PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology?
Abstract
The first clinical description of Parkinson’s disease (PD) will embrace its two century anniversary in 2017. For the past 30 years, mitochondrial dysfunction has been hypothesized to play a central role in the pathobiology of this devastating neurodegenerative disease. The identifications of mutations in genes encoding PINK1 (PTEN-induced kinase 1) and Parkin (E3 ubiquitin ligase) in familial PD and their functional association with mitochondrial quality control provided further support to this hypothesis. Recent research focused mainly on their key involvement in the clearance of damaged mitochondria, a process known as mitophagy. It has become evident that there are many other aspects of this complex regulated, multifaceted pathway that provides neuroprotection. As such, numerous additional factors that impact PINK1/Parkin have already been identified including genes involved in other forms of PD. A great pathogenic overlap amongst different forms of familial, environmental and even sporadic disease is emerging that potentially converges at the level of mitochondrial quality control. Tremendous efforts now seek to further detail the roles and exploit PINK1 and Parkin, their upstream regulators and downstream signaling pathways for future translation. This review summarizes the latest findings on PINK1/Parkin-directed mitochondrial quality control, its integration and cross-talk with other disease factors and pathways as well as the implications for idiopathic PD. In addition, we highlight novel avenues for the development of biomarkers and disease-modifying therapies that are based on a detailed understanding of the PINK1/Parkin pathway.
THE ROLE OF MITOCHONDRIAL DYSFUNCTION IN PD PATHOGENESIS
Mitochondria are energy producing organelles that are enclosed by a double membrane and form a complex and highly dynamic network regulated by constant fission and fusion [1]. An electrochemical gradient (ΔΨm) across the inner mitochondrial membrane (IMM) is important for ATP production [2, 3]. Other important functions of mitochondria include Ca2+ buffering and the regulation of cellular apoptosis [4, 5]. While mitochondria harbor their own genome, it only encodes few subunits of the mitochondrial respiratory chain complexes [6]. The majority of mitochondrial proteins are encoded by the nuclear genome, thus requiring a coordinated transcription, translation, import into, and protein complex assembly inside mitochondria [7]. The importance of intact, functional mitochondria is evident by a variety of symptoms that are associated with classical mitochondrial diseases [1]. Especially vulnerable are tissues with a high energy demand such as brain, muscle, heart, kidney, and liver. Neurological symptoms include developmental delays, declining mental abilities, seizures, blindness, and loss of motor skills.
In addition, an increasing body of evidence suggests that mitochondrial dysfunction is associated with several age-related diseases and the aging process itself. Initial studies showed that dysfunctional mitochondria accumulate during aging [8], which is a major risk factor for many neurodegenerative diseases including PD [9]. The characteristic motor and non-motor symptoms of PD result from striatal depletion of the neurotransmitter dopamine (DA), secondary to the relatively selective degeneration of DA neurons in the substantia nigra pars compacta (SNpc). DA neurons are characterized by distinctive physiological features that could contribute to their vulnerability and sensitivity toward mitochondrial dysfunction [10–12]. One of the theories explaining the susceptibility of DA neurons is based on their high metabolic activities resulting in the increased production of harmful reactive oxygen species (ROS), which subsequently induce mitochondrial damage [13]. The deleterious effects could then spread within the entire mitochondrial network, eventually resulting in neuronal cell death [14].
The first direct evidence linking mitochondrial dysfunction to PD appeared with the observation that accidental exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) caused parkinsonism and DA neuron degeneration in patients [15]. The product of MPTP oxidation, MPP+, was shown to selectively inhibit complex I, a component of the mitochondrial respiratory chain, which subsequently induces DA neuronal death [16, 17]. Later, reduced activity of complex I was also observed in different tissues of sporadic PD patients, such as the SNpc, skeletal muscle and platelets [18–20]. The exposure to other toxins such as the pesticide rotenone and the herbicide paraquat that inhibit complex I or induce ROS, respectively, also result in loss of nigrostriatal DA neurons and have been linked to parkinsonian phenotypes in humans and in animal models [21].
THE RECESSIVE PD GENES PINK1 AND PARKIN
Among others, the identification of the two genes associated with PD, PINK1 (PARK6) [22] and Parkin (PARK2) [23], became milestones in PD research. Homozygous or compound heterozygous mutations in both genes are the most common causes of recessive early-onset PD (usually below age of 40) [24]. Though both genes are ubiquitously expressed, complete loss of either function results in selective loss of DA neurons and manifestation of tremor, bradykinesia, and rigidity. Besides earlier disease onset, PINK1 and Parkin associated PD develops generally more benign than idiopathic disease, with a slower disease progression as well as a good and sustained response to levodopa treatment [25, 26]. Aside from the cardinal symptoms of PD, PINK1 patients more frequently show psychiatric symptoms (depression, anxiety, and psychosis) [27]. Neuropathological examination has casted some doubts as to whether Parkin-related PD has the same disease mechanism as sporadic disease, since several studies described the absence of Lewy bodies, the pathognomonic sign of PD. However, to date, at least five studies reported the presence of Lewy bodies in Parkin cases [28–32]. In addition, two out of three PINK1 cases that were studied so far reported Lewy bodies [33–35]. The reason for the neuropathological heterogeneity is unclear but might relate to some specific mutations within PINK1 or Parkin and/or the age of the patients when they decease.
While a mitochondrial function had been suggested for PINK1 from the beginning simply based on its mitochondrial targeting sequence (MTS), a mitochondrial role for Parkin remained elusive for several years. The first studies supporting the involvement of both in regulating mitochondrial function were performed in Drosophila [36–38]. PINK1-/- and Parkin-/- mutant flies exhibited similar mitochondrial morphological abnormalities, locomotor deficits, muscle degeneration, male sterility as well as neuronal loss [37, 38]. The PINK1-/- phenotype was rescued by Parkin overexpression, but not vice versa, suggesting that PINK1 acts upstream of Parkin in a common, linear pathway [37–39]. Mitochondrial abnormalities and rescue of PINK1 loss by Parkin, but not PD-associated mutations were confirmed in human cell lines and primary fibroblasts [40]. However, PINK1 or Parkin knockout mice showed only subtle phenotypes with some mitochondrial dysfunction, yet without overt pathological changes in ultrastructure [41, 42].
In a breakthrough study in 2008, massive Parkin translocation from the cytosol to damaged mitochondria was observed after treatment with the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), a chemical that dissipates ΔΨm [43]. Strikingly, Parkin recruitment to mitochondria was demonstrated shortly after to depend on the presence of catalytically active PINK1 by several laboratories [44–47]. Since PINK1 and Parkin were shown to coordinate the selective clearance of damaged mitochondria via the autophagy/lysosome pathway (mitophagy), this also provided a link between the two major cellular dysfunctions implicated in PD pathogenesis, namely alterations in mitochondria and in cellular degradation [48]. Importantly, PD-associated mutations in either gene abrogated this sequential process at different steps and through distinct pathomechanisms, underscoring the etiological importance of this pathway [44, 49, 50].
THE MITOCHONDRIAL KINASE PINK1
PINK1 was initially identified in an expression analysis of genes that were upregulated upon introduction of the tumor growth suppressor phosphatase and tensin homolog (PTEN) and was named accordingly (PTEN-induced putative kinase 1) [51]. PINK1, a 581 amino acid protein, contains an N-terminal MTS, a transmembrane domain (TM), a highly conserved serine/threonine kinase domain, and a C-terminal auto-regulatory domain (Fig. 1A). The overall frequency of PINK1 mutations in early-onset PD ranges between 1–9% depending on the ethnicity of the subjects [52]. Although crystal structures of PINK1 are currently unavailable, in silico analysis and structural modeling indicated some similarities between its kinase domain and members of the calmodulin-dependent kinase family [53–56].
Two critical regulatory regions within PINK1 are the cleavage sites of the mitochondrial processing peptidase (MPP) and the presenilin-associated rhomboid-like protease (PARL) [57–59]. PINK1 activity is determined by autophosphorylation on three residues (Ser228, Thr257, and Ser402) in the activation loop [60–62]. While a complete loss of kinase activity is unequivocally linked to early-onset PD, as seen in, for instance, homozygous p.Q456X carriers [63], a single PINK1 mutation that only causes partial reduction in enzymatic activity could also result in a milder phenotype or contribute to disease vulnerability later in life. While this is a matter of debate, mild PD symptoms were observed in heterozygous individuals carrying the p.W437X or p.Q456X mutation [64–66]. In addition, data from our laboratory suggested that the heterozygous PINK1 p.G411S mutation increases risk for PD by a dominant-negative mechanism [67]. Though not all PINK1 mutations appear to be alike, these studies highlight the disease effects of particular variants and encourage a more detailed genetic and functional analysis of heterozygous mutations in recessive PD genes.
THE CYTOSOLIC E3 UBIQUITIN LIGASE PARKIN
Parkin, a 465 amino acid protein, is a RING-in-between-RING (RBR)-type E3 ubiquitin (Ub) ligase [68] that catalyzes (multi-) mono- and poly-ubiquitylation of numerous substrates that are structurally and functionally divers, including itself [68, 69]. Together with specific co-enzymes, Parkin adds the small modifier protein Ub (76 amino acids) to lysine residues of substrate proteins including Ub that itself contains seven internal lysines. Consecutive rounds of conjugation result in growth of poly-Ub chains that, depending on the linkage type, present with distinct topologies and thus biological functions. Structurally, Parkin consists of an N-terminal Ub-like (UBL) domain followed by a flexible linker and four cysteine-rich regions that each coordinate two Zn2+ atoms (Fig. 1B). Of those, three are really interesting new gene (RING) domains (RING0, RING1 and RING2), the last two of which are separated by an in-between-RING (IBR) domain. Parkin mutations are the primary cause of familial early-onset PD that are found in nearly 50% of all young PD patients (≤40 years old) [70, 71]. To date, over 150 PD-related Parkin mutations have been identified across various ethnic groups [24]. In addition to genetic mutations in familial PD, Parkin inactivation by post-translational modifications in sporadic disease has been suggested [72].
Initially Parkin was identified as a classical RING-type E3 Ub ligase [73] that bridges the interaction between the E2 Ub-conjugating enzyme and a substrate without physically receiving Ub. More recently it was shown that Parkin has a catalytic cysteine residue (Cys431) in its RING2 domain that forms a transient thioester with Ub before further transfer onto substrate protein, similar to E6-AP carboxyl terminus (HECT)-type enzymes [74]. Indeed, the PD-associated mutation p.C431F abolishes the E3 Ub ligase activity of Parkin as it can no longer bind the Ub moiety [75, 76]. This novel RING/HECT-hybrid mechanism lead to its re-classification as a member of the RBR-type family of E3 Ub ligases that are auto-inhibited through several intra-molecular interactions [68]. While a role of the UBL domain was already suggested earlier [77], elucidation of Parkin’s inactive structure provided a complete picture of the auto-inhibitory mode [78–82]. In addition to the N-terminus that keeps Parkin in a closed conformation, its activity is further repressed by the REP (repressor of Parkin) region that blocks the E2 binding site in its canonical RING1 (Fig. 1C). Moreover, RING0 intersects between RING1 and RING2 and thereby buries Cys431 resulting in a spatial separation between the active sites of E2 and Parkin, which disrupts Ub transfer. Targeted mutations in the auto-inhibitory regions indeed activated Parkin and increased its auto-ubiquitylation activity [79]. Based on the closed structure, it has become apparent that Parkin must get activated and has to undergo major structural rearrangements in order to gain enzymatic E3 Ub ligase activity [82, 83].
