Epigenetic Biomarkers for Parkinson’s Disease: From Diagnostics to Therapeutics
Abstract
Parkinson’s disease (PD) is a prevalent neurodegenerative illness that is often diagnosed after significant pathology and neuronal cell loss has occurred. Biomarkers of PD are greatly needed for early diagnosis, as well as for the prediction of disease progression and treatment outcome. In this regard, the epigenome, which is partially dynamic, holds considerable promise for the development of molecular biomarkers for PD. Epigenetic marks are modified by both DNA sequence and environmental factors associated with PD, and such marks could serve as a unifying predictor of at-risk individuals. Epigenetic abnormalities have been detected in PD and other age-dependent neurodegenerative diseases, some of which were reported to occur early on and were reversible by PD medications. Emerging reports indicate that certain epigenetic differences observed in the PD brain are detectable in more easily accessible tissues. In this review, we examine epigenetic-based strategies for the development of PD biomarkers. Despite the complexities and challenges faced, the epigenome offers a new source of biomarkers with potential etiological relevance to PD, and may expand opportunities for personalized therapies.
INTRODUCTION
Biomarkers are molecules that represent a pathological signature that is easily identified and quantified for a particular disease or disorder [1]. In recent years biomarkers have proven essential in identifying individuals at risk for disease, tracking disease progression, and selection of therapeutic intervention [2]. Despite their clinical importance and extensive genetic studies, there are currently no definitive biomarkers for PD available for use in research or diagnostics.
It is estimated that by the time a patient with PD sees a physician for the hallmark motor symptoms of rigidity, resting tremor, bradykinesia, and postural instability, up to 70% of the patient’s dopaminergic neurons have already been lost [1]. One of the major aims of biomarker research in PD is to provide both medical professionals and researchers with tools that enable accurate diagnosis before extensive neuronal death has occurred [1]. In addition to monitoring at-risk individuals for early diagnosis, biomarkers may expedite drug discovery and development. Current clinical trials for PD use relatively subjective measures such as motor activity, behavior, and mood to assess treatment efficacy [3]. This makes such trials extremely expensive and labor-intensive, especially considering that more than 90% of drugs entering clinical trials for brain diseases do not yield marketable compounds [4]. Adding biomarkers to clinical trials may reduce this drug failure rate. In clinical trials for cancers, biomarker-based studies have had considerable success, to the point that there are now biomarker-driven “basket” trials underway, which enroll patients based on genetic biomarkers independent of tumor histology [5, 6]. The use of biomarkers rather histopathological diagnosis in these basket trials has generated enormous interest, because they implement a hypothesis-driven strategy for incorporating precision medicine into clinical trials. In PD, accurate and measurable biomarkers have the potential to add great value to clinical trials by identifying patient subgroups that are more likely to be treatment-responsive and by providing quantifiable measures of disease outcome and treatment response.
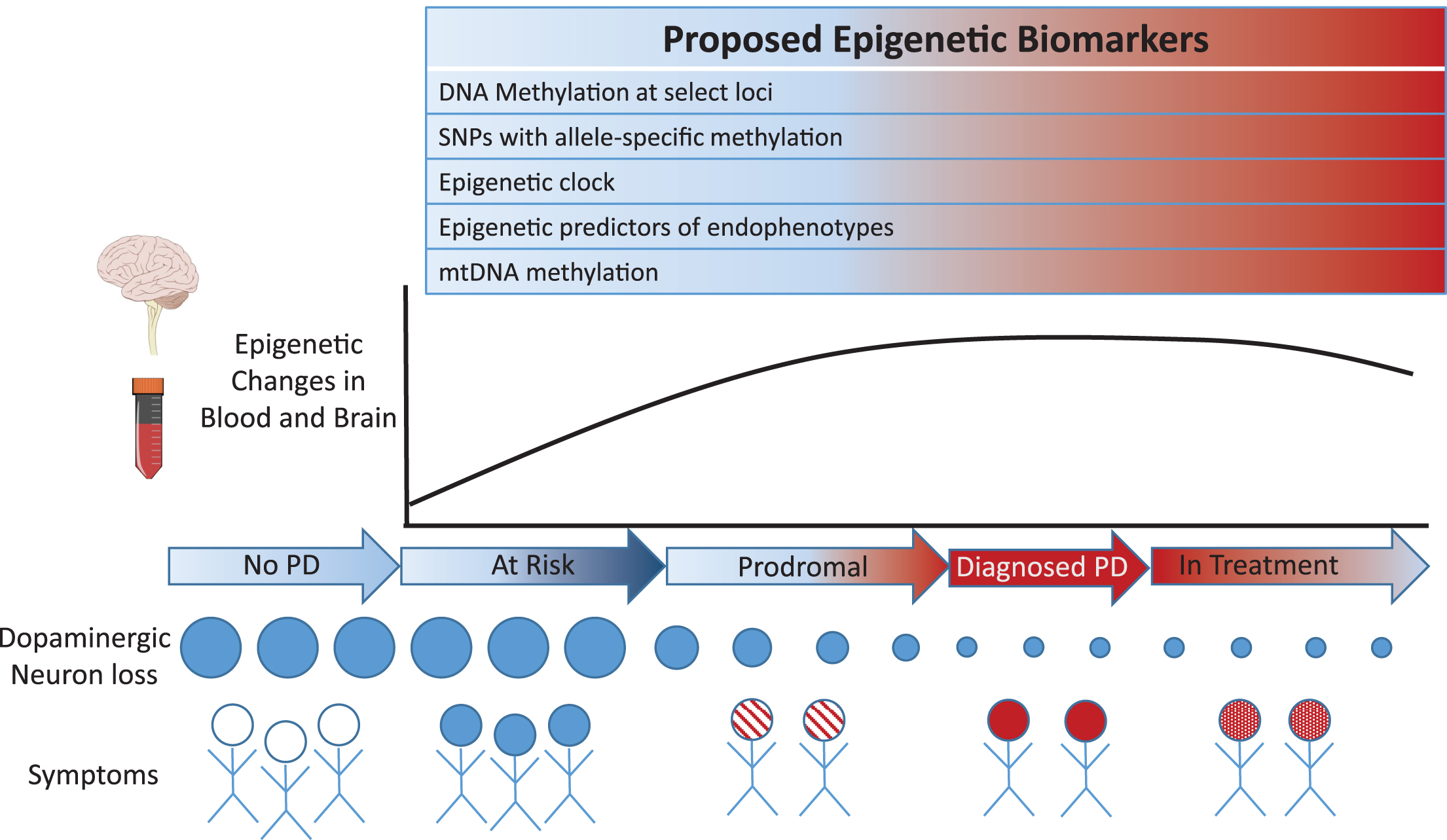
Recent investigations suggest that epigenetic marks may be a new source of biomarkers for PD (see Fig. 1). Epigenetics refers to heritable and acquired alterations in gene activity and expression, without changes in DNA sequence. The epigenome is partially dynamic, such that stable epigenetic changes can occur in fully differentiated, post-mitotic cells in response to environmental signals [7]. Epigenetic marks, such as biochemical modifications of DNA and of histone proteins, regulate gene expression by controlling DNA accessibility and signaling to the transcriptional machinery [8, 9]. To date, DNA modifications, which includes DNA methylation and other covalent cytosine modifications, have been the most commonly investigated epigenetic marks for biomarker discovery studies. Historically, DNA modifications were thought to occur exclusively within CpG (cytosine-phosphate-guanine) sites, but it is now established that DNA modification occurs at non-CpG sites as well, particularly in brain neurons [10–12]. Other epigenetic marks, such as post-translational modification of histones and noncoding RNAs, frequently act in concert with DNA modifications, affecting chromatin accessibility and structure as well as the recruitment of various transcription factors and protein complexes [13–15]. By modifying genomic activities, epigenetic mechanisms impact downstream protein amounts, cellular function, and phenotypic outcome. The epigenome also exhibits considerable heterogeneity between tissues, tissue subregions, and cell types within an organism, which enables the divergent biological roles of tissues and cell types [13]. In the brain, epigenetic mechanisms are central to neurodevelopment, synaptic transmission, and plasticity [16–18]. Epigenetic abnormalities have been implicated in the mechanism of numerous brain illnesses, including PD [19].
