
The early development and application of FTIR difference spectroscopy to membrane proteins: A personal perspective
Abstract
Membrane proteins facilitate some of the most important cellular processes including energy conversion, ion transport and signal transduction. While conventional infrared absorption provides information about membrane protein secondary structure, a major challenge is to develop a dynamic picture of the functioning of membrane proteins at the molecular level. The introduction of FTIR difference spectroscopy around 1980 to study structural changes in membrane proteins along with a number of associated techniques including protein isotope labeling, site-directed mutagenesis, polarization dichroism, attenuated total reflection and time-resolved spectroscopy have led to significant progress towards this goal. It is now possible to routinely detect conformational changes of individual amino acid residues, backbone peptides, binding ligands, chromophores and even internal water molecules under physiological conditions with time-resolution down to nanoseconds. The advent of ultrafast pulsed-IR lasers has pushed this time-resolution down to femtoseconds. The early development of FTIR difference spectroscopy as applied to membrane proteins with special focus on bacteriorhodopsin is reviewed from a personal perspective.
1.Introduction
The purpose of this review is to present a personal perspective of the early development of the field of Fourier transform infrared (FTIR) difference spectroscopy and research that led up to and followed its first application to membrane proteins. The review mainly covers research done during the period 1972–1995 at MIT and at the Molecular Biophysics Laboratory at Boston University with specific focus on bacteriorhodopsin, a light-driven proton pump. For a broader and more comprehensive discussion of the application of FTIR to biomolecules, the reader is referred to several excelled reviews including [23,45,84,109,237].
1.1.Early application of infrared spectroscopy to proteins
Infrared (IR) spectroscopy has a long history that traces back to the discovery of solar IR radiation by the astronomer Sir William Herschel in 1800 [115]. In 1905 William Coblentz, who worked as a researcher at Cornell University and later at the National Bureau of Standards (now NIST), reported the IR spectra of various chemical compounds [58,223]. This led to the realization that different types of molecular groups displayed characteristic bands in the IR absorbance spectrum which can serve as identifying “fingerprints”. However, it wasn’t until after WWII and the development of the double beam dispersive IR spectrometer that IR spectroscopy became a common analytical tool for the chemist [66,245].
Eliot and Ambrose in the 1950s were among the earliest researchers to use IR spectroscopy to investigate the structure of proteins [79,80]. Subsequent studies of model compounds such as N-methylacetamide by Miyazawa provided the foundation for future IR studies of proteins [27,167]. In addition, early characterization of amino acids and peptides using Raman spectroscopy by John Edsall at Harvard [78] and polyatomic molecules and proteins by Richard Lord at MIT [145,146] helped establish vibrational spectroscopy as an important tool for studying biomolecules (see also [179]).
The introduction of commercial Fourier transform infrared (FTIR) instruments in the mid-1970s also served as a major impetus for spectroscopic studies of biomolecules (see for example references [103,224]). The design of the FTIR instrument is based on the classic Michelson interferometer. Several advantages ensue from the use of an interferometer compared to dispersive instruments [45] including: (i) the Felligett’s multiplex advantage which relates to the ability to measure all wavelengths of light simultaneously resulting in higher signal-noise; (ii) the Jacquinot’s throughput advantage, which eliminates entrance and exist slits [103]; and (iii) Cone’s frequency advantage which stems from the more accurate wavelength calibration that accrues from using a He–Ne laser. However, practical use of FTIR was not possible until the advent of inexpensive compact computers that could perform fast Fourier transforms such as the Cooley and Tukey algorithm developed at Bell Labs [224].
Early infrared studies of the secondary structure of proteins was based on the analysis of the amide I and II bands which appear in the 1500–1600 cm−1 and 1600–1700 cm−1 regions, respectively. These bands arise from the C=O stretching (amide I) and NH bending (amide II) modes of polypeptides. Methods developed to resolve the subcomponents of these bands including Fourier self-deconvolution (FSD) and second-derivative analysis along with improvements in normal mode calculations contributed significantly to using IR to analyze quantitatively the secondary structure composition of proteins [53,74,131,166,233,234]. Although most studies were performed on proteins in D2O in order to avoid the strong overlapping absorption of water, subtraction methods also were developed which enabled measurements in bulk H2O solution [70].
1.2.Membrane proteins and the need to probe at the level of single chemical groups
The Davson–Danielli model of the cellular plasma membrane introduced in the 1930s held sway for almost 40 years [186]. It envisioned proteins as forming extended fixed β-sheets bound to the membrane surface and later in a modification of the model formed also pores for ions to cross the hydrophobic lipid bilayer [186].
In contrast, in 1972 Singer and Nicolson proposed that membrane proteins existed in a globular state similar to soluble proteins but were able due to their hydrophobic core region that contacted the acyl chains of the lipid bilayer to span the membrane [222]. Further, it was postulated that these membrane proteins constituted the basic units of energy conversion, ion transport and signal transduction critical to all cells.
Fig. 1.
Early “permion” model proposed by Rothschild and Stanley in 1972 to account for membrane protein ion transport and selectivity. Small conformational changes involving a few charged residues were postulated to provide a basis for control of ion permeation and selectivity (adapted from [211]).
![Early “permion” model proposed by Rothschild and Stanley in 1972 to account for membrane protein ion transport and selectivity. Small conformational changes involving a few charged residues were postulated to provide a basis for control of ion permeation and selectivity (adapted from [211]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g001.jpg)
Expanding on this picture I proposed in 1972 along with Gene Stanley (my PhD thesis advisor at MIT) the “permion” model (see Fig. 1) [211]. This model postulated that ion transport proteins contained a water-filled channel and a barrier consisting of charged residues which acted to control selectivity and modulated ion transport through small conformational changes. This model, for which many elements have subsequently been found to be true for ion transport proteins, served as an important impetus in my subsequent search for experimental techniques to probe membrane protein structure and conformational changes. Some of this work is described below.
2.Initial studies of model ion carriers and channels
In addition to my PhD advisor Gene Stanley, a leader in the field of statistical physics, I was fortunate to interact with other scientists at MIT including Richard Lord, one of the pioneers of vibrational spectroscopy of biomolecules and Alex Rich, a leader in the early development of molecular biology. My interest in ion transport was also greatly stimulated by my interactions with Alvin Essig and his group, in the Department of Physiology at the Boston University School of Medicine. This collaboration, which included Sam Elias, who was also a PhD student at MIT, subsequently led to a theoretical analysis of the non-equilibrium behavior of biological systems and prediction of a multidimensional inflection point in enzyme-mediated reactions [203].
My initial foray into the world of vibrational spectroscopy and its application to the membrane proteins was a Raman spectroscopic study of valinomycin which constituted part of my PhD thesis. These studies were facilitated by Evangelos Anastasskakis, a Raman spectroscopist then in the Physics Department at Northeastern University. Valinomycin is a natural antibiotic which exhibits the ability to selectively transport K+ across lipid bilayers with 10,000 times higher affinity for K+ compared to Na+. It was known that valinomycin acted as a cation carrier by surrounding ions inside a hydrophobic cage which undergoes a conformational change at the bilayer surface, thereby acting to release the cation. However, the details of this conformational change and the basis of cation selectivity was unknown.
In a series of experiments that also involved Irv Asher, then a postdoctoral research at MIT, the amide I and ester carbonyl frequencies of uncomplexed and cation complexed VM under different conditions was studied [8,9,195,196,212]. We deduced from these measurements that there existed two conformations, closed and open, of VM depending on whether the molecule was in a hydrophobic or hydrophilic environments, helping to explain the mechanism of cation release when VM was located at the membrane surface. We also studied the interaction of the ester carbonyl groups with K+ offering insight into the nature of the cation selectivity [8].
Soon after, I began Raman studies of the antibiotic gramicidin A, which was considered a prototype for membrane proteins cation channels [212]. Dan Urry’s group at University of Minnesota Twin Cities had predicted that gramicidin A could form a cation channel that had helical structure which was fundamentally different than α-helices and displayed intra- and intermolecular beta-sheet type hydrogen bond [241]. Our Raman studies confirmed the existence of β-type hydrogen bonding when gramicidin A was solubilized in a hydrophobic solvent similar to the hydrophobic interior of lipid bilayers. Many subsequent vibrational studies have been made on both VM and Gramicin A [60,169,244,248].
3.Initial studies on animal rhodopsin
As a post-doctoral Fellow in the Harvard-MIT Program in Health Sciences and Technology from 1975–1976, I began a life-long collaboration with Wim DeGrip from the University of Nijmegen in Holland. These studies were aimed at studying the structure and conformational changes of animal rhodopsins. At the time Wim was a visiting scholar in the Harvard University laboratory of George Wald, a world leader in the rhodopsin field. Together under the tutelage of Paul Brown, a distinguished member of the Wald group [50,51], we removed enzymatically the retinal chromophore from bovine rhodopsin (known as opsin) by bleaching in the presence of reduced nicotinamide adenine dinucleotide phosphate which activates the photoreceptor retinal oxidoreductase. The complete removal of the retinylidene chromophore, allowed us to measure the first non-resonance Raman spectrum of a membrane protein in its native membrane [193] (Fig. 2). Normally, the presence of retinal produces resonance enhanced of the vibrations of this chromophore thereby precluding the detection of protein vibrations [137,138,177,212]. The non-resonance Raman spectra revealed that the frequency of the amide I and III bands of opsin were characteristic of a predominantly α-helical structure [193]. This conclusion was somewhat surprising at the time since earlier models of opsin postulated that opsin had a structure similar to gramicin A [181].
![Raman spectrum of calf opsin photoreceptor membrane (adapted from Ref. [193]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g002.jpg)
4.Probing membrane protein orientation
4.1.The discovery of oriented transmembrane alpha-helices
In addition to the Raman studies on rhodopsin described above, Richard Henderson and Nigel Unwin at the Molecular Research Council at Cambridge England reported the low-resolution 3D structure of bacteriorhodopsin from 2D crystalline native purple membrane (PM) based on electron diffraction [114]. This structure revealed 7 tubes of electron density running transversely through the protein (e.g. perpendicular to the membrane plane) and suggested that each tube consist of α-helical segments. An important question was whether most membrane proteins consisted of such transversely oriented α-helices.
Fig. 3.
Polarized IR of purple membrane. Dried purple membrane films deposited on Irtran-4 were tilted relative to the IR beam direction at 0, 15, 30, 45 and 60° as shown in spectra 2A–E, respectively. (Adopted from [201].)
![Polarized IR of purple membrane. Dried purple membrane films deposited on Irtran-4 were tilted relative to the IR beam direction at 0, 15, 30, 45 and 60° as shown in spectra 2A–E, respectively. (Adopted from [201].)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g003.jpg)
In collaboration with Noel Clark, who at the time was a faculty member at Harvard University, we developed a method that utilized polarized infrared dichroism to probe the orientation of α-helical structure in membrane proteins. As seen in Fig. 3, polarized FTIR measurements of dried films of PM deposited on the surface of IR transmitting windows revealed significant dichroism of the amide I, II bands which arise from the alignment of the amide carbonyl (C=O) and amide NH groups in α-helices which are in turn have a net orientation in the already oriented PM sheets. Dichroism measurements allowed us to calculate the average orientation of α-helices relative to the membrane plane. The result agreed well with the presence of an oriented bundle of α-helices consistent with the Unwin–Henderson electron diffraction derived 3D picture [201].
Due to the flat geometry of 2D crystalline patches of PM approximately 1/2 micron in diameter, BR is particularly amenable to produce highly oriented multilamellar films by drying. However, most membranes proteins do not produce such patches and are more difficult to incorporate into well order multilamellar films. For this purpose, we developed a method termed isopotential spin-drying (ISD) [57,201]. As shown in Fig. 4, ISD utilizes simultaneous centrifugation and drying on an isopotential surface to produce highly oriented layers of membrane sheets on a flat substrate [57]. Studies on PM using linear visible dichroism and freeze fracture electron microscopy revealed that the mosaic spread obtained by ISD approached the maximum orientation calculated theoretically on the basis of considering membrane fragment steric interactions, finite size, elasticity and thermal fluctuations [57].
Fig. 4.
Comparison of different methods of orienting membrane sheets including: (a) drying without centrifugation, (b) isopotential spin-drying (ISD) where both drying and centrifugation occur and (c) example of ISD where biological membrane fragments are less dense than the suspending medium (adapted from [57]).
![Comparison of different methods of orienting membrane sheets including: (a) drying without centrifugation, (b) isopotential spin-drying (ISD) where both drying and centrifugation occur and (c) example of ISD where biological membrane fragments are less dense than the suspending medium (adapted from [57]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g004.jpg)
The first polarized FTIR measurements using ISD were performed on native rhodopsin photoreceptor membrane. These measurements indicated that rhodopsin also had the same overall TM orientation of α-helices [202,209].
Since this original work on BR and rhodopsin membranes, FTIR dichroism has been used extensively to study the orientation and structure of several membrane proteins and other biomembrane systems including Gramicidin A [169], cytochrome oxidase [25], phospholipids [46], antibiotic peptides [163], fusagenic peptides [100,160], C-terminus of A1 domain of shinga toxin [165], and H+, K+-ATPase [185]. In our laboratory, John Hunt from the Don Engelman’s group at Yale University (and now at Columbia University) analyzed the insertion and orientation of individually chemically synthesized polypeptides corresponding to the 7-TM segments in BR in dimyristoyl phosphatidylcholine (DMPC) bilayers membranes [120]. The discovery of a spontaneous pH-dependent membrane insertion of a trans-bilayer α-helix [121] has led to the development of pH (low) insertion peptide (pHLIP) technology as a tool for translocating cargo molecules attached to the plasma membrane [238]. The membrane bound state of the 178-residue channel forming peptide colicin E1 in DMPC liposomes was also studied using this approach in collaboration with William Cramer at Purdue University [182]. In another study with the Englelman group using polarized attenuated total reflection (ATR) FTIR, the orientation of phospholambin, a small membrane protein involved in regulation of calcium in sarcoplasmic reticulum membranes, was found to have an α-helical structure oriented perpendicular to the membrane plane.
The ISD method also has proved useful for a variety of biophysical measurements besides polarized FTIR including CD [119,202], ESR [75,76,97], solid-state NMR [104] and X-ray scattering measurements of oriented biomembrane films [106]. In collaboration with Sol Gruner, who was then at Princeton University, ISD prepared films of native photoreceptor membrane were studied as a function of hydration using X-ray diffraction [106]. An unusual phase separation was discovered upon drying with the co-existence of multi-layer lamellae interspersed with microdomains of hexagonally packed tubes of lipids in the HII configuration. In collaboration with Cyrus Safinya’s group at UC Santa Barbara we also showed using synchrotron X-ray scattering that in dehydrated multilamellar films of PM, the BR structure displayed remarkable stability up to 140°C [217].
5.Probing membrane protein structural changes using FTIR difference spectroscopy
5.1.Initial application of FTIR difference spectroscopy
Although conventional FTIR spectroscopy can provide useful information about membrane protein structure and the orientation of secondary structure, this approach cannot easily reveal details of protein mechanisms. For this purpose, structural changes at the level of individual molecular group need to be probed.
Fig. 5.
Principles underlying FTIR difference spectroscopy. Top figure shows example of a protein which undergoes a structural change from state X to Y which involves a single carboxylate or carboxylic acid group (carbonyl shown in red circle). Bottom figure shows spectral differences expected in the 1700–1800 cm−1 arising from the hypothesized protonation of a carboxylate group (yellow band) or hydrogen bond change involving the hypothesized carboxylic acid group (blue bands). Overall magnitude of these changes is expected to be less than 1 mOD.
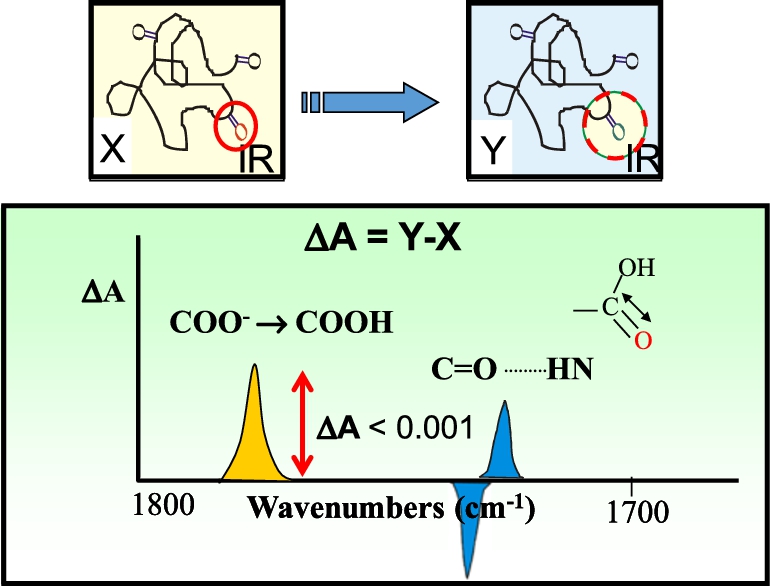
As shown in Fig. 5, even a small change in a protein involving a single carboxylic group, should in principle be detected in the FTIR difference spectrum of a protein. However, despite many early attempts, it was not until 1981 that we first succeeded in obtaining a high/signal noise FTIR differences of a membrane protein [213]. This was accomplished using BR in its native PM, which as described above can be incorporated into a highly order multilamellar film using the ISD method. Furthermore, unlike rhodopsin, BR exhibits a photocycle which allows repeated differences to be acquired and signal averaged (Fig. 6).