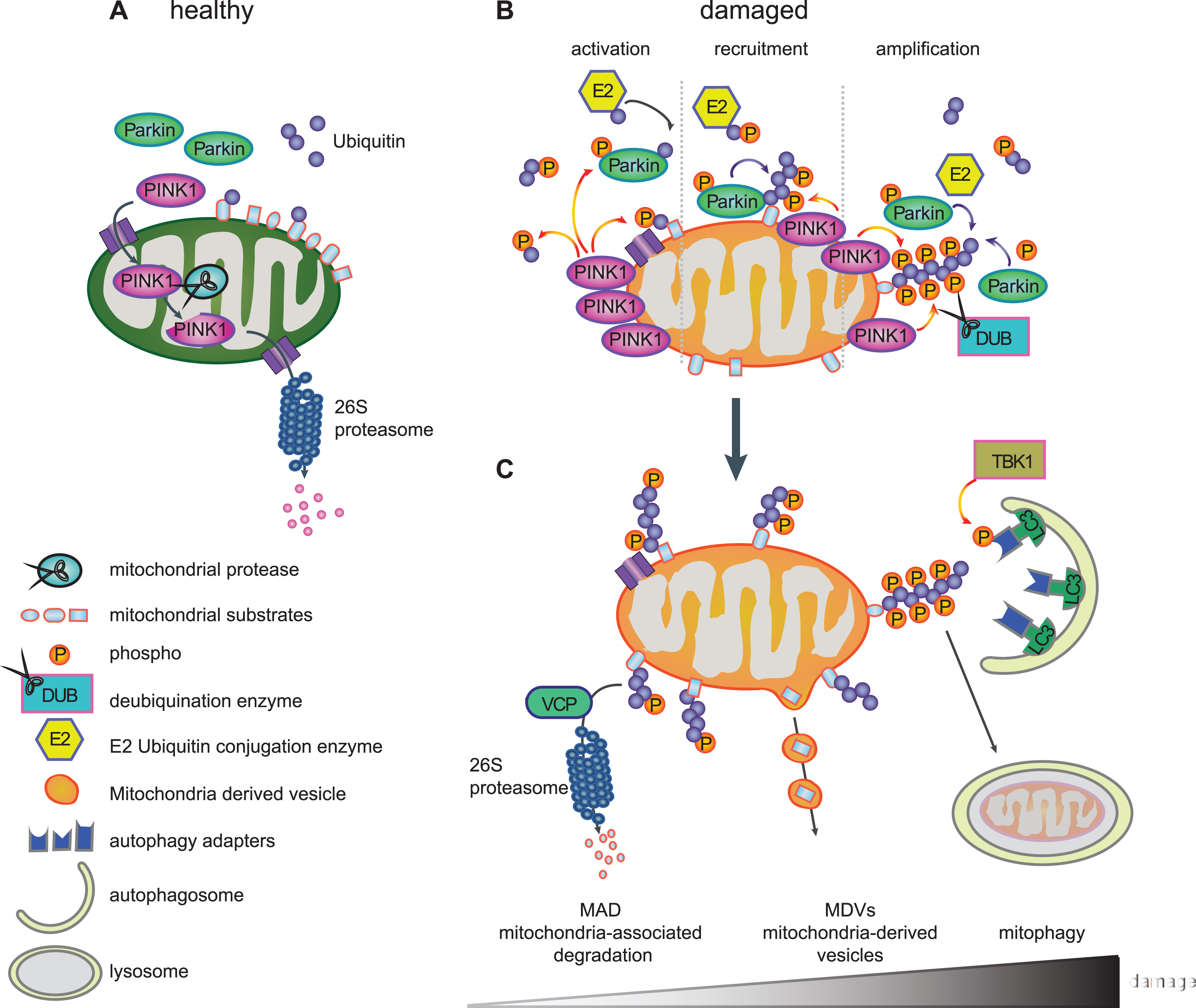
THE LIFE CYCLE OF PINK1 – A SENSOR FOR MITOCHONDRIAL DAMAGE
PINK1 is imported into healthy mitochondria through the general import machinery, the translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM) [84]. Inside mitochondria, PINK1 undergoes consecutive cleavages by the mitochondrial proteases MPP that removes the MTS in the matrix and by PARL that cleaves the TM domain in the IMM [57–59]. As a result of both events, cleaved PINK1 is re-exported and subsequently degraded by the N-end rule pathway through the Ub/proteasome system resulting in low levels of endogenous PINK1 in healthy mitochondria [85] (Fig. 2A). However, inside mitochondria, PINK1 has been implicated in the phosphorylation of different proteins, although for some of them it is unclear whether this occurs directly. PINK1-mediated phosphorylation of inner mitochondrial proteins is generally thought to be protective. Among the reported PINK1 substrates are the PD-linked serine protease HTRA2/Omi (PARK13) [86], the chaperone TRAP1 [87] in the intermembrane space and the complex I subunit NDUFA10 [88] located on the IMM. In flies, overexpression of TRAP1, for which direct phosphorylation by PINK1 has been shown, completely rescued phenotypes of PINK1, but not Parkin, deficiency [89, 90]. The phospho-mimetic version of NDUFA10 reversed PINK1 loss-of-function induced deficits in mouse cells and fly models [88].
Import of proteins, including PINK1, into mitochondria requires an active proton gradient and is inhibited upon mitochondrial damage induced by mitochondrial depolarizing agents (e.g. CCCP) [91]. Consequently, full-length PINK1 accumulates on the outer mitochondrial membrane (OMM) [91] (Fig. 2B), where it is stabilized by autophosphorylation [61] and the formation of a large 700kDa multimeric protein complex with the TOM machinery [84, 92]. Components of the TOM complex are essential for the alternative insertion of PINK1 into the OMM [93, 94]. The dynamic nature of PINK1 protein levels and its dual mitochondrial localization explain how PINK1 serves as a sensor of mitochondrial impairment and initiates the degradation of selectively damaged mitochondria.
PINK1-MEDIATED PARKIN ACTIVATION
When full-length PINK1 is stabilized on the OMM of damaged mitochondria, its kinase domain is facing the cytosol. Several cytosolic and OMM proteins that modulate mitochondrial dynamics or apoptotic signaling have also been suggested as candidate substrates for PINK1 [95–97]. PINK1 was demonstrated to phosphorylate Parkin at serine-65 (Ser65) in the N-terminal UBL domain [60, 98, 99]. PINK1-mediated phosphorylation of Parkin was shown to activate its enzymatic functions and to induce its translocation to damaged mitochondria (Fig. 2B) [98, 99]. Using molecular modeling and dynamics simulation, we suggested that phosphorylation of Parkin releases its auto-inhibited structure and initiates opening conformations that could lead to its enzymatic activation [82]. Second, PINK1 was recently identified as the first Ub kinase, which phosphorylates not only the E3 Ub ligase Parkin but also the Ub itself at this conserved residue (p-Ser65-Ub) [100–103]. Binding of p-Ser65-Ub to the RING1 domain of Parkin leads to straightening of a helix that results in an initial release of the auto-inhibitory UBL domain [104–106]. In turn, this facilitates phosphorylation of Parkin, which causes further major structural rearrangements, de-repression of enzymatic activity, and maintenance of an active conformation [107–109]. While p-Ser65-Ub also serves as the receptor for Parkin on damaged mitochondria [106], both PINK1-mediated phosphorylation events together fully activate its enzymatic functions.
Recruitment of Parkin to damaged mitochondria then initiates a feedforward loop (Fig. 2B), wherein activated Parkin further conjugates Ub moieties onto OMM proteins and thereby provides more Ub substrates for PINK1-dependent phosphorylation, which in turn amplifies Parkin activation, recruitment, and E3 Ub ligase activity [101, 102, 110]. As a result of the concerted action of PINK1 and Parkin, damaged mitochondria are coated with p-Ser65-Ub chains. While the detection of endogenous, activated PINK1 and Parkin has long been hampered by lack of sensitive tools, especially in primary cells, we have recently developed phospho-specific Ub antibodies that allow the detection of the PINK1/Parkin pathway in patients’ fibroblasts, neurons, and post-mortem brains [111, 112]. The observed increase of p-Ser65-Ub signal with mitochondrial stress, age, and disease, highlights its potential as an important disease biomarker and a therapeutictarget.
PINK1/PARKIN-DIRECTED MITOCHONDRIAL QUALITY CONTROL
The PINK1/Parkin pathway is a complex regulated, sequential process that coordinates several aspects of mitochondrial quality control, likely dependent on the extent of mitochondrial damage (Fig. 2C). Thereby, PINK1 and Parkin modify a wide range of substrate proteins and mediate their clearance [69, 113, 114]. The best characterized ones include the mitochondrial fusion proteins Mitofusin-1 and Mitofusin-2 that are targeted to prevent refusion of damaged mitochondria with the healthy network. Mitofusins are eliminated by valosin-containing protein (VCP/p97) through mitochondria-associated degradation (MAD) via the 26 S proteasome [115, 116]. This process further leads to fragmentation of damaged mitochondria [117], which also facilitates their later engulfment by autophagosomes [118, 119]. Other substrates involved in mitochondrial dynamics are the OMM proteins Miro-1 and Miro-2, which anchor mitochondria to microtubules via the kinesin motor protein and thus play significant roles in their trafficking. Degradation of these transporter proteins leads to inhibition of mitochondrial movement and further promotes segregation of damaged mitochondria [95]. In addition, numerous other proteins from all mitochondrial subcompartments have been identified as targets for PINK1 and Parkin [69, 115], but additional functional roles other than their pure elimination remain unclear. Intriguingly, distinct poly-Ub chains linkages have been identified on substrates suggesting a more complex regulation of fates mediated by those topologically diverse chains [120].
Intriguingly, recent studies identified an additional role for the PINK1/Parkin pathway in the formation of so-called mitochondria-derived vesicles (MDVs) that appear to mediate an alternative route for degradation (Fig. 2C). Mass spectrometry in Drosophila showed a slower turnover of mitochondrial respiratory chain proteins in PINK1–/– and Parkin–/– mutants compared to autophagy mutant Atg7–/– flies [121]. While this suggested a mechanism of mitochondrial proteins turnover dependent on PINK1 and Parkin, but independent from autophagy, mitochondrial components were shown to be degraded through an MDV pathway [122]. MDVs selectively transport oxidized protein and lipid cargo to peroxisomes [123] or other specific cargos to lysosomes via multi-vesicular bodies [124]. The so far identified cargoes and destinations of MDVs might only represent the tip of the iceberg and it is conceivable that other routes such as exosome-mediated secretion are yet to be discovered [125]. Of note, at least some of these MDVs are generated by the vacuolar protein sorting 35 homolog gene (VPS35) [123], which is linked to late-onset autosomal dominant PD (PARK17) [126, 127]. This component of the retromer complex, which guides retrograde protein sorting from the endosome-lysosome pathway [128], has also been found localized on mitochondria. Interestingly, VPS35 expression in flies rescued phenotypes associated with loss of Parkin, but not PINK1 [129].