Epigenetic investigations of PD may yield biomarkers involved in disease pathogenesis. PD has numerous complex, non-Mendelian features that are consistent with the dynamic nature of the epigenome. Such features includes the prevalence of sporadic, non- familial PD cases (∼90%); the low concordance rates (only 11%) between monozygotic twins for sporadic PD [20]; the 1.5-2 times higher risk of PD in males relative to females [21]; diurnal fluctuations in symptom severity (sundowning syndrome) [22]; and the various agricultural and industrial chemicals associated with increased PD risk [23–25]. The epigenome is well known to vary between monozygotic twins [26], respond to sex hormones [27], exhibit circadian fluctuations [28] and affect neuronal functions following exposure to PD-associated environmental toxins [29–31]. Moreover, epigenetic mechanisms may contribute to the greatest risk factor for PD: aging. Epigenetic variation accumulates in aging cells, even between genetically-identical individuals, a phenomenon often referred to as “epigenetic drift”. In most tissues, including the brain, there are rapid changes in DNA modifications in the early life period, but these changes gradually slow over the life span [12, 32–34]. Epigenetic aging affects genomic locations differentially: promoter-associated CpG islands tend to increase in DNA methylation with age, while areas with high DNA methylation (i.e. repetitive elements in intergenic regions) tend to lose methylation with age [33, 35–37] Epigenomic alterations during aging are mediated not only by external/environmental factors but by genetic factors as well. There are numerous sites in the genome that exhibit allele-specific epigenetic differences, which have been shown to be haplotype-dependent, highly tissue-specific, and prevalent in the brain [38–40]. Different DNA haplotypes can demonstrate markedly different epigenetic changes with age, which affects phenotypic outcome [41]. Thus, the epigenome can serve as a convergence point for genetic and environmental risk factors of disease, making it an attractive means of detecting heritable and nonheritable disease risk, disease progression, and treatment efficacy in biomarker applications.
CANDIDATE BIOMARKER LOCI
Recent studies have begun to search for epigenetic-based biomarkers using candidate gene and genome-wide approaches. DNA modifications, particularly DNA methylation, are being used as successful biomarkers for several types of cancer [42, 43], and they show the most promise for epigenetic biomarker development in neurodegenerative disease. Effective PD biomarkers based on DNA methylation status will greatly depend on the concordance of this epigenetic mark between brain and more easily accessible tissues, such as blood. This is challenging, because epigenetic patterns are significantly different between tissue types (such as brain and blood) [44]. Indeed, data from the NIH Epigenomics Roadmap and ENCODE projects demonstrate tissue and cell-type specific DNA methylation patterns at key genes implicated in PD [13, 45]. Epigenetic divergence between blood and brain can be further amplified by aging, environmental, and stochastic factors. Moreover, capacity to detect reliable epigenetic patterns within blood (and other tissues) can be skewed by variability in cellular composition. This is an important consideration for developing epigenetic biomarkers to examine whole blood, because the sensitivity and reliability of the test needs to exceed circadian fluctuations [46] and inter-individual differences in blood leukocyte populations [47]. Thus, the reliability and extent to which peripheral tissues are able to mirror the non-dividing cells of the brain represents a major hurdle for epigenetic biomarker strategies. Nonetheless, several studies suggest that DNA methylation profiles at certain genes in blood can distinguish control subjects from PD cases, opening a new source for biomarker discovery in PD.
At present, the most studied epigenetic-based biomarker for PD is DNA methylation in the α-synuclein gene. α-Synuclein is a presynaptic neuronal protein that has been linked to familial and sporadic cases of PD. α-Synuclein is the principal component of Lewy bodies and Lewy neurites, the hallmark protein inclusions of PD. Increases in α-synuclein levels lead to its abnormal aggregation and to neuronal degeneration in vivo [48, 49] Elevated α-synuclein mRNA has been observed in individual laser-captured dopaminergic neurons in the substantia nigra of sporadic PD cases [50]. DNA methylation in the promoter element of the α-synuclein gene (a CpG island located in intron 1) was reported to be essential for the regulation of α- synuclein transcription [51]. Moreover, DNA methylation at the α-synuclein intron 1 promoter was reduced in postmortem brain tissue (particularly the substantia nigra) of patients with sporadic PD [51, 52]. The change in DNA methylation at α-synuclein intron 1 in the substantia nigra appears to be specific to PD, as it was not observed in cohorts including individuals with Lewy body dementia [52, 53]. Loss of DNA methylation at the α-synuclein promoter could explain the increase of α-synuclein mRNA in sporadic PD, which in turn leads to Lewy body formation and neurotoxicity. However, this conclusion merits caution, as the tissues analysed in these epigenetic studies have the potentially confounding factors of neuronal loss, medication differences, and limited sample size.
Encouragingly, recent studies of blood have supported the reduction in DNA methylation at the α-synuclein gene in PD cases when compared with healthy controls. A relatively large study of the peripheral blood of 490 sporadic PD patients and 485 healthy controls observed significant hypomethylation at the α-synuclein intron 1 promoter in the PD cases [54]. This was replicated in three additional, independent studies [55–57]. The α-synuclein DNA methylation patterns in blood mirrored those of the brain of PD patients [56], suggesting that α-synuclein DNA methylation signatures in blood may be a good proxy for the brain changes at this loci in PD.
However, there has been some conflicting reports on the detection of DNA methylation differences at the α-synuclein intron 1 promoter in PD [58, 59]. Though certain studies in the PD substantia nigra reported large DNA methylation differences (>30%) at select CpGs [51, 52], studies in blood have detected much smaller DNA methylation differences between PD cases and controls (∼5%) (52,53, 54, 55). Low sample numbers (n≤50) may account for why some studies did not identify DNA methylation differences at α-synuclein in PD blood [58, 59]. Discrepancies in the selection of CpG sites examined at the α-synuclein promoter and differences in PD clinical subtypes have likely also contributed to the conflicting findings [55, 58, 59]. Overall, modest DNA methylation differences in peripheral tissues and genomic target selection represents significant challenges for epigenetic biomarkers. The robustness and suitability of α-synuclein-based epigenetic biomarkers may be improved by further work examining patient and clinical variables, including PD subtypes, symptoms, genetic risk carriers, and age groups.