Fig. 6.
The BR photocycle: After absorption of a photon (yellow arrow) by light-adapt BR570 the photocycle consists of a series of intermediate states that ultimately returns back to the original light-adapted BR570 state. By dehydrating purple membrane films, the decay of the M412 intermediate is significantly slowed to over 30 seconds. Subscripts designates the wavelength of maximum visible absorption (adapted from [126]).
![The BR photocycle: After absorption of a photon (yellow arrow) by light-adapt BR570 the photocycle consists of a series of intermediate states that ultimately returns back to the original light-adapted BR570 state. By dehydrating purple membrane films, the decay of the M412 intermediate is significantly slowed to over 30 seconds. Subscripts designates the wavelength of maximum visible absorption (adapted from [126]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g006.jpg)
Fig. 7.
FTIR difference spectrum of BR → M transition of dried purple membrane (PM) containing bacteriorhodopsin (BR) reported in 1981. (a) Absolute infrared absorbance of PM film deposited on AgCl using ISD, (b) RRS of PM suspension using 5145 Å excitation with positive intensity of bands reversed. (c) FTIR-differences calculated as difference between spectrum recorded immediately after illumination for one minute from one recorded immediately before flash excitation. Note largest band in the difference spectrum near 1527 cm−1 (inside red box) arises from the ethylenic C=C stretch and reflects isomerization of the retinal chromophore. The positive band at 1762 cm−1 (inside red box) was assigned to protonation of an Asp or Glu residue containing a carboxylate group which becomes protonated during formation of the M412 intermediate. A negative band at 1640 cm−1 is assigned to the Schiff Base C=N stretching mode (adapted from Ref. [213]).
![FTIR difference spectrum of BR → M transition of dried purple membrane (PM) containing bacteriorhodopsin (BR) reported in 1981. (a) Absolute infrared absorbance of PM film deposited on AgCl using ISD, (b) RRS of PM suspension using 5145 Å excitation with positive intensity of bands reversed. (c) FTIR-differences calculated as difference between spectrum recorded immediately after illumination for one minute from one recorded immediately before flash excitation. Note largest band in the difference spectrum near 1527 cm−1 (inside red box) arises from the ethylenic C=C stretch and reflects isomerization of the retinal chromophore. The positive band at 1762 cm−1 (inside red box) was assigned to protonation of an Asp or Glu residue containing a carboxylate group which becomes protonated during formation of the M412 intermediate. A negative band at 1640 cm−1 is assigned to the Schiff Base C=N stretching mode (adapted from Ref. [213]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g007.jpg)
For the first experiment, we recorded the difference between the unphotolyzed dried BR and the light-flash induced M412 intermediate which for dried BR films exhibits a significantly slowed return back to the unphotolyzed state (Fig. 6) [213]. The difference spectrum consisted of a subtraction of the spectrum recorded of the BR570 immediately before light exposure from one recorded during accumulation of the M412 states. Each spectrum was measured using a Nicolet MX-1 FTIR instrument with 1-minute total acquisition time consisting of 32 scans and an effective resolution of 8 cm−1. As seen in Fig. 7, the difference spectra exhibited bands less than 1 mOD. While this was much smaller than the absolute IR absorbance of the most intense amide I and II bands seen in Fig. 7, the difference bands were highly reproducible. Furthermore, many of the negative bands in the difference spectrum matched major bands in the resonance Raman spectrum of BR which reflect almost exclusively vibrations of the retinylidene chromophore (Fig. 7). As a further confirmation that these differences reflected real conformational changes, the RRS was compared with the FTIR difference spectrum for BR with N15 and deuterium stable isotope substitutions at all non-exchangeable positions obtained from H. Crespi of the National Argonne Laboratories (see Fig. 2 of Ref. [213]).
One of the most important findings from this initial experiment was the appearance of a positive band at 1762 cm−1 which was assigned to the C=O stretch mode of a carboxylic group from either an Asp or Glu residue. As discussed below, subsequent studies combining FTIR difference spectroscopy and site-mutagenesis identified this residue as Asp-85 which serves as the primary BR C=N Schiff base (SB) counterion and proton acceptor, a key feature of the BR proton pump mechanism.
5.2.Low-temperature FTIR difference spectroscopy
This initial room temperature study was followed in 1982 by low-temperature measurements of the primary phototransition (BR570 to K630) where K is stable and can be photoreversed at 77 K [192]. It was confirmed that the light-adapted BR570 state has protonated SB which remains protonated in the K intermediate but is highly perturbed [192]. It was also established that the protonation detected at 1762 cm−1 had not yet occurred by formation of the K intermediate. In collaboration with the Rich Mathies’ group we also later reported a remarkable similarity between the FTIR difference and RRS difference for BR570 → K630 recorded at 77 K [207] for peaks assigned to the retinylidene chromophore.
Fig. 8.
FTIR difference spectra for different steps in the bovine rhodopsin bleaching sequence (adapted from [65]).
![FTIR difference spectra for different steps in the bovine rhodopsin bleaching sequence (adapted from [65]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g008.jpg)
In the case of animal rhodopsin, the first low-temperature FTIR difference spectra were reported by us in 1983 on bovine rhodopsin for the Rho → Batho (primary phototransition) and Rho → Meta II transition (the active state of rhodopsin) [200] (Fig. 8). This work confirmed that the 11-cis to all-trans isomerization of the chromophore occurs during the primary phototransition and that by formation of Meta II conformational changes has spread to the water-accessible regions of the protein and again involved protonation changes of glutamic and/or aspartic acid residues.
In general, the conformational changes that rhodopsin undergoes during the bleaching sequence (Rho → Bath → Lumi → Meta I → Meta II → Meta III) can be measured by recording the difference spectrum while controlling the temperature of the sample which blocks specific transitions in the sequence [62] (Fig. 8). This data reveals structural changes occurring in both protein and the retinylidene chromophore. For example, amide I and II difference bands recorded at room temperature reveals that major structural changes occur upon formation of the Meta II intermediate which act to activate a G-protein enzymatic cascade resulting in activation of rod cells [200]. Small structural changes are detected even at the bathorhodopsin stage as indicated by difference bands in the 1730–1775 cm−1 region assigned to Asp and or Glu carboxylic acid groups and bands in the amide I and II region as revealed by rhodopsin regenerated with isotopic labeled retinals [64].
5.3.Addition studies
During this early period several other groups also began using IR and infrared difference spectroscopy to study BR and rhodopsin. As a precursor to this work, the Dennis Chapman group used infrared difference spectroscopy of aqueous model and biological membranes to subtract out water contributions using a Perkin–Elmer infrared data station combined with an IR spectrometer (model 298) [55]. They emphasized the potential of IR-difference spectroscopy to study protein conformational changes. Along with our initial work in 1981, Laura Eisenstein at the University of Illinois used FTIR difference spectroscopy starting in 1982 to study BR [16,17] and later rhodopsin [18,19]. In the case of BR, this work confirmed that the SB in the BR570 and the K630 intermediate are both protonated. and deprotonated in the M state. Fritz Siebert and Werner Mäntele at the University of Freiburg, reported in 1980 a flash-induced kinetic infrared method based on an IR dispersive spectrometer to obtained difference spectra from rhodopsin [157,220,221]. However, this approach required a new sample for each spectral data point [220]. The Siebert group subsequently switched to FTIR difference spectroscopy and reported in 1983 the difference spectrum for both BR [218] and rhodopsin [219]. James Alben was also an early adopter of FTIR difference spectroscopy and in 1980 reported changes in the SH vibration of human hemoglobin due to binding of different heme ligands [3].
6.Assigning bands in the FTIR difference spectrum
While FTIR difference spectroscopy has the potential to provide information about protein structural changes at the level of individual amino acid, a major challenge is to assign the hundreds of difference bands that typically appear in the mid-IR region. For this purpose, we utilized stable isotope labeling and later site-directed mutagenesis. We also developed a new method termed site-specific isotope labeling (SDIL) which is based on in vitro translation (IVT) of proteins along with charging of suppressor tRNAs with isotope labeled amino acids. The application of these methods to FTIR-deference spectroscopy is reviewed below:
6.1.Stable isotope labeling of membrane proteins
Our application of stable isotope labeling to FTIR difference spectroscopy originated from earlier RRS studies of BR performed in collaboration with Judy Herzfeld (then at the Biophysical Laboratory at Harvard Medical School) and H.G. Khorana in the Chemistry and Biology Departments at MIT [194]. In one experiment reported in 1981, lysines in BR were substituted with lysines where the ε-amino group was either completely or partial labeled with 15N [6]. The results disproved the previously suggested [140] hypothesis [18] that two different lysine residues interacted with the retinylidene Schiff base (SB). In a second experiment ε-amino N15 lysine labeling was combined with proteolytic fragmentation and recombination of intact BR from these fragments [194]. This work showed that the retinylidene chromophore remains attached to Lys-216 and does not migrate to a different lysine during the BR photocycle as previously suggested [47].
Stable isotope labeling was first applied by us in 1986 in conjunction with FTIR difference spectroscopy in a collaboration with Judy Herzfeld to study the role of tyrosines in the BR proton pump mechanism [188,190,208]. We later used this same approach to study tyrosine changes occurring during dark-adaptation and the primary photointemediate of BR548 [191]. Band shifts were detected at the BR → K, L and M stage of the photocycle leading to the conclusion that one or more tyrosines undergo a partial change in their environment and/or protonation state during these steps. In the BR → K transition the appearance of several 2H2O insensitive negative bands characteristic of a tyrosinate group were identified based on model compound studies. For example, a highly reproducible negative band at 1277 cm−1 was observed (Fig. 9). These results suggested that a tyrosinate group is present in the unphotolyzed light-adapted form of BR (BR570) possibly due to the existence of a strong hydrogen bond and/or ionic interaction with a nearby protein group. In fact, subsequent studies using site-directed mutagenesis (see below) implicated Asp-212 [198], the counterion to the SB as this group in agreement with later high-resolution X-ray diffraction measurements [85]. The conversion of a tyrosinate to tyrosine was also detected by the Eisenstein group using a similar approach [69].
Fig. 9.
Comparison of the BR → K difference spectrum for native (solid line) and BR-[2H4]tyrosine (dashed line). Shaded region shows bands characteristic of tyrosinate (adapted from [208]).
![Comparison of the BR → K difference spectrum for native (solid line) and BR-[2H4]tyrosine (dashed line). Shaded region shows bands characteristic of tyrosinate (adapted from [208]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g009.jpg)
Many other examples of combining selective amino acid isotope labeling with FTIR difference spectroscopy contributed to understanding of the BR proton pump mechanism including isotope labeling of Asp/Glu residues [81,86], proline [206], lysine [77,96,236]; threonine [143]; tryptophan [154,189]. However, in many cases biosynthesis of the particular amino acid and/or chemical scavenging pathways cause incomplete labeling or scramble the isotope labels. In addition, this approach does not identify the particular amino acid residues that undergo structural change except in rare cases where a single residue of a particular amino acid is present in the amino acid sequence.
In addition to isotope labeling of amino acids, it is possible to incorporate isotope labels into the retinylidene chromophore of BR by regenerating bacteriooopsin (BO) with the isotope labeled retinal as had been done previously for RRS studies of BR [39]. This approach was combined with FTIR difference spectroscopy in 1984 in collaboration with Johan Lugtenburg’s group in the Department of Chemistry at the University of Leiden, Holland. For example, isotope induced band shifts allowed us to assign a positive band at 1609 cm−1 to the C=N vibration of the protonated SB in the K630 intermediate (Fig. 6). The shift of the C=N frequency from 1639 to 1609 cm−1 revealed that the SB undergoes a substantial change due to in its environment during the all-trans to 13-cis retinal isomerization.
6.2.Site-directed mutagenesis of BR
Site-directed mutagenesis and FTIR difference spectroscopy are commonly combined to assign difference bands to specific amino acid residues in a protein sequence. The first application of this approach was demonstrated by us in collaboration with H.G. Khorana’s laboratory in 1988. Mark Braiman (now at Syracuse University), worked as a joint post-doctoral Fellow in my group at BU and H.G. Khorana’s at MIT. He conducted extensive low-temperature FTIR difference measurements on a series of BR mutants expressed in E. coli and subsequently refolded by adding retinal and H. salinarum polar lipids into reconstituted membranes [42,43]. For the case of wild type (WT) BR this resulted in FTIR difference spectra almost identical to native purple membrane. Later studies utilized mutants expressed directly in the native H. salinarum and isolated in the form of PM.
Fig. 10.
BR → K difference spectrum of WT and 11 Tyr → Phe substitutions in the BR amino acid sequence (adapted from Ref. [43]).
![BR → K difference spectrum of WT and 11 Tyr → Phe substitutions in the BR amino acid sequence (adapted from Ref. [43]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g010.jpg)
One study reported in 1988 involved the substitution of Tyr → Phe at each of the 11 tyrosine positions in the BR sequence. Measurements of the BR → K and BR → M difference spectra revealed that only one substitution at Tyr-185 produced changes (Fig. 10). In this case, the set of bands previously associated with protonation changes in a Tyr disappeared (see Section 6.1). This result combined with results of other FTIR difference/site-directed mutagenesis experiments indicated that the Tyr-185/Pro-186 region of the F helix in BR and possibly other rhodopsins [199] may serve as a “hinge” and responds to light induced all-trans to 13-cis isomerization of the BR retinal as early as formation of the K intermediate by undergoing a tilt in its orientation.
Fig. 11.
Comparison of FTIR difference spectra for WT and Asp → Asn, Glu substations in the BR amino acid sequence for the BR → K, L and M transitions recorded at low-temperature. Addition low-temperature studies on the BR → N [36] and BR → O transitions [37], as well as time-resolved FTIR difference measurements [37,40,41], led to an early model of the BR proton pump mechanism. (Adapted from [42].)
![Comparison of FTIR difference spectra for WT and Asp → Asn, Glu substations in the BR amino acid sequence for the BR → K, L and M transitions recorded at low-temperature. Addition low-temperature studies on the BR → N [36] and BR → O transitions [37], as well as time-resolved FTIR difference measurements [37,40,41], led to an early model of the BR proton pump mechanism. (Adapted from [42].)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g011.jpg)
In a second experiment reported in 1988, several Asp residues in the BR sequence at positions 85, 96, 115 and 212 were replaced by Asn and Glu resides in order to assign their possible contributions to the BR → K, L and M FTIR difference spectra [42] (see Fig. 11). Subsequent FTIR difference studies were also performed on mutants of Tyr [37], Pro [206], Leu [232] and Trp [1,205] residues as well time-resolved FTIR difference [37,40,41] and low-temperature UV-Visible difference spectroscopy on wild-type and mutants BR [1,2,72,73,229,232].
6.3.Site-directed isotope labeling and cell-free expression of BR
![Major steps in site-directed isotope labeling (SDIL) of proteins (adapted from [227]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g012.jpg)
A third approach to assigning bands in the FTIR difference spectrum to individual chemical groups involves elements of methods used for stable isotope labeling and site-directed mutagenesis. Termed, site-directed isotope labeling (SDIL) [227], this approach was developed by us in collaboration with H.G. Khorana and U.L. RajBhandry’s groups at MIT. As shown schematically in Fig. 12 for the particular case of SDIL of a tyrosine incorporated at position 185 in BR, the method involves several major steps. These include: (i) enzymatic amino acylation of a suppressor tRNA (e.g. E. coli Tyr sup-tRNA as shown in Fig. 1) with the cognate isotope labeled amino acid ([ring-2H4]); (ii) insertion via mutagenesis of the suppressed codon (e.g. amber codon UAG) at position in gene sequence where labeled amino acid is to be incorporated; (iii) mRNA translation of the target gene in a cell-free system in the presence of the suppressor tRNA, In addition, in the case of a membrane protein the cell-free expressed protein must be refolded into an intact membrane in a functional form.
The first application of SDIL and the application of FTIR difference spectroscopy was demonstrated in 1994 [226]. Two site-directed tyrosine labels incorporated into BR at positions Tyr-147 and Tyr-185 were measured by FTIR difference spectroscopy and in agreement with site-directed mutagenesis, only the isotope label at Tyr-185 produced shifts in the BR → K difference spectrum [44]. Subsequent studies also used this approach in combination with ATR-FTIR difference spectroscopy (see below) to probe a specific chemical group orientation [148] and to study the changes that occur during the BR → M transition [144]. A similar approach was used later to study of the human c-Ha-Ras protein by NMR [247].
In general, a key requirement of SDIL is the cell-free expression of proteins. Unlike, site-directed incorporation of non-native amino acids into proteins which can be accomplished in vivo and has been used in successfully in conjunction with FTIR difference spectroscopy [249], aminoacyl synthetases and tRNAs cannot be engineered so that they can distinguish between a non-isotope and isotope labelled amino acid. In this regard, significant progress has been made since the introduction of SDIL in the area of cell-free protein expression. For example, it is possible to develop high-yield cell-free protein expression systems including membrane proteins [101,127,128,204,215].