While these additional roles of PINK1 and Parkin are just beginning to emerge, the best characterized functions of the pathway relate to mitophagy (Fig. 2C). Here, accumulated p-Ser65-Ub on the mitochondrial surface appears to act as the mitophagy tag that leads to recruitment of autophagy receptors. Five Ub-binding proteins, including Optineurin (OPTN), nuclear dot protein 52 (NDP52), p62/SQSTM1, neighbor of BRCA1 gene 1 (NBR1) and Tax1-binding protein 1 (TAX1BP1) [130], bind to poly-Ub chains and to the microtubule-associated proteins 1A/1B light chain 3 (LC3)/GABAA receptor-associated protein. Autophagosomal membrane attachment is mediated through the LC3-interacting region, which targets damaged mitochondria to phagophores for clearance in the lysosome [131]. It has been suggested that Optineurin and NDP52 are primary receptors in this pathway [130, 132], at least in HeLa cells, possibly due to preferential binding to p-Ser65-Ub [130, 133]. Recent studies have shown that the tank binding kinase 1 (TBK1) enhances Optineurin binding through phosphorylation during mitophagy[133, 134].
MITOCHONDRIAL HEALTH AND CELLULAR DEATH
In addition to the clearance of individual component and whole mitochondria, Parkin also plays a role in mitochondrial biogenesis that counteracts its degradative functions. For instance, Parkin has been shown to ubiquitylate the transcriptional repressor PARIS (ZNF746) in vitro, and the low basal activity of Parkin (i.e. in the absence of CCCP) was reported sufficient to target it for proteasomal degradation [135]. Elimination of PARIS facilitates the expression of the transcriptional coactivator PGC-1α, one of the major transcriptional regulators of mitochondrial biogenesis [136]. Therefore, loss of Parkin may contribute to cell death by impairing mitochondrial biogenesis and consequently mitochondrial health [137].
Parkin protects cells against a variety of stimuli when overexpressed and renders them sensitive when knocked down/out. Following stress, it has been shown that Parkin opposes the translocation of the pro-apoptotic Bax protein to mitochondria [138–142]. This might be facilitated by direct ubiquitylation of Bax by Parkin [138]. As a consequence, Parkin inhibits cytochrome c release into the cytosol and the induction of apoptosis [138–142]. Depending on the stressor, Parkin-mediated induction of mitochondrial quality control and Bax-dependent inhibition of apotosis might act together to prevent cell death [140–142].
MODIFIERS OF MITOCHONDRIAL QUALITY CONTROL
Along the multiple steps and aspects of mitochondrial quality control, a variety of processes and modifiers have been identified that collaboratively regulate the PINK1/Parkin pathway in either positive or negative manner. Genome-wide RNAi screens have uncovered numerous regulators of Parkin translocation and/or mitophagy [94, 143–145]. For instance, Parkin is controlled by several different enzymes of the Ub system. Distinct E2 Ub-conjugating enzymes were shown to regulate the activation and translocation of Parkin as well as its enzymatic activity on damaged mitochondria [146–148]. UBE2D and UBE2L3 directly charge Parkin with Ub on its catalytic center; UBE2N mediates clustering of damaged mitochondria at a later step and through different Ub linkages; UBE2R1, on the other hand, negatively modifies Parkin activation [146]. In addition to E2 enzymes, Parkin is also regulated through other E3 Ub ligases. For instance, F-box only protein 7 (FBXO7), a component of a multimeric E3 Ub ligase complex, interacts with both PINK1 and Parkin and contributes to Parkin recruitment to dysfunctional mitochondria [149]. The association of FBXO7 with familial PD (PARK15) thus ties in a third recessive disease gene and further underlines the disease relevance of thepathway.
Parkin’s enzymatic functions are antagonized by de-ubiquitylation enzymes (DUBs) including USP30 [150] and USP15 [151] that cleave poly-Ub chains on mitochondrial substrates. In contrast, another DUB, USP8, selectively removes K6-linked Ub chains on Parkin and thereby facilitates its recruitment [152]. Together with the opposing roles of some E2 enzymes, this further highlights the complex regulation of Parkin and mitochondrial quality control mediated by different members within the same enzyme family. Moreover, some of the PINK1/Parkin substrates have been suggested to regulate the activation of and/or the progression through the pathway [49, 91, 144, 153]. Other modifiers of the PINK1/Parkin pathway are related to autophagy and apoptosis. Ambra1 binds to Parkin and promotes mitophagy [154], while Beclin1 binds to PINK1 to promote autophagy [155]. Members of the anti-apoptotic Bcl-2 family, Bcl-xL and Mcl-1, are localized in the OMM, inhibit Parkin translocation and thereby suppress mitophagy [156]. In addition to protein modifiers, we recently also found certain microRNAs to be significantly induced under chronic mitophagic flux and to act as molecular suppressors of PINK1 expression, suggesting further negative feedback regulation [157]. In summary, numerous molecules and cellular processes appear to impact PINK1/Parkin, the mitochondrial quality control as well as several other PD genes.
CROSS-TALK OF PINK1/PARKIN WITH OTHER PD GENES
In addition to PINK1, Parkin and FBXO7, mitochondrial and/or autophagic/lysosomal dysfunctions have also been associated with further familial PD genes. Loss of DJ-1 (PARK7) in primary cortical neurons and mouse embryonic fibroblast resulted in fragmented mitochondria and increased ROS production [158]. DJ-1 deficiency is associated with reduced mitochondrial membrane potential, ATP production and aberrant complex I assembly [159]. The regulation of DJ-1 on mitochondrial function and autophagy was suggested to operate in a pathway parallel to PINK1/Parkin [160]. In addition, phospholipase A2 group VI (PLA2G6, PARK14) knockout flies and mice display swollen mitochondria with disrupted morphology [161] and autophagic dysfunction [162]. ATPase type 13A2 (ATP13A2, PARK9) deficiency results in the fragmentation of the mitochondrial network, increased ROS production and a decrease in the autophagic events [163]. Mutations in the MTS of the coiled-coil-helix-coiled-coil-helix domain containing protein 2 (CHCHD2, PARK22) further support mitochondrial dysfunctions in PD [164]. Most recently, VPS13C (PARK23) was added to that list, however, its loss-of-function resulted in exacerbated PINK1/Parkin-dependent mitophagy [165].
A mitochondrial function of LRRK2 (PARK8), which plays a role in familial and sporadic PD, has been described as well, although the mitochondrial localization of LRRK2 is contradictory [166–169]. Two reports showed that endogenous LRRK2 interacts with and phosphorylates the fission regulator dynamin related protein 1 (Drp1) at the OMM in murine primary neurons and human neuroblastoma cells [170, 171]. Overexpression of LRRK2 wild-type or expression of kinase hyper-active G2019S LRRK2 mutation enhanced mitochondrial fragmentation and could be rescued by Drp1 inhibition [171, 172]. In contrast with previous models overexpressing LRRK2 G2019S, LRRK2 G2019S knock-in mice showed deformed mitochondria, an increase of swollen mitochondria with abnormal cristae and less Drp1 phosphorylation, which could be explained by compensatory effects to restore mitochondrial imbalance in this preclinical model [173]. In dopamine neurons and fibroblasts of PD patients harboring LRRK2 G2019S mutation, LRRK2 was shown to induce fragmentation of mitochondria, which increased vulnerability of cells to stressors and impaired mitochondrial and cellular health [174–176]. A recent phosphoproteomic study on LRRK2 G2019S or A2016T mutant knock-in mice identified several LRRK2 substrates belonging to the Rab GTPase family, which are involved in protein transport and regulate the intracellular trafficking [177]. Mitochondrial members of the Rab family were not among the identified substrates. This finding may support an indirect role of LRRK2 on mitochondria through the endosome-mitochondria transport pathway.
Importantly, alpha-synuclein (SNCA, PARK1/4) was also shown to bind to mitochondrial membranes and to inhibit complex I function. Such effect was more pronounced in the presence of the PD-associated A53T mutation and was also observed in the SNpc of PD patients [178]. Additionally, mitochondrial abnormalities were observed in transgenic mouse models overexpressing mutant SNCA. Expression of SNCA A53T induced mtDNA damage and increased sensitivity towards MPTP and paraquat [179]. In cultured cells, nematode and fly models, expression of SNCA caused prominent mitochondrial fragmentation, which could be rescued by co-expression of PINK1, Parkin, or DJ-1 [180–182]. In line with this, downregulation or deletion of either PINK1 or Parkin significantly increased mitochondrial pathologies and enhanced the sensitivity towards SNCA-induced neurotoxicity [183, 184]. Intriguingly, a recent study showed direct binding of SNCA to TOM20, which resulted in impaired protein import into mitochondria and thus could possibly alter PINK1/Parkin signaling, thereby providing a link to sporadic disease [185].
CONCLUDING REMARKS
Since the initial identification of PINK1/Parkin-directed mitophagy, many details of this complex pathway have been worked out with enormous pace. While the clearance of terminally damaged organelles seems to be the last resort, it has become evident that PINK1 and Parkin regulate many aspects of a stress-activated, mitochondrial quality control including MAD, MDVs and organelle biogenesis. However, more research is required to fully understand all facets of this signaling on the structural, molecular and cellular levels. It is important to mention that most of the pathway has been worked out in vitro and in cell culture and that translation of this knowledge into model organisms is needed. While fly models generally seem to support the findings [121], a limited number of studies using mouse models report ambiguous findings on PINK1/Parkin-dependent mitochondrial quality control [186, 187]. Yet, dysfunctions in mitochondrial quality control have already advanced to the center stage in PD pathogenesis. Indeed, mitochondrial dysfunction is emerging as a common feature suggesting a great pathogenic overlap among the individual familial, environmental and even sporadic disease pathways with a potential convergence at the level of PINK1/Parkin [188]. Many, if not all, PD genes have already been directly linked to impairments either of mitochondria and/or of lysosomal/autophagic pathways. Regardless of the exact disturbance, each of those would impact mitochondrial quality control, though at different steps of the sequential process.
Along those thoughts, sporadic disease could result from a complex interaction of exposure to environmental mitochondrial toxins and genetic factors that impinges on activation, progression or execution of mitochondrial quality control. Given the direct link of a few PD genes and the involvement of numerous proteins along the pathway, genetic variants with weaker effects could play in concert to diminish protection through PINK1/Parkin signaling. In such context, it is important to mention that even carriers of heterozygous mutations in PINK1 or Parkin are at an increased risk for PD [67, 189]. However, the detection of multigenic forms of disease is further complicated by individual stress levels that will vary, depending on genetic pre-disposition and environmental mitochondrial toxins exposure, from patient to patient. Nevertheless, future genetic studies that analyze the synergistic effects of variants in PINK1/Parkin-related functional assays should lead to nomination of additional disease genes, which may help, at least in part, to further understand idiopathic disease.
