and 8 are labelled at the top whereas novel variants 5, 9, 11 are below. Synonymous variants 7 and 10 are not shown. See Table 2 for variant identities. Bottom Panel: Sanger sequencing traces for validation of three novel variants; 5, 9 and 11.
![Top panel: Protein domains of human CtIP. The N-terminus of human CtIP contains the dimerization domain (amino acids 40–165) [30]. In vitro CtIP shows DNA-binding activity, which could play a role in recruitment to DSBs (amino acids 509–557) [12]. Interaction with the MRN complex has been shown to occur at both the N-terminal region (amino acids 22–45) and the C- terminal region (amino acids 650–897) of CtIP [13, 31]. Signalling via two ATM phosphorylation sites (S664 and S745) are conserved in vertebrates [32]. The location of non-synonymous variants are shown. Known variants 1–4, 6 and 8 are labelled at the top whereas novel variants 5, 9, 11 are below. Synonymous variants 7 and 10 are not shown. See Table 2 for variant identities. Bottom Panel: Sanger sequencing traces for validation of three novel variants; 5, 9 and 11.](https://content.iospress.com:443/media/blc/2015/1-1/blc-1-1-blc150007/blc-1-1-blc150007-g002.jpg)
Fig.3
Kaplan Meier curves of cancer-specific survival for patients with at least one CtIP variant. A) Of 160 patients, 68 patients carried at least one of 11 detected variants in the CtIP gene within the exons and 15 bp intronic shoulder regions. Curve ‘0’ represents patients wild-type for all 11 variants, and Curve ‘at least 1’ represents all patients with carriage of at least one variant allele. B) Fifteen patients carried one ‘rare variant’, defined as minor allele frequency <0.01 or novel variant, but excluding rs34780140 (MAF 0.005) which resulted in the synonymous change Asp548Asp.
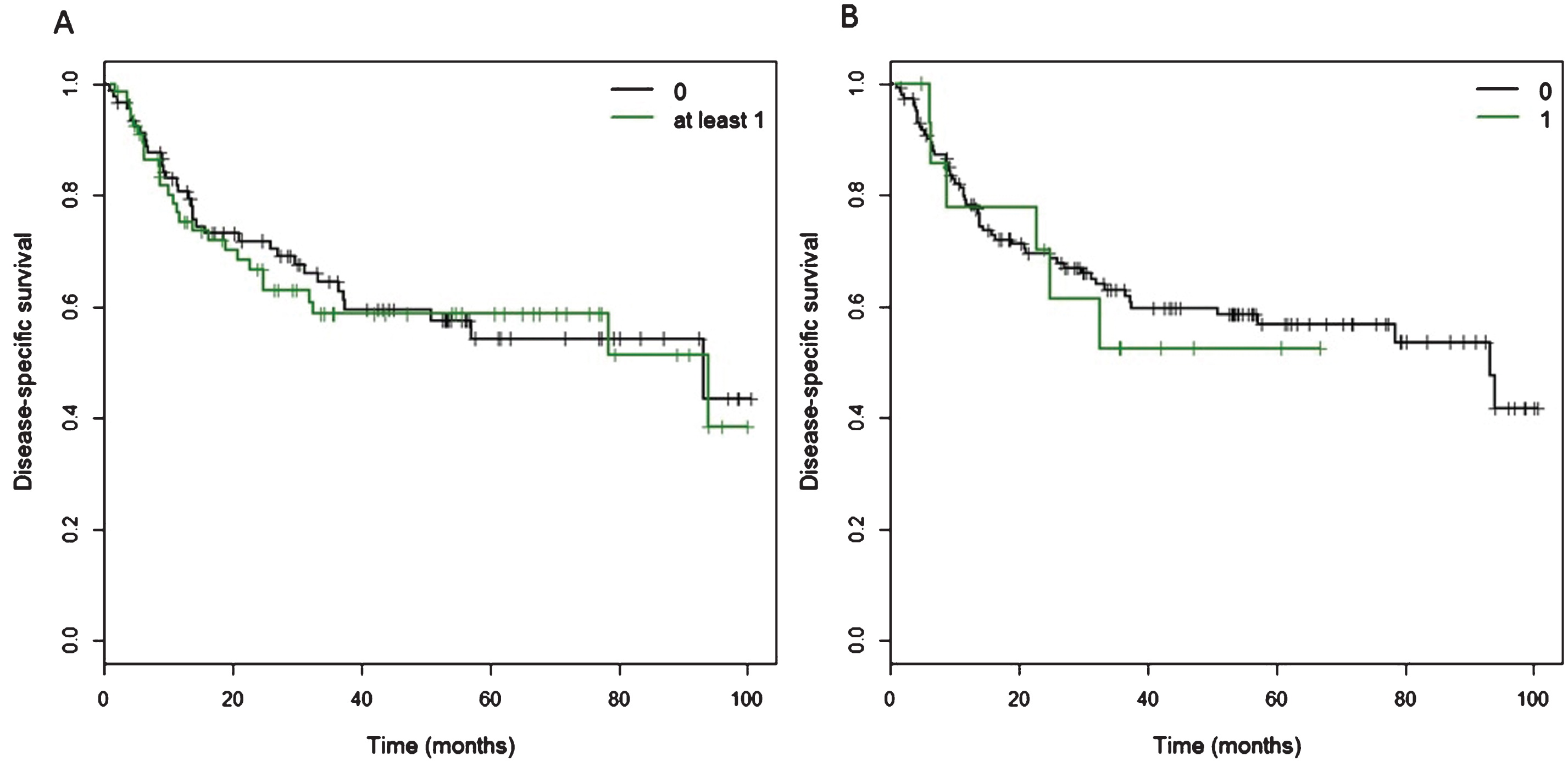
Fig.4