In a recent collaboration with M. Coleman at UC Davis, formerly a PhD student in my group, the rapid production of solubilized and functional BR in nanodiscs was demonstrated and characterized using FTIR difference spectroscopy [54]. This was accomplished by simultaneous cell-free expression of an apolipoprotein and BO in the presence of lipids and all-trans retinals, leading to the self-assembly of BR-containing nanolipoprotein particles (NLPs) [54]. In addition, high-yield cell-free synthesis of BR in the presence of small unilamellar liposomes has been demonstrated [122].
6.4.Early model of the BR proton pump mechanism
The results from the research described above on BR as well as a variety of other spectroscopic studies [7,10,39,63,82,108,139,161,162,225]) led by 1990 to the “standard” model of the BR proton pump mechanism” shown in Fig. 13. This model is supported by subsequent high-resolution X-ray studies of BR crystals trapped in the BR570 unphotolyzed “ground state” and other intermediates in the photocycle [134,135,150,151].
Fig. 13.
“Standard” model of BR proton pump mechanism. FTIR difference studies conducted in the Rothschild and Khorana laboratories [37,40,41] led to the proposal of a proton pump mechanism. Key aspects of this model are supported by high resolution crystallographic X-ray diffraction studies of various BR photointermediates [150,151]. Overall structure shown is based on results of high resolution electron diffraction [113]. (Adapted from [226].)
![“Standard” model of BR proton pump mechanism. FTIR difference studies conducted in the Rothschild and Khorana laboratories [37,40,41] led to the proposal of a proton pump mechanism. Key aspects of this model are supported by high resolution crystallographic X-ray diffraction studies of various BR photointermediates [150,151]. Overall structure shown is based on results of high resolution electron diffraction [113]. (Adapted from [226].)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g013.jpg)
Proton translocation can be divided into two distinct phases corresponding to proton release, which occurs during the early phase of the photocycle (∼40 µs), and proton uptake, which occurs in the slower late phase of the photocycle (∼5 ms). Proton release involves a proton transfer from the Schiff base (SB) to Asp-85 (L → M) [42] (see step 2 of Fig. 13) and an almost simultaneous ejection of a proton into the outer medium [178]. This involves the initial movement of Arg-82 away from the active site due to proton transfer from the Schiff base to counterion Asp-85, and towards an extracellular proton release group [42] subsequently identified as Glu-194 and Glu-204 [48,49,68]. Proton uptake involves the reprotonation of the Schiff base via transfer of a proton from Asp-96 through a proton pathway (M → N) and the reprotonation of Asp-96 via uptake of a proton from the cytoplasmic medium (N → O). A switch in the Schiff base accessibility from the extracellular to cytoplasmic medium is expected to occur [125,240] during substates of the M intermediate and may involve a hinge near Tyr-185/Pro186 which is conserved in all microbial rhodopsins and causes a tilting of the F helix (red arrow) [148]. Changes in the state of Tyr-185 are detected during formation of the K intermediate (step 1) and thus global changes in the protein may be initially triggered soon after retinal isomerization [44,168].
Although a detailed crystallographic high resolution structural of model of BR was not achieved until around 1998, many of the key events in the BR proton pump mechanism were elucidated using FTIR difference spectroscopy in combination of various methods of bands assignment as described in the previous sections [210].
7.ATR FTIR difference spectroscopy of biomembranes
7.1.Application to bacteriorhodopsin
N.J. Harrick helped popularize the use of attenuated total reflection (ATR) IR spectroscopy with his classic 1967 book “Internal Reflection Spectroscopy” [110]. This approach allows biological samples to be studied in the presence of bulk aqueous solutions. Normally, the strong water band near 1650 cm−1 interferes with accurate measurement of weaker protein bands such as the amide I mode. For this reason, IR measurements in aqueous solution are often performed using a combination of small path length cells (<10 µm), in D2O and/or partially dehydration.
Fig. 14.
Cutaway diagram of ATR cell first used to obtain FTIR difference spectra of purple membrane. Clamp with light access hole (C2), plastic or quartz window (W), rubber gasket (G), liquid chamber made of Teflon (L), liquid input tubing (Teflon) (F), germanium ATR crystal (X), sample (S) and holding clamp (C1). Bottom schematic shows IR optical path including detector (D), mirrors (M1, M2), polarizer (P), and optical fiber (O). (Adapted from Ref. [158].)
![Cutaway diagram of ATR cell first used to obtain FTIR difference spectra of purple membrane. Clamp with light access hole (C2), plastic or quartz window (W), rubber gasket (G), liquid chamber made of Teflon (L), liquid input tubing (Teflon) (F), germanium ATR crystal (X), sample (S) and holding clamp (C1). Bottom schematic shows IR optical path including detector (D), mirrors (M1, M2), polarizer (P), and optical fiber (O). (Adapted from Ref. [158].)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g014.jpg)
ATR-IR avoids this problem by limiting the effective sample absorbance to a thin layer near the surface of an internal reflection element (IRE) [110]. As shown in Fig. 14, a sample (e.g. biomembranes) is bound to the surface of an IRE crystal which is in turn exposed to a bulk aqueous solution. This allows precise control of the pH and ionic composition of the buffer as the ability to change the solution by flowing it through the chamber. Depending on several factors including the relative refractive index of the IRE crystal and the biological material, wavelength of radiation and angle of incidence, a portion of the reflected IR light penetrates as an evanescent wave into the biological material. The actual penetration depth is typically about ∼5 µm at 1000 cm−1 for biological materials using a Ge IRE crystal. Thus under these conditions most of the infrared absorption will occur in the biomembrane film and not the bulk aqueous solution.
Fig. 15.
Polarized ATR FTIR difference spectrum for BR570 → M412 recorded on PM film bathed in a pH 6.8 solution of 0.065 M LaCl3 at room temperature. Difference spectra are shown for FTIR difference spectra recorded using vertical polarization (thick line) and horizontal polarization (thin line) (see Fig. 14). (Adapted from Ref. [158].)
![Polarized ATR FTIR difference spectrum for BR570 → M412 recorded on PM film bathed in a pH 6.8 solution of 0.065 M LaCl3 at room temperature. Difference spectra are shown for FTIR difference spectra recorded using vertical polarization (thick line) and horizontal polarization (thin line) (see Fig. 14). (Adapted from Ref. [158].)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g015.jpg)
In 1987, Hector Marrero, a graduate student in my group (now at University of Puerto Rico at Rio Piedras), succeeded to applying ATR-FTIR difference spectroscopy to bacteriorhodopsin in PM. In one study, FTIR differences were obtained for the BR570 → M412 transition (Fig. 15) as well as light to dark adaptation [158]. PM was bound to the surface of a Ge crystal and directly exposed to a bulk aqueous solution. The composition and pH of the bulk solution could be precisely controlled so the BR photocycle could be measured under different conditions. Polarized ATR-FTIR was also measured with this apparatus (see Fig. 14), revealing information about the orientation of specific molecular groups relative to the membrane plane.
In a second study [159], FTIR-ATR pH difference spectra were recorded to study the acid blue transition which occurs a low pH. It was found that protein structural changes which occur during this transition are very similar to those associated with the BR570 → M412 transition and most likely related to the protonation of the SB counterion Asp-85 which occurs in both cases.
Since this early work, ATR FTIR difference spectroscopy has been applied to BR and various other rhodopsins. In 1994, Pal Ormos and coworkers combined this approach with site-directed mutagenesis to probe the pKa of specific aspartic residues in BR [235]. In 1995, Anders Nilsson, a post-doctoral fellow in my laboratory (now at Bruker Inc., Sweden) applied this method to study the mutant D85N of BR [171]. Additional studies in our group facilitated by ATR FTIR difference spectroscopy include measurements of the alkaline form of the mutant D85N, which exhibits reverse proton pumping activity [171] and changes upon removal of the retinal chromophore [149] (see Fig. 16).
Fig. 16.
Simplified model showing conformational changes occurring in BR during the photocycle and during conversion to BO (adapted from Ref. [147]).
![Simplified model showing conformational changes occurring in BR during the photocycle and during conversion to BO (adapted from Ref. [147]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g016.jpg)
7.2.Hydrogen–deuterium exchange using ATR-FTIR
An important use of IR spectroscopy is to monitor the kinetics and extent of hydrogen–deuterium (H–D) exchange in proteins [71,83]. This is possible because the amide II mode downshifts from around 1545 to 1445 cm−1 due to NH to ND exchange of backbone peptide groups [35]. The ability of ATR-FTIR to measure biomembranes under near physiological solutions and to rapidly switch from H2O for D2O solutions by flowing liquid in and out of the ATR chamber (see Fig. 14) makes this approach extremely useful for detecting H–D changes induced by a stimulus such as light.
Fig. 17.
Comparison of kinetics H–D exchange in bovine rhodopsin disc membrane after photo bleaching (indicated by dashed arrow) measured by transmittance (top) and ATR-FTIR (bottom) (see Ref. [183] for additional details).
![Comparison of kinetics H–D exchange in bovine rhodopsin disc membrane after photo bleaching (indicated by dashed arrow) measured by transmittance (top) and ATR-FTIR (bottom) (see Ref. [183] for additional details).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g017.jpg)
In one study by us that utilized this approach performed by Parshuram Rath, a postdoctoral fellow in my group, ATR FTIR difference spectroscopy was used to monitor the rate of H–D exchange in unphotolyzed and bleached rhodopsin [183]. As shown in Fig. 17, photoactivation causes an increased H–D exchange indicating that light causes a conformational change which exposes buried peptide groups to the solvent medium.
7.3.ATR-FTIR differences of nicotinic acetylcholine receptor
Although FTIR difference spectroscopy of biomembranes is most conveniently applied to systems activated by light such as BR, such measurements can also be made on non-light activated systems. For example, this is possible by using caged-substrates whose light-activated release triggers target activation (e.g. a membrane bound receptor or an ATP driven ion pumps) [21,132,164].
Fig. 18.
FTIR difference spectra of affinity purified and reconstituted nAChR membranes. Top, spectra recorded in the presence and absence of 50 µM carbamylcholine, and Bottom, consecutive spectra recorded with buffer flowing past the nAChR film. Both difference spectra are the average of 20 1,000-scan difference spectra, each recorded at 22.5°C and at a resolution of 8 cm−1 (adapted from [11]).
![FTIR difference spectra of affinity purified and reconstituted nAChR membranes. Top, spectra recorded in the presence and absence of 50 µM carbamylcholine, and Bottom, consecutive spectra recorded with buffer flowing past the nAChR film. Both difference spectra are the average of 20 1,000-scan difference spectra, each recorded at 22.5°C and at a resolution of 8 cm−1 (adapted from [11]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g018.jpg)
Alternatively, the receptor substrate can be introduced into solution using an ATR-FTIR flow system such as shown in Fig. 14. The approach was utilized in 1992 by John Baenziger, a post-doctoral fellow working at the time in my laboratory (now at University of Ottawa), to study structural changes that occur when the ligands bind to the nicotinic acetylcholine receptor (nAChR). Previously work had already established the usefulness of ATR-IR for studying biomembranes systems. For example, an early application of IR-ATR was by Fringeli to study the structure of lipids and proteins including pore formation in lipid membranes by alamethicin [90,91]. In the first study of nAChR, performed in collaboration with Keith Miller’s group at Harvard Medical School and MGH, reconstituted nAChR membrane was deposited in a thin film on the surface of a Ge IRE [12,13]. High signal/noise FTIR difference spectra were obtained by sequentially flowing a buffer with and without the nAChR agonist carbamylcholine (Carb) past the film surface (Fig. 18). Several of the FTIR difference bands were characteristic of protein vibrations including the amide I and amide I vibrations from backbone peptides, Tyr and Asp/Glu groups. In addition, bands due to binding of Carb were detected. Using the fluorescence probe ethidium bromide, it was also demonstrated that nAChR underwent the “resting-to-desensitized” state transition monitored by ATR-FTIR difference spectroscopy under the same conditions [12,13].
In a subsequent study [14], different agonists such as α-bungarotoxin and antagonists, some of which were isotope labelled, were utilized to assign bands in the “resting-to-desensitized” FTIR difference spectrum. It was shown that bands can be assigned to bound agonists, providing a new means to study the interaction of the ligand with the receptor. Other bands are assigned to perturbations of side-chain residues in nAChR include Tyr and Asp; and/or Glu residues. Subsequent studies by the Baenziger group have extended this work. For example structural changes as a function of membrane lipid composition [61] and changes in α-helical orientation upon desensitization using polarization and H–D exchange have been demonstrated [117]. In addition, nAChR has been studied using transmittance FTIR difference spectroscopy using a “caged” carbamylcholine [102].
8.Time-resolved FTIR difference spectroscopy
Elucidating the basis for membrane protein function requires a dynamic picture of structural changes at a molecular level. While X-ray diffraction can provide high-spatial resolution and potentially high temporal resolution using recently developed methods such as coherent X-ray laser diffraction [180], the restraints imposed by a crystalline lattice (either 2D or 3D) can interfere with native conformational changes which the proteins normally undergo in their native environment. In contrast, FTIR difference spectroscopy provides an approach which is highly amenable to measuring dynamics under non-crystalline conditions. This stems largely from the ability to record absorption of all wavelengths in the infrared absorption simultaneously (Felligett’s multiplex advantage). Accordingly, a number of approaches have been developed which span the time range from milliseconds to sub-nanoseconds including rapid-scan stroboscopic, and step-scan FTIR. Complementing this approach, improvements in pulse IR lasers have made it possible to record changes in infrared spectra of biomolecules down to femtosecond level.
8.1.Millisecond rapid-scan
This approach which was applied in our lab to BR by Mark Braiman allows FTIR difference spectra of a sample to be recorded with millisecond-time resolution. This is facilitated by the ability of many commercial FTIR instruments to achieve high mirror velocities (∼10 cm/s), thus enabling the collection of an interferograms in millisecond time periods [45].
Fig. 19.
Timing diagram for rapid-scan techniques as applied to PM. See Ref. [40] for details (adapted from [40]).
![Timing diagram for rapid-scan techniques as applied to PM. See Ref. [40] for details (adapted from [40]).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g019.jpg)
One of the first applications of rapid-scan FTIR difference spectroscopy to a biological system was reported by our group in 1987 [40]. Using a Nicolet Instruments 60SX, 512-point interferograms were recorded of PM within 5 ms immediately before and following a laser flash. The timing diagram for the rapid-scan experiment is shown in Fig. 19. This enable us to record 8 cm−1 time-resolved difference spectra. By averaging 1000 s of such differences, high signal-to-noise BR570 → M412 difference spectra were obtained. Comparison with static FTIR difference spectrum recorded at −23°C revealed that the backbone structural changes at lower temperature are partially blocked. This method has been used routinely in our laboratory to study a wide-range of microbial rhodopsins including sensory rhodopsins [31,32], proteorhodopsins [33,129] and channelrhodopsins [175].
8.2.Microsecond stroboscopic time-resolved
A major limitation of the rapid-scan technique is that the total duration of the time for acquiring an interferograms is normally in the milliseconds even when one-sided or even one-half of the inteferogram is utilized. This effectively limits measurements of kinetic events that occur on a much shorter time-interval such as the early BR photocycle (see Fig. 6). For example, during a 5 ms scan of the moving mirror need to acquire a single interferograms, the sample is undergoing time-dependent structural alterations. Thus, each point in the interferograms could correspond to a different time-point in the samples response.
In order to circumvent this problem, the stroboscopic FTIR-technique utilizes points from multiple interferograms that correspond to a single time point in the samples response. This is made possible by collecting a set of interferograms which are initiated at variable delays relative to the initiation (e.g. laser flash) of the sample response. In this way, a set of “reshuffled” interferograms can be constructed, each corresponding to a specific time point in the sample response.
Fig. 20.
Stroboscopic time-resolved FTIR difference spectra of the BR photocycle. (Adapted from [41]; see reference for further details.)
![Stroboscopic time-resolved FTIR difference spectra of the BR photocycle. (Adapted from [41]; see reference for further details.)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g020.jpg)
In one of the first application of the stroboscopic approach to biological systems demonstrated in our lab by Mark Braiman and Olaf Bousche, 50 µs time-resolved FTIR difference were recorded of the BR photocycle with 4 cm−1 resolution and a detection limit of
8.3.Step-scan FTIR difference spectroscopy
This approach has been used in conjunction with FTIR difference spectroscopy of light-activated molecules, especially those which exhibit a cyclic activity such as BR [20,99,187]. The group of Fritz Siebert has pioneered the use of this technique to study a variety of microbial rhodopsins [26,107,112,172,187].
In comparison to rapid-scan and stroboscopic FTIR, the moving mirror of the interferometer is typically “locked” at discrete positions where transient changes in absorption are measured after a rapid stimulus (e.g. nanosecond laser-flash). By repeating these measurements at each discrete mirror position that comprises the interferogram, a time-resolved set of interferograms can be assembled. Fourier transformation of this data yields a compete time-resolved FTIR difference spectrum of the sample. Depending on the time-resolution of the detector and speed of the electronics, sub-nanosecond time resolution can be obtained.
8.4.Sub-picosecond IR-difference spectroscopy
Advances in infrared lasers have made it possible to perform ultrafast sub-picosecond transient infrared (TIR) studies on photoactive proteins. Examples include studies on BR by Diller and coworkers [67,105], photoactive yellow protein [170,214,243], myoglobin [141,142,239] and green fluorescent protein [136,231,242]. Although this approach does not require an FTIR spectrometer, it has many advantages which have pushed the time-resolution for biological systems to femtoseconds.