Despite the exquisite sensitivity of DA neurons and the great relevance to PD, PINK1/Parkin signaling appears as a fundamental, cell biological mechanism and could have more far-reaching implications. This is supported by the involvement of several other disease genes including, but not limited to, factors involved in Ub binding and substrate degradation. For instance, VCP/p97, p62/SQSTM1, OPTN1 and TBK1 have all been genetically linked to the disease continuum of amyotrophic lateral sclerosis and frontotemporal dementia [190, 191]. This is further consistent with the broad neuroprotective roles of PINK1 and Parkin, whose overexpression in cell and animal models provided protection against various genetic and toxic insults [192]. Furthermore, mitochondrial quality control appears to play a role not only in age-related diseases, but potentially the aging process itself. While PINK1 and Parkin mutant flies showed decreased life span [36, 39], overexpression of Parkin was found to extend life span [193] as was induction of mitophagy in C. elegans [194]. This is in line with increased accumulation of cellular damage and concomitant decline in degradative capacities with age in general.
Many breakthroughs in the field have provided novel opportunities towards the development of targeted therapeutics and selective biomarkers over the past few years. Amongst those are the identification of PINK1 as the first Ub kinase and the establishment of p-Ser65-Ub as a highly selective label for mitochondrial quality control. Using novel antibodies, we have demonstrated PINK1/Parkin signaling under endogenous conditions in primary neurons and in post-mortem brains [111, 112]. Detection of activated PINK1 or Parkin had been hampered thus far by the unavailability of sensitive tools, however, p-Ser65-Ub is amplified through the concerted actions of both enzymes upon stress, age, and disease. Yet, as a specific label of damaged mitochondria targeted for destruction, p-Ser65-Ub levels can be expected to increase with impairments at either end of this quality control pathway. In addition to further exploiting this mitophagy tag, modifying the activity or abundance of other proteins involved in the Ub pathway may represent promising strategies to modulate the PINK1/Parkin pathway. First small molecule DUB inhibitors as well as Parkin activators are already under development and could furnish the next generation drugs for disease-modifying therapies.
FUNDING
W.S. is partially supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) [R01 #NS085070], the Michael J. Fox Foundation for Parkinson’s Research and the Foundation for Mitochondrial Medicine, the Mayo Clinic Center for Regenerative Medicine (CRM), Center for Individualized Medicine (CIM), and Center for Biomedical Discovery (CBD), the Marriott Family Foundation, and a Gerstner Family Career Development Award. F.C.F. is supported by the American Parkinson Disease Association (APDA) and the Younkin Scholar Program.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest.
REFERENCES
[1] | Chan DC ((2006) ) Mitochondria: Dynamic organelles in disease, aging, and development. Cell, 125: , 1241–1252. |
[2] | Davis RE & Williams M ((2012) ) Mitochondrial function and dysfunction: An update. J Pharmacol Exp Ther, 342: , 598–607. |
[3] | Marchi S , Giorgi C , Suski JM , Agnoletto C , Bononi A , Bonora M , De Marchi E , Missiroli S , Patergnani S , Poletti F , Rimessi A , Duszynski J , Wieckowski MR & Pinton P ((2012) ) Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct, 2012: , 329635. |
[4] | Finkel T , Menazza S , Holmstrom KM , Parks RJ , Liu J , Sun J , Liu J , Pan X & Murphy E ((2015) ) The ins and outs of mitochondrial calcium. Circ Res, 116: , 1810–1819. |
[5] | Gillies LA & Kuwana T ((2014) ) Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem, 115: , 632–640. |
[6] | Schon EA , DiMauro S & Hirano M ((2012) ) Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat Rev Genet, 13: , 878–890. |
[7] | Quiros PM , Mottis A & Auwerx J ((2016) ) Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol, 17: , 213–226. |
[8] | Beal MF ((1995) ) Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol, 38: , 357–366. |
[9] | de Rijk MC , Breteler MM , Graveland GA , Ott A , Grobbee DE , van der Meche FG & Hofman A ((1995) ) Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study. Neurology, 45: , 2143–2146. |
[10] | Sulzer D ((2007) ) Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci, 30: , 244–250. |
[11] | Surmeier DJ , Guzman JN & Sanchez-Padilla J ((2010) ) Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium, 47: , 175–182. |
[12] | Surmeier DJ , Guzman JN , Sanchez-Padilla J & Goldberg JA ((2010) ) What causes the death of dopaminergic neurons in Parkinson’s disease? Prog Brain Res, 183: , 59–77. |
[13] | Haddad D & Nakamura K ((2015) ) Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett, 589: , 3702–3713. |
[14] | Blesa J , Trigo-Damas I , Quiroga-Varela A & Jackson-Lewis VR ((2015) ) Oxidative stress and Parkinson’s disease. Front Neuroanat, 9: , Article 91. |
[15] | Langston JW , Ballard P , Tetrud JW & Irwin I ((1983) ) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science, 219: , 979–980. |
[16] | Javitch JA , D’Amato RJ , Strittmatter SM & Snyder SH ((1985) ) Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A, 82: , 2173–2177. |
[17] | Ramsay RR & Singer TP ((1986) ) Energy-dependent uptake of N-methyl-4-phenylpyridinium, the neurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria. J Biol Chem, 261: , 7585–7587. |
[18] | Mizuno Y , Ohta S , Tanaka M , Takamiya S , Suzuki K , Sato T , Oya H , Ozawa T & Kagawa Y ((1989) ) Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun, 163: , 1450–1455. |
[19] | Parker WD Jr , Boyson SJ & Parks JK ((1989) ) Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol, 26: , 719–723. |
[20] | Schapira AH , Cooper JM , Dexter D , Jenner P , Clark JB & Marsden CD ((1989) ) Mitochondrial complex I deficiency in Parkinson’s disease. Lancet, 1: , 1269. |
[21] | Bove J & Perier C ((2012) ) Neurotoxin-based models of Parkinson’s disease. Neuroscience, 211: , 51–76. |
[22] | Valente EM , Abou-Sleiman PM , Caputo V , Muqit MM , Harvey K , Gispert S , Ali Z , Del Turco D , Bentivoglio AR , Healy DG , Albanese A , Nussbaum R , Gonzalez-Maldonado R , Deller T , Salvi S , Cortelli P , Gilks WP , Latchman DS , Harvey RJ , Dallapiccola B , Auburger G & Wood NW ((2004) ) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science, 304: , 1158–1160. |
[23] | Kitada T , Asakawa S , Hattori N , Matsumine H , Yamamura Y , Minoshima S , Yokochi M , Mizuno Y & Shimizu N ((1998) ) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392: , 605–608. |
[24] | Corti O , Lesage S & Brice A ((2011) ) What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev, 91: , 1161–1218. |
[25] | Li Y , Tomiyama H , Sato K , Hatano Y , Yoshino H , Atsumi M , Kitaguchi M , Sasaki S , Kawaguchi S , Miyajima H , Toda T , Mizuno Y & Hattori N ((2005) ) Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology, 64: , 1955–1957. |
[26] | Kim HJ , Kim HJ , Lee JY , Yun JY , Kim SY , Park SS & Jeon BS ((2011) ) Phenotype analysis in patients with early onset Parkinson’s disease with and without parkin mutations. J Neurol, 258: , 2260–2267. |
[27] | Ephraty L , Porat O , Israeli D , Cohen OS , Tunkel O , Yael S , Hatano Y , Hattori N & Hassin-Baer S ((2007) ) Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to PINK1 mutations. Mov Disord, 22: , 566–569. |
[28] | Farrer M , Chan P , Chen R , Tan L , Lincoln S , Hernandez D , Forno L , Gwinn-Hardy K , Petrucelli L , Hussey J , Singleton A , Tanner C , Hardy J & Langston JW ((2001) ) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol, 50: , 293–300. |
[29] | Pramstaller PP , Schlossmacher MG , Jacques TS , Scaravilli F , Eskelson C , Pepivani I , Hedrich K , Adel S , Gonzales-McNeal M , Hilker R , Kramer PL & Klein C ((2005) ) Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol, 58: , 411–422. |
[30] | Doherty KM & Hardy J ((2013) ) Parkin disease and the Lewy body conundrum. Mov Disord, 28: , 702–704. |
[31] | Miyakawa S , Ogino M , Funabe S , Uchino A , Shimo Y , Hattori N , Ichinoe M , Mikami T , Saegusa M , Nishiyama K , Mori H , Mizuno Y , Murayama S & Mochizuki H ((2013) ) Lewy body pathology in a patient with a homozygous Parkin deletion. Mov Disord, 28: , 388–391. |
[32] | Cornejo-Olivas MR , Torres L , Mata IF , Mazzetti P , Rivas D , Cosentino C , Inca-Martinez M , Cuba JM , Zabetian CP & Leverenz JB ((2015) ) A Peruvian family with a novel PARK2 mutation: Clinical and pathological characteristics. Parkinsonism Relat Disord, 21: , 444–448. |
[33] | Samaranch L , Lorenzo-Betancor O , Arbelo JM , Ferrer I , Lorenzo E , Irigoyen J , Pastor MA , Marrero C , Isla C , Herrera-Henriquez J & Pastor P ((2010) ) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain, 133: , 1128–1142. |
[34] | Steele JC , Guella I , Szu-Tu C , Lin MK , Thompson C , Evans DM , Sherman HE , Vilarino-Guell C , Gwinn K , Morris H , Dickson DW & Farrer MJ ((2015) ) Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol, 77: , 458–468. |
[35] | Takanashi M , Li YZ & Hattori N ((2016) ) Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology, 86: , 2212–2213. |