Phasing of variants 20513374 and 20514119 in Patient 1 and variants 20513374 and 20513432 in Patient 2. Sanger sequencing traces shown for patient 1: allele 1 mutant 20513374 and wildtype 20514119 vs. allele 2 wildtype 20513374 and mutant 20514119. Patient 2: allele 1 mutant 20513374 and wildtype 20513432 vs. allele 2 wildtype 20513374 and mutant 20513432.
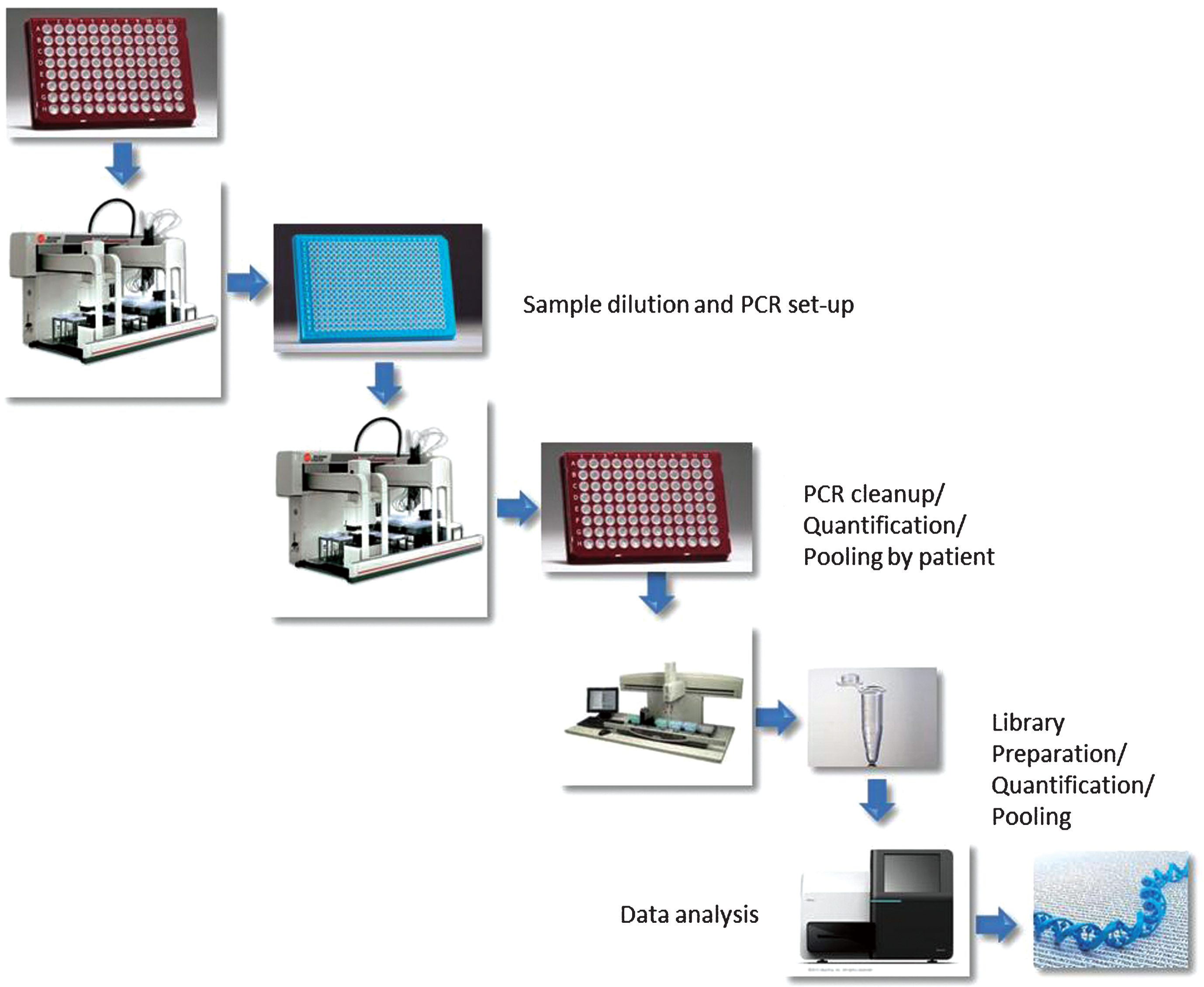
SupplementaryFigure S1
Schematic representation of the workflow followed for this study. Briefly, liquid handling platforms were programmed to perform sample transfers, PCR reaction set up, clean up and pooling, library construction. The final pooled library was sequenced on a single MiSeq run.
SupplementaryFigure S2
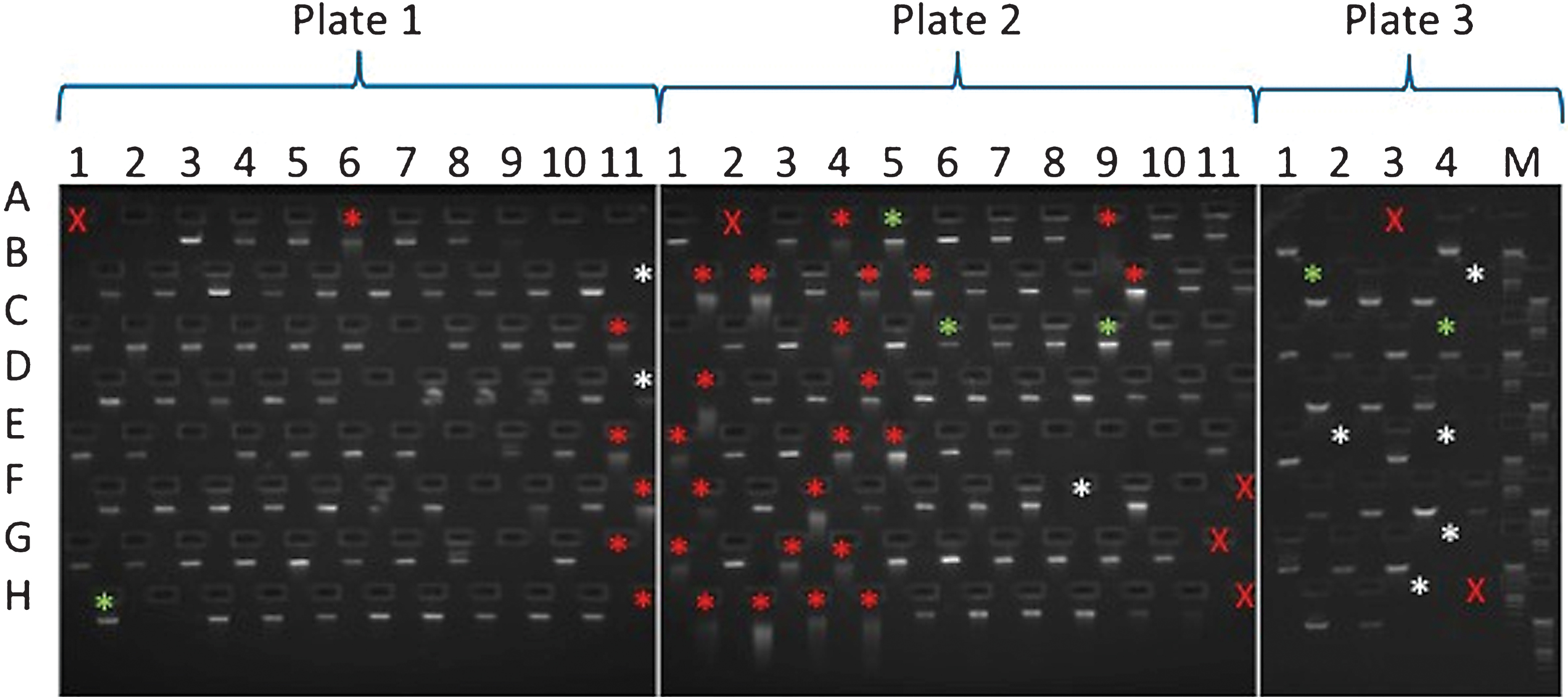
E-gel run of sample DNAs. A red X is used where a sample was not loaded into that plate position. Asterisks mark samples which failed to produce one or more amplicons. Asterisk colours denote the following features: green, no obvious issues