In one study performed in our laboratory by Jason Amsden (now at Duke University) in collaboration with Shyam Erramilli and Larry Ziegler at the Boston University Photonics Center, and John Spudich at the U. Texas Health Science Center, the early structural changes in the protein backbone of green-absorbing proteorhodopsin (GPR), proton pump similar to BR, found in marine proteobacteria [5] was investigated. Measurements were made over the first 30 ps of the photocycle with a time-resolution of ∼200 fs. In addition, both chromophore and protein isotope labeling (see Section 6.1) were used to assign bands in the IR-difference spectrum. The sample which was sandwiched in a liquid form between two circular 32 mm × 3 mm circular CaF2 windows was rotated and translated perpendicular to the laser beam in order to refresh the sample area probed for each laser shot. An example of IR differences measured in the amide II region are shown in Fig. 21. In addition to changes assigned to the retinylidene chromophore ethylenic stretch region, total-15N and retinal C15D (retinal with a deuterium on carbon 15) isotope labeling demonstrated that the protein backbone responds almost concomitant with retinal all-trans to 13-cis isomerization [5].
Fig. 21.
Transient infrared spectrum of unlabeled green proteorhodopsin (GPR) in the ethylenic region after ∼100 nJ 520 nm photoexcitation (adapted [5]; see reference for further details).
![Transient infrared spectrum of unlabeled green proteorhodopsin (GPR) in the ethylenic region after ∼100 nJ 520 nm photoexcitation (adapted [5]; see reference for further details).](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g021.jpg)
9.Structural changes of internal water molecules
In addition to the vibrations of individual protein groups in the FTIR difference spectra of membrane proteins, vibrations of internal water molecules are also detected. For example, the OH stretching mode of water, appears in the 3200–3600 cm−1 region. The frequency is extremely sensitive to hydrogen bonding and thus changes can be detected in the difference spectrum and provides a window on the dynamics of internal water molecules.
Such bands were first detected in the FTIR difference spectra of BR by the A. Maeda’s group in the OD stretching region using D2O18 substitution [155] and by W. Fischer working in my group in the OH stretching region using H2O18 substitution [87,88]. The earliest changes detected are in the BR570 → K630 difference spectrum recorded at 77 K with a negative band at 3642 cm−1 down-shifting 12 cm−1 due to an increase in hydrogen bonding of a single water molecule [87,88]. The high frequency of this OH stretch is indicative of “dangling” OH groups which lacks the normal hydrogen bonding found in bulk water. Based on the high-resolution X-ray diffraction structure of BR [150], the high frequency water bands has been assigned to W401 [15] which appears to have a dangling hydrogen bond (see Fig. 22). Several other internal waters appear in the vicinity of the BR SB which together along with the negatively charged residues Asp-85 and Asp-212 form a pentagonal cluster (Fig. 22). In general, site-directed mutagenesis can provide information about the approximate location of these waters in the BR structure [87,123,153,156] while polarized FTIR difference spectroscopy can provide information about thee orientation of the OH dipole moments [111].
Fig. 22.
X-ray crystallographic structure of BR from PDB coordinates 13CW [151] showing position of water molecules oriented as shown in Fig. 2 of [15]. (Adapted from [56].)
![X-ray crystallographic structure of BR from PDB coordinates 13CW [151] showing position of water molecules oriented as shown in Fig. 2 of [15]. (Adapted from [56].)](https://content.iospress.com:443/media/bsi/2016/5-3/bsi-5-3-bsi148/bsi-5-bsi148-g022.jpg)
In addition to the internal water molecules that give rise to specific OH vibrational water bands, FTIR differences also reveal broad continuum IR absorbance that is associated with networks of strongly hydrogen bonded water and other OH groups [251]. In the case of BR, the detection of such broad absorbance bands has been associated with specific steps in proton transport [89,93–95]. In a recent rapid-scan FTIR difference study of the light-driven proton pump from Halorubrum sodomense known as archaerhodopsins-3 broad continuum IR absorbance was discovered that is very similar to BR [56]. Although, AR3 does not exhibit the same pattern of high frequency OH stretch bands seen in BR, this similarity indicates that there exists a conserved network of hydrogen bonds in both proteins which might serve as a proton wire [56].
10.Beyond bacteriorhodopsin
In addition to the application of FTIR difference spectroscopy and associated techniques to BR described in this review, our group has been active since 1988 in using these techniques to investigate the molecular basis of the function of several other microbial rhodopsins. These studies were all conducted in collaboration with John Spudich, a leader in the microbial rhodopsin field at the University of Texas Health Science Center at Houston. They included FTIR difference studies along with RRS on halorhodopsin (HR) [197], sensory rhodopsins (SRI, SRII) from archaebacterial [29,31,34,38,184], fungal neurospora rhodopsin (NO) [30], anabaena sensory rhodopsin from cyanobacteria (ASR) [32], green and blue proteorhodopsin proton pumps from marine bacteria (PRs) [4,5,24,28,33,129,130]; archaerhodopsins-3 (AR3), a BR-like protein [56,216] and most recently channelrhodopsins (ChRs) from algae [173,174,176,250]. These proteins span a range of functions which are representative of diverse biomembranes process and include active anion transport (HRs), signal transduction (SRI, SRII and ASR), and light-gated ion channels (ChRs).
Fig. 23.
Growth in application of FTIR difference spectroscopy to study membrane proteins and other photoactive proteins or proteins that can be photoactivated. Graph shows the approximate number of publications/year. Yellow boxes indicate the approximate time that FTIR difference spectroscopy was first applied for the particular protein.
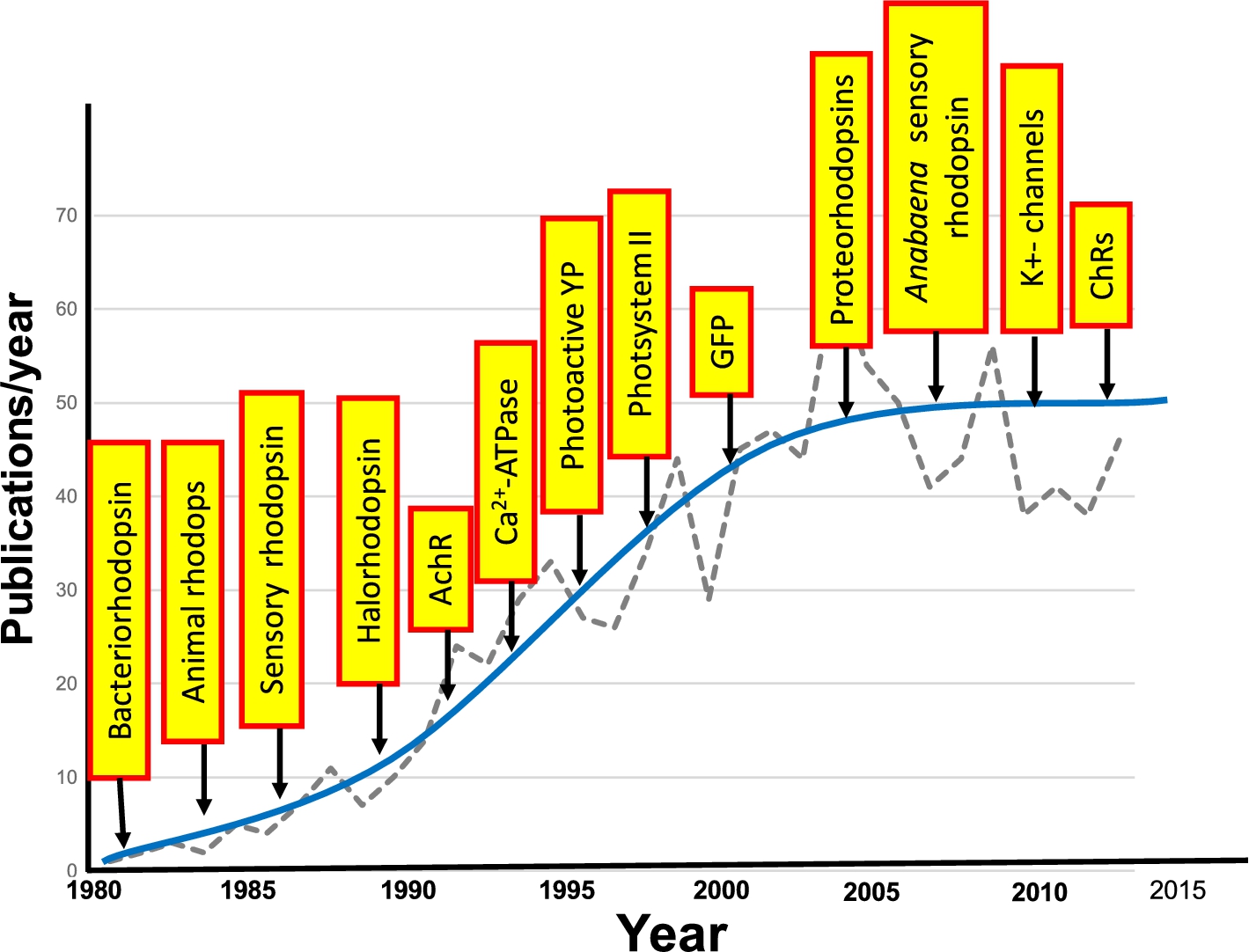
From a broader perspective, FTIR difference spectroscopy has become a useful tool for investigating the structural changes which underlie the function of proteins. In the case of membrane proteins and photoactive proteins, a variety of systems have been investigated using the approaches described in this review and first introduced for bacteriorhodopsin. A few examples are the photosystem II [116,152], photoactive yellow protein (PYP) [59,118,124,246]; Ca2+-ATPase [21,22,52,98,133]; green fluorescent protein (GFP) and potassium channels [92,230]. One measure of the growth of FTIR difference spectroscopy of biomolecules is shown in Fig. 23.
Acknowledgements
The work reviewed here would not have been possible without the critical contributions of the many graduate students and postdoctoral associates who worked in the Molecular Biophysics Laboratory at Boston University. I am also grateful to have had the opportunity to work closely with many collaborators who are leaders in their fields including N. Clark, W. DeGrip, J. Herzfeld, H.G. Khorana, L. Lugtenburg and J.L. Spudich. This work was also made possible by the strong support of a number of sponsors including the National Science Foundation, National Institutes of Health, Army Research Office and the American Heart Association. Thanks to Adrian Yi for proof reading this review. I wish to also thank my many colleagues at Boston University and the BU Photonics Center who have provided me a stimulating environment to conduct this research.
References
[1] | P.L. Ahl, L.J. Stern, D. During, T. Mogi, H.G. Khorana and K.J. Rothschild, Effects of amino acid substitutions in the F helix of bacteriorhodopsin. Low temperature ultraviolet/visible difference spectroscopy, J. Biol. Chem. 263: ((1988) ), 13594–13601. |
[2] | P.L. Ahl, L.J. Stern, T. Mogi, H.G. Khorana and K.J. Rothschild, Substitution of amino acids in helix F of bacteriorhodopsin: Effects on the photochemical cycle, Biochemistry 28: ((1989) ), 10028–10034. doi:10.1021/bi00452a022. |
[3] | J.O. Alben and G.H. Bare, Ligand-dependent heme-protein interactions in human hemoglobin studied by Fourier transform infrared spectroscopy. Effects of quaternary structure on alpha chain Tertiary structure measured at the alpha-104(G11) cysteine sulfhydryl, J. Biol. Chem. 255: ((1980) ), 3892–3897. |
[4] | J.J. Amsden, J.M. Kralj, V.B. Bergo, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Different structural changes occur in blue- and green-proteorhodopsins during the primary photoreaction, Biochemistry 47: ((2008) ), 11490–11498. doi:10.1021/bi800945t. |
[5] | J.J. Amsden, J.M. Kralj, L.R. Chieffo, X. Wang, S. Erramilli, E.N. Spudich, J.L. Spudich, L.D. Ziegler and K.J. Rothschild, Subpicosecond protein backbone changes detected during the green-absorbing proteorhodopsin primary photoreaction, J. Phys. Chem. B 111: ((2007) ), 11824–11831. doi:10.1021/jp073490r. |
[6] | P.V. Argade, K.J. Rothschild, A.H. Kawamoto, J. Herzfeld and W.C. Herlihy, Resonance Raman spectroscopy of specifically [ä-15N]-lysine-labeled bacteriorhodopsin, Proc. Natl. Acad. Sci. USA 78: ((1981) ), 1643–1646. doi:10.1073/pnas.78.3.1643. |
[7] | P.V.R. Argade and J. Kenneth, Quantitative analysis of resonance Raman spectra of purple membrane from Halobacterium halobium: L550 intermediate, Biochemistry 22: ((1983) ), 3460–3466. doi:10.1021/bi00283a024. |
[8] | I.M. Asher, K.J. Rothschild and H.E. Stanley, Raman spectroscopic study of the valinomycin–KSCN complex, J. Mol. Biol. 89: ((1974) ), 205–222. doi:10.1016/0022-2836(74)90171-5. |
[9] | I.M. Asher, K.J. Rothschild, H.E. Stanley and E. Anastassakes, Raman spectroscopy of uncomplexed valinomycin and its components in the solid state, J. Am. Chem. Soc. 99: ((1977) ), 2024–2032. doi:10.1021/ja00449a002. |
[10] | B. Aton, A.G. Doukas, R.H. Callender, B. Becher and T.G. Ebrey, Resonance Raman studies of the purple membrane, Biochemistry 16: ((1977) ), 2995–2999. doi:10.1021/bi00632a029. |
[11] | J.B. Baenziger, K.W. Miller and K.J. Rothschild, Fourier transform infrared difference spectroscopy of the nicotinic acetylcholine receptor: Evidence for specific protein structural changes upon desensitization, Biochemistry 32: ((1993) ), 5448–5454. doi:10.1021/bi00071a022. |
[12] | J.E. Baenziger, K.W. Miller, M.P. McCarthy and K.J. Rothschild, Probing conformational changes in the nicotinic acetylcholine receptor by Fourier transform infrared difference spectroscopy, Biophysical Journal 62: ((1992) ), 64–66. doi:10.1016/S0006-3495(92)81780-0. |
[13] | J.E. Baenziger, K.W. Miller and K.J. Rothschild, Incorporation of the nicotinic acetylcholine receptor into planar multilamellar films: Characterization by fluorescence and Fourier transform infrared difference spectroscopy, Biophysical Journal 61: ((1992) ), 983–992. doi:10.1016/S0006-3495(92)81905-7. |
[14] | J.E. Baenziger, K.W. Miller and K.J. Rothschild, Fourier transform infrared difference spectroscopy of the nicotinic acetylcholine receptor: Evidence for specific protein structural changes upon desensitization, Biochemistry 32: ((1993) ), 5448–5454. doi:10.1021/bi00071a022. |
[15] | M. Baer, G. Mathias, I.F. Kuo, D.J. Tobias, C.J. Mundy and D. Marx, Spectral signatures of the pentagonal water cluster in bacteriorhodopsin, ChemPhysChem 9: ((2008) ), 2703–2707. doi:10.1002/cphc.200800473. |
[16] | K. Bagley, G. Dollinger, L. Eisenstein, M. Hong, J. Vittitow and L. Zimanyi, Infrared studies of the photocycle of bacteriorhodopsin, Prog. Clin. Biol. Res. 164: ((1984) ), 27–37. |
[17] | K. Bagley, G. Dollinger, L. Eisenstein, A.K. Singh and L. Zimanyi, Fourier transform infrared difference spectroscopy of bacteriorhodopsin and its photoproducts, Proc. Natl. Acad. Sci. USA 79: ((1982) ), 4972–4976. doi:10.1073/pnas.79.16.4972. |
[18] | K.A. Bagley, V. Balogh-Nair, A.A. Croteau, G. Dollinger, T.G. Ebrey, L. Eisenstein, M.K. Hong, K. Nakanishi and J. Vittitow, Fourier-transform infrared difference spectroscopy of rhodopsin and its photoproducts at low temperature, Biochemistry 24: ((1985) ), 6055–6071. doi:10.1021/bi00343a006. |
[19] | K.A. Bagley, L. Eisenstein, T.G. Ebrey and M. Tsuda, A comparative study of the infrared difference spectra for octopus and bovine rhodopsins and their bathorhodopsin photointermediates, Biochemistry 28: ((1989) ), 3366–3373. doi:10.1021/bi00434a036. |
[20] | A. Barth, Infrared spectroscopy of proteins, Biochim. Biophys. Acta 1767: ((2007) ), 1073–1101. doi:10.1016/j.bbabio.2007.06.004. |
[21] | A. Barth, W. Kreutz and W. Mantele, Molecular changes in the sarcoplasmic reticulum calcium ATPase during catalytic activity. A Fourier transform infrared (FTIR) study using photolysis of caged ATP to trigger the reaction cycle, FEBS Lett. 277: ((1990) ), 147–150. doi:10.1016/0014-5793(90)80830-C. |
[22] | A. Barth, W. Kreutz and W. Mantele, Changes of protein structure, nucleotide microenvironment, and Ca(2+)-binding states in the catalytic cycle of sarcoplasmic reticulum Ca(2+)-ATPase: Investigation of nucleotide binding, phosphorylation and phosphoenzyme conversion by FTIR difference spectroscopy, Biochim. Biophys. Acta 1194: ((1994) ), 75–91. doi:10.1016/0005-2736(94)90205-4. |
[23] | A. Barth and C. Zscherp, What vibrations tell us about proteins, Quarterly Reviews of Biophysics 35: ((2002) ), 369–430. doi:10.1017/S0033583502003815. |
[24] | H. Bayraktar, A.P. Fields, J.M. Kralj, J.L. Spudich, K.J. Rothschild and A.E. Cohen, Ultrasensitive measurements of microbial rhodopsin photocycles using photochromic FRET, Photochem. Photobiol. 88: ((2012) ), 90–97. doi:10.1111/j.1751-1097.2011.01011.x. |
