
Frequency and Phenotype of Myotubular Myopathy Amongst Danish Patients with Congenital Myopathy Older than 5 Years
Abstract
Background: Centronuclear myopathy (CNM) is one of four main subtypes of congenital myopathy. X-linked myotubular myopathy (XLMTM) is considered one of the most severe forms, but survivors past infancy have been described. However, detailed information on XLMTM phenotypes in patients who survive infancy is scarce.
Objective: The aim of the study was to report the genetic findings in patients with a predominant centronuclear finding on muscle biopsy and describe the prevalence, phenotypes and the course of the disease in patients with XLMTM in a Danish cohort of patients with congenital myopathies older than five years.
Methods: Ninety-four out of 119 invited patients older than five years were included in the study and assessed by muscle tests, functional tests, muscle biopsy, plasma creatine kinase levels and genetic testing. Genes related to CNM were sequenced in patients who had centronuclear findings on muscle histology. In patients with MTM1 mutations, medical records from local hospitals were reviewed to obtain information on birth history and course of disease.
Results: Sixteen of 94 patients had CNM on muscle biopsy; three male patients, aged 14–25 years, carried a pathogenic MTM1 mutation, six patients carried a pathogenic DNM2 mutation and two carried pathogenic RYR1 mutations. The mutations have all been described before to cause CNM. The MTM phenotypes ranged from severe (classical) to mild; one patient had always been non-ambulant, one had lost ambulation, and one was still ambulant at 25 years.
Conclusions: Our findings show that CNM caused by DNM2 mutations is the most common form of CNM in Danish patients older than 5 years, but XLMTM is not negligible even past age 5 years, and the phenotype may be much milder than generally described - also in patients with the classically described infantile form of the disease.
INTRODUCTION
Congenital myopathies (CM) encompass a group of neuromuscular disorders in which symptoms typically appear at birth or in infancy. The diseases are caused by mutations in genes that often encode proteins involved in the contractile function of muscle. The diseases are classically grouped into four morphological subclasses based on features seen on muscle biopsy [1].
Centronuclear myopathy (CNM) is characterized by a predominance of central nuclei seen on muscle biopsy and encompasses dominant CNM, recessive CNM and X-linked myotubular myopathy (XLMTM). The dominant and recessive forms can be caused by mutations in various genes with the most common forms affecting the dynamin (DNM2), Ryanodine (RYR1) and more rarely the amphiphysin 2 (BIN1) [2] and the titin (TTN) genes [3]. XLMTM is considered the severest form of CM. The disease is caused by mutations in the MTM1 gene located on Xq28 [4], and affects males, although manifesting female carriers have been reported [5–7]. The prevalence and incidence of XLMTM are unknown, but the incidence has been estimated at one in 50.000 newborn males [8]. The classical XLMTM phenotype is severe and typically presents with profound muscle weakness, marked hypotonia and respiratory failure at birth, often leading to death in the first year of life, but wider ranges of XLMTM phenotypes and survival into adulthood have been reported, [9–12]. The phenotypes and course of disease have generally been poorly described, because they appeared in papers focusing on the molecular aspects of the disease.
In this study, we describe the genetic findings in patients with a predominant centronuclear finding on muscle biopsy, which were identified in a national cohort of 94 patients with CM older than five years, and we present the course of disease and phenotypes in patients with MTM1 mutations who have survived infancy and reached adolescence/adulthood.