with sample; white, sample of insufficient amount; red, degraded sample. Samples are arranged in the following locations on 96 well plates: columns 1–11 and rows A–H. All the DNAs used for this study were arranged across 3 plates (plate 1–3). For some of the samples that did not show a band on the gel, additional material was used for the amplicon generation. However, in many cases, apparently undetectable DNA successfully produced amplicons.
SupplementaryFigure S3
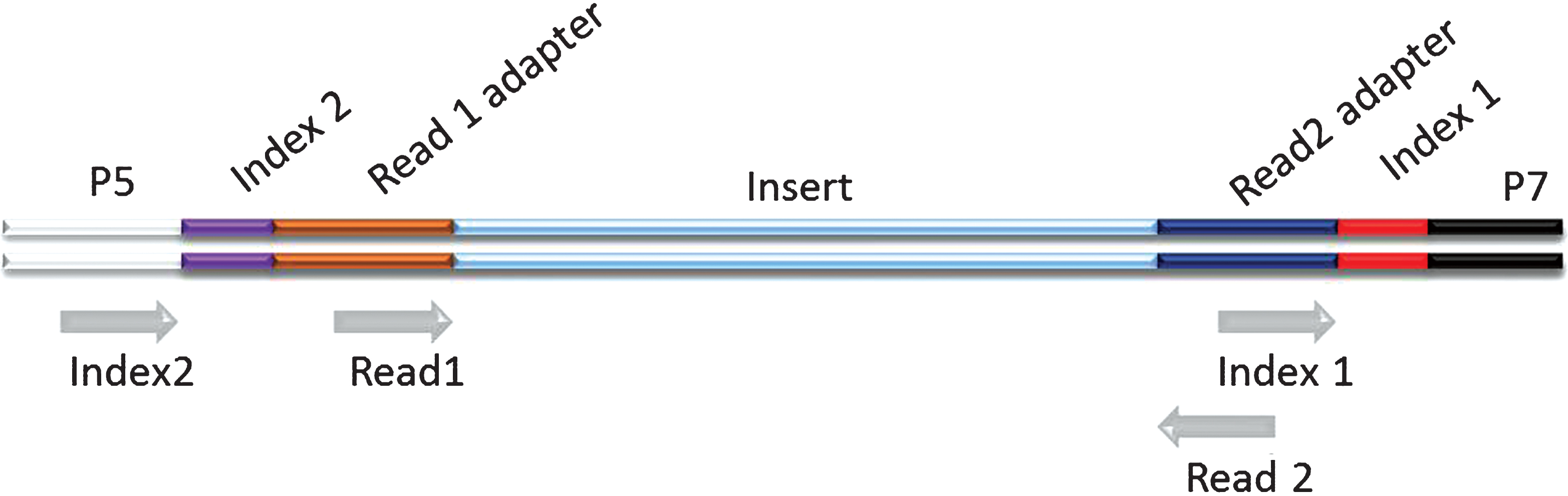
Schematic representation of a dual indexed Illumina library. The different components of the adapters are shown flanking an insert sequence. Grey arrows indicate priming sites for the generation of the different sequence reads on Illumina platforms. The order by which the reads are generated is: Read 1, Index 1, Index 2, Read 2. (Colours are visible in the online version of the article; http://dx.doi.org/10.3233/BLC-150007).