[25] | M.D. Bazzi and R.W. Woody, Oriented secondary structure in integral membrane proteins: I. Circular dichroism and infrared spectroscopy of cytochrome oxidase in multilamellar films, Biophysical Journal 48: ((1985) ), 957–966. doi:10.1016/S0006-3495(85)83859-5. |
[26] | A. Becker, C. Taran, W. Uhmann and F. Siebert, Time-resolved FTIR spectroscopy using a step-scan interferometer, SPIE 1145: ((1989) ), 512–513. |
[27] | L.J. Bellamy, The Infrared Spectra of Complex Molecules, Chapman and Hall, London, (1968) . |
[28] | V. Bergo, J.J. Amsden, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Structural changes in the photoactive site of proteorhodopsin during the primary photoreaction, Biochemistry 43: ((2004) ), 9075–9083. doi:10.1021/bi0361968. |
[29] | V. Bergo, E.N. Spudich, K.L. Scott, J.L. Spudich and K.J. Rothschild, FTIR analysis of the SII540 intermediate of sensory rhodopsin II: Asp73 is the Schiff base proton acceptor, Biochemistry 39: ((2000) ), 2823–2830. doi:10.1021/bi991676d. |
[30] | V. Bergo, E.N. Spudich, J.L. Spudich and K.J. Rothschild, A Fourier transform infrared study of Neurospora rhodopsin: Similarities with archaeal rhodopsins, Photochem. Photobiol. 76: ((2002) ), 341–349. doi:10.1562/0031-8655(2002)076<0341:AFTISO>2.0.CO;2. |
[31] | V. Bergo, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Conformational changes detected in a sensory rhodopsin II-transducer complex, J. Biol. Chem. 278: ((2003) ), 36556–36562. doi:10.1074/jbc.M303719200. |
[32] | V.B. Bergo, M. Ntefidou, V.D. Trivedi, J.J. Amsden, J.M. Kralj, K.J. Rothschild and J.L. Spudich, Conformational changes in the photocycle of Anabaena sensory rhodopsin: Absence of the Schiff base counterion protonation signal, J. Biol. Chem. 281: ((2006) ), 15208–15214. doi:10.1074/jbc.M600033200. |
[33] | V.B. Bergo, O.A. Sineshchekov, J.M. Kralj, R. Partha, E.N. Spudich, K.J. Rothschild and J.L. Spudich, His-75 in proteorhodopsin, a novel component in light-driven proton translocation by primary pumps, J. Biol. Chem. 284: ((2009) ), 2836–2843. doi:10.1074/jbc.M803792200. |
[34] | V.B. Bergo, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Active water in protein–protein communication within the membrane: The case of SRII-HtrII signal relay, Biochemistry 48: ((2009) ), 811–813. doi:10.1021/bi802180a. |
[35] | E.R. Blout, C. de Lozé and A. Asadourian, The deuterium exchange of water-soluble polypeptides and proteins as measured by infrared spectroscopy, Journal of American Chemical Society 83: ((1961) ), 1895–1900. doi:10.1021/ja01469a028. |
[36] | O. Bousche, M. Braiman, Y.W. He, T. Marti, H.G. Khorana and K.J. Rothschild, Vibrational spectroscopy of bacteriorhodopsin mutants. Evidence that ASP-96 deprotonates during the M–N transition, J. Biol. Chem. 266: ((1991) ), 11063–11067. |
[37] | O. Bousche, S. Sonar, M.P. Krebs, H.G. Khorana and K.J. Rothschild, Time-resolved Fourier transform infrared spectroscopy of the bacteriorhodopsin mutant Tyr-185 → Phe: Asp-96 reprotonates during O formation; Asp-85 and Asp-212 deprotonate during O decay, Photochem. Photobiol. 56: ((1992) ), 1085–1095. doi:10.1111/j.1751-1097.1992.tb09732.x. |
[38] | O. Bousche, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Conformational changes in sensory rhodopsin I: Similarities and differences with bacteriorhodopsin, halorhodopsin, and rhodopsin, Biochemistry 30: ((1991) ), 5395–5400. doi:10.1021/bi00236a010. |
[39] | M. Braiman and R. Mathies, Resonance Raman spectra of bacteriorhodopsin’s primary photoproduct: Evidence for a distorted 13-cis retinal chromophore, Proc. Natl. Acad. Sci. USA 79: ((1982) ), 403–407. doi:10.1073/pnas.79.2.403. |
[40] | M.S. Braiman, P.L. Ahl and K.J. Rothschild, Millisecond Fourier-transform infrared difference spectra of bacteriorhodopsin’s M412 photoproduct, Proc. Natl. Acad. Sci. USA 84: ((1987) ), 5221–5225. doi:10.1073/pnas.84.15.5221. |
[41] | M.S. Braiman, O. Bousche and K.J. Rothschild, Protein dynamics in the bacteriorhodopsin photocycle: Submillisecond Fourier transform infrared spectra of the L, M, and N photointermediates, Proc. Natl. Acad. Sci. USA 88: ((1991) ), 2388–2392. doi:10.1073/pnas.88.6.2388. |
[42] | M.S. Braiman, T. Mogi, T. Marti, L.J. Stern, H.G. Khorana and K.J. Rothschild, Vibrational spectroscopy of bacteriorhodopsin mutants: Light-driven proton transport involves protonation changes of aspartic acid residues 85, 96, and 212, Biochemistry 27: ((1988) ), 8516–8520. doi:10.1021/bi00423a002. |
[43] | M.S. Braiman, T. Mogi, L.J. Stern, N.R. Hackett, B.H. Chao, H.G. Khorana and K.J. Rothschild, Vibrational spectroscopy of bacteriorhodopsin mutants: I. Tyrosine-185 protonates and deprotonates during the photocycle, Proteins 3: ((1988) ), 219–229. doi:10.1002/prot.340030403. |
[44] | M.S. Braiman, T. Mogi, L.J. Stern, N.R. Hackett, B.H. Chao, H.G. Khorana and K.J. Rothschild, Vibrational spectroscopy of bacteriorhodopsin mutants. I. Tyrosine-185 protonates and deprotonates during the photocycle, Proteins: Struct., Funct., Genet. 3: ((1988) ), 219–229. doi:10.1002/prot.340030403. |
[45] | M.S. Braiman and K.J. Rothschild, Fourier transform infrared techniques for probing membrane protein structure, Annu. Rev. Biophys. Biophys. Chem. 17: ((1988) ), 541–570. doi:10.1146/annurev.bb.17.060188.002545. |
[46] | K. Brandenburg and U. Seydel, Orientation measurements on ordered multibilayers of phospholipids and sphingolipids from synthetic and natural origin by ATR Fourier transform infrared spectroscopy, Zeitschrift fur Naturforschung. C, Journal of Biosciences 41: ((1986) ), 453–467. |
[47] | J. Bridgen and I.D. Walker, Photoreceptor protein from the purple membrane of Halobacterium halobium. Molecular weight and retinal binding site, Biochemistry 15: ((1976) ), 792–798. doi:10.1021/bi00649a010. |
[48] | L.S. Brown, A.K. Dioumaev, R. Needleman and J.K. Lanyi, Local-access model for proton transfer in bacteriorhodopsin, Biochemistry 37: ((1998) ), 3982–3993. doi:10.1021/bi9728396. |
[49] | L.S. Brown, J. Sasaki, H. Kandori, A. Maeda, R. Needleman and J.K. Lanyi, Glutamic Acid 204 is the terminal proton release group at the extracellular surface of bacteriorhodopsin, The Journal of Biological Chemistry 270: (45) ((1995) ), 27122–27126. doi:10.1074/jbc.270.45.27122. |
[50] | P.K. Brown, Rhodopsin rotates in the visual receptor membrane, Nature (London), New Biol. 236: ((1972) ), 35–38. doi:10.1038/newbio236035a0. |
[51] | P.K. Brown and R.H. White, Rhodopsin of the larval mosquito, J. Gen. Physiol. 59: ((1972) ), 401–414. doi:10.1085/jgp.59.4.401. |
[52] | R. Buchet, I. Jona and A. Martonosi, Ca2+ release from caged-Ca2+ alters the FTIR spectrum of sarcoplasmic reticulum, Biochim. Biophys. Acta 1069: ((1991) ), 209–217. doi:10.1016/0005-2736(91)90126-S. |
[53] | M.D. Byler and H. Susi, Examination of the secondary structure of proteins by deconvolved FTIR spectra, Biopolymers 25: ((1986) ), 469–487. doi:10.1002/bip.360250307. |
[54] | J.A. Cappuccio, C.D. Blanchette, T.A. Sulchek, E.S. Arroyo, J.M. Kralj, A.K. Hinz, E.A. Kuhn, B.A. Chromy, B.W. Segelke, K.J. Rothschild, J.E. Fletcher, F. Katzen, T.C. Peterson, W.A. Kudlicki, G. Bench, P.D. Hoeprich and M.A. Coleman, Cell-free co-expression of functional membrane proteins and apolipoprotein, forming soluble nanolipoprotein particles, Mol. Cell. Proteomics 7: ((2008) ), 2246–2253. doi:10.1074/mcp.M800191-MCP200. |
[55] | D. Chapman, J.C. Gomez-Fernandez, F.M. Goni and M. Barnard, Difference infrared spectroscopy of aqueous model and biological membranes using an infrared data station, J. Biochem. Biophys. Methods 2: ((1980) ), 315–323. doi:10.1016/S0165-022X(80)90048-2. |
[56] | E.C. Clair, J.I. Ogren, S. Mamaev, J.M. Kralj and K.J. Rothschild, Conformational changes in the archaerhodopsin-3 proton pump: Detection of conserved strongly hydrogen bonded water networks, J. Biol. Phys. 38: ((2012) ), 153–168. doi:10.1007/s10867-011-9246-4. |
[57] | N.A. Clark, K.J. Rothschild, D.A. Luippold and B.A. Simon, Surface-induced lamellar orientation of multilayer membrane arrays. Theoretical analysis and a new method with application to purple membrane fragments, Biophysical Journal 31: ((1980) ), 65–96. doi:10.1016/S0006-3495(80)85041-7. |
[58] | W.W. Coblentz, Investigation of Infrared Spectra, Carnegie Institute Publications, Carnegie Institute, Washington, D.C., (1905) . |
[59] | R. Cordfunke, R. Kort, A. Pierik, B. Gobets, G.J. Koomen, J.W. Verhoeven and K.J. Hellingwerf, Trans/cis (Z/E) photoisomerization of the chromophore of photoactive yellow protein is not a prerequisite for the initiation of the photocycle of this photoreceptor protein, Proc. Natl. Acad. Sci. USA 95: ((1998) ), 7396–7401. doi:10.1073/pnas.95.13.7396. |
[60] | M. Cortijo, A. Alonso, J.C. Gomez-Fernandez and D. Chapman, Intrinsic protein-lipid interactions. Infrared spectroscopic studies of gramidicin A, bacteriorhodopsin, and calcium-ATPase in biomembranes and reconstituted systems, J. Mol. Biol. 157: ((1982) ), 597–618. doi:10.1016/0022-2836(82)90501-0. |
[61] | C.J. daCosta and J.E. Baenziger, A lipid-dependent uncoupled conformation of the acetylcholine receptor, J. Biol. Chem. 284: ((2009) ), 17819–17825. doi:10.1074/jbc.M900030200. |
[62] | W.J. de Grip and K.J. Rothschild, Structure and mechanism of vertebrate visual pigments, in: Molecular Mechanisms in Visual Transduction, D.G. Stavenga, W.J. DeGrip and E.N. Pugh, eds, Elsevier, Amsterdam, (2000) , pp. 1–54. |
[63] | H.J.M. De Groot, V. Copie, S.O. Smith, P.J. Allen, C. Winkel, J. Lugtenburg, J. Herzfeld and R.G. Griffin, Magic-angle-sample-spinning NMR difference spectroscopy, J. Magn. Reson. 77: (72) ((1988) ), 251–257. |
[64] | W.J. DeGrip, D. Gray, J. Gillespie, P.H.M. Bovee, E.M.M. Van den Berg, J. Lugtenburg and K.J. Rothschild, Photoexcitation of rhodopsin: Conformation changes in the chromophore, protein and associated lipids as determined by FTIR difference spectroscopy, Photochem. Photobiol. 48: ((1988) ), 497–504. doi:10.1111/j.1751-1097.1988.tb02852.x. |
[65] | W.J. DeGrip and K.J. Rothschild, Structure and mechanism of vertebrate visual pigments, in: Molecular Mechanisms Oin Visual Transduction, D.G. Stravenga, W.J. de Grip and E.N. Pugh, eds, Elsevier Science B.V., Amsterdam, (2000) , pp. 1–54. doi:10.1016/S1383-8121(00)80004-4. |
[66] | M.R. Derrick, D. Stulik and J.M. Landry, Infrared Spectroscopy in Conservation Science, J. Paul Getty Trust, (1955) . |
[67] | R. Diller, R. Jakober, C. Schumann, F. Peters, J.P. Klare and M. Engelhard, The trans-cis isomerization reaction dynamics in sensory rhodopsin II by femtosecond time-resolved midinfrared spectroscopy: Chromophore and protein dynamics, Biopolymers 82: ((2006) ), 358–362. doi:10.1002/bip.20475. |
[68] | A.K. Dioumaev, H.T. Richter, L.S. Brown, M. Tanio, S. Tuzi, H. Saito, Y. Kimura, R. Needleman and J.K. Lanyi, Existence of a proton transfer chain in bacteriorhodopsin: Participation of Glu-194 in the release of protons to the extracellular surface, Biochemistry 37: ((1998) ), 2496–2506. doi:10.1021/bi971842m. |
[69] | G. Dollinger, L. Eisenstein, S.L. Lin, K. Nakanishi and J. Termini, Fourier transform infrared difference spectroscopy of bacteriorhodopsin and its photoproducts regenerated with deuterated tyrosine, Biochemistry 25: ((1986) ), 6524–6533. doi:10.1021/bi00369a028. |
[70] | A. Dong, P. Huang and W.S. Caughey, Protein secondary structures in water from second-derivative amide I infrared spectra, Biochemistry 29: ((1990) ), 3303–3308. doi:10.1021/bi00465a022. |
[71] | N.W. Downer, T.J. Bruchman and J.H. Hazzard, Infrared spectroscopic study of photoreceptor membrane and purple membrane. Protein secondary structure and hydrogen deuterium exchange, J. Biol. Chem. 261: ((1986) ), 3640–3647. |
[72] | M. Dunach, S. Berkowitz, T. Marti, Y.W. He, S. Subramaniam, H.G. Khorana and K.J. Rothschild, Ultraviolet-visible transient spectroscopy of bacteriorhodopsin mutants. Evidence for two forms of tyrosine-185 – phenylalanine, J. Biol. Chem. 265: ((1990) ), 16978–16984. |
[73] | M. Dunach, T. Marti, H.G. Khorana and K.J. Rothschild, Uv-visible spectroscopy of bacteriorhodopsin mutants: Substitution of Arg-82, Asp-85, Tyr-185, and Asp-212 results in abnormal light-dark adaptation, Proc. Natl. Acad. Sci. USA 87: ((1990) ), 9873–9877. doi:10.1073/pnas.87.24.9873. |
[74] | A.M. Dwivedi and S. Krimm, Vibrational analysis of peptides, polypeptides, and proteins. 19. Force fields for à-Helix and á-Sheet structures in a side-chain point-mass approximation, J. Phys. Chem. 88: ((1984) ), 620–627. doi:10.1021/j150647a056. |
[75] | B. Dzikovski, K. Earle, S. Pachtchenko and J. Freed, High-field ESR on aligned membranes: A simple method to record spectra from different membrane orientations in the magnetic field, Journal of Magnetic Resonance 179: ((2006) ), 273–279. doi:10.1016/j.jmr.2005.12.015. |
[76] | B.G. Dzikovski, P.P. Borbat and J.H. Freed, Spin-labeled gramicidin a: Channel formation and dissociation, Biophysical Journal 87: ((2004) ), 3504–3517. doi:10.1529/biophysj.104.044305. |
[77] | T.N. Earnest, J. Herzfeld and K.J. Rothschild, Polarized Fourier transform infrared spectroscopy of bacteriorhodopsin. Transmembrane alpha helices are resistant to hydrogen/deuterium exchange, Biophysical Journal 58: ((1990) ), 1539–1546. doi:10.1016/S0006-3495(90)82498-X. |
[78] | D. Eisenberg, John Edsall and protein science, Protein Science: A Publication of the Protein Society 1: ((1992) ), 1399–1401. doi:10.1002/pro.5560011101. |
[79] | A. Elliott and E.J. Ambrose, Structure of synthetic polypeptides, Nature 165: ((1950) ), 921–922. doi:10.1038/165921a0. |
[80] | A. Elliott, E.J. Ambrose and C. Robinson, Chain configurations in natured and denatured insulin: Evidence from infrared spectra, Nature 166: ((1950) ), 194. doi:10.1038/166194a0. |