Age-dependent changes in DNA methylation have also been observed at the α- synuclein intron 1 promoter [54, 60]. In sporadic PD patients, age of onset was positively correlated with DNA methylation atα-synuclein intron 1, where the greater the methylation levels, the later the occurrence of motor symptoms [54]. Age-dependent differences in DNA methylation at α-synuclein intron 1 in blood are, however, relatively low (<10%). Interestingly, across ages, lower levels of DNA methylation at α-synuclein intron 1 were detected in males relative to females [54]. Further studies will be needed to determine whether this could contribute to the known higher incidence of PD in males [21].
Finally, levodopa treatment, the mainstay treatment for PD [61], reverted the age- dependent hypomethylation at the α-synuclein intron 1 promoter in a dose-dependent manner [54]. Levodopa also increased DNA methylation at this promoter in drug-naive blood cells cultured from PD patients [54]. Though levodopa altered DNA methylation status at numerous other genes (3% of genes assayed) in blood samples from PD patients, these findings do suggest that DNA methylation at the α-synuclein gene may be a useful biomarker for predicting treatment response and for aiding the discovery of new drugs. In addition, the ability of levodopa to reverse hypomethylation at the α-synuclein gene suggests that studies of treated PD individuals may have underestimated α-synuclein hypomethylation. Consequently, this biomarker may be more appropriate for early diagnostics, i.e., prior to levodopa treatment.
Supporting this idea, DNA methylation at the α-synuclein intron 1 promoter did not have good sensitivity for discerning treated PD patients from healthy controls; DNA methylation status at α-synuclein intron 1 identified only about 40% of PD patients [54]. However, the specificity of DNA methylation at α-synuclein intron 1, i.e., its capacity to correctly identify healthy individuals, was ∼76% [54], which is a higher specificity than the current gold standard for determining Parkinsonian syndromes, the DaTscan method (DaTscan specificity is 67% and sensitivity is 98%) [62]. Thus, α-synuclein methylation may best serve as biomarker for early diagnosis in untreated individuals and as a predictor of responsiveness to levodopa treatment.
In neuronal cells, α-synuclein has been shown to sequester the DNA methyltransferase DNMT1 from the nucleus into the cytoplasm, resulting in a global loss (30%) of DNA methylation in the brains of PD patients [63]. This indicates that many more genomic sites could have DNA methylation changes in PD, and that there may be many more epigenetic-based biomarkers. Genome-wide scans have the potential to expedite the discovery of such epigenetic biomarkers. One approach is to perform a genome-wide search for differentially methylated sites in PD patients relative to controls, and determine whether there are sites that show concordant methylation differences in both blood and brain. Investigation of blood and cortical samples from the same individuals found 124 sites that were differentially methylated in PD and had concordant cross-tissue changes in DNA methylation [64]. Many of the cross-tissue, differentially methylated sites had previously been implicated in PD in transcriptional or genome- wide association studies (GWAS) [64, 65]. Epigenetic abnormalities common across tissues in PD suggest that the epigenetic risk at these loci was inherited or acquired in early development.
Another genome-wide strategy for biomarker identification is to examine easily accessible tissues of living Parkinson’s patients at an early or prodromal stage in the disease. A study examining drug-naive, sporadic Parkinson’s patients at the onset of motor symptoms found transcriptomic changes in the chromatin remodeling and methylation machineries [66]. This suggests that widespread epigenetic differences occur early in PD, prior to pharmacological treatment, and that such differences may be useful biomarkers for prodromal PD cases.
Familial and sporadic PD has been shown to exhibit many common DNA methylation abnormalities, particularly at gene enhancer elements in stem cell-derived dopaminergic neurons [67]. As an extension to this, a genome-wide search for common DNA methylation disturbances across diseases with overlapping neuropathological features could identify new, pathogenically-relevant biomarkers. In addition to PD, α-synuclein inclusions have been detected in dementia with Lewy bodies, Alzheimer’s disease, and Down syndrome [68, 69]. Whole-genome DNA methylation analysis found more than 700 loci with DNA methylation abnormalities that were common across multiple neurodegenerative diseases [70]. This study also detected 1400 sites in common between PD and Lewy body dementia [70]. Though limited by sample size, this study represents a starting point for follow-up research seeking biomarkers that are shared across neurodegenerative illnesses.
EPIGENETIC STRATEGIES FOR REFINING GENETIC BIOMARKERS
A strength of genetic biomarkers of disease risk is their ease of detection in peripheral tissues, such as saliva and blood, as well as the certainty that they are detectable prior to symptomatic onset. Idiopathic PD is mediated by mutations in genes such as α-synuclein, parkin, PTEN-induced kinase 1, and leucine-rich repeat kinase 2 (LRRK2), but these account for only about 10% of all cases [71]. The majority of PD cases are sporadic. Genetic variants have been implicated in sporadic PD [72], although the contributions of these risk variants are not well defined. Recent GWAS meta-analyses examining 7.8 million single nucleotide polymorphisms (SNPs) in 13,708 PD cases and 95,282 controls reported 28 independent risk variants for PD [73]. The overwhelming majority (>90%) of identified risk SNPs are located in intergenic regions, and as a result their roles in gene function and disease pathogenesis are not evident [74]. However, several of the top GWAS risk SNPs for PD demonstrated allelic differences in nearby DNA methylation levels [73]. Furthermore, a GWAS risk SNP in intron 1 of the α-synuclein gene (rs3756063) was found to modify DNA methylation at the overlapping α- synuclein promoter in the blood and brain of PD patients [54, 56]. Epigenetic analysis can therefore provide insight into the functional effects of SNPs, which aids in prioritizing candidate sites for biomarker development.
Disease-associated SNPs are, for the most part, in linkage disequilibrium with many other SNPs. This means that SNPs identified in GWAS are often not the true disease risk factor, but rather one of the co-inherited SNPs is likely responsible for disease risk. As a result it may be impossible to identify true causal variants using GWAS alone. Epigenetic profiling of regions surrounding co-inherited SNPs could be a useful approach to revealing true risk variants. This was shown in an elegant recent study by Soldner et al. [75], who identified a SNP allele that can up-regulate the α-synuclein gene by 10–20%. This SNP was identified by intersecting PD- associated SNPs in the α-synuclein locus (463 SNPs) with publicly available epigenetic data generated by the NIH Roadmap. This effort revealed two SNPs in α-synuclein intron 4 that overlapped histone marks characteristic of enhancers. Enhancers are distal regulatory elements that increase the expression of genes, and enhancer activity is determined by epigenetic status. The authors then tested the effects of the SNP alleles using the CRISPR-Cas9 gene editing technique in human pluripotent stem cell-derived neurons. The G-allele of SNP rs356168 was found to significantly increase α-synuclein mRNA expression and was associated with greater PD disease risk. This is one example of a DNA variant that modifies disease risk via epigenetically-controlled regulatory elements. However, there are certainly many more, because studies of the adult brain have found that PD-associated SNPs are enriched in distal enhancers [76]. Analysis of active enhancers overlapping PD risk loci have also implicated tissues outside of the central nervous system [45]. Such research indicates that epigenetic analysis at PD-associated SNPs can generate molecular biomarkers for detection of disease vulnerability. In addition, combined epigenetic–genetic analysis can pave the way for the development of genetically-modified cell lines useful for drug candiadate screening and therapeutic discovery.