MATERIAL AND METHODS
Patients
All patients aged ≥5 years (n = 119) registered in August 2009 by the Danish National Centre for Neuromuscular Diseases (RCFM) and/or Copenhagen Neuromuscular Center, Rigshospitalet with CM were invited to participate. RCFM registers patients with a neuromuscular diagnosis referred to the Centre by local hospitals across Denmark; Rigshospitalet registers primarily patients from the eastern part of Denmark. In combination, the two centers register the vast majority of patients with neuromuscular diseases in Denmark. The age limit of 5 years was chosen, because the patients were assessed by a number of tests validated only for persons from five years of age. Also, our ability to identify patients below age 5 years was poor, because XLMTM could not be traced according to diagnosis in departments of pediatrics across the country. The Ethics Committee of Copenhagen approved the study, and all patients/parents signed a consent form. 107 patients accepted the invitation and registered information on disease onset, symptoms, motor development, previous surgery and use of respiratory aids on a form. Thirteen patients were excluded; one patient due to co-morbidity (severe hypothyreosis), one patient due to cognitive impairment and eleven patients who could not fulfil three of the following four diagnostic criteria; childhood disease onset, biopsy without prominent dystrophic features, slow or no progression and normal or only slightly elevated creatine kinase (CK). Hence 94 patients (5–72 years; 47 females) were included in the study.
Predominant centronuclear findings on muscle biopsy were found in 16 patients, of whom three already had a genetic diagnosis of XLMTM (n = 1) or DNM2 (n = 2). The remaining 13 patients were tested for mutations in the four main genes known to cause CNM; RYR1, BIN1, DNM2 and MTM1.
Assessments
All patients had a cardiac evaluation including electrocardiogram (ECG), echocardiography and 48-hours Holter-monitoring and were assessed for cranial nerve territory abnormalities, musculoskeletal malformations, anthropometrics, forced vital capacity (FVC), muscle strength and functional tests, including Manual Muscle Test (MMT) of 40 muscle groups, using the modified Medical Research Council (MRC) scale from 0–10, [13] and Motor function measure (MFM) [14].
MRC % score was calculated as total score (40 muscle groups), proximal score (shoulder and hip = 16 muscle groups) and distal score (hands and ankles = 12 muscle groups) by the fraction: (sum of graded scores)/(number of muscles tested×10)×100.
The MFM scale has 32 items in three domains of function: standing and transfer, axial/proximal dimension and distal dimension. Each item is scored from 0 to 3; the higher the score, the higher function. The MFM score is calculated as a percentage of highest possible score for each dimension and/or for alldimensions.
In patients with a confirmed mutation of MTM1, medical records from local hospitals were reviewed to confirm the patient’s information on birth history and motor development.
RESULTS
Genetic findings
Pathogenic mutations were identified in 11 of 16 patients. Three male patients had MTM1 mutations corresponding to a prevalence of 6% of all male patients in the cohort. Six patients (four women) had single pathogenic DNM2 mutations, and two females had two pathogenic RYR1 mutations each. In five patients, mutations could not be found in the four tested genes. All identified mutations are described in the Leiden database [15]. Two of the MTM1 mutations were originally described according to old nomenclature as T728C (present c.674T>C) and G(1407+1)A (present c.1353+1G>A) [5]. Two of six DNM2 patients had other affected family members: patient no. 8 was third generation in a family with several members (mother was patient no. 7).
The two patients with RYR1 mutations were siblings and had three aberrations in the RYR1 gene: a previously pathogenic mutation c.325C>T, a non-pathogenic change c.1453A>G and a pathogenic non-sense mutation c.7308_7309delTG. The parents each carried one of the two pathogenic mutations.Clinical and genetic findings are summarized in Table 1.
Heart examination
None of the patients had signs of cardiac involvement on ECG, 48-hours Holter-monitoring or echocardiography.
Clinical presentations
Patients with MTM1 mutations
All three patients (Fig. 1) were hypotonic from birth with respiratory distress and need for respiratory support and all had recurrent severe lung infections during childhood; present clinical signs and symptoms are summarized in Table 2. Muscle strength in individual muscle groups at time of assessment is illustrated in Fig. 2.
None of the mothers were symptomatic or had had any miscarriages.
Patient 1 was the second of two siblings; he was delivered in the 43th week of pregnancy by vaginal birth. At birth he had profound muscle weakness, hypotonia, elongated face with tent-shaped mouth, pigeon chest, weak cry and respiratory insufficiency. In his first week of life, he was on 24-h ventilation and tube fed. At eight months, he could lift limbs against gravity, but not his head. He crawled at 15 months and walked independently at 18 months, but with frequent falls. He could rise from floor (with support) until he was eight years and walk independently until age nine years. During childhood, he was hospitalized numerous times due to respiratory infections; from age six years, he was treated with non-invasive ventilation (NIV) at night, which reduced the respiratory problems significantly. Notable ptosis was always present, and externalophthalmoplegia was likely present from early age, but not diagnosed until he was 7 years old.