[81] | M. Engelhard, K. Gerwert, B. Hess, W. Kreutz and F. Siebert, Light-driven protonation changes of internal aspartic acids of bacteriorhodopsin: An investigation by static and time-resolved infrared difference spectroscopy using [4-13C]aspartic acid labeled purple membrane, Biochemistry 24: ((1985) ), 400–407. doi:10.1021/bi00323a024. |
[82] | M. Engelhard, B. Hess, G. Metz, W. Kreutz, F. Siebert, J. Soppa and D. Oesterhelt, High resolution 13C-solid state NMR of bacteriorhodopsin: Assignment of specific aspartic acids and structural implications of single site mutations, Eur. Biophysical Journal 18: ((1990) ), 17–24. doi:10.1007/BF00185416. |
[83] | J.J. Englander, N.W. Downer and S.W. Englander, Reexamination of rhodopsin structure by hydrogen exchange, J. Biol. Chem. 257: ((1982) ), 7982–7986. |
[84] | H. Fabian and W. Mantele, Infrared Spectroscopy of Proteins: Handbook of Vibrational Spectroscopy, (2006) . |
[85] | M.T. Facciotti, S. Rouhani, F.T. Burkard, F.M. Betancourt, K.H. Downing, R.B. Rose, G. McDermott and R.M. Glaeser, Structure of an early intermediate in the M-state phase of the bacteriorhodopsin photocycle, Biophysical Journal 81: ((2001) ), 3442–3455. doi:10.1016/S0006-3495(01)75976-0. |
[86] | K. Fahmy, O. Weidlich, M. Engelhard, H. Sigrist and F. Siebert, Aspartic acid-212 of bacteriorhodopsin is ionized in the M and N photocycle intermediates: An FTIR study on specifically 13C-labeled reconstituted purple membranes, Biochemistry 32: ((1993) ), 5862–5869. doi:10.1021/bi00073a020. |
[87] | W. Fischer, S. Sonar, T. Marti, H.G. Khorana and K.J. Rothschild, Detection of a water molecule in the active site of bacteriorhodopsin: Hydrogen bonding changes during the primary photoreaction, Biochemistry 33: ((1994) ), 12757–12762. doi:10.1021/bi00209a005. |
[88] | W.B. Fischer and K.J. Rothschild, Water molecules are active during the primary photoreaction of bacteriorhodopsin, Society of Photo-Optical Instrumentation Engineers Proceedings Series 2089: ((1993) ), 118. |
[89] | E. Freier, S. Wolf and K. Gerwert, Proton transfer via a transient linear water-molecule chain in a membrane protein, Proc. Natl. Acad. Sci. USA 108: ((2011) ), 11435–11439. doi:10.1073/pnas.1104735108. |
[90] | U.P. Fringeli, The structure of lipids and proteins studied by attenuated total reflection (ATR) infrared spectroscopy. II. Oriented layers of a homologous series: Phosphatidylethanolamine to phosphatidylcholine, Z. Naturforsch. C 32: ((1977) ), 20–45. |
[91] | U.P. Fringeli and M. Fringeli, Pore formation in lipid membranes by alamethicin, Proc. Natl. Acad. Sci. USA 76: ((1979) ), 3852–3856. doi:10.1073/pnas.76.8.3852. |
[92] | Y. Furutani, H. Shimizu, Y. Asai, T. Fukuda, S. Oiki and H. Kandori, ATR-FTIR spectroscopy revealing the different vibrational modes of the selectivity filter interacting with K(+) and Na(+) in the open and collapsed conformations of the KcsA potassium channel, J. Phys. Chem. Lett. 3: ((2012) ), 3806–3810. doi:10.1021/jz301721f. |
[93] | F. Garczarek, L.S. Brown, J.K. Lanyi and K. Gerwert, Proton binding within a membrane protein by a protonated water cluster, Proc. Natl. Acad. Sci. USA 102: ((2005) ), 3633–3638. doi:10.1073/pnas.0500421102. |
[94] | F. Garczarek and K. Gerwert, Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy, Nature 439: ((2006) ), 109–112. doi:10.1038/nature04231. |
[95] | F. Garczarek, J. Wang, M.A. El-Sayed and K. Gerwert, The assignment of the different infrared continuum absorbance changes observed in the 3000–1800-cm−1 region during the bacteriorhodopsin photocycle, Biophysical Journal 87: ((2004) ), 2676–2682. doi:10.1529/biophysj.104.046433. |
[96] | Y. Gat, M. Grossjean, I. Pinevsky, H. Takei, Z. Rothman, H. Sigrist, A. Lewis and M. Sheves, Participation of bacteriorhodopsin active-site lysine backbone in vibrations associated with retinal photochemistry, Proc. Natl. Acad. Sci. USA 89: ((1992) ), 2434–2438. doi:10.1073/pnas.89.6.2434. |
[97] | M. Ge, D.E. Budil and J.H. Freed, ESR studies of spin-labeled membranes aligned by isopotential spin-dry ultracentrifugation: Lipid-protein interactions, Biophysical Journal 67: ((1994) ), 2326–2344. doi:10.1016/S0006-3495(94)80719-2. |
[98] | H. Georg, A. Barth, W. Kreutz, F. Siebert and W. Mantele, Structural changes of sarcoplasmic reticulum Ca(2+)-ATPase upon Ca2+ binding studied by simultaneous measurement of infrared absorbance changes and changes of intrinsic protein fluorescence, Biochim. Biophys. Acta 1188: ((1994) ), 139–150. doi:10.1016/0005-2728(94)90032-9. |
[99] | K. Gerwert, Molecular reaction mechanisms of proteins monitored by time-resolved FTIR-spectroscopy, Biological Chemistry 380: ((1999) ), 931–935. |
[100] | J.K. Ghosh, S.G. Peisajovich, M. Ovadia and Y. Shai, Structure-function study of a heptad repeat positioned near the transmembrane domain of Sendai virus fusion protein which blocks virus-cell fusion, The Journal of Biological Chemistry 273: ((1998) ), 27182–27190. doi:10.1074/jbc.273.42.27182. |
[101] | S. Gite, M. Lim and K.J. Rothschild, Cell-free protein synthesis systems: Biotechnological applications, Biotechnol. Genet. Eng. Rev. 22: ((2006) ), 151–169. doi:10.1080/02648725.2006.10648069. |
[102] | U. Gorne-Tschelnokow, F. Hucho, D. Naumann, A. Barth and W. Mantele, Fourier transform infrared (FTIR) spectroscopic investigation of the nicotinic acetylcholine receptor (nAChR). Investigation of agonist binding and receptor conformational changes by flash-induced release of ‘caged’ carbamoylcholine, FEBS Lett. 309: ((1992) ), 213–217. doi:10.1016/0014-5793(92)81097-6. |
[103] | P.R. Griffiths and J.A. de Hasenth, Fourier Transform Spectrometry, 2nd edn, John Wiley and Sons, Inc., (2007) . |
[104] | G. Grobner, A. Taylor, P.T. Williamson, G. Choi, C. Glaubitz, J.A. Watts, W.J. de Grip and A. Watts, Macroscopic orientation of natural and model membranes for structural studies, Analytical Biochemistry 254: ((1997) ), 132–138. doi:10.1006/abio.1997.2415. |
[105] | R. Gross, C. Schumann, M.M. Wolf, J. Herbst, R. Diller, N. Friedman and M. Sheves, Ultrafast protein conformational alterations in bacteriorhodopsin and its locked analogue BR5.12, The Journal of Physical Chemistry B 113: ((2009) ), 7851–7860. |
[106] | S.M. Gruner, K.J. Rothschild and N.A. Clark, X-ray diffraction and electron microscope study of phase separation in rod outer segment photoreceptor membrane multilayers, Biophysical Journal 39: ((1982) ), 241–251. doi:10.1016/S0006-3495(82)84514-1. |
[107] | C. Hackmann, J. Guijarro, I. Chizhov, M. Engelhard, C. Rodig and F. Siebert, Static and time-resolved step-scan Fourier transform infrared investigations of the photoreaction of halorhodopsin from Natronobacterium pharaonis: Consequences for models of the anion translocation mechanism, Biophysical Journal 81: ((2001) ), 394–406. doi:10.1016/S0006-3495(01)75708-6. |
[108] | G.S. Harbison, S.O. Smith, J.A. Pardoen, P.P.J. Mulder, J. Lugtenburg, J. Herzfeld, R. Mathies and R.G. Griffin, Solid-state carbon-13 NMR studies of retinal in bacteriorhodopsin, Biochemistry 23: ((1984) ), 2662–2667. doi:10.1021/bi00307a019. |
[109] | P.I. Haris and D. Chapman, The conformational analysis of peptides using Fourier transform IR spectroscopy, Biopolymers 37: ((1995) ), 251–263. doi:10.1002/bip.360370404. |
[110] | N.J. Harrick, Internal Reflection Spectroscopy, Interscience Publishers, New York, (1967) . |
[111] | M. Hatanaka, H. Kandori and A. Maeda, Localization and orientation of functional water molecules in bacteriorhodopsin as revealed by polarized Fourier transform infrared spectroscopy, Biophysical Journal 73: ((1997) ), 1001–1006. doi:10.1016/S0006-3495(97)78133-5. |
[112] | M. Hein, A.A. Wegener, M. Engelhard and F. Siebert, Time-resolved FTIR studies of Sensory Rhodopsin II (NpSRII) from Natronobacterium pharaonis: Implications for proton transport and receptor activation, Biophysical Journal 84: ((2003) ), 1208–1217. doi:10.1016/S0006-3495(03)74935-2. |
[113] | R. Henderson, J.M. Baldwin, T.A. Ceska, F. Zemlin, E. Beckman and K.H. Downing, Model for the structure of bacteriorhodopsin based on high-resolution electron cryomicroscopy, J. Mol. Biol. 213: ((1990) ), 899–929. doi:10.1016/S0022-2836(05)80271-2. |
[114] | R. Henderson and P.N.T. Unwin, Three-dimensional model of purple membrane obtained by electron microscopy, Nature 257: ((1975) ), 28–31. doi:10.1038/257028a0. |
[115] | W. Herschel, Observations tending to investigate the nature of the sun, in order to find the causes or symptoms of its variable emission of light and heat; with remarks on the use that may possibly be drawn from solar observations, Philosophical Transactions of the Royal Society of London XCII: ((1801) ), 265–318. doi:10.1098/rstl.1801.0015. |
[116] | R. Hienerwadel, A. Boussac, J. Breton, B.A. Diner and C. Berthomieu, Fourier transform infrared difference spectroscopy of photosystem II tyrosine D using site-directed mutagenesis and specific isotope labeling, Biochemistry 36: ((1997) ), 14712–14723. doi:10.1021/bi971521a. |
[117] | D.G. Hill and J.E. Baenziger, The net orientation of nicotinic receptor transmembrane alpha-helices in the resting and desensitized states, Biophysical Journal 91: ((2006) ), 705–714. doi:10.1529/biophysj.106.082693. |
[118] | W.D. Hoff, A. Xie, I.H. Van Stokkum, X.J. Tang, J. Gural, A.R. Kroon and K.J. Hellingwerf, Global conformational changes upon receptor stimulation in photoactive yellow protein, Biochemistry 38: ((1999) ), 1009–1017. doi:10.1021/bi980504y. |
[119] | T.L. Hsiao and K.J. Rothschild, Circular dichroism of oriented photoreceptor membrane film, Biochem. Biophys. Res. Commun. 94: ((1980) ), 618–624. doi:10.1016/0006-291X(80)91277-2. |
[120] | J.F. Hunt, T.N. Earnest, O. Bousche, K. Kalghatgi, K. Reilly, C. Horvath, K.J. Rothschild and D.M. Engelman, A biophysical study of integral membrane protein folding, Biochemistry 36: ((1997) ), 15156–15176. doi:10.1021/bi970146j. |
[121] | J.F. Hunt, P. Rath, K.J. Rothschild and D.M. Engelman, Spontaneous, pH-dependent membrane insertion of a transbilayer alpha-helix, Biochemistry 36: ((1997) ), 15177–15192. doi:10.1021/bi970147b. |
[122] | R. Kalmbach, I. Chizhov, M.C. Schumacher, T. Friedrich, E. Bamberg and M. Engelhard, Functional cell-free synthesis of a seven helix membrane protein: In situ insertion of bacteriorhodopsin into liposomes, J. Mol. Biol. 371: ((2007) ), 639–648. doi:10.1016/j.jmb.2007.05.087. |
[123] | H. Kandori, Role of internal water molecules in bacteriorhodopsin, Biochim. Biophys. Acta 1460: ((2000) ), 177–191. doi:10.1016/S0005-2728(00)00138-9. |
[124] | H. Kandori, T. Iwata, J. Hendriks, A. Maeda and K.J. Hellingwerf, Water structural changes involved in the activation process of photoactive yellow protein, Biochemistry 39: ((2000) ), 7902–7909. doi:10.1021/bi000357f. |
[125] | M. Kataoka, H. Kamikubo, F. Tokunga, L.S. Brown, Y. Yamazaki, A. Maeda, M. Sheves, R. Needleman and J.K. Lanyi, Energy coupling in an ion pump the reprotonation switch of bacteriorhodopsin, J. Mol. Biol. 243: ((1994) ), 621–638. doi:10.1016/0022-2836(94)90037-X. |
[126] | H.G. Khorana, Two light-transducing membrane proteins: Bacteriorhodopsin and the mammalian rhodopsin, Proc. Natl. Acad. Sci. USA 90: ((1993) ), 1166–1171. doi:10.1073/pnas.90.4.1166. |
[127] | C. Klammt, D. Schwarz, F. Lohr, B. Schneider, V. Dotsch and F. Bernhard, Cell-free expression as an emerging technique for the large scale production of integral membrane protein, The FEBS Journal 273: ((2006) ), 4141–4153. doi:10.1111/j.1742-4658.2006.05432.x. |
[128] | A. Koglin, C. Klammt, N. Trbovic, D. Schwarz, B. Schneider, B. Schafer, F. Lohr, F. Bernhard and V. Dotsch, Combination of cell-free expression and NMR spectroscopy as a new approach for structural investigation of membrane proteins, Magnetic Resonance in Chemistry: MRC 44: Spec No ((2006) ), S17–S23. |
[129] | J.M. Kralj, V.B. Bergo, J.J. Amsden, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Protonation state of Glu142 differs in the green- and blue-absorbing variants of proteorhodopsin, Biochemistry 47: ((2008) ), 3447–3453. doi:10.1021/bi7018964. |
[130] | J.M. Kralj, E.N. Spudich, J.L. Spudich and K.J. Rothschild, Raman spectroscopy reveals direct chromophore interactions in the Leu/Gln105 spectral tuning switch of proteorhodopsins, J. Phys. Chem. B 112: ((2008) ), 11770–11776. doi:10.1021/jp802629e. |
[131] | S. Krimm and J. Bandekar, Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins, in: Advances in Protein Chemistry, Academic Press, (1986) , pp. 181–365. |
[132] | S. Kumar, Infrared spectroscopy: Method development and ligand binding studies, Stockholm University, 2010. |
[133] | S. Kumar, C. Li, C. Montigny, M. le Maire and A. Barth, Conformational changes of recombinant Ca2+-ATPase studied by reaction-induced infrared difference spectroscopy, The FEBS Journal 280: ((2013) ), 5398–5407. doi:10.1111/febs.12131. |
[134] | J.K. Lanyi, Bacteriorhodopsin, Annual Review of Physiology 66: ((2004) ), 665–688. doi:10.1146/annurev.physiol.66.032102.150049. |
[135] | J.K. Lanyi, Proton transfers in the bacteriorhodopsin photocycle, Biochim. Biophys. Acta 1757: ((2006) ), 1012–1018. doi:10.1016/j.bbabio.2005.11.003. |
[136] | S.P. Laptenok, A. Lukacs, A. Gil, R. Brust, I.V. Sazanovich, G.M. Greetham, P.J. Tonge and S.R. Meech, Complete proton transfer cycle in GFP and its T203V and S205V mutants, Angew. Chem. Int. Ed. Engl. 54: ((2015) ), 9303–9307. doi:10.1002/anie.201503672. |
[137] | A. Lewis, Tunable laser resonance Raman spectroscopic investigations of the transduction process in vertebrate rod cells, Fed. Proc., Fed. Am. Soc. Exp. Biol. 35: ((1976) ), 51–53. |
[138] | A. Lewis, Primary photophysical and photochemical processes in visual excitation, Biophys. Struct. Mech. 3: (2) ((1977) ), 97–100. doi:10.1007/BF00535800. |
[139] | A. Lewis, The structure of the retinylidene chromophore in bathorhodopsin, Biophysical Journal 24: ((1978) ), 249–254. doi:10.1016/S0006-3495(78)85366-1. |