MITOCHONDRIAL EPIGENETIC TARGETS
Although mitochondrial DNA (mtDNA) represents only 1% of the total cellular DNA, mitochondrial gene products are essential for normal cell function. Thus, it stands to reason that mtDNA would be studied for epigenetic changes, yet only recently have research efforts moved beyond nuclear DNA (nDNA) and into mtDNA. While methylation of nDNA is well established, methylation of mtDNA has been a controversial matter [77]. Arguments against mtDNA methylation have included the idea that methylases could not access the mitochondria in eukaryotes, and that mtDNA is devoid of histones as it is arranged in nucleoid clusters that adhere to the mitochondrial membrane [78]. Recently, however, both methylated mtDNA and the DNA methyltransferase DNMT1 have been found within the mitochondria [79, 80]. In the central nervous system of humans, mitochondria also contain the de novo methyltransferase DNMT3a [81]. Changes in mtDNA methylation have been associated with environmental toxins, oxidative stress, drug treatment, disease, and aging [82]. Methylation levels of mtDNA were analyzed in the brain of 4- and 24-month old mice, and it was found that hydroxymethylation, but not methylation levels in mtDNA decreased with age in the frontal cortex [83].
Recently, mitochondrial methylation and hydroxymethylation were examined in the D- loop region (which regulates mitochondrial transcription and replication [84]), and the NADH dehydrogenase 6 (MT-ND6) gene in the substantia nigra of PD cases and healthy controls. Strikingly, the D-loop region of mtDNA in the PD brain showed a loss of methylation in nearly all CpG and non-CpG sites relative to control samples [85]. Methylation levels in the MT-ND6 gene and hydroxymethylation in the D-loop were unchanged in PD cases relative to controls [85]. This supports the hypothesis that reduced methylation levels in the D-loop was not the result of diminished neuronal content in the PD substantia nigra, but rather that there was epigenetic misregulation of a site important to mitochrondrial function in PD. If further research shows concordance between blood and brain, the methylation status of the D-loop of mtDNA could become a diagnostic biomarker for PD.
PRODROMAL BIOMARKERS AND ENDOPHENOTYPES
In order to develop disease-modifying and neuroprotective therapies, biomarkers that can discern the prodromal phase of PD will be necessary. Prodromal PD refers to the stage before the disease has fully manifested, when very early signs and symptoms of the disease are present, but diagnosis using the current motor symptom criteria is not yet possible [86]. Prodromal PD involves a range of non-motor symptoms – such as sleep dysfunction, severe constipation, late-onset hyposmia, and episodes of major depression – that often pre-date the motor symptoms by years. Identifying biomarkers for the prodromal period is complicated because the period in which these early symptoms dominate is poorly defined, with estimates ranging from 5–20 years [87]. Furthermore, there is extensive inter-individual variability for many of the non- motor symptoms. At present, there is no accepted combination of symptoms to assist clinicians in definitively diagnosing prodromal PD.
One method of predicting whether individuals presenting non-motor symptoms may develop the full illness is by examining their ‘epigenetic clock’. It has been repeatedly shown that DNA methylation status at specific CpG sites in the genome reliably changes with age such that it can be used to accurately predict chronological age [36, 88, 89]. In a study by Hovarth, DNA methylation datasets for 8,000 samples from various tissues were used to construct and evaluate a predictor of DNA methylation age [89]. Astonishingly, this age prediction method shows chronological accuracy across both sexes and in most cell and tissue types, including blood, breast, kidney, liver, and brain [89, 90]. It detected accelerated epigenetic aging due to progeria [89], obesity, Down syndrome [91], and HIV infection [92], and it predicted all-cause mortality even after adjusting for various risk factors [93]. In the blood of PD patients, the epigenetic clock tool found accelerated epigenetic aging [94]. In addition, genes linked to accelerated epigenetic aging in the brain had significant overlap with those implicated in PD and other neurodegenerative diseases [95]. Analysis of epigenetic aging in PD also identified striking differences in blood cell type composition between PD cases and controls. Specifically, blood from PD patients contained more granulocytes and fewer T-helper and B cells than did control samples [94]. Accelerated epigenetic aging in combination with altered immune cell counts may precede the onset of motor and cognitive symptoms in PD, which could be a useful biomarker for predicting progression to PD in prodromal individuals.
Emerging evidence suggests that disruption of circadian rhythms may not only be a consequence of neuronal loss in PD, but may itself contribute to the neurodegenerative process [96]. Circadian rhythms are physiological and behavioral cycles generated by an endogenous biological clock in the suprachiasmatic nucleus [97]. Circadian rhythms regulate many physiological and behavioral functions, such as 24-h rhythmicity in rest-activity behavior, body temperature, hormone levels, and homeostasis [97]. Numerous studies have reported that the symptoms and behaviors associated with PD, such as disruption in motor activity, sleep dysfunction, and responsiveness to dopaminergic treatments, show diurnal fluctuation [46]. These observations indicate there are circadian influences on the expression of PD’s clinical features. Genes that control circadian rhythms are known as clock genes. Several clock genes have been identified including period (PER1, PER2, and PER3), cryptochrome (CRY1 and CRY2), CLOCK, BMAL1, and NPAS2 [46]. Histone modifications and DNA modifications regulate many of these clock genes and can exhibit circadian fluctuations [28, 98–100]. In PD, a circadian regulator, the NPAS2 gene promoter, was shown to have a 13% decrease in DNA methylation relative to controls [100]. Clock genes are known to greatly interact through complex feedback loops to generate and sustain circadian rhythms. Hence, aberrant DNA methylation of key clock genes in the PD brain may potentiate widespread circadian deregulation and neuronal dysfunction.
CONCLUSION
While the epigenome has promise for both prognostic and diagnostic biomarkers for PD, it is not without its limitations. The ability to detect these biomarkers using noninvasive means will be crucial, and it is known that epigenetic marks, such as DNA methylation, vary widely across tissues. Another critical challenge is that the size of the epigenetic differences observed in patients will have to substantially exceed the variation within populations and cell composition of the assay tissue. Detection of the epigenetic signal will also have to reliably surpass the technical noise of the assay. Although there is now a wide range of tools to measure epigenetic marks, sensitivity and specificity come at a price. Many of the current platforms require specialized, expensive equipment that would make the use of these tests cost prohibitive. Furthermore, determining which specific genomic locations are most appropriate for epigenetic biomarker development is challenging. Detection of histone marks, though not as streamlined and practical for clinical biomarker purposes, could be used to predict which genomic sites have biomarker potential. Since there are many types of histone modifications, researchers could use this diversity of histone marks to determine which sites in the genome are most homologous between tissues, such as blood and brain. Sites demonstrating consistently similar histone modification profiles between brain and peripheral tissues are likely more reliable for epigenetic (and genetic) biomarker applications. As such, analysis of histone modification patterns can refine the discovery and development of DNA modification biomarkers for PD.