[36] | Greene JC , Whitworth AJ , Kuo I , Andrews LA , Feany MB & Pallanck LJ ((2003) ) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A, 100: , 4078–4083. |
[37] | Clark IE , Dodson MW , Jiang C , Cao JH , Huh JR , Seol JH , Yoo SJ , Hay BA & Guo M ((2006) ) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature, 441: , 1162–1166. |
[38] | Park J , Lee SB , Lee S , Kim Y , Song S , Kim S , Bae E , Kim J , Shong M , Kim JM & Chung J ((2006) ) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature, 441: , 1157–1161. |
[39] | Yang Y , Gehrke S , Imai Y , Huang Z , Ouyang Y , Wang JW , Yang L , Beal MF , Vogel H & Lu B ((2006) ) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A, 103: , 10793–10798. |
[40] | Exner N , Treske B , Paquet D , Holmstrom K , Schiesling C , Gispert S , Carballo-Carbajal I , Berg D , Hoepken HH , Gasser T , Kruger R , Winklhofer KF , Vogel F , Reichert AS , Auburger G , Kahle PJ , Schmid B & Haass C ((2007) ) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci, 27: , 12413–12418. |
[41] | Kahle PJ & Haass C ((2004) ) How does parkin ligate ubiquitin to Parkinson’s disease? EMBO Rep, 5: , 681–685. |
[42] | Heeman B , Van den Haute C , Aelvoet SA , Valsecchi F , Rodenburg RJ , Reumers V , Debyser Z , Callewaert G , Koopman WJH , Willems PHGM & Baekelandt V ((2011) ) Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci, 124: , 1115–1125. |
[43] | Narendra D , Tanaka A , Suen DF & Youle RJ ((2008) ) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol, 183: , 795–803. |
[44] | Geisler S , Holmstrom KM , Skujat D , Fiesel FC , Rothfuss OC , Kahle PJ & Springer W ((2010) ) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol, 12: , 119–131. |
[45] | Matsuda N , Sato S , Shiba K , Okatsu K , Saisho K , Gautier CA , Sou YS , Saiki S , Kawajiri S , Sato F , Kimura M , Komatsu M , Hattori N & Tanaka K ((2010) ) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol, 189: , 211–221. |
[46] | Vives-Bauza C , Zhou C , Huang Y , Cui M , de Vries RL , Kim J , May J , Tocilescu MA , Liu W , Ko HS , Magrane J , Moore DJ , Dawson VL , Grailhe R , Dawson TM , Li C , Tieu K & Przedborski S ((2010) ) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A, 107: , 378–383. |
[47] | Narendra DP , Jin SM , Tanaka A , Suen DF , Gautier CA , Shen J , Cookson MR & Youle RJ ((2010) ) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol, 8: , e1000298. |
[48] | Springer W & Kahle PJ ((2011) ) Regulation of PINK1-Parkin-mediated mitophagy. Autophagy, 7: , 266–278. |
[49] | Geisler S , Holmstrom KM , Treis A , Skujat D , Weber SS , Fiesel FC , Kahle PJ & Springer W ((2010) ) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy, 6: , 871–878. |
[50] | Fiesel FC , Caulfield TR , Moussaud-Lamodiere EL , Ogaki K , Dourado DF , Flores SC , Ross OA & Springer W ((2015) ) Structural and functional impact of Parkinson disease-associated mutations in the E3 ubiquitin ligase parkin. Hum Mutat, 36: , 774–786. |
[51] | Unoki M & Nakamura Y ((2001) ) Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene, 20: , 4457–4465. |
[52] | Klein C & Westenberger A ((2012) ) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med, 2: , a008888. |
[53] | Beilina A , Van Der Brug M , Ahmad R , Kesavapany S , Miller DW , Petsko GA & Cookson MR ((2005) ) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A, 102: , 5703–5708. |
[54] | Mills RD , Sim CH , Mok SS , Mulhern TD , Culvenor JG & Cheng HC ((2008) ) Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1). J Neurochem, 105: , 18–33. |
[55] | Cardona F , Sanchez-Mut JV , Dopazo H & Perez-Tur J ((2011) ) Phylogenetic and in silico structural analysis of the Parkinson disease-related kinase PINK1. Hum Mutat, 32: , 369–378. |
[56] | Sim CH , Gabriel K , Mills RD , Culvenor JG & Cheng HC ((2012) ) Analysis of the regulatory and catalytic domains of PTEN-induced kinase-1 (PINK1). Hum Mutat, 33: , 1408–1422. |
[57] | Jin SM , Lazarou M , Wang C , Kane LA , Narendra DP & Youle RJ ((2010) ) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol, 191: , 933–942. |
[58] | Deas E , Plun-Favreau H , Gandhi S , Desmond H , Kjaer S , Loh SH , Renton AE , Harvey RJ , Whitworth AJ , Martins LM , Abramov AY & Wood NW ((2011) ) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet, 20: , 867–879. |
[59] | Greene AW , Grenier K , Aguileta MA , Muise S , Farazifard R , Haque ME , McBride HM , Park DS & Fon EA ((2012) ) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep, 13: , 378–385. |
[60] | Kondapalli C , Kazlauskaite A , Zhang N , Woodroof HI , Campbell DG , Gourlay R , Burchell L , Walden H , Macartney TJ , Deak M , Knebel A , Alessi DR & Muqit MM ((2012) ) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol, 2: , 120080. |
[61] | Okatsu K , Oka T , Iguchi M , Imamura K , Kosako H , Tani N , Kimura M , Go E , Koyano F , Funayama M , Shiba-Fukushima K , Sato S , Shimizu H , Fukunaga Y , Taniguchi H , Komatsu M , Hattori N , Mihara K , Tanaka K & Matsuda N ((2012) ) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun, 3: , 1016. |
[62] | Aerts L , Craessaerts K , De Strooper B & Morais VA ((2015) ) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem, 290: , 2798–2811. |
[63] | Siuda J , Jasinska-Myga B , Boczarska-Jedynak M , Opala G , Fiesel FC , Moussaud-Lamodiere EL , Scarffe LA , Dawson VL , Ross OA , Springer W , Dawson TM & Wszolek ZK ((2014) ) Early-onset Parkinson’s disease due to PINK1 p.Q456X mutation–clinical and functional study. Parkinsonism Relat Disord, 20: , 1274–1278. |
[64] | Criscuolo C , Volpe G , De Rosa A , Varrone A , Marongiu R , Mancini P , Salvatore E , Dallapiccola B , Filla A , Valente EM & De Michele G ((2006) ) PINK1 homozygous W437X mutation in a patient with apparent dominant transmission of parkinsonism. Mov Disord, 21: , 1265–1267. |
[65] | Ricciardi L , Petrucci S , Guidubaldi A , Ialongo T , Serra L , Ferraris A , Spano B , Bozzali M , Valente EM & Bentivoglio AR ((2014) ) Phenotypic variability of PINK1 expression: 12 Years’ clinical follow-up of two Italian families. Mov Disord, 29: , 1561–1566. |
[66] | Eggers C , Schmidt A , Hagenah J , Bruggemann N , Klein JC , Tadic V , Kertelge L , Kasten M , Binkofski F , Siebner H , Neumaier B , Fink GR , Hilker R & Klein C ((2010) ) Progression of subtle motor signs in PINK1 mutation carriers with mild dopaminergic deficit. Neurology, 74: , 1798–1805. |
[67] | Puschmann A , Fiesel FC , Caulfield TR , Hudec R , Ando M , Truban D , Hou X , Ogaki K , Heckman MG , James ED , Swanberg M , Jimenez-Ferrer I , Hansson O , Opala G , Siuda J , Boczarska-Jedynak M , Friedman A , Koziorowski D , Aasly JO , Lynch T , Mellick GD , Mohan M , Silburn PA , Sanotsky Y , Vilarino-Guell C , Farrer MJ , Chen L , Dawson VL , Dawson TM , Wszolek ZK , Ross OA & Springer W ((2016) ) Heterozygous PINK1 p. G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain. doi: 10.1093/brain/aww261 |
[68] | Spratt DE , Walden H & Shaw GS ((2014) ) RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem J, 458: , 421–437. |
[69] | Sarraf SA , Raman M , Guarani-Pereira V , Sowa ME , Huttlin EL , Gygi SP & Harper JW ((2013) ) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature, 496: , 372–376. |
[70] | Lucking CB , Durr A , Bonifati V , Vaughan J , De Michele G , Gasser T , Harhangi BS , Meco G , Denefle P , Wood NW , Agid Y & Brice A ((2000) ) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med, 342: , 1560–1567. |
[71] | Puschmann A ((2013) ) Monogenic Parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord, 19: , 407–415. |
[72] | Dawson TM & Dawson VL ((2014) ) Parkin plays a role in sporadic Parkinson’s disease. Neurodegener Dis, 13: , 69–71. |
[73] | Shimura H , Hattori N , Kubo S , Mizuno Y , Asakawa S , Minoshima S , Shimizu N , Iwai K , Chiba T , Tanaka K & Suzuki T ((2000) ) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet, 25: , 302–305. |
[74] | Wenzel DM , Lissounov A , Brzovic PS & Klevit RE ((2011) ) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature, 474: , 105–108. |
[75] | Maruyama M , Ikeuchi T , Saito M , Ishikawa A , Yuasa T , Tanaka H , Hayashi S , Wakabayashi K , Takahashi H & Tsuji S ((2000) ) Novel mutations, pseudo-dominant inheritance, and possible familial affects in patients with autosomal recessive juvenile parkinsonism. Ann Neurol, 48: , 245–250. |
[76] | Lazarou M , Narendra DP , Jin SM , Tekle E , Banerjee S & Youle RJ ((2013) ) PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol, 200: , 163–172. |
[77] | Chaugule VK , Burchell L , Barber KR , Sidhu A , Leslie SJ , Shaw GS & Walden H ((2011) ) Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J, 30: , 2853–2867. |
[78] | Riley BE , Lougheed JC , Callaway K , Velasquez M , Brecht E , Nguyen L , Shaler T , Walker D , Yang Y , Regnstrom K , Diep L , Zhang Z , Chiou S , Bova M , Artis DR , Yao N , Baker J , Yednock T & Johnston JA ((2013) ) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun, 4: , 1982. |
[79] | Trempe JF , Sauve V , Grenier K , Seirafi M , Tang MY , Menade M , Al-Abdul-Wahid S , Krett J , Wong K , Kozlov G , Nagar B , Fon EA & Gehring K ((2013) ) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science, 340: , 1451–1455. |
[80] | Wauer T & Komander D ((2013) ) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J, 32: , 2099–2112. |