[140] | A. Lewis, M.A. Marcus, B. Ehrenberg and H. Crespi, Experimental evidence for secondary protein-chromophore interactions at the Schiff base linkage in bacteriorhodopsin: Molecular mechanism for proton pumping, Proc. Natl. Acad. Sci. USA 75: ((1978) ), 4642–4646. doi:10.1073/pnas.75.10.4642. |
[141] | M. Lim and P.A. Anfinrud, Ultrafast time-resolved IR studies of protein-ligand interactions, Methods Mol. Biol. 305: ((2005) ), 243–260. |
[142] | M. Lim, T.A. Jackson and P.A. Anfinrud, Ultrafast rotation and trapping of carbon monoxide dissociated from myoglobin, Nat. Struct. Biol. 4: ((1997) ), 209–214. doi:10.1038/nsb0397-209. |
[143] | X. Liu, M.J. Lee, M. Coleman, P. Rath, A. Nilsson, W.B. Fischer, M. Bizounok, J. Herzfeld, W.F. Karstens, J. Raap, J. Lugtenburg and K.J. Rothschild, Detection of threonine structural changes upon formation of the M-intermediate of bacteriorhodopsin: Evidence for assignment to Thr-89, Biochim. Biophys. Acta 1365: ((1998) ), 363–372. doi:10.1016/S0005-2728(98)00088-7. |
[144] | X.M. Liu, S. Sonar, C.P. Lee, M. Coleman, U.L. RajBhandary and K.J. Rothschild, Site-directed isotope labeling and FTIR spectroscopy: Assignment of tyrosine bands in the bR → M difference spectrum of bacteriorhodopsin, Biophys. Chem. 56: ((1995) ), 63–70. doi:10.1016/0301-4622(95)00016-Q. |
[145] | R.C. Lord and N.T. Yu, Laser-excited Raman spectroscopy of biomolecules. II. Native ribonuclease and alpha-chymotrypsin, J. Mol. Biol. 51: ((1970) ), 203–213. doi:10.1016/0022-2836(70)90137-3. |
[146] | R.C. Lord and N.T. Yu, Laser-excited Raman spectroscopy of biomolecules. I. Native lysozyme and its constituent amino acids, J. Mol. Biol. 50: ((1970) ), 509–524. doi:10.1016/0022-2836(70)90208-1. |
[147] | C.F. Ludlam, I.T. Arkin, X.M. Liu, M.S. Rothman, P. Rath, S. Aimoto, S.O. Smith, D.M. Engelman and K.J. Rothschild, Fourier transform infrared spectroscopy and site-directed isotope labeling as a probe of local secondary structure in the transmembrane domain of phospholamban, Biophysical Journal 70: ((1996) ), 1728–1736. doi:10.1016/S0006-3495(96)79735-7. |
[148] | C.F. Ludlam, S. Sonar, C.P. Lee, M. Coleman, J. Herzfeld, U.L. RajBhandary and K.J. Rothschild, Site-directed isotope labeling and ATR-FTIR difference spectroscopy of bacteriorhodopsin: The peptide carbonyl group of Tyr 185 is structurally active during the bR → N transition, Biochemistry 34: ((1995) ), 2–6. doi:10.1021/bi00001a001. |
[149] | G.J. Ludlam and K.J. Rothschild, Similarity of bacteriorhodopsin structural changes triggered by chromophore removal and light-driven proton transport, FEBS Lett. 407: ((1997) ), 285–288. doi:10.1016/S0014-5793(97)00351-7. |
[150] | H. Luecke, B. Schobert, H.T. Richter, J.P. Cartailler and J.K. Lanyi, Structural changes in bacteriorhodopsin during ion transport at 2 angstrom resolution [see comments], Science 286: ((1999) ), 255–261. doi:10.1126/science.286.5438.255. |
[151] | H. Luecke, B. Schobert, H.T. Richter, J.P. Cartailler and J.K. Lanyi, Structure of bacteriorhodopsin at 1.55 A resolution, J. Mol. Biol. 291: ((1999) ), 899–911. doi:10.1006/jmbi.1999.3027. |
[152] | G.M. MacDonald and B.A. Barry, Difference FT-IR study of a novel biochemical preparation of photosystem II, Biochemistry 31: ((1992) ), 9848–9856. doi:10.1021/bi00155a043. |
[153] | A. Maeda, H. Kandori, Y. Yamazaki, S. Nishimura, M. Hatanaka, Y.S. Chon, J. Sasaki, R. Needleman and J.K. Lanyi, Intramembrane signaling mediated by hydrogen-bonding of water and carboxyl groups in bacteriorhodopsin and rhodopsin, J. Biochem. 121: ((1997) ), 399–406. doi:10.1093/oxfordjournals.jbchem.a021602. |
[154] | A. Maeda, J. Sasaki, Y.J. Ohkita, M. Simpson and J. Herzfeld, Tryptophan perturbation in the L intermediate of bacteriorhodopsin: Fourier transform infrared analysis with indole-15N shift, Biochemistry 31: ((1992) ), 12543–12545. doi:10.1021/bi00165a001. |
[155] | A. Maeda, J. Sasaki, Y. Shichida and T. Yoshizawa, Water structural changes in the bacteriorhodopsin photocycle: Analysis by Fourier transform infrared spectroscopy, J. Biochem. 31: ((1992) ), 462–467. doi:10.1021/bi00117a023. |
[156] | A. Maeda, F.L. Tomson, R.B. Gennis, H. Kandori, T.G. Ebrey and S.P. Balashov, Relocation of internal bound water in bacteriorhodopsin during the photoreaction of M at low temperatures: An FTIR study, Biochemistry 39: ((2000) ), 10154–10162. doi:10.1021/bi000190q. |
[157] | W. Mantele, F. Siebert and W. Kreutz, Kinetic properties of rhodopsin and bacteriorhodopsin measured by kinetic infrared spectroscopy (KIS), Methods Enzymol. 88: ((1982) ), 729–741. doi:10.1016/0076-6879(82)88090-7. |
[158] | H. Marrero and K.J. Rothschild, Conformational changes in bacteriorhodopsin studied by infrared attenuated total reflection, Biophysical Journal 52: ((1987) ), 629–635. doi:10.1016/S0006-3495(87)83254-X. |
[159] | H. Marrero and K.J. Rothschild, Bacteriorhodopsin’s M412 and Br605 protein conformations are similar, FEBS Lett. 223: ((1987) ), 289–293. doi:10.1016/0014-5793(87)80306-X. |
[160] | I. Martin, H. Schaal, A. Scheid and J.M. Ruysschaert, Lipid membrane fusion induced by the human immunodeficiency virus type 1 gp41 N-terminal extremity is determined by its orientation in the lipid bilayer, Journal of Virology 70: ((1996) ), 298–304. |
[161] | R. Mathies, T.B. Freedman and L. Stryer, Resonance Raman studies of the conformation of retinal in rhodopsin and isorhodopsin, J. Mol. Biol. 109: ((1977) ), 367–372. doi:10.1016/S0022-2836(77)80040-5. |
[162] | R.A. Mathies, From femtoseconds to biology: Mechanism of bacteriorhodopsin’s light-driven proton pump, Proc. – Indian Acad. Sci., Chem. Sci. 103: ((1991) ), 283–293. |
[163] | K. Matsuzaki, T. Shioyama, E. Okamura, J. Umemura, T. Takenaka, Y. Takaishi, T. Fujita and K. Miyajima, A comparative study on interactions of alpha-aminoisobutyric acid containing antibiotic peptides, trichopolyn I and hypelcin A with phosphatidylcholine bilayers, Biochimica et Biophysica Acta 1070: ((1991) ), 419–428. doi:10.1016/0005-2736(91)90082-J. |
[164] | McCray and D.R. Trentham, Properties and uses of photoreactive caged compounds, Annu. Rev. Biophys. Biophys. Chem. 18: ((1989) ), 239–270. doi:10.1146/annurev.bb.18.060189.001323. |
[165] | A. Menikh, M.T. Saleh, J. Gariepy and J.M. Boggs, Orientation in lipid bilayers of a synthetic peptide representing the C-terminus of the A1 domain of shiga toxin. A polarized ATR-FTIR study, Biochemistry 36: ((1997) ), 15865–15872. doi:10.1021/bi970944+. |
[166] | T. Miyazawa and E.R. Blout, The infrared spectra of polypeptides in various conformations: Amide I and amide II bands, J. Am. Chem. Soc. 83: ((1961) ), 712–719. doi:10.1021/ja01464a042. |
[167] | T. Miyazawa, T. Shimanouchi and S. Mizushima, Normal vibrations of N-methylacetamide, J. Chem. Phys. 29: ((1958) ), 611–616. doi:10.1063/1.1744547. |
[168] | T. Mogi, L.J. Stern, N.R. Hackett and H.G. Khorana, Bacteriorhodopsin mutants containing single tyrosine to phenylalanine substitutions are all active in proton translocation, Proc. Natl. Acad. Sci. USA 84: ((1987) ), 5595–5599. doi:10.1073/pnas.84.16.5595. |
[169] | E. Nabedryk, M.P. Gingold and J. Breton, Orientation of gramicidin A transmembrane channel. Infrared dichroism study of gramicidin in vesicles, Biophysical Journal 38: ((1982) ), 243–249. doi:10.1016/S0006-3495(82)84555-4. |
[170] | R. Nakamura and N. Hamada, Vibrational energy flow in photoactive yellow protein revealed by infrared pump-visible probe spectroscopy, The Journal of Physical Chemistry B 119: ((2015) ), 5957–5961. |
[171] | A. Nilsson, P. Rath, J. Olejnik, M. Coleman and K.J. Rothschild, Protein conformational changes during the bacteriorhodopsin photocycle. A Fourier transform infrared/resonance Raman study of the alkaline form of the mutant Asp-85 → Asn, J. Biol. Chem. 270: ((1995) ), 29746–29751. doi:10.1074/jbc.270.50.29746. |
[172] | K. Noelker, O. Weidlich and F. Siebert, Chromophore and protein reactions of bacteriorhodopsin studied by sub-microsecond time-resolved step-scan FTIR spectroscopy, in: Time-Resolved Vib. Spectrosc. V, Springer Proc. Phys., Vol. 68: , (1992) , pp. 57–60. doi:10.1007/978-3-642-84771-4_17. |
[173] | J.I. Ogren, S. Mamaev, D. Russano, H. Li, J.L. Spudich and K.J. Rothschild, Retinal chromophore structure and Schiff base interactions in red-shifted channelrhodopsin-1 from Chlamydomonas augustae, Biochemistry 53: ((2014) ), 3961–3970. doi:10.1021/bi500445c. |
[174] | J.I. Ogren, A. Yi, S. Mamaev, H. Li, J. Lugtenburg, W.J. DeGrip, J.L. Spudich and K.J. Rothschild, Comparison of the structural changes occurring during the primary phototransition of two different Channelrhodopsins from Chlamydomonas algae, Biochemistry 54: ((2015) ), 377–388. doi:10.1021/bi501243y. |
[175] | J.I. Ogren, A. Yi, S. Mamaev, H. Li, J.L. Spudich and K.J. Rothschild, Proton transfers in a Channelrhodopsin-1 studied by FTIR difference spectroscopy and site-directed mutagenesis, J. Biol. Chem. 290: ((2015) ), 12719–12739. doi:10.1074/jbc.M114.634840. |
[176] | J.I. Ogren, A. Yi, S. Mamaev, H. Li, J.L. Spudich and K.J. Rothschild, Proton transfers in a channelrhodopsin-1 studied by Fourier transform infrared (FTIR) difference spectroscopy and site-directed mutagenesis, J. Biol. Chem. 290: ((2015) ), 12719–12730. doi:10.1074/jbc.M114.634840. |
[177] | A.R. Oseroff and R.H. Callender, Resonance Raman spectroscopy of rhodopsin in retinal disk membranes, Biochemistry 13: ((1974) ), 4243–4248. doi:10.1021/bi00717a027. |
[178] | H. Otto, T. Marti, M. Holz, T. Mogi, L.J. Stern, F. Engel, H.G. Khorana and M.P. Heyn, Substitution of amino acids Asp-85, Asp-212, and Arg-82 in bacteriorhodopsin affects the proton release phase of the pump and the pK of the Schiff base, Proc. Natl. Acad. Sci. USA 87: ((1990) ), 1018–1022. doi:10.1073/pnas.87.3.1018. |
[179] | F.S. Parker, Applications of Infrared, Raman and Resonance Raman Spectroscopy in Biochemistry, Plenum Press, New York, (1983) . |
[180] | B. Pedrini, C.J. Tsai, G. Capitani, C. Padeste, M.S. Hunter, N.A. Zatsepin, A. Barty, W.H. Benner, S. Boutet, G.K. Feld, S.P. Hau-Riege, R.A. Kirian, C. Kupitz, M. Messerschmitt, J.I. Ogren, T. Pardini, B. Segelke, G.J. Williams, J.C. Spence, R. Abela, M. Coleman, J.E. Evans, G.F. Schertler, M. Frank and X.D. Li, 7 A resolution in protein two-dimensional-crystal X-ray diffraction at Linac Coherent Light Source, Philos. Trans. R. Soc. Lond. B Biol. Sci. 369: ((2014) ), 20130500. doi:10.1098/rstb.2013.0500. |
[181] | M. Poo and R.A. Cone, Lateral diffusion of rhodopsin in the photoreceptor membrane, Nature 247: ((1974) ), 438–441. doi:10.1038/247438a0. |
[182] | P. Rath, O. Bousché, A.R. Merrill, W.A. Cramer and K.J. Rothschild, Fourier transform infrared evidence for a predominantly alpha-helical structure of the membrane bound channel forming COOH-terminal peptide of colicin E1, Biophysical Journal 59: ((1991) ), 516–522. doi:10.1016/S0006-3495(91)82268-8. |
[183] | P. Rath, W.J. DeGrip and K.J. Rothschild, Photoactivation of rhodopsin causes an increased hydrogen–deuterium exchange of buried peptide groups, Biophysical Journal 74: ((1998) ), 192–198. doi:10.1016/S0006-3495(98)77779-3. |
[184] | P. Rath, E. Spudich, D.D. Neal, J.L. Spudich and K.J. Rothschild, Asp76 is the Schiff base counterion and proton acceptor in the proton-translocating form of sensory rhodopsin I, Biochemistry 35: ((1996) ), 6690–6696. doi:10.1021/bi9600355. |
[185] | V. Raussens, H. de Jongh, M. Pezolet, J.M. Ruysschaert and E. Goormaghtigh, Secondary structure of the intact H+, K+-ATPase and of its membrane-embedded region. An attenuated total reflection infrared spectroscopy, circular dichroism and Raman spectroscopy study, European Journal of Biochemistry/FEBS 252: ((1998) ), 261–267. doi:10.1046/j.1432-1327.1998.2520261.x. |
[186] | C. Ricci, Dynamics of the cell membranes, Annali Sclavo; rivista di microbiologia e di immunologia 17: ((1975) ), 1–16. |
[187] | C. Rodig, I. Chizhov, O. Weidlich and F. Siebert, Time-resolved step-scan Fourier transform infrared spectroscopy reveals differences between early and late M intermediates of bacteriorhodopsin [in process citation], Biophysical Journal 76: ((1999) ), 2687–2701. doi:10.1016/S0006-3495(99)77421-7. |
[188] | P. Roepe, P.L. Ahl, S.K. Das Gupta, J. Herzfeld and K.J. Rothschild, Tyrosine and carboxyl protonation changes in the bacteriorhodopsin photocycle. 1. M412 and L550 intermediates, Biochemistry 26: ((1987) ), 6696–6707. doi:10.1021/bi00395a020. |
[189] | P. Roepe, D. Gray, J. Lugtenburg, E.M.M. van den Berg, J. Herzfeld and K.J. Rothschild, FTIR evidence for tryptophan perturbations during the bacteriorhodopsin photocycle, J. Am. Chem. Soc. 110: ((1988) ), 7223–7224. doi:10.1021/ja00229a052. |
[190] | P. Roepe, P. Scherrer, P.L. Ahl, S.K. Das Gupta, R.A. Bogomolni, J. Herzfeld and K.J. Rothschild, Tyrosine and carboxyl protonation changes in the bacteriorhodopsin photocycle. 2. Tyrosines-26 and -64, Biochemistry 26: ((1987) ), 6708–6717. doi:10.1021/bi00395a021. |
[191] | P.D. Roepe, P.L. Ahl, J. Herzfeld, J. Lugtenburg and K.J. Rothschild, Tyrosine protonation changes in bacteriorhodopsin. A Fourier transform infrared study of bR548 and its primary photoproduct, J. Biol. Chem. 263: ((1988) ), 5110–5117. |
[192] | K. Rothschild and H. Marrero, Infrared evidence that the Schiff base of bacteriorhodopsin is protonated: bR570 and K intermediates, Proc. Natl. Acad. Sci. USA 79: ((1982) ), 4045–4049. doi:10.1073/pnas.79.13.4045. |
[193] | K.J. Rothschild, J. Andrew, W.J. DeGrip and H.E. Stanley, Opsin structure probed by Raman spectroscopy of photorecptor membranes, Science 191: ((1976) ), 1176–1178. doi:10.1126/science.1257742. |