Despite its current limitations, epigenetics represents an auspicious target for PD biomarkers. Both stool- and blood-based epigenetic tests are already commercially available for early-stage colorectal cancer, and there are many more epigenetic based biomarkers in clinical studies [42]. Since DNA methylation patterns at specific genomic sites in the blood of PD patients can mirror those of brain, there is promise for these types of tests for PD. Not only could epigenetic marks serve to predict and diagnose patients, but epigenetic information could also help determine which patient subgroups would benefit most from a treatment. For example, in patients newly diagnosed with glioblastoma, MGMT promoter methylation is predictive of a favorable response to temozolomide chemotherapy [43]. Epigenetic biomarkers therefore can greatly expand the potential for personalized therapeutics.
Integrating epigenetic information with existing PD diagnostic tools may enhance early detection, the confidence of diagnosis and therapeutic approaches. For example, neuroimaging techniques such as DaTscan, which is used to detect the density of dopaminergic transporters in the brain, helps clinicians distinguish PD from atypical parkinsonian disorders. Patients, however, are typically symptomatic before this tool is used [101]. Epigenetic-based biomarkers could rapidly discern individuals at greater risk, which would prompt clinical monitoring and neuroimaging earlier; enhancing detection of prodromal PD cases. In addition, the combination of DaTscan and epigenetic biomarkers could also predict which patients will be most responsive to the main drug for PD, levodopa, given that dopaminergic treatments affect DNA methylation at the α-synuclein gene [54]. Epigenetic biomarkers may also predict therapeutic utility of the newer treatments targeting α-synuclein which are currently in clinical trials [102]. Finally, epigenetic biomarkers could be used in combination with genetic screens to identify individuals at risk for familial and sporadic forms of PD. Recent studies suggest that phenotypic effects of sequence variants can be influenced by accompanying epigenetic signatures, via allele-specific methylation. Studies demonstrating the abundance of allele-specific methylation in the brain [39, 103] and its presence at PD risk genes [75] may lead to the development of novel combinatorial genetic-epigenetic biomarkers for PD. Though still at a very early stage, epigenetic research in PD may unify the aging, environmental and genetic risk factors, and thus change the strategies used in PD diagnosis and treatment.
CONFLICT OF INTEREST
The authors have no conflict of interest.
ACKNOWLEDGMENTS
Viviane Labrie is supported by grants from the Alzheimer’s Society of Canada (16 15) and Scottish Rite Charitable Foundation of Canada (15110).
REFERENCES
[1] | Graeber MB ((2009) ) Biomarkers for Parkinson’s disease. Exp Neurol, 216: , 249–253. |
[2] | Wu Y , Le W , & Jankovic J ((2011) ) Preclinical biomarkers of Parkinson disease. Arch Neurol, 68: , 22–30. |
[3] | Venuto CS , Potter NB , Ray Dorsey E , & Kieburtz K ((2016) ) A review of disease progression models of Parkinson’s disease and applications in clinical trials. Mov Disord, 31: , 947–956. |
[4] | OECD. ((2015) ) Addressing Dementia: The OECD Response, OECD Publishing, Paris. doi: 10.1787/9789264231726-en |
[5] | Herbst RS , Gandara DR , Hirsch FR , Redman MW , LeBlanc M , Mack PC , Schwartz LH , Vokes E , Ramalingam SS , Bradley JD , Sparks D , Zhou Y , Miwa C , Miller VA , Yelensky R , Li Y , Allen JD , Sigal EV , Wholley D , Sigman CC , Blumenthal GM , Malik S , Kelloff GJ , Abrams JS , Blanke CD , & Papadimitrakopoulou VA ((2015) ) Lung master protocol (Lung-MAP)-a biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin Cancer Res, 21: , 1514–1524. |
[6] | Brower V ((2015) ) NCI-MATCH pairs tumor mutations with matching drugs. Nat Biotechnol, 33: , 790–791. |
[7] | Turner BM ((2009) ) Epigenetic responses to environmental change and their evolutionary implications. Philos Trans R Soc Lond B Biol Sci, 364: , 3403–3418. |
[8] | Bannister AJ , & Kouzarides T ((2011) ) Regulation of chromatin by histone modifications. Cell Res, 21: , 381–395. |
[9] | Berger SL ((2007) ) The complex language of chromatin regulation during transcription. Nature, 447: , 407–412. |
[10] | Schultz MD , He Y , Whitaker JW , Hariharan M , Mukamel EA , Leung D , Rajagopal N , Nery JR , Urich MA , Chen H , Lin S , Lin Y , Jung I , Schmitt AD , Selvaraj S , Ren B , Sejnowski TJ , Wang W , & Ecker JR ((2015) ) Human body epigenome maps reveal noncanonical DNA methylation variation. Nature, 523: , 212–216. |
[11] | Guo JU , Su Y , Shin JH , Shin J , Li H , Xie B , Zhong C , Hu S , Le T , Fan G , Zhu H , Chang Q , Gao Y , Ming GL , & Song H ((2014) ) Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci, 17: , 215–222. |
[12] | Lister R , Mukamel EA , Nery JR , Urich M , Puddifoot CA , Johnson ND , Lucero J , Huang Y , Dwork AJ , Schultz MD , Yu M , Tonti-Filippini J , Heyn H , Hu S , Wu JC , Rao A , Esteller M , He C , Haghighi FG , Sejnowski TJ , Behrens MM , & Ecker JR ((2013) ) Global epigenomic reconfiguration during mammalian brain development. Science, 341: , 1237905. |
[13] | Roadmap Epigenomics C , Kundaje A , Meuleman W , Ernst J , Bilenky M , Yen A , Heravi-Moussavi A , Kheradpour P , Zhang Z , Wang J , Ziller MJ , Amin V , Whitaker JW , Schultz MD , Ward LD , Sarkar A , Quon G , Sandstrom RS , Eaton ML , Wu YC , Pfenning AR , Wang X , Claussnitzer M , Liu Y , Coarfa C , Harris RA , Shoresh N , Epstein CB , Gjoneska E , Leung D , Xie W , Hawkins RD , Lister R , Hong C , Gascard P , Mungall AJ , Moore R , Chuah E , Tam A , Canfield TK , Hansen RS , Kaul R , Sabo PJ , Bansal MS , Carles A , Dixon JR , Farh KH , Feizi S , Karlic R , Kim AR , Kulkarni A , Li D , Lowdon R , Elliott G , Mercer TR , Neph SJ , Onuchic V , Polak P , Rajagopal N , Ray P , Sallari RC , Siebenthall KT , Sinnott-Armstrong NA , Stevens M , Thurman RE , Wu J , Zhang B , Zhou X , Beaudet AE , Boyer LA , De Jager PL , Farnham PJ , Fisher SJ , Haussler D , Jones SJ , Li W , Marra MA , McManus MT , Sunyaev S , Thomson JA , Tlsty TD , Tsai LH , Wang W , Waterland RA , Zhang MQ , Chadwick LH , Bernstein BE , Costello JF , Ecker JR , Hirst M , Meissner A , Milosavljevic A , Ren B , Stamatoyannopoulos JA , Wang T , & Kellis M ((2015) ) Integrative analysis of 111 reference human epigenomes. Nature, 518: , 317–330. |