[81] | Spratt DE , Martinez-Torres RJ , Noh YJ , Mercier P , Manczyk N , Barber KR , Aguirre JD , Burchell L , Purkiss A , Walden H & Shaw GS ((2013) ) A molecular explanation for the recessive nature of parkin-linked Parkinson’s disease. Nat Commun, 4: , 1983. |
[82] | Caulfield TR , Fiesel FC , Moussaud-Lamodiere EL , Dourado DF , Flores SC & Springer W ((2014) ) Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput Biol, 10: , e1003935. |
[83] | Caulfield TR , Fiesel FC & Springer W ((2015) ) Activation of the E3 ubiquitin ligase Parkin. Biochem Soc Trans, 43: , 269–274. |
[84] | Lazarou M , Jin SM , Kane LA & Youle RJ ((2012) ) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell, 22: , 320–333. |
[85] | Yamano K & Youle RJ ((2013) ) PINK1 is degraded through the N-end rule pathway. Autophagy, 9: , 1758–1769. |
[86] | Plun-Favreau H , Klupsch K , Moisoi N , Gandhi S , Kjaer S , Frith D , Harvey K , Deas E , Harvey RJ , McDonald N , Wood NW , Martins LM & Downward J ((2007) ) The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol, 9: , 1243–1252. |
[87] | Pridgeon JW , Olzmann JA , Chin LS & Li L ((2007) ) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol, 5: , e172. |
[88] | Morais VA , Haddad D , Craessaerts K , De Bock PJ , Swerts J , Vilain S , Aerts L , Overbergh L , Grunewald A , Seibler P , Klein C , Gevaert K , Verstreken P & De Strooper B ((2014) ) PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science, 344: , 203–207. |
[89] | Costa AC , Loh SH & Martins LM ((2013) ) Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson’s disease. Cell Death Dis, 4: , e467. |
[90] | Zhang L , Karsten P , Hamm S , Pogson JH , Muller-Rischart AK , Exner N , Haass C , Whitworth AJ , Winklhofer KF , Schulz JB & Voigt A ((2013) ) TRAP1 rescues PINK1 loss-of-function phenotypes. Hum Mol Genet, 22: , 2829–2841. |
[91] | Narendra D , Kane LA , Hauser DN , Fearnley IM & Youle RJ ((2010) ) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy, 6: , 1090–1106. |
[92] | Becker D , Richter J , Tocilescu MA , Przedborski S & Voos W ((2012) ) Pink1 kinase and its membrane potential (Deltapsi)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem, 287: , 22969–22987. |
[93] | Bertolin G , Ferrando-Miguel R , Jacoupy M , Traver S , Grenier K , Greene AW , Dauphin A , Waharte F , Bayot A , Salamero J , Lombes A , Bulteau AL , Fon EA , Brice A & Corti O ((2013) ) The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy, 9: , 1801–1817. |
[94] | Hasson SA , Kane LA , Yamano K , Huang CH , Sliter DA , Buehler E , Wang C , Heman-Ackah SM , Hessa T , Guha R , Martin SE & Youle RJ ((2013) ) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature, 504: , 291–295. |
[95] | Wang X , Winter D , Ashrafi G , Schlehe J , Wong YL , Selkoe D , Rice S , Steen J , LaVoie MJ & Schwarz TL ((2011) ) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell, 147: , 893–906. |
[96] | Chen Y & Dorn GW 2nd ((2013) ) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science, 340: , 471–475. |
[97] | Arena G , Gelmetti V , Torosantucci L , Vignone D , Lamorte G , De Rosa P , Cilia E , Jonas EA & Valente EM ((2013) ) PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ, 20: , 920–930. |
[98] | Iguchi M , Kujuro Y , Okatsu K , Koyano F , Kosako H , Kimura M , Suzuki N , Uchiyama S , Tanaka K & Matsuda N ((2013) ) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem, 288: , 22019–22032. |
[99] | Shiba-Fukushima K , Imai Y , Yoshida S , Ishihama Y , Kanao T , Sato S & Hattori N ((2012) ) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep, 2: , 1002. |
[100] | Kane LA , Lazarou M , Fogel AI , Li Y , Yamano K , Sarraf SA , Banerjee S & Youle RJ ((2014) ) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol, 205: , 143–153. |
[101] | Kazlauskaite A , Kondapalli C , Gourlay R , Campbell DG , Ritorto MS , Hofmann K , Alessi DR , Knebel A , Trost M & Muqit MM ((2014) ) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J, 460: , 127–139. |
[102] | Koyano F , Okatsu K , Kosako H , Tamura Y , Go E , Kimura M , Kimura Y , Tsuchiya H , Yoshihara H , Hirokawa T , Endo T , Fon EA , Trempe JF , Saeki Y , Tanaka K & Matsuda N ((2014) ) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature, 510: , 162–166. |
[103] | Zhang C , Lee S , Peng Y , Bunker E , Giaime E , Shen J , Zhou Z & Liu X ((2014) ) PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr Biol, 24: , 1854–1865. |
[104] | Wauer T , Simicek M , Schubert A & Komander D ((2015) ) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature, 524: , 370–374. |
[105] | Sauve V , Lilov A , Seirafi M , Vranas M , Rasool S , Kozlov G , Sprules T , Wang J , Trempe JF & Gehring K ((2015) ) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J, 34: , 2492–2505. |
[106] | Okatsu K , Koyano F , Kimura M , Kosako H , Saeki Y , Tanaka K & Matsuda N ((2015) ) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol, 209: , 111–128. |
[107] | Yamano K , Queliconi BB , Koyano F , Saeki Y , Hirokawa T , Tanaka K & Matsuda N ((2015) ) Site-specific Interaction Mapping of Phosphorylated Ubiquitin to Uncover Parkin Activation. J Biol Chem, 290: , 25199–25211. |
[108] | Kazlauskaite A , Martinez-Torres RJ , Wilkie S , Kumar A , Peltier J , Gonzalez A , Johnson C , Zhang J , Hope AG , Peggie M , Trost M , van Aalten DM , Alessi DR , Prescott AR , Knebel A , Walden H & Muqit MM ((2015) ) Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep, 16: , 939–954. |
[109] | Kumar A , Aguirre JD , Condos TE , Martinez-Torres RJ , Chaugule VK , Toth R , Sundaramoorthy R , Mercier P , Knebel A , Spratt DE , Barber KR , Shaw GS & Walden H ((2015) ) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J, 34: , 2506–2521. |
[110] | Ordureau A , Sarraf SA , Duda DM , Heo JM , Jedrychowski MP , Sviderskiy VO , Olszewski JL , Koerber JT , Xie T , Beausoleil SA , Wells JA , Gygi SP , Schulman BA & Harper JW ((2014) ) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell, 56: , 360–375. |
[111] | Fiesel FC , Ando M , Hudec R , Hill AR , Castanedes-Casey M , Caulfield TR , Moussaud-Lamodiere EL , Stankowski JN , Bauer PO , Lorenzo-Betancor O , Ferrer I , Arbelo JM , Siuda J , Chen L , Dawson VL , Dawson TM , Wszolek ZK , Ross OA , Dickson DW & Springer W ((2015) ) (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep, 16: , 1114–1130. |
[112] | Fiesel FC & Springer W ((2015) ) Disease relevance of phosphorylated ubiquitin (p-S65-Ub). Autophagy, 11: , 2125–2126. |
[113] | Chan NC , Salazar AM , Pham AH , Sweredoski MJ , Kolawa NJ , Graham RL , Hess S & Chan DC ((2011) ) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet, 20: , 1726–1737. |
[114] | Cunningham CN , Baughman JM , Phu L , Tea JS , Yu C , Coons M , Kirkpatrick DS , Bingol B & Corn JE ((2015) ) USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol, 17: , 160–169. |
[115] | Tanaka A , Cleland MM , Xu S , Narendra DP , Suen DF , Karbowski M & Youle RJ ((2010) ) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol, 191: , 1367–1380. |
[116] | Xu S , Peng G , Wang Y , Fang S & Karbowski M ((2011) ) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell, 22: , 291–300. |
[117] | Poole AC , Thomas RE , Yu S , Vincow ES & Pallanck L ((2010) ) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One, 5: , e10054. |
[118] | Gomes LC & Scorrano L ((2013) ) Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta, 1833: , 205–212. |
[119] | Ziviani E , Tao RN & Whitworth AJ ((2010) ) Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc Natl Acad Sci U S A, 107: , 5018–5023. |
[120] | Swatek KN & Komander D ((2016) ) Ubiquitin modifications. Cell Res, 26: , 399–422. |
[121] | Vincow ES , Merrihew G , Thomas RE , Shulman NJ , Beyer RP , MacCoss MJ & Pallanck LJ ((2013) ) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad SciU S A, 110: , 6400–6405. |
[122] | McLelland GL , Soubannier V , Chen CX , McBride HM & Fon EA ((2014) ) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J, 33: , 282–295. |
[123] | Braschi E , Goyon V , Zunino R , Mohanty A , Xu L & McBride HM ((2010) ) Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol, 20: , 1310–1315. |
[124] | Soubannier V , McLelland GL , Zunino R , Braschi E , Rippstein P , Fon EA & McBride HM ((2012) ) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol, 22: , 135–141. |
[125] | Roberts RF , Tang MY , Fon EA & Durcan TM ((2016) ) Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int J Biochem Cell Biol, 79: , 427–436. |
[126] | Vilarino-Guell C , Wider C , Ross OA , Dachsel JC , Kachergus JM , Lincoln SJ , Soto-Ortolaza AI , Cobb SA , Wilhoite GJ , Bacon JA , Behrouz B , Melrose HL , Hentati E , Puschmann A , Evans DM , Conibear E , Wasserman WW , Aasly JO , Burkhard PR , Djaldetti R , Ghika J , Hentati F , Krygowska-Wajs A , Lynch T , Melamed E , Rajput A , Rajput AH , Solida A , Wu RM , Uitti RJ , Wszolek ZK , Vingerhoets F & Farrer MJ ((2011) ) VPS35 mutations in Parkinson disease. Am J Hum Genet, 89: , 162–167. |
[127] | Zimprich A , Benet-Pages A , Struhal W , Graf E , Eck SH , Offman MN , Haubenberger D , Spielberger S , Schulte EC , Lichtner P , Rossle SC , Klopp N , Wolf E , Seppi K , Pirker W , Presslauer S , Mollenhauer B , Katzenschlager R , Foki T , Hotzy C , Reinthaler E , Harutyunyan A , Kralovics R , Peters A , Zimprich F , Brucke T , Poewe W , Auff E , Trenkwalder C , Rost B , Ransmayr G , Winkelmann J , Meitinger T & Strom TM ((2011) ) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet, 89: , 168–175. |