At time of assessment (18 years old), he used electrical wheelchair and needed assistance for most activities of daily living. He was able to eat independently, but needed additional time to chew his food. He used non-invasive ventilation (BIPAP) at night and in short periods during the day. Strongest muscles groups were wrists and finger flexors, weakest muscle groups were neck, trunk, hip muscles and knee extensors. He attended normal school.
Patient 2 was the third of four siblings. He was delivered at term by acute caesarean section after abnormal pregnancy with polyhydramnios and decreased fetal movements; he had birth asphyxia and survived due to epinephrine injected directly into the heart. He was hypotonic with decreased mimics, no spontaneous movements and in his first two week of life he was on 24-h ventilation, which gradually was changed to intermittent non-invasive nasal ventilation (CPAP). He was discharged from the hospital with non-invasive nasal ventilation (CPAP), but subsequently hospitalized for long periods due to recurrent lung infections. From age three years, he was ventilated by tracheotomy, which stabilized his respiratory function and reduced the number and intensity of lung infections. After that he made some motor improvements and could lift his arms and legs against gravity. He was never able to lift his head against gravity, but could sit unsupported for some years with a poor balance and a curved back; he never achieved ambulation. Ophthalmoplegia and myopia was diagnosed when he was elevenyears old.
At time of assessment (14 years old) he used electrical wheelchair and had 24-h assistance. He still had recurrent lung infections. He had always been fed by tube and had bladder dysfunction and urinary incontinence. Strongest muscles groups were wrist and fingers; weakest muscle groups were neck, trunk, hip muscles and knee extensors. He attended normal school.
Patient 3 was the first of two siblings; he was delivered in the 42th week of an uncomplicated pregnancy by vaginal birth. At birth he had marked hypotonia, long and slender extremities and was cyanotic with short and intermittent breath and very weak cry. He was put in an incubator with respiratory support and fed by tube. Within 24 hours he could breathe independently, and tube feeding was suspended after four days. Three months old, he could jerk his arms and leg albeit with less agility that normal, but could not raise his head from prone or supine. He satindependently at eight months and walked independently at 18 months. During childhood, he had recurrent lung infections and pneumonias and had NIV at night when he was nine years old. He was operated for undescended testes at three years. Ophthalmoplegia and myopia was diagnosed when he was five years old.
At time of assessment (25 years old) he was ambulant with an electric mobility device for longer distances. Strongest muscles groups were wrist and finger flexors; weakest muscle groups were neck flexors, shoulder girdle and hip muscles. He lived in his own apartment and was a university student.
All three patients had notable ptosis and ophthalmoplegia (Fig. 1). All had contractures in jaws; the two non-ambulant patients also had contractures in their elbows, hips and knee corresponding to theirsitting position. Finger joints were hyper-mobile in all patients.
Patients with DMN2 mutations and RYR1 mutations
All patients with DNM2 mutations had reached ambulation before age 18 months, but none obtained the same gross motor function as healthy children. All, but one patient (no. 9), considered their course of disease as stable. All DNM2 patients had impaired mimics, but to varying degrees; patients 4, 7 and 8 had ophthalmoplegia, patients 5 and 6 had ptosis when they were tired. Patient 9 had persistent and pronounced ptosis.
The two sisters with RYR1 mutations were born with marked hypotonia and treated in incubators in the first days of life. The younger sister had normal motor milestones as child, and considered her disease course stable, but she needed support to rise from a chair and walk outdoors from her mid twenties. The older sister achieved independent ambulation at 22 month, but never became a steady walker and lost ambulation at 13 years. Both patients had ophthalmoplegia from an early age, but no ptosis.