SupplementaryFigure S4
Kaplan-Meier curves for overall survival in patients with one or more CtIP variant. A) Of 160 patients, 68 patients carried at least one of 11 detected variants in the CtIP gene within the exons and 15 bp intronic shoulder regions. Curve ‘0’ represents patients wild-type for all 11 variants, and Curve ‘at least 1’ represents all patients with carriage of at least one variant allele. B) Fifteen patients carried one ‘rare variant’, defined as minor allele frequency <0.01 or novel variant, but excluding rs34780140 (MAF 0.005) which resulted in the synonymous change Asp548Asp.
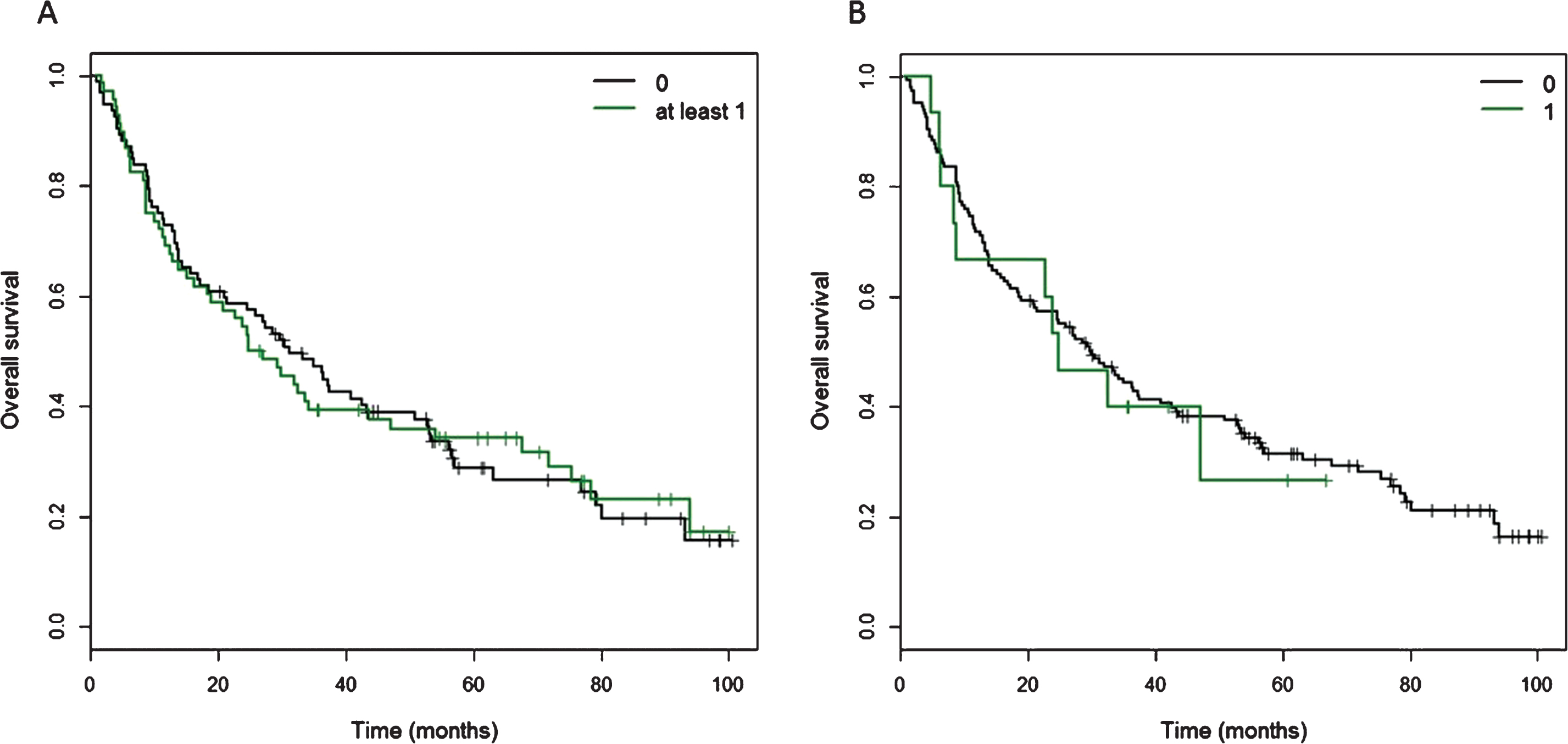
Table 1
Clinical demographics of the patients with successful sequencing
| N | 160 |
| Median age at treatment (years) | 78.8 |
| Age at treatment (years) - range | 55.70–92.50 |
| Median survival time (months) | 28.7 |
| Survival time (months) - range | 0.89–100.50 |
| Mortality | |
| Overall | 116 (72.5% ) |
| Disease-specific | 62 (38.8% ) |
| Sex | |
| Male | 120 (75.0% ) |
| Female | 40 (25.0% ) |
| Tumour stage | |
| a | 2 (1.3% ) |
| 1 | 5 (3.1% ) |
| 2 | 85 (53.1% ) |
| 3 | 58 (36.2% ) |
| 4 | 10 (6.3% ) |
| Nodal stage | |
| N0 | 156 (97.5% ) |
| N1 | 1 (0.6% ) |
| N2 | 2 (1.3% ) |
| Nx | 1 (0.6% ) |
| Histological grade | |
| 1 | 0 (0% ) |
| 2 | 13 (8.2% ) |
| 3 | 144 (91.1% ) |
| Missing | 1 (0.6% ) |
| Hydronephrosis | |
| No | 116 (72.5% ) |
| Yes | 22 (27.5% ) |
| Median radiotherapy dose (Gy) | 55 |
| Radiotherapy dose (Gy) range: | 50–60 |
| Neoadjuvant chemotherapy | |
| Not received | 152 (95.0% ) |
| Received | 8 (5.0% ) |
| Concurrent chemotherapy | |
| Not received | 153 (95.6% ) |
| Received | 7 (4.4% ) |
Table 2
Summary of CtIP variant discovery, location and frequency, with SIFT and PolyPhen-2 prediction for disruption of protein function
| No | rs identifier | Position |
Chromosome location | Variant |
MAF frequency |