[128] | Haft CR , de la Luz Sierra M , Bafford R , Lesniak MA , Barr VA & Taylor SI ((2000) ) Human orthologs of yeast vacuolar protein sorting proteins Vps26, 29, and 35: Assembly into multimeric complexes. Mol Biol Cell, 11: , 4105–4116. |
[129] | Malik BR , Godena VK & Whitworth AJ ((2015) ) VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum Mol Genet, 24: , 6106–6117. |
[130] | Lazarou M , Sliter DA , Kane LA , Sarraf SA , Wang C , Burman JL , Sideris DP , Fogel AI & Youle RJ ((2015) ) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature, 524: , 309–314. |
[131] | Stolz A , Ernst A & Dikic I ((2014) ) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol, 16: , 495–501. |
[132] | Wong YC & Holzbaur EL ((2014) ) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A, 111: , E4439–E4448. |
[133] | Heo JM , Ordureau A , Paulo JA , Rinehart J & Harper JW ((2015) ) The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell, 60: , 7–20. |
[134] | Richter B , Sliter DA , Herhaus L , Stolz A , Wang C , Beli P , Zaffagnini G , Wild P , Martens S , Wagner SA , Youle RJ & Dikic I ((2016) ) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A, 113: , 4039–4044. |
[135] | Shin JH , Ko HS , Kang H , Lee Y , Lee YI , Pletinkova O , Troconso JC , Dawson VL & Dawson TM ((2011) ) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell, 144: , 689–702. |
[136] | Wu Z , Puigserver P , Andersson U , Zhang C , Adelmant G , Mootha V , Troy A , Cinti S , Lowell B , Scarpulla RC & Spiegelman BM ((1999) ) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell, 98: , 115–124. |
[137] | Stevens DA , Lee Y , Kang HC , Lee BD , Lee YI , Bower A , Jiang H , Kang SU , Andrabi SA , Dawson VL , Shin JH & Dawson TM ((2015) ) Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A, 112: , 11696–11701. |
[138] | Johnson BN , Berger AK , Cortese GP & LaVoie MJ ((2012) ) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A, 109: , 6283–6288. |
[139] | Khan SH , Zhao D , Shah SZ , Hassan MF , Zhu T , Song Z , Zhou X & Yang L ((2016) ) Parkin Overexpression ameliorates PrP106-126-induced neurotoxicity via enhanced autophagy in N2a cells. Cell Mol Neurobiol. doi: 10.1007/s10571-016-0407-7 |
[140] | Pascucci B , D’Errico M , Romagnoli A , De Nuccio C , Savino M , Pietraforte D , Lanzafame M , Calcagnile AS , Fortini P , Baccarini S , Orioli D , Degan P , Visentin S , Stefanini M , Isidoro C , Fimia GM & Dogliotti E ((2016) ) Overexpression of parkin rescues the defective mitochondrial phenotype and the increased apoptosis of Cockayne Syndrome A cells. Oncotarget. doi: 10.18632/oncotarget.9913 |
[141] | Swiader A , Nahapetyan H , Faccini J , D’Angelo R , Mucher E , Elbaz M , Boya P & Vindis C ((2016) ) Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget, 7: , 28821–28835. |
[142] | Zhou H , Forveille S , Sauvat A , Sica V , Izzo V , Durand S , Muller K , Liu P , Zitvogel L , Rekdal O , Kepp O & Kroemer G ((2015) ) The oncolytic peptide LTX-315 kills cancer cells through Bax/Bak-regulated mitochondrial membrane permeabilization. Oncotarget, 6: , 26599–26614. |
[143] | Ivatt RM , Sanchez-Martinez A , Godena VK , Brown S , Ziviani E & Whitworth AJ ((2014) ) Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc Natl Acad Sci U S A, 111: , 8494–8499. |
[144] | McCoy MK , Kaganovich A , Rudenko IN , Ding J & Cookson MR ((2014) ) Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet, 23: , 145–156. |
[145] | Lefebvre V , Du Q , Baird S , Ng AC , Nascimento M , Campanella M , McBride HM & Screaton RA ((2013) ) Genome-wide RNAi screen identifies ATPase inhibitory factor 1 (ATPIF1) as essential for PARK2 recruitment and mitophagy. Autophagy, 9: , 1770–1779. |
[146] | Fiesel FC , Moussaud-Lamodiere EL , Ando M & Springer W ((2014) ) A specific subset of E2 ubiquitin-conjugating enzymes regulate Parkin activation and mitophagy differently. J Cell Sci, 127: , 3488–3504. |
[147] | Geisler S , Vollmer S , Golombek S & Kahle PJ ((2014) ) The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for Parkin-dependent mitophagy. J Cell Sci, 127: , 3280–3293. |
[148] | Haddad DM , Vilain S , Vos M , Esposito G , Matta S , Kalscheuer VM , Craessaerts K , Leyssen M , Nascimento RM , Vianna-Morgante AM , De Strooper B , Van Esch H , Morais VA & Verstreken P ((2013) ) Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol Cell, 50: , 831–843. |
[149] | Burchell VS , Nelson DE , Sanchez-Martinez A , Delgado-Camprubi M , Ivatt RM , Pogson JH , Randle SJ , Wray S , Lewis PA , Houlden H , Abramov AY , Hardy J , Wood NW , Whitworth AJ , Laman H & Plun-Favreau H ((2013) ) The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci, 16: , 1257–1265. |
[150] | Bingol B , Tea JS , Phu L , Reichelt M , Bakalarski CE , Song Q , Foreman O , Kirkpatrick DS & Sheng M ((2014) ) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature, 510: , 370–375. |
[151] | Cornelissen T , Haddad D , Wauters F , Van Humbeeck C , Mandemakers W , Koentjoro B , Sue C , Gevaert K , De Strooper B , Verstreken P & Vandenberghe W ((2014) ) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet, 23: , 5227–5242. |
[152] | Durcan TM , Tang MY , Perusse JR , Dashti EA , Aguileta MA , McLelland GL , Gros P , Shaler TA , Faubert D , Coulombe B & Fon EA ((2014) ) USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J, 33: , 2473–2491. |
[153] | Sun Y , Vashisht AA , Tchieu J , Wohlschlegel JA & Dreier L ((2012) ) Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem, 287: , 40652–40660. |
[154] | Van Humbeeck C , Cornelissen T , Hofkens H , Mandemakers W , Gevaert K , De Strooper B & Vandenberghe W ((2011) ) Parkin interacts with Ambra1 to induce mitophagy. J Neurosci, 31: , 10249–10261. |
[155] | Michiorri S , Gelmetti V , Giarda E , Lombardi F , Romano F , Marongiu R , Nerini-Molteni S , Sale P , Vago R , Arena G , Torosantucci L , Cassina L , Russo MA , Dallapiccola B , Valente EM & Casari G ((2010) ) The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ, 17: , 962–974. |
[156] | Hollville E , Carroll RG , Cullen SP & Martin SJ ((2014) ) Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell, 55: , 451–466. |
[157] | Kim J , Fiesel FC , Belmonte KC , Hudec R , Wang WX , Kim C , Nelson PT , Springer W & Kim J ((2016) ) miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol Neurodegener, 11: , 55. |
[158] | Irrcher I , Aleyasin H , Seifert EL , Hewitt SJ , Chhabra S , Phillips M , Lutz AK , Rousseaux MW , Bevilacqua L , Jahani-Asl A , Callaghan S , MacLaurin JG , Winklhofer KF , Rizzu P , Rippstein P , Kim RH , Chen CX , Fon EA , Slack RS , Harper ME , McBride HM , Mak TW & Park DS ((2010) ) Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet, 19: , 3734–3746. |
[159] | Hao LY , Giasson BI & Bonini NM ((2010) ) DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A, 107: , 9747–9752. |
[160] | Thomas KJ , McCoy MK , Blackinton J , Beilina A , van der Brug M , Sandebring A , Miller D , Maric D , Cedazo-Minguez A & Cookson MR ((2011) ) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet, 20: , 40–50. |
[161] | Kinghorn KJ , Castillo-Quan JI , Bartolome F , Angelova PR , Li L , Pope S , Cocheme HM , Khan S , Asghari S , Bhatia KP , Hardy J , Abramov AY & Partridge L ((2015) ) Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain, 138: , 1801–1816. |
[162] | Zhou Q , Yen A , Rymarczyk G , Asai H , Trengrove C , Aziz N , Kirber MT , Mostoslavsky G , Ikezu T , Wolozin B & Bolotina VM ((2016) ) Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson’s disease. Nat Commun, 7: , 10332. |
[163] | Gusdon AM , Zhu J , Van Houten B & Chu CT ((2012) ) ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis, 45: , 962–972. |
[164] | Ogaki K , Koga S , Heckman MG , Fiesel FC , Ando M , Labbe C , Lorenzo-Betancor O , Moussaud-Lamodiere EL , Soto-Ortolaza AI , Walton RL , Strongosky AJ , Uitti RJ , McCarthy A , Lynch T , Siuda J , Opala G , Rudzinska M , Krygowska-Wajs A , Barcikowska M , Czyzewski K , Puschmann A , Nishioka K , Funayama M , Hattori N , Parisi JE , Petersen RC , Graff-Radford NR , Boeve BF , Springer W , Wszolek ZK , Dickson DW & Ross OA ((2015) ) Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders. Neurology, 85: , 2016–2025. |
[165] | Lesage S , Drouet V , Majounie E , Deramecourt V , Jacoupy M , Nicolas A , Cormier-Dequaire F , Hassoun SM , Pujol C , Ciura S , Erpapazoglou Z , Usenko T , Maurage CA , Sahbatou M , Liebau S , Ding J , Bilgic B , Emre M , Erginel-Unaltuna N , Guven G , Tison F , Tranchant C , Vidailhet M , Corvol JC , Krack P , Leutenegger AL , Nalls MA , Hernandez DG , Heutink P , Gibbs JR , Hardy J , Wood NW , Gasser T , Durr A , Deleuze JF , Tazir M , Destee A , Lohmann E , Kabashi E , Singleton A , Corti O & Brice A ((2016) ) Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/parkin-dependent mitophagy. Am J Hum Genet, 98: , 500–513. |
[166] | Biskup S , Moore DJ , Celsi F , Higashi S , West AB , Andrabi SA , Kurkinen K , Yu SW , Savitt JM , Waldvogel HJ , Faull RL , Emson PC , Torp R , Ottersen OP , Dawson TM & Dawson VL ((2006) ) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol, 60: , 557–569. |
[167] | West AB , Moore DJ , Biskup S , Bugayenko A , Smith WW , Ross CA , Dawson VL & Dawson TM ((2005) ) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A, 102: , 16842–16847. |