[14] | Holoch D , & Moazed D ((2015) ) RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet, 16: , 71–84. |
[15] | Domcke S , Bardet AF , Adrian Ginno P , Hartl D , Burger L , & Schubeler D ((2015) ) Competition between DNA methylation and transcription factors determines binding of NRF1. Nature, 528: , 575–579. |
[16] | Song J , Zhong C , Bonaguidi MA , Sun GJ , Hsu D , Gu Y , Meletis K , Huang ZJ , Ge S , Enikolopov G , Deisseroth K , Luscher B , Christian KM , Ming GL , & Song H ((2012) ) Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature, 489: , 150–154. |
[17] | Yu H , Su Y , Shin J , Zhong C , Guo JU , Weng YL , Gao F , Geschwind DH , Coppola G , Ming GL , & Song H ((2015) ) Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat Neurosci, 18: , 836–843. |
[18] | Halder R , Hennion M , Vidal RO , Shomroni O , Rahman RU , Rajput A , Centeno TP , van Bebber F , Capece V , Garcia Vizcaino JC , Schuetz AL , Burkhardt S , Benito E , Navarro Sala M , Javan SB , Haass C , Schmid B , Fischer A , & Bonn S ((2016) ) DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci, 19: , 102–110. |
[19] | Landgrave-Gomez J , Mercado-Gomez O , & Guevara-Guzman R ((2015) ) Epigenetic mechanisms in neurological and neurodegenerative diseases. Front Cell Neurosci, 9: , 58. |
[20] | Wirdefeldt K , Gatz M , Reynolds CA , Prescott CA , Pedersen NL ((2011) ) Heritability of Parkinson disease in Swedish twins: A longitudinal study. Neurobiol Aging, 32: , 1923 e1921–1928. |
[21] | Van Den Eeden SK , Tanner CM , Bernstein AL , Fross RD , Leimpeter A , Bloch DA , & Nelson LM ((2003) ) Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am J Epidemiol, 157: , 1015–1022. |
[22] | Bliwise DL , Watts RL , Watts N , Rye DB , Irbe D , & Hughes M ((1995) ) Disruptive nocturnal behavior in Parkinson’s disease and Alzheimer’s disease. J Geriatr Psychiatry Neurol, 8: , 107–110. |
[23] | Wirdefeldt K , Adami HO , Cole P , Trichopoulos D , Mandel J. ((2011) ) Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur J Epidemiol, 26: (Suppl 1), S1–S58. |
[24] | Agim ZS , & Cannon JR ((2015) ) Dietary factors in the etiology of Parkinson’s disease. Biomed Res Int, 2015: , 672838. |
[25] | Gorell JM , Johnson CC , Rybicki BA , Peterson EL , Kortsha GX , Brown GG , & Richardson RJ ((1997) ) Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology, 48: , 650–658. |
[26] | Kaminsky ZA , Tang T , Wang SC , Ptak C , Oh GH , Wong AH , Feldcamp LA , Virtanen C , Halfvarson J , Tysk C , McRae AF , Visscher PM , Montgomery GW , Gottesman II , Martin NG , & Petronis A ((2009) ) DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet, 41: , 240–245. |
[27] | Nugent BM , Wright CL , Shetty AC , Hodes GE , Lenz KM , Mahurkar A , Russo SJ , Devine SE , & McCarthy MM ((2015) ) Brain feminization requires active repression of masculinization via DNA methylation. Nat Neurosci, 18: , 690–697. |
[28] | Lim AS , Srivastava GP , Yu L , Chibnik LB , Xu J , Buchman AS , Schneider JA , Myers AJ , Bennett DA , & De Jager PL ((2014) ) 24-hour rhythms of DNA methylation and their relation with rhythms of RNA expression in the human dorsolateral prefrontal cortex. PLoS Genet, 10: , e1004792. |
[29] | Song C , Kanthasamy A , Jin H , Anantharam V , & Kanthasamy AG ((2011) ) Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology, 32: , 586–595. |
[30] | Song C , Kanthasamy A , Anantharam V , Sun F , & Kanthasamy AG ((2010) ) Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: Relevance to epigenetic mechanisms of neurodegeneration. Mol Pharmacol, 77: , 621–632. |
[31] | Wright RO , & Baccarelli A ((2007) ) Metals and neurotoxicology. J Nutr, 137: , 2809–2813. |
[32] | Salehi F , Scheithauer BW , Kovacs K , Horvath E , Syro LV , Sharma S , Manoranjan B , Cusimano M ((2012) ) O-6-methylguanine-DNA methyltransferase (MGMT) immunohistochemical expression in pituitary corticotroph adenomas. Neurosurgery, 496: , 491–496. discussion. |
[33] | Horvath S , Zhang Y , Langfelder P , Kahn RS , Boks MP , van Eijk K , van den Berg LH , & Ophoff RA ((2012) ) Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol, 13: , R97. |
[34] | Numata S , Ye T , Hyde TM , Guitart-Navarro X , Tao R , Wininger M , Colantuoni C , Weinberger DR , Kleinman JE , & Lipska BK ((2012) ) DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet, 90: , 260–272. |
[35] | Rakyan VK , Down TA , Maslau S , Andrew T , Yang TP , Beyan H , Whittaker P , McCann OT , Finer S , Valdes AM , Leslie RD , Deloukas P , & Spector TD ((2010) ) Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res, 20: , 434–439. |
[36] | Florath I , Butterbach K , Muller H , Bewerunge-Hudler M , & Brenner H ((2014) ) Cross- sectional and longitudinal changes in DNA methylation with age: An epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet, 23: , 1186–1201. |
[37] | Heyn H , Li N , Ferreira HJ , Moran S , Pisano DG , Gomez A , Diez J , Sanchez-Mut JV , Setien F , Carmona FJ , Puca AA , Sayols S , Pujana MA , Serra-Musach J , Iglesias-Platas I , Formiga F , Fernandez AF , Fraga MF , Heath SC , Valencia A , Gut IG , Wang J , & Esteller M ((2012) ) Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A, 109: , 10522–10527. |
[38] | Kuleshov V , Xie D , Chen R , Pushkarev D , Ma Z , Blauwkamp T , Kertesz M , & Snyder M ((2014) ) Whole-genome haplotyping using long reads and statistical methods. Nat Biotechnol, 32: , 261–266. |
[39] | Do C , Lang CF , Lin J , Darbary H , Krupska I , Gaba A , Petukhova L , Vonsattel JP , Gallagher MP , Goland RS , Clynes RA , Dwork A , Kral JG , Monk C , Christiano AM , & Tycko B ((2016) ) Mechanisms and disease associations of haplotype-dependent allele-specific DNA methylation. Am J Hum Genet, 98: , 934–955. |
[40] | Leung D , Jung I , Rajagopal N , Schmitt A , Selvaraj S , Lee AY , Yen CA , Lin S , Lin Y , Qiu Y , Xie W , Yue F , Hariharan M , Ray P , Kuan S , Edsall L , Yang H , Chi NC , Zhang MQ , Ecker JR , & Ren B ((2015) ) Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature, 518: , 350–354. |
[41] | Labrie V , Buske OJ , Oh E , Jeremian R , Ptak C , Gasiunas G , Maleckas A , Petereit R , Zvirbliene A , Adamonis K , Kriukiene E , Koncevicius K , Gordevicius J , Nair A , Zhang A , Ebrahimi S , Oh G , Siksnys V , Kupcinskas L , Brudno M , & Petronis A ((2016) ) Lactase nonpersistence is directed by DNA-variation-dependent epigenetic aging. Nat Struct Mol Biol, 23: , 566–573. |