DISCUSSION
In this study we present eleven patients with genetically confirmed CNM. Three patients had MTM1 mutations; this corresponds to 19% of patients with CNM and 6% of all male patients in a representative Danish population of patients with CM older than five years. Considering that this was found in patients who had survived into adolescence, indicates a higher prevalence of the disease than the 2% that was reported by Maggi et al. [16] in children below age 1.5 years, and by Amburgey et al. [17] in a pediatric population.
The observation of patients with MTM1 mutations having survived infancy is not new; several studies [9–12] have reported patients with milder phenotypes who survived into adulthood, but in this study we report detailed information on phenotypes and courses of disease from birth to adulthood, besides the prevalence data. Two of our XLMTM patients were notgenetically diagnosed until age 14 and 25 years despite the fact that muscle biopsy had shown central nuclei at an early age. This delay in diagnosis could relate to the common notion that XLMTM is a severe congenital myopathy, incompatible with ambulation and life beyond 1 year and to the fact that these boys were seen at local hospitals; given the incidence of XLMTM reported by Jungbluth et al. [8] the estimated incidence in Denmark should be 1 case every second year, which leaves the local child neurologist with little experience in this rare disease; as a consequence, floppy infants may not be correctly diagnosed. Our study demonstrates that one third of male patients, 5 years or older, with centronuclear findings on muscle biopsy have MTM1 mutations, and this diagnosis should be considered even in patients with a much milder phenotype than generally believed for XLMTM. Since it was not possible to include patients below the age of five, we cannot elaborate on the overall incidence of XLMTM in Denmark.
Mutations in the MTM1 gene have been reported by different nomenclatures [4], which complicated our search for previous findings of the mutations. In the Leiden Open Variation Database [15] sequence variations are described as variants at DNA level as recommended by the Human Genome Variation Society (HGVS) and the database additionally list the variant as reported originally (old nomenclature). By searching the database we found that all three mutations had been reported previously.
McEntagart et al. [11] classified the XLMTM phenotypes as mild, intermediate and severe based on the need of ventilator support. Following this classification, patient 1 had an intermediate phenotype. Despite a spontaneous improvement in physical function, the disease has been progressive with an early loss of ambulation and a decrease in pulmonary function resulting in NIV from an early age. The progressive decline in motor function may be explained in the boy’s growth, since an increase in weight and height influences balance and the capability to lift limbs against gravity. The mutation (c.674T>C) has been described as both severe and moderate in otherstudies [4, 5].
The mutation in patient 2 (c.1353+1G>A), has previously been reported as “severe”, but without further clinical details [5]. The severe phenotype was confirmed in our patient, who had respiratory failure at birth and survived due to prolonged respiratory support and early invasive ventilation by tracheotomy. The mutation in patient 3 (c.1037G>C), has previously been reported as “mild or moderate”, again without further clinical details [4]. This was confirmed in our patient, who is still ambulant at age 25 years, but with a need for ventilator support duringnight time.
Marked ophthalmoplegia and ptosis are common in patients with XLMTMs, which was also the case in our patients. Additionally, our patients also had myopia, a finding also illustrated by Herman et al. [9], but not reported as a frequent finding in the literature. We found that proximal muscles were more impaired than distal (Fig. 2); especially hand function was well preserved, even in the non-ambulant patients. Additionally, we found that very weak neck flexors were an early and persistent finding. Two of our patient had spinal surgery and all three patients had surgical elongation of tight tibial anterior muscles. Other contractures were in general related to ambulation status, but jaw contractures were found in all three patients. In addition, all three patients had hypermobile finger joints. Limitation of jaw movement has previously been reported in one patient with CNM by Zanoteli et al. [18], but has not been reported in other studies.
Pulmonary function was significantly impaired in all three patients, who all relied on day and/or night-time ventilation. Without this intervention, patients 1 and 2 would not have reached adolescence. This could potentially explain why we did not identify patients older than 30 years, which is the time period in which XLMTM patients have been offered assisted ventilation in Denmark. The finding of overgrowth and retention testes is a well-described characteristic of the disorder [8, 9, 11]. Besides bladder dysfunction and urinary incontinence reported by patient 2, our patients did not report any of the potential medical complications described by Herman et al. [9] such as gall stones and liver dysfunction.