Number of patients |
SIFT (Score 0-1) |
PolyPhen-2 (Score 0-1) |
| 1 | rs7227168 | 5’UTR | 18:20513374 | C->T | 0.1422 | 47 | ||
| 2 | rs116097101 | 5’UTR | 18:20513432 | C->T | 0.0589 | 2 | ||
| 3 | rs140562665 | 5’UTR | 18:20514119 | G->T | 0.0006 | 1 | ||
| 4 | rs181038099 | 5’UTR | 18:20516758 | T->C | 0.0016 | 2 | ||
| 5 | rs372643826 | Exon 4 | 18:20548848 | C->T | (novel)*NA | 1 | damaging (0.00) | possibly damaging (0.646) |
| 6 | rs149842490 | Exon 4 | 18:20548849 | G->A | 0.0002 | 2 | damaging (0.03) | probably damaging: (0.978) |
| 7 | rs34780140 | Exon 10 | 18:20573434 | T->C | 0.005 | 8 | tolerated (1.00) | |
| 8 | rs111445733 | Exon 10 | 18:20573556 | G->A | 0.0026 | 7 | tolerated (0.14) | benign (0.000) |
| 9 | n/a | Exon 10 | 18:20573562 | G->C | (novel) | 1 | damaging [low confidence] (0.05) | benign (0.152) |
| 10 | rs17852769 | Exon 13 | 18:20577669 | G->A | 0.1687 | 43 | tolerated (1.00) | |
| 11 | n/a | Exon 14 | 18:20581662 | G->A | (novel) | 1 | tolerated (0.11) | benign (0.376) |
The 11 CtIP variants identified are summarised showing their reference SNP (rs) identifiers, exon and chromosome positions, and base substitution according to dbSNP. The minor allele frequency (MAF) was obtained from 1000 Genomes data (as reported in dbSNP), and the number of patients in this study carrying the variant is shown. SIFT and PolyPhen-2 predictions for disruption of protein function are presented. *note that rs372643826 was not present in the 1000 Genomes database but was reported in http://evs.gs.washington.edu/EVS/ and no MAF data were available in dbSNP (NA). The SIFT score (range 0-1, low value means more likely to be damaging) and PolyPhen score (range 0-1, high value means more likely to be damaging). SIFT scores indicate that three of the variants (18:20548848, 18:20548849 and 18:20573562) we identified have a potential impact on protein function.