[168] | Alegre-Abarrategui J , Ansorge O , Esiri M & Wade-Martins R ((2008) ) LRRK2 is a component of granular alpha-synuclein pathology in the brainstem of Parkinson’s disease. Neuropathol Appl Neurobiol, 34: , 272–283. |
[169] | Alegre-Abarrategui J , Christian H , Lufino MM , Mutihac R , Venda LL , Ansorge O & Wade-Martins R ((2009) ) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet, 18: , 4022–4034. |
[170] | Niu J , Yu M , Wang C & Xu Z ((2012) ) Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J Neurochem, 122: , 650–658. |
[171] | Wang X , Yan MH , Fujioka H , Liu J , Wilson-Delfosse A , Chen SG , Perry G , Casadesus G & Zhu X ((2012) ) LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet, 21: , 1931–1944. |
[172] | Su YC & Qi X ((2013) ) Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet, 22: , 4545–4561. |
[173] | Yue M , Hinkle KM , Davies P , Trushina E , Fiesel FC , Christenson TA , Schroeder AS , Zhang L , Bowles E , Behrouz B , Lincoln SJ , Beevers JE , Milnerwood AJ , Kurti A , McLean PJ , Fryer JD , Springer W , Dickson DW , Farrer MJ & Melrose HL ((2015) ) Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis, 78: , 172–195. |
[174] | Nguyen HN , Byers B , Cord B , Shcheglovitov A , Byrne J , Gujar P , Kee K , Schule B , Dolmetsch RE , Langston W , Palmer TD & Pera RR ((2011) ) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell, 8: , 267–280. |
[175] | Reinhardt P , Schmid B , Burbulla LF , Schondorf DC , Wagner L , Glatza M , Hoing S , Hargus G , Heck SA , Dhingra A , Wu G , Muller S , Brockmann K , Kluba T , Maisel M , Kruger R , Berg D , Tsytsyura Y , Thiel CS , Psathaki OE , Klingauf J , Kuhlmann T , Klewin M , Muller H , Gasser T , Scholer HR & Sterneckert J ((2013) ) Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell, 12: , 354–367. |
[176] | Smith GA , Jansson J , Rocha EM , Osborn T , Hallett PJ & Isacson O ((2016) ) Fibroblast biomarkers of sporadic Parkinson’s disease and LRRK2 kinase inhibition. Mol Neurobiol, 53: , 5161–5177. |
[177] | Steger M , Tonelli F , Ito G , Davies P , Trost M , Vetter M , Wachter S , Lorentzen E , Duddy G , Wilson S , Baptista MA , Fiske BK , Fell MJ , Morrow JA , Reith AD , Alessi DR & Mann M ((2016) ) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife, 5: , pii: e12813. |
[178] | Devi L , Raghavendran V , Prabhu BM , Avadhani NG & Anandatheerthavarada HK ((2008) ) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem, 283: , 9089–9100. |
[179] | Norris EH , Uryu K , Leight S , Giasson BI , Trojanowski JQ & Lee VM ((2007) ) Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol, 170: , 658–666. |
[180] | Kamp F , Exner N , Lutz AK , Wender N , Hegermann J , Brunner B , Nuscher B , Bartels T , Giese A , Beyer K , Eimer S , Winklhofer KF & Haass C ((2010) ) Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J, 29: , 3571–3589. |
[181] | Nakamura K , Nemani VM , Azarbal F , Skibinski G , Levy JM , Egami K , Munishkina L , Zhang J , Gardner B , Wakabayashi J , Sesaki H , Cheng Y , Finkbeiner S , Nussbaum RL , Masliah E & Edwards RH ((2011) ) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem, 286: , 20710–20726. |
[182] | Todd AM & Staveley BE ((2012) ) Expression of Pink1 with alpha-synuclein in the dopaminergic neurons of Drosophila leads to increases in both lifespan and healthspan. Genet Mol Res, 11: , 1497–1502. |
[183] | Chen L , Xie Z , Turkson S & Zhuang X ((2015) ) A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci, 35: , 890–905. |
[184] | Oliveras-Salva M , Macchi F , Coessens V , Deleersnijder A , Gerard M , Van der Perren A , Van den Haute C & Baekelandt V ((2014) ) Alpha-synuclein-induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol Aging, 35: , 2625–2636. |
[185] | Di Maio R , Barrett PJ , Hoffman EK , Barrett CW , Zharikov A , Borah A , Hu X , McCoy J , Chu CT , Burton EA , Hastings TG & Greenamyre JT ((2016) ) alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med, 8: , 342ra378. |
[186] | Sterky FH , Lee S , Wibom R , Olson L & Larsson NG ((2011) ) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A, 108: , 12937–12942. |
[187] | Pickrell AM , Huang CH , Kennedy SR , Ordureau A , Sideris DP , Hoekstra JG , Harper JW & Youle RJ ((2015) ) Endogenous parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron, 87: , 371–381. |
[188] | Ryan BJ , Hoek S , Fon EA & Wade-Martins R ((2015) ) Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem Sci, 40: , 200–210. |
[189] | Huttenlocher J , Stefansson H , Steinberg S , Helgadottir HT , Sveinbjornsdottir S , Riess O , Bauer P & Stefansson K ((2015) ) Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum Mol Genet, 24: , 5637–5643. |
[190] | Majcher V , Goode A , James V & Layfield R ((2015) ) Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci, 66: , 43–52. |
[191] | Freischmidt A , Wieland T , Richter B , Ruf W , Schaeffer V , Muller K , Marroquin N , Nordin F , Hubers A , Weydt P , Pinto S , Press R , Millecamps S , Molko N , Bernard E , Desnuelle C , Soriani MH , Dorst J , Graf E , Nordstrom U , Feiler MS , Putz S , Boeckers TM , Meyer T , Winkler AS , Winkelman J , de Carvalho M , Thal DR , Otto M , Brannstrom T , Volk AE , Kursula P , Danzer KM , Lichtner P , Dikic I , Meitinger T , Ludolph AC , Strom TM , Andersen PM & Weishaupt JH ((2015) ) Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci, 18: , 631–636. |
[192] | Feany MB & Pallanck LJ ((2003) ) Parkin A multiurpose neuroprotective agent? Neuron, 38: , 13–16. |
[193] | Rana A , Rera M & Walker DW ((2013) ) Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A, 110: , 8638–8643. |
[194] | Schiavi A , Maglioni S , Palikaras K , Shaik A , Strappazzon F , Brinkmann V , Torgovnick A , Castelein N , De Henau S , Braeckman BP , Cecconi F , Tavernarakis N & Ventura N ((2015) ) Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr Biol, 25: , 1810–1822. |
Figures and Tables
Fig.1
PINK1 and Parkin domain structures and PD-related mutations. (A-B) Given are schematic, color-coded domain representations of PINK1 and Parkin. PD-associated missense and nonsense mutations from the PD Mutation Database (http://www.molgen.vib-ua.be/PDMutDB/) are displayed on top of each structure with their respective locations. Mutations in red have been experimentally verified as loss-of-function mutations and are considered pathogenic, while functional defects for variants shown in black remain unclear. Underlined mutations are common variants based on the ExAC database (http://exac.broadinstitute.org) with allele frequencies greater than 1:10 000. (A) Domain structure of PINK1 (581 amino acids): mitochondrial targeting sequence (MTS, orange), transmembrane region (TM, red), N-terminal regulatory region (NT, gray), N-lobe of the kinase domain (cyan), C-lobe of the kinase domain (purple) and the C-terminal domain (CTD, blue). PD-associated mutations are listed on the top. Mitochondrial protease (MPP and PARL) cleavage sites and PINK1 auto-phosphorylation sites are displayed at the bottom. (B) Domain structure of Parkin (465 amino acids): ubiquitin-like domain (UBL, red), linker (gray), really-interesting-new-gene (RING)/unique Parkin domain (R0/UPD, green), RING1 (R1, cyan), in-between-RING (IBR, purple), repressor element of Parkin (REP, yellow), and RING2 (R2, pink). E2 co-enzyme and p-Ser65-Ub binding sites as well as Ser65 phosphorylation and Cys431 catalytic sites are displayed at the bottom. (C) Closed, inactive conformation of full-length human Parkin (left: front view and right: back view). The structure is shown in colored ribbons that correspond to the respective domain colors. The solvent-accessible surface area of each domain is shown in semi-transparent rendering in the same color. Ser65 is highlighted in Van der Waal representation with standard atom coloring (hydrogen: white, oxygen: red, nitrogen: blue). The zinc-finger motifs of Parkin are rendered in licorice stick with standard atom coloring and the corresponding zinc ions as spheres (cyan).
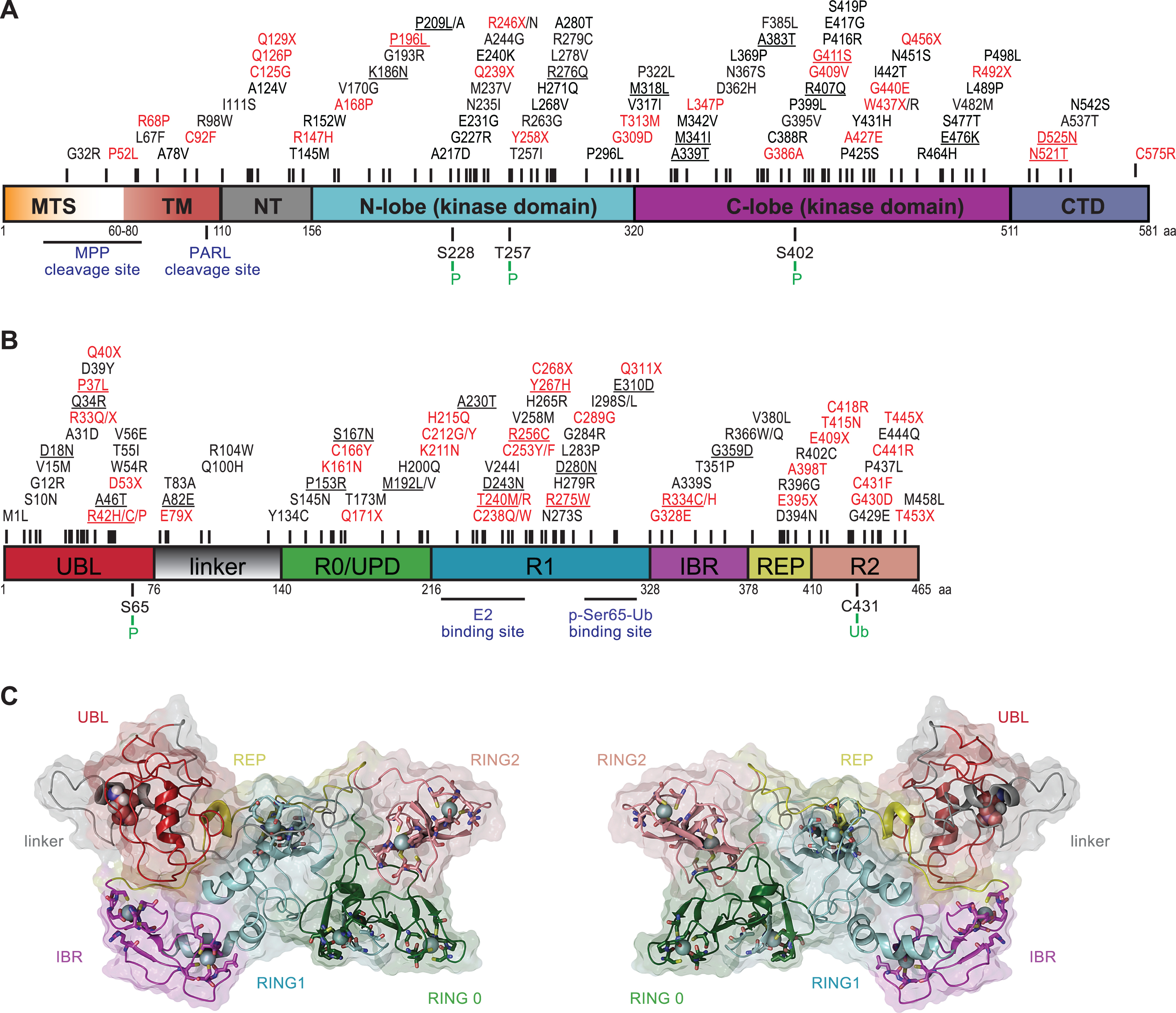
Fig.2
PINK1/Parkin-directed mitochondria quality control. Shown is a cartoon of the PINK1/Parkin pathway(s). (A) In healthy mitochondria, PINK1 is imported and it undergoes cleavage by the mitochondrial proteases MPP and PARL. N-terminally cleaved PINK1 is then subsequently degraded by the Ub/proteasome system. Parkin remains inactive in the cytosol. (B) Upon mitochondrial damage, PINK1 is no longer imported, but accumulates on the outer mitochondrial membrane (OMM) to activate Parkin. PINK1-mediated phosphorylation of Ub and Parkin enable its E3 Ub ligase functions in concert with E2 Ub-conjugating enzymes. p-Ser65-Ub on OMM substrates acts as the Parkin receptor for its recruitment from the cytosol. Together, PINK1 and Parkin then engage a feed-forward loop amplification. Formed poly-Ub chains can be cleaved by DUBs to reverse PINK1/Parkin functions. (C) Individual OMM proteins decorated with poly-Ub can be extracted from the membrane and degraded by the 26 S proteasome (mitochondria-associated degradation; MAD). Greater, but localized mitochondrial damage is sequestered by the formation of mitochondria-derived vesicles (MDV). Severely damaged mitochondria are decorated with high levels of p-Ser65-Ub that serves as the mitophagy tag for autophagy receptors that direct their degradation in lysosomes.