[194] | K.J. Rothschild, P.V. Argade, T.N. Earnest, K.S. Huang, E. London, M.J. Liao, H. Bayley, H.G. Khorana and J. Herzfeld, The site of attachment of retinal in bacteriorhodopsin. A resonance Raman study, J. Biol. Chem. 257: ((1982) ), 8592–8595. |
[195] | K.J. Rothschild, I.M. Asher, E. Anastassakis and H.E. Stanley, Raman spectroscopic evidence for two conformations of uncomplexed valinomycin in the solid state, Science 182: ((1973) ), 384–386. doi:10.1126/science.182.4110.384. |
[196] | K.J. Rothschild, I.M. Asher and H.E. Stanley, Raman spectroscopy of uncomplexed valinomycin in polar and non-polar solutions, J. Am. Chem. Soc. 99: ((1977) ), 2032–2039. doi:10.1021/ja00449a003. |
[197] | K.J. Rothschild, O. Bousche, M.S. Braiman, C.A. Hasselbacher and J.L. Spudich, Fourier transform infrared study of the halorhodopsin chloride pump, Biochemistry 27: ((1988) ), 2420–2424. doi:10.1021/bi00407a026. |
[198] | K.J. Rothschild, M.S. Braiman, Y.W. He, T. Marti and H.G. Khorana, Vibrational spectroscopy of bacteriorhodopsin mutants. Evidence for the interaction of aspartic acid 212 with tyrosine 185 and possible role in the proton pump mechanism, J. Biol. Chem. 265: ((1990) ), 16985–16991. |
[199] | K.J. Rothschild, M.S. Braiman, T. Mogi, L.J. Stern and H.G. Khorana, Conserved amino acids in F-helix of bacteriorhodopsin form part of a retinal binding pocket, FEBS Lett. 250: ((1989) ), 448–452. doi:10.1016/0014-5793(89)80774-4. |
[200] | K.J. Rothschild, W.A. Cantore and H. Marrero, Fourier transform infrared difference spectra of intermediates in rhodopsin bleaching, Science 219: ((1983) ), 1333–1335. doi:10.1126/science.6828860. |
[201] | K.J. Rothschild and N.A. Clark, Polarized infrared spectroscopy of oriented purple membrane, Biophysical Journal 25: ((1979) ), 473–487. doi:10.1016/S0006-3495(79)85317-5. |
[202] | K.J. Rothschild, N.A. Clark, K.M. Rosen, R. Sanches and T.L. Hsiao, Spectroscopic study of photoreceptor membrane incorporated into a multilamellar film, Biochem. Biophys. Res. Commun. 92: ((1980) ), 1266–1272. doi:10.1016/0006-291X(80)90423-4. |
[203] | K.J. Rothschild, S.A. Ellias, A. Essig and H.E. Stanley, Nonequilibrium linear behavior of biological systems. Existence of enzyme-mediated multidimensional inflection points, Biophysical Journal 30: ((1980) ), 209–230. doi:10.1016/S0006-3495(80)85090-9. |
[204] | K.J. Rothschild, S. Gite, S. Mamaev and J. Olejnik, Building photonic proteins, in: CRC Handbook of Organic Photochemistry and Photobiology, F. Lenci, ed., CRC, (2003) , pp. 1–21. |
[205] | K.J. Rothschild, D. Gray, T. Mogi, T. Marti, M.S. Braiman, L.J. Stern and H.G. Khorana, Vibrational spectroscopy of bacteriorhodopsin mutants: Chromophore isomerization perturbs tryptophan-86, Biochemistry 28: ((1989) ), 7052–7059. doi:10.1021/bi00443a041. |
[206] | K.J. Rothschild, Y.W. He, D. Gray, P.D. Roepe, S.L. Pelletier, R.S. Brown and J. Herzfeld, Fourier transform infrared evidence for proline structural changes during the bacteriorhodopsin photocycle, Proc. Natl. Acad. Sci. USA 86: ((1989) ), 9832–9835. doi:10.1073/pnas.86.24.9832. |
[207] | K.J. Rothschild, H. Marrero, M. Braiman and R. Mathies, Primary photochemistry of bacteriorhodopsin: Comparison of Fourier transform infrared difference spectra with resonance Raman spectra, Photochem. Photobiol. 40: ((1984) ), 675–679. doi:10.1111/j.1751-1097.1984.tb05359.x. |
[208] | K.J. Rothschild, P. Roepe, P.L. Ahl, T.N. Earnest, R.A. Bogomolni, S.K. Das Gupta, C.M. Mulliken and J. Herzfeld, Evidence for a tyrosine protonation change during the primary phototransition of bacteriorhodopsin at low temperature, Proc. Natl. Acad. Sci. USA 83: ((1986) ), 347–351. doi:10.1073/pnas.83.2.347. |
[209] | K.J. Rothschild, R. Sanches, T.L. Hsiao and N.A. Clark, A spectroscopic study of rhodopsin alpha-helix orientation, Biophysical Journal 31: ((1980) ), 53–64. doi:10.1016/S0006-3495(80)85040-5. |
[210] | K.J. Rothschild and S. Sonar, Bacteriorhodopsin: New biophysical perspectives, in: CRC Handbook of Organic Photochemistry and Photobiology, W.M. Horspool and P.-S. Song, eds, CRC Press Inc., (1995) , pp. 1521–1544. |
[211] | K.J. Rothschild and H.E. Stanley, Globular membrane proteins as functional units of ionic transport, in: Biomedical Physics and Biomaterials Science, H.E. Stanley, ed., MIT Press, Cambridge, (1972) , pp. 3–23. |
[212] | K.J. Rothschild and H.E. Stanley, Models of ionic transport in biological membranes. Raman spectroscopy as a probe of valinomycin, gramicidin A’, and rhodopsin conformations, Am. J. Clin. Pathol. 63: ((1975) ), 695–713. doi:10.1093/ajcp/63.5.695. |
[213] | K.J. Rothschild, M. Zagaeski and W.A. Cantore, Conformational changes of bacteriorhodopsin detected by Fourier transform infrared difference spectroscopy, Biochem. Biophys. Res. Commun. 103: ((1981) ), 483–489. doi:10.1016/0006-291X(81)90478-2. |
[214] | A.B. Rupenyan, J. Vreede, I.H. van Stokkum, M. Hospes, J.T. Kennis, K.J. Hellingwerf and M.L. Groot, Proline 68 enhances photoisomerization yield in photoactive yellow protein, The Journal of Physical Chemistry B 115: ((2011) ), 6668–6677. |
[215] | R. Sachse, S.K. Dondapati, S.F. Fenz, T. Schmidt and S. Kubick, Membrane protein synthesis in cell-free systems: From bio-mimetic systems to bio-membranes, FEBS Lett. 588: ((2014) ), 2774–2781. doi:10.1016/j.febslet.2014.06.007. |
[216] | E.C. Saint Clair, J.I. Ogren, S. Mamaev, D. Russano, J.M. Kralj and K.J. Rothschild, Near-IR resonance Raman spectroscopy of archaerhodopsin 3: Effects of transmembrane potential, J. Phys. Chem. B 116: ((2012) ), 14592–14601. doi:10.1021/jp309996a. |
[217] | Y. Shen, C.R. Safinya, K.S. Liang, A.F. Ruppert and K.J. Rothschild, Stabilization of the membrane protein bacteriorhodopsin to 140°C in two-dimensional films, Nature 366: ((1993) ), 48–50. doi:10.1038/366048a0. |
[218] | F. Siebert and W. Maentele, Investigation of the primary photochemistry of bacteriorhodopsin by low-temperature Fourier-transform infrared spectroscopy, Eur. J. Biochem. 130: ((1983) ), 565–573. doi:10.1111/j.1432-1033.1983.tb07187.x. |
[219] | F. Siebert, W. Maentele and K. Gerwert, Fourier-transform infrared spectroscopy applied to rhodopsin. The problem of the protonation state of the retinylidene Schiff base reinvestigated, Eur. J. Biochem. 136: ((1983) ), 119–127. doi:10.1111/j.1432-1033.1983.tb07714.x. |
[220] | F. Siebert and W. Mantele, Investigations of the rhodopsin/meta I and rhodopsin/meta II transitions of bovine rod outer segments by means of kinetic infrared spectroscopy, Biophysics of Structure and Mechanism 6: ((1980) ), 147–164. doi:10.1007/BF00535751. |
[221] | F. Siebert, W. Mantele and W. Kreutz, Flash-induced kinetic infrared spectroscopy applied to biochemical systems, Biophysics of Structure and Mechanism 6: ((1980) ), 139–146. doi:10.1007/BF00535750. |
[222] | S.J. Singer and G.L. Nicolson, The fluid mosaic model of the structure of cell membranes, Science 175: ((1972) ), 720–731. doi:10.1126/science.175.4023.720. |
[223] | A.L. Smith, Applied Infrared Spectroscopy: Fundamentals, Techniques, and Analytical Problem Solving, John Wiley and Sons, (1979) . |
[224] | B.C. Smith, Fundamentals of Fourier Transform Infrared Spectroscopy, CRC Press, (1996) . |
[225] | S.O. Smith, M.S. Braiman, A.B. Myers, J.A. Pardoen, J.M.L. Courtin, C. Winkel, J. Lugtenburg and R.A. Mathies, Vibrational analysis of the all-trans-retinal chromophore in light-adapted bacteriorhodopsin, J. Am. Chem. Soc. 109: ((1987) ), 3108–3125. doi:10.1021/ja00244a038. |
[226] | S. Sonar, C.P. Lee, M. Coleman, N. Patel, X. Liu, T. Marti, H.G. Khorana, U.L. RajBhandary and K.J. Rothschild, Site-directed isotope labelling and FTIR spectroscopy of bacteriorhodopsin, Nat. Struct. Biol. 1: ((1994) ), 512–517. doi:10.1038/nsb0894-512. |
[227] | S. Sonar, C.P. Lee, C.F. Ludlam, X.-M. Liu, M. Coleman, T. Marti, U.L. RajBhandary and K.J. Rothschild, Site-directed isoltope labeling of membrane proteins: A new tool for spectroscopists, in: Techniques in Protein Chemistry VII, D.R. Marshak, ed., Academic Press, (1996) , pp. 152–159. |
[228] | G. Souvignier and K. Gerwert, Proton uptake mechanism of bacteriorhodopsin as determined by time-resolved stroboscopic-FTIR-spectroscopy, Biophysical Journal 63: ((1992) ), 1393–1405. doi:10.1016/S0006-3495(92)81722-8. |
[229] | L.J. Stern, P.L. Ahl, T. Marti, T. Mogi, M. Dunach, S. Berkowitz, K.J. Rothschild and H.G. Khorana, Substitution of membrane-embedded aspartic acids in bacteriorhodopsin causes specific changes in different steps of the photochemical cycle, Biochemistry 28: ((1989) ), 10035–10042. doi:10.1021/bi00452a023. |
[230] | P. Stevenson, C. Gotz, C.R. Baiz, J. Akerboom, A. Tokmakoff and A. Vaziri, Visualizing KcsA conformational changes upon ion binding by infrared spectroscopy and atomistic modeling, The Journal of Physical Chemistry B 119: ((2015) ), 5824–5831. |
[231] | D. Stoner-Ma, A.A. Jaye, K.L. Ronayne, J. Nappa, S.R. Meech and P.J. Tonge, An alternate proton acceptor for excited-state proton transfer in green fluorescent protein: Rewiring GFP, J. Am. Chem. Soc. 130: ((2008) ), 1227–1235. doi:10.1021/ja0754507. |
[232] | S. Subramaniam, D.A. Greenhalgh, P. Rath, K.J. Rothschild and H.G. Khorana, Replacement of leucine-93 by alanine or threonine slows down the decay of the N and O intermediates in the photocycle of bacteriorhodopsin: Implications for proton uptake and 13-cis-retinal – All-trans-retinal reisomerization, Proc. Natl. Acad. Sci. USA 88: ((1991) ), 6873–6877. doi:10.1073/pnas.88.15.6873. |
[233] | H. Susi and D.M. Byler, Resolution-enhanced Fourier transform infrared spectroscopy of enzymes, Methods Enzymol. 130: ((1986) ), 290–311. doi:10.1016/0076-6879(86)30015-6. |
[234] | H. Susi, S.N. Timasheff and L. Stevens, Infrared spectra and protein conformations in aqueous solutions. I. The amide I band in H2O and D2O solutions, J. Biol. Chem. 242: ((1967) ), 5460–5466. |
[235] | S. Szára, D. Oesterhelt and P. Ormos, pH-induced structural changes in bacteriorhodopsin studied by Fourier transform infrared spectroscopy, Biophysical Journal 67: ((1994) ), 1706–1712. doi:10.1016/S0006-3495(94)80644-7. |
[236] | H. Take, Y. Gat, Z. Rothman, A. Lewis and M. Sheves, Active site lysine backbone undergoes conformational changes in the bacteriorhodopsin photocycle, J. Biol. Chem. 269: ((1994) ), 7387–7389. |
[237] | L.K. Tamm and S.A. Tatulian, Infrared spectroscopy of proteins and peptides in lipid bilayers, Quarterly Reviews of Biophysics 30: ((1997) ), 365–429. doi:10.1017/S0033583597003375. |
[238] | T.T. Tapmeier, A. Moshnikova, J. Beech, D. Allen, P. Kinchesh, S. Smart, A. Harris, A. McIntyre, D.M. Engelman, O.A. Andreev, Y.K. Reshetnyak and R.J. Muschel, The pH low insertion peptide pHLIP variant 3 as a novel marker of acidic malignant lesions, Proc. Natl. Acad. Sci. USA 112: ((2015) ), 9710–9715. doi:10.1073/pnas.1509488112. |
[239] | M.C. Thielges, J.Y. Axup, D. Wong, H.S. Lee, J.K. Chung, P.G. Schultz and M.D. Fayer, Two-dimensional IR spectroscopy of protein dynamics using two vibrational labels: A site-specific genetically encoded unnatural amino acid and an active site ligand, The Journal of Physical Chemistry B 115: ((2011) ), 11294–11304. |
[240] | J. Tittor, U. Schweiger, D. Oesterhelt and E. Bamberg, Inversion of proton translocation in bacteriorhodopsin mutants D85N, D85T, and D85, 96N, Biophysical Journal 67: ((1994) ), 1682–1690. doi:10.1016/S0006-3495(94)80642-3. |
[241] | D.W. Urry, A molecular theory of ion-conducting channels: A field-dependent transition between conducting and nonconducting conformations, Proc. Natl. Acad. Sci. USA 69: ((1972) ), 1610–1614. doi:10.1073/pnas.69.6.1610. |
[242] | J.J. van Thor, G.Y. Georgiev, M. Towrie and J.T. Sage, Ultrafast and low barrier motions in the photoreactions of the green fluorescent protein, J. Biol. Chem. 280: ((2005) ), 33652–33659. doi:10.1074/jbc.M505473200. |
[243] | L.J. van Wilderen, M.A. van der Horst, I.H. van Stokkum, K.J. Hellingwerf, R. van Grondelle and M.L. Groot, Ultrafast infrared spectroscopy reveals a key step for successful entry into the photocycle for photoactive yellow protein, Proc. Natl. Acad. Sci. USA 103: ((2006) ), 15050–15055. doi:10.1073/pnas.0603476103. |
[244] | F. Wang, C. Zhao and P.L. Polavarapu, A study of the conformations of valinomycin in solution phase, Biopolymers 75: ((2004) ), 85–93. doi:10.1002/bip.20103. |
[245] | N. Wright and L.W. Hersher, Journal of the Optical Society of America 37: ((1947) ), 211. doi:10.1364/JOSA.37.000211. |
[246] | A. Xie, W.D. Hoff, A.R. Kroon and K.J. Hellingwerf, Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein, Biochemistry 35: ((1996) ), 14671–14678. doi:10.1021/bi9623035. |
[247] | T. Yabuki, T. Kigawa, N. Dohmae, K. Takio, T. Terada, Y. Ito, E.D. Laue, J.A. Cooper, M. Kainosho and S. Yokoyama, Dual amino acid-selective and site-directed stable-isotope labeling of the human c-Ha-Ras protein by cell-free synthesis, Journal of Biomolecular NMR 11: ((1998) ), 295–306. doi:10.1023/A:1008276001545. |
[248] | S. Yamamoto, M. Straka, H. Watarai and P. Bour, Formation and structure of the potassium complex of valinomycin in solution studied by Raman optical activity spectroscopy, Phys. Chem. Chem. Phys. 12: ((2010) ), 11021–11032. doi:10.1039/c003277h. |
[249] | S. Ye, T. Huber, R. Vogel and T.P. Sakmar, FTIR analysis of GPCR activation using azido probes, Nature Chemical Biology 5: ((2009) ), 397–399. doi:10.1038/nchembio.167. |
[250] | A. Yi, N. Mamaeva, H. Li, J.L. Spudich and K.J. Rothschild, Resonance Raman study of an anion Channelrhodopsin: Effects of mutations near the retinylidene Schiff base, Biochemistry 55: ((2016) ), 2371–2380. doi:10.1021/acs.biochem.6b00104. |
[251] | G. Zundel, Hydrogen-bonded chains with large proton polarizability as charge conductors in proteins bacteriorhodopsin and the F subunit of E. coli, Journal of Molecular Structure 322: ((1994) ), 33–42. doi:10.1016/0022-2860(94)87019-5. |