[42] | Gyparaki MT , Basdra EK , & Papavassiliou AG ((2013) ) DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. J Mol Med (Berl), 91: , 1249–1256. |
[43] | Dunn J , Baborie A , Alam F , Joyce K , Moxham M , Sibson R , Crooks D , Husband D , Shenoy A , Brodbelt A , Wong H , Liloglou T , Haylock B , & Walker C ((2009) ) Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer, 101: , 124–131. |
[44] | Varley KE , Gertz J , Bowling KM , Parker SL , Reddy TE , Pauli-Behn F , Cross MK , Williams BA , Stamatoyannopoulos JA , Crawford GE , Absher DM , Wold BJ , & Myers RM ((2013) ) Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res, 23: , 555–567. |
[45] | Coetzee SG , Pierce S , Brundin P , Brundin L , Hazelett DJ , & Coetzee GA ((2016) ) Enrichment of risk SNPs inregulatory regions implicate diverse tissues in Parkinson’s disease etiology. Sci Rep, 6: , 30509. |
[46] | Videnovic A , Lazar AS , Barker RA , & Overeem S ((2014) ) ‘The clocks that time us’–circadian rhythms in neurodegenerative disorders. Nat Rev Neurol, 10: , 683–693. |
[47] | Chahine LM , Stern MB , & Chen-Plotkin A ((2014) ) Blood-based biomarkers for Parkinson’s disease. Parkinsonism Relat Disord, 20: (Suppl 1), S99–S103. |
[48] | Masliah E , Rockenstein E , Veinbergs I , Mallory M , Hashimoto M , Takeda A , Sagara Y , Sisk A , & Mucke L ((2000) ) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science, 287: , 1265–1269. |
[49] | Emmer KL , Waxman EA , Covy JP , & Giasson BI ((2011) ) E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem, 286: , 35104–35118. |
[50] | Grundemann J , Schlaudraff F , Haeckel O , & Liss B ((2008) ) Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res, 36: , e38. |
[51] | Jowaed A , Schmitt I , Kaut O , & Wullner U ((2010) ) Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci, 30: , 6355–6359. |
[52] | Matsumoto L , Takuma H , Tamaoka A , Kurisaki H , Date H , Tsuji S , & Iwata A ((2010) ) CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One, 5: , e15522. |
[53] | de Boni L , Tierling S , Roeber S , Walter J , Giese A , & Kretzschmar HA ((2011) ) Next-generation sequencing reveals regional differences of the alpha-synuclein methylation state independent of Lewy body disease. Neuromolecular Med, 13: , 310–320. |
[54] | Schmitt I , Kaut O , Khazneh H , deBoni L , Ahmad A , Berg D , Klein C , Frohlich H , & Wullner U ((2015) ) L-dopa increases alpha-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov Disord, 30: , 1794–1801. |
[55] | Tan YY , Wu L , Zhao ZB , Wang Y , Xiao Q , Liu J , Wang G , Ma JF , & Chen SD ((2014) ) Methylation of alpha-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Parkinsonism Relat Disord, 20: , 308–313. |
[56] | Pihlstrom L , Berge V , Rengmark A , & Toft M ((2015) ) Parkinson’s disease correlates with promoter methylation in the alpha-synuclein gene. Mov Disord, 30: , 577–580. |
[57] | Ai SX , Xu Q , Hu YC , Song CY , Guo JF , Shen L , Wang CR , Yu RL , Yan XX , & Tang BS ((2014) ) Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J Neurol Sci, 337: , 123–128. |
[58] | Richter J , Appenzeller S , Ammerpohl O , Deuschl G , Paschen S , Bruggemann N , Klein C , & Kuhlenbaumer G ((2012) ) No evidence for differential methylation of alpha-synuclein in leukocyte DNA of Parkinson’s disease patients. Mov Disord, 27: , 590–591. |
[59] | Song Y , Ding H , Yang J , Lin Q , Xue J , Zhang Y , Chan P , & Cai Y ((2014) ) Pyrosequencing analysis of SNCA methylation levels in leukocytes from Parkinson’s disease patients. Neurosci Lett, 569: , 85–88. |
[60] | de Boni L , Riedel L , Schmitt I , Kraus TF , Kaut O , Piston D , Akbarian S , Wullner U ((2015) ) DNA methylation levels of alpha-synuclein intron 1 in the aging brain. Neurobiol Aging, 36: , 3334 e3337–3311. |
[61] | Smith Y , Wichmann T , Factor SA , & DeLong MR ((2012) ) Parkinson’s disease therapeutics: New developments and challenges since the introduction of levodopa. Neuropsychopharmacology, 37: , 213–246. |
[62] | de la Fuente-Fernandez R ((2012) ) Role of DaTSCAN and clinical diagnosis in Parkinson disease. Neurology, 78: , 696–701. |
[63] | Desplats P , Spencer B , Coffee E , Patel P , Michael S , Patrick C , Adame A , Rockenstein E , & Masliah E ((2011) ) Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem, 286: , 9031–9037. |
[64] | Masliah E , Dumaop W , Galasko D , & Desplats P ((2013) ) Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics, 8: , 1030–1038. |
[65] | Moore K , McKnight AJ , Craig D , & O’Neill F ((2014) ) Epigenome-wide association study for Parkinson’s disease. Neuromolecular Med, 16: , 845–855. |
[66] | Calligaris R , Banica M , Roncaglia P , Robotti E , Finaurini S , Vlachouli C , Antonutti L , Iorio F , Carissimo A , Cattaruzza T , Ceiner A , Lazarevic D , Cucca A , Pangher N , Marengo E , di Bernardo D , Pizzolato G , & Gustincich S ((2015) ) Blood transcriptomics of drug-naive sporadic Parkinson’s disease patients. BMC Genomics, 16: , 876. |
[67] | Fernandez-Santiago R , Carballo-Carbajal I , Castellano G , Torrent R , Richaud Y , Sanchez-Danes A , Vilarrasa-Blasi R , Sanchez-Pla A , Mosquera JL , Soriano J , Lopez-Barneo J , Canals JM , Alberch J , Raya A , Vila M , Consiglio A , Martin-Subero JI , Ezquerra M , & Tolosa E ((2015) ) Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med, 7: , 1529–1546. |
[68] | Galpern WR , & Lang AE ((2006) ) Interface between tauopathies and synucleinopathies: A tale of two proteins. Ann Neurol, 59: , 449–458. |
[69] | Lippa CF , Schmidt ML , Lee VM , & Trojanowski JQ ((1999) ) Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol, 45: , 353–357. |
[70] | Sanchez-Mut JV , Heyn H , Vidal E , Moran S , Sayols S , Delgado-Morales R , Schultz MD , Ansoleaga B , Garcia-Esparcia P , Pons-Espinal M , de Lagran MM , Dopazo J , Rabano A , Avila J , Dierssen M , Lott I , Ferrer I , Ecker JR , & Esteller M ((2016) ) Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry, 6: , e718. |
[71] | Gasser T ((2009) ) Mendelian forms of Parkinson’s disease. Biochim Biophys Acta, 1792: , 587–596. |
[72] | Trinh J , & Farrer M ((2013) ) Advances in the genetics of Parkinson disease. Nat Rev Neurol, 9: , 445–454. |