Table 3
Cox proportional hazards model for disease-specific survival.histological variables
| Covariate | HR | 95% CI | p-value |
| Carriage of at least one common variant | 0.97 | (0.55, 1.70) | 0.92 |
| Carriage of one rare variant | 1.21 | (0.51, 2.90) | 0.66 |
| Sex | 0.79 | (0.40, 1.55) | 0.49 |
| Age at treatment | 1.01 | (0.97, 1.04) | 0.68 |
| Concurrent chemotherapy | 0.24 | (0.03, 1.79) | 0.16 |
| Neoadjuvant chemotherapy | 0.32 | (0.04, 2.40) | 0.27 |
| Histology | 0.99 | (0.75, 1.31) | 0.95 |
| Grade | 1.19 | (0.47, 3.01) | 0.71 |
| Hydronephrosis | 1.62 | (0.88, 3.01) | 0.12 |
| Previous superficial bladder cancer | 1.34 | (0.72, 2.48) | 0.36 |
| Radiotherapy dose | 0.88 | (0.74, 1.06) | 0.18 |
HR: hazard ratio, 95% CI: 95% confidence interval.
Supplementary Table S1
Seven CtIP long range PCR primer pair sequences and corresponding amplicon size and exon coverage
| Amplicon | Amplicon Size (bp) | Exons | Forward Primer Sequence | Reverse Primer Sequence |
| 1 | 4215 | 5’UTR, 1 | GCTTAAGCTAGACACAGTGTACAG | GGACACAAAGAGGGGAACAA |
| 2 | 4306 | 2, 3 | CCATAGGCAGTGGTCTTCTCTT | CAAATGCTGGAAGCCTTTTC |
| 3 | 7437 | 4, 5 | GAGACGGTGTTTACTGTATTGGC | ACACAAGAGGAAGTGGCACA |
| 4 | 3761 | 6, 7 | ATTTCCTTGCCTCCTGTCCT | CGGGCTTGCTAATTTCTCTG |
| 5 | 9375 | 8–13 | CTGCTTAGCACAATACCTGGAA | CTCAACTCTGCAGCTGTATGGA |
| 6 | 5755 | 14, 15 | CCTGCAGCCCTCATCATAA | ACTCTGACAAAGACAGCTCGAC |
| 7 | 10030 | 16–18, 3’UTR | GAATTTGCAGGTCACTTTGGA | GACACTGATATGACCTAGAGAAGAG |
Supplementary Table S2
Costs involved in processing samples
| Number of samples | Cost (GBP) | Cost per sample (GBP) |
| 205 samples for amplicon generation: | ||
| Primers | 14.56 | 0.07 |
| Polymerase | 509.43 | 2.49 |
| Ampure XP | 240.00 | 1.17 |
| Amplicon QC | 428.80 | 2.09 |
| Labware (tips, plates, tubes) | 174.82 | 0.85 |
| 300 cycles of reagents for MiSeq | 638.35 | |
| 160 samples for Library prep: | ||
| Library prep | 714.67 | 4.47 |
| Library QC | 44.98 | 0.28 |
| Labware (tips, plates, tubes) | 120.93 | 0.59 |
| Total cost | 2886.54 | ÷ 160 = 18.04 per sample |
Supplementary Table S3
Results from univariate Cox proportional hazards model for each variant. Univariate models were fitted on each of the variants for which there was at least one event
| Variant | HR | 95% CI | p-value |
| 20513374 | 0.94 | (0.54, 1.64) | 0.84 |
| 20548848 | 2.81 | (0.39, 20.40) | 0.31 |
| 20573562 | 2.19 | (0.30, 15.86) | 0.44 |
| 20573556 | 1.82 | (0.66, 5.03) | 0.25 |
| 20577669 | 0.93 | (0.52, 1.64) | 0.80 |
| 20573434 | 1.03 | (0.32, 3.30) | 0.96 |
Supplementary Table S4
Coordinates for the variants in three patients carrying three or more variants
| Variant 1 | Variant 2 | Variant 3 | Variant 4 | |
| Patient 1 | 18:20577669 (synonymous) | 18:20513374 (5’UTR) | 18:20513432 (5’UTR) | 18:20581622 (exon 14) |
| Patient 2 | 18:20577669 (synonymous) | 18:20513374 (5’UTR) | 18:20514119 (5’UTR) | |
| Patient 3 | 18:20577669 (synonymous) | 18:20513374 (5’UTR) | 18:20548849 (exon 4) |