[73] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R , International Parkinson’s Disease Genomics Consortium (IPDGC), Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI), 23andMe, GenePD, NeuroGenetics Research Consortium (NGRC), Hussman Institute of Human Genomics (HIHG), Ashkenazi Jewish Dataset Investigator, Cohorts for Health, Aging Research in Genetic Epidemiology (CHARGE), North American Brain Expression Consortium (NABEC), United Kingdom Brain Expression Consortium (UKBEC), Greek Parkinson’s Disease Consortium, Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , & Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet, 46: , 989–993. |
[74] | Ramanan VK , & Saykin AJ ((2013) ) Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am J Neurodegener Dis, 2: , 145–175. |
[75] | Soldner F , Stelzer Y , Shivalila CS , Abraham BJ , Latourelle JC , Barrasa MI , Goldmann J , Myers RH , Young RA , & Jaenisch R ((2016) ) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature, 533: , 95–99. |
[76] | Vermunt MW , Reinink P , Korving J , de Bruijn E , Creyghton PM , Basak O , Geeven G , Toonen PW , Lansu N , Meunier C , van Heesch S , Netherlands Brain B , Clevers H , de Laat W , Cuppen E , & Creyghton MP ((2014) ) Large-scale identification of coregulated enhancer networks in the adult human brain. Cell Rep, 9: , 767–779. |
[77] | Groot GS , & Kroon AM ((1979) ) Mitochondrial DNA from various organisms does not contain internally methylated cytosine in -CCGG-sequences. Biochim Biophys Acta, 564: , 355–357. |
[78] | Satoh M , & Kuroiwa T ((1991) ) Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp Cell Res, 196: , 137–140. |
[79] | Shock LS , Thakkar PV , Peterson EJ , Moran RG , & Taylor SM ((2011) ) DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A, 108: , 3630–3635. |
[80] | Liu B , Du Q , Chen L , Fu G , Li S , Fu L , Zhang X , Ma C , & Bin C ((2016) ) CpG methylation patterns of human mitochondrial DNA. Sci Rep, 6: , 23421. |
[81] | Chestnut BA , Chang Q , Price A , Lesuisse C , Wong M , & Martin LJ ((2011) ) Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci, 31: , 16619–16636. |
[82] | Byun HM , & Baccarelli AA ((2014) ) Environmental exposure and mitochondrial epigenetics: Study design and analytical challenges. Hum Genet, 133: , 247–257. |
[83] | Dzitoyeva S , Chen H , & Manev H ((2012) ) Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging, 33: , 2881–2891. |
[84] | Iacobazzi V , Castegna A , Infantino V , & Andria G ((2013) ) Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab, 110: , 25–34. |
[85] | Blanch M , Mosquera JL , Ansoleaga B , Ferrer I , & Barrachina M ((2016) ) Altered Mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Pathol, 186: , 385–397. |
[86] | Berg D , Postuma RB , Adler CH , Bloem BR , Chan P , Dubois B , Gasser T , Goetz CG , Halliday G , Joseph L , Lang AE , Liepelt-Scarfone I , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , & Deuschl G ((2015) ) MDS research criteria for prodromal Parkinson’s disease. Mov Disord, 30: , 1600–1611. |
[87] | Hawkes CH ((2008) ) The prodromal phase of sporadic Parkinson’s disease: Does it exist and if so how long is it? Mov Disord, 23: , 1799–1807. |
[88] | Hannum G , Guinney J , Zhao L , Zhang L , Hughes G , Sadda S , Klotzle B , Bibikova M , Fan JB , Gao Y , Deconde R , Chen M , Rajapakse I , Friend S , Ideker T , & Zhang K ((2013) ) Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell, 49: , 359–367. |
[89] | Horvath S ((2013) ) DNA methylation age of human tissues and cell types. Genome Biol, 14: , R115. |
[90] | Spiers H , Hannon E , Schalkwyk LC , Smith R , Wong CC , O’Donovan MC , Bray NJ , & Mill J ((2015) ) Methylomic trajectories across human fetal brain development. Genome Res, 25: , 338–352. |
[91] | Horvath S , Garagnani P , Bacalini MG , Pirazzini C , Salvioli S , Gentilini D , Di Blasio AM , Giuliani C , Tung S , Vinters HV , & Franceschi C ((2015) ) Accelerated epigenetic aging in Down syndrome. Aging Cell, 14: , 491–495. |
[92] | Horvath S , & Levine AJ ((2015) ) HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis, 212: , 1563–1573. |
[93] | Marioni RE , Shah S , McRae AF , Chen BH , Colicino E , Harris SE , Gibson J , Henders AK , Redmond P , Cox SR , Pattie A , Corley J , Murphy L , Martin NG , Montgomery GW , Feinberg AP , Fallin MD , Multhaup ML , Jaffe AE , Joehanes R , Schwartz J , Just AC , Lunetta KL , Murabito JM , Starr JM , Horvath S , Baccarelli AA , Levy D , Visscher PM , Wray NR , & Deary IJ ((2015) ) DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol, 16: , 25. |
[94] | Horvath S , & Ritz BR ((2015) ) Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY), 7: , 1130–1142. |
[95] | Lu AT , Hannon E , Levine ME , Hao K , Crimmins EM , Lunnon K , Kozlenkov A , Mill J , Dracheva S , & Horvath S ((2016) ) Genetic variants near MLST8 and DHX57 affect the epigenetic age of the cerebellum. Nat Commun, 7: , 10561. |
[96] | Videnovic A , & Willis GL ((2016) ) Circadian system - A novel diagnostic and therapeutic target in Parkinson’s disease? Mov Disord, 31: , 260–269. |
[97] | Hastings M , O’Neill JS , & Maywood ES ((2007) ) Circadian clocks: Regulators of endocrine and metabolic rhythms. J Endocrinol, 195: , 187–198. |
[98] | Koike N , Yoo SH , Huang HC , Kumar V , Lee C , Kim TK , & Takahashi JS ((2012) ) Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science, 338: , 349–354. |
[99] | Duong HA , & Weitz CJ ((2014) ) Temporal orchestration of repressive chromatin modifiers by circadian clock Period complexes. Nat Struct Mol Biol, 21: , 126–132. |
[100] | Lin Q , Ding H , Zheng Z , Gu Z , Ma J , Chen L , Chan P , & Cai Y ((2012) ) Promoter methylation analysis of seven clock genes in Parkinson’s disease. Neurosci Lett, 507: , 147–150. |
[101] | Seifert KD , & Wiener JI ((2013) ) The impact of DaTscan on the diagnosis and management of movement disorders: A retrospective study. Am J Neurodegener Dis, 2: , 29–34. |
[102] | Games D , Valera E , Spencer B , Rockenstein E , Mante M , Adame A , Patrick C , Ubhi K , Nuber S , Sacayon P , Zago W , Seubert P , Barbour R , Schenk D , & Masliah E ((2014) ) Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci, 34: , 9441–9454. |
[103] | Gagliano SA , Ptak C , Mak DY , Shamsi M , Oh G , Knight J , Boutros PC , & Petronis A ((2016) ) Allele-skewed DNA modification in the brain: Relevance to a schizohrenia GWAS. Am J Hum Genet, 98: , 956–962. |
Figures and Tables
Fig.1
Proposed epigenetic-based biomarkers involving sites exhibiting concordant and epigenetic changes in blood and brain of PD patients.