DNM2 mutations were the most common cause of CNM (38% ) in persons older than 5 years; this finding is a little less than the findings described by Romero [19] who found that DNM2-related CNM accounted for about 50% of CNM cases. The phenotypes were relatively mild with a stable course of disease, except one patient who always had more impaired motor function and a progressive course of disease.
Two of our patients (13% ) had RYR1 mutations; thus contrary to Wilmshurst et al. [20], we did not find that aberrations in RYR1 are common causes of CNM in Danish CM patients older than 5 years. The phenotypes were intermediate as described by Jungbluth [8], however, highly variable considering the patients shared the same mutations.
Five patients with predominantly centronuclear findings on muscle biopsy were unsolved. Our mutations analyses did not include genes assumed to be a rare cause of CNM such as the TTN, CCDC78 and SPEG genes [3, 21, 22].
This study demonstrates that the prevalence of patients with XLMTM among male patients with congenital myopathies, who have survived past age 5 years, is not negligible, and that MTM1 mutations may be the cause of disease also in adult patients. Even in patients with a severe phenotype, muscle strength and motor function may improve if intensive medical intervention is initiated.
CONFLICT OF INTEREST
The study has not received any external funding. The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank all the patients for participating in the study.
REFERENCES
1 | North K(2008) What’s new in congenital myopathies? Neuromuscul Disord18: 6433442 |
2 | Jungbluth H(2013) ENMC International Workshop: 7th Workshop on Centronu425 clear (Myotubular) myopathies, 31st May – 2nd June 2013, 426 Naarden, The NetherlandsNeuromuscul Disord23: 1210331043 |
3 | Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC, Soemedi R, Vasli N, Iannaccone ST, Shieh PB, Shur N, Dennison JM, Lawlor MW, Laporte J, Markianos K, Fairbrother WG, Granzier H, Beggs AH(2013) Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathyNeurology81: 1412051214 |
4 | Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, Wallgren-Pettersson C, Herger F, Buj-Bello A, Blondeau F, Liechti-Gallati S, Mandel JL(2000) MTM1 mutations in X-linked myotubular myopathyHum Mutat15: 5393409 |
5 | Tanner SM, Schneider V, Thomas NS, Clarke A, Lazarou L, Liechti-Gallati S(1999) Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients Neuromuscul Disord9: 14149 |
6 | Jungbluth H, Sewry CA, Buj-Bello A, Kristiansen M, Ørstavik KH, Kelsey A, Manzur AY, Mercuri E, Wallgren-Pettersson C, Muntoni F(2003) Early and severe presentation of X-linked myotubular myopathy in a girl with skewed X-inactivationNeuromuscul Disord13: 15559 |
7 | Pénisson-Besnier I, Biancalana V, Reynier P, Cossée M, Dubas F(2007) Diagnosis of myotubular myopathy in the oldest known manifesting female carrier: A clinical and genetic studyNeuromuscul Disord17: 2180185 |
8 | Jungbluth H, Wallgren-Pettersson C, Laporte J(2008) Centronuclear (myotubular) myopathy25: 326 |
9 | Herman GE, Finegold M, Zhao W, de Gouyon B, Metzenberg A(1999) Medical complications in long-term survivors with X-linked myotubular myopathyJ Pediatr134: 2206214 |
10 | Biancalana V, Caron O, Gallati S, Baas F, Kress W, Novelli G, D’Apice MR, Lagier-Tourenne C, Buj-Bello A, Romero NB, Mandel JL(2003) Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotypeHum Genet112: 2135142 |
11 | McEntagarta M(2002) Gretchen Parsonsb, Anna Buj-Belloc, Valérie Biancalanad, Iain Fentona, Mark LittleGenotype–phenotype correlations in X-linked myotubular myopathy. Neuromuscul DisordKrawczak M, Thomas N, Herman G, Clarke A, Wallgren-Pettersson C12: 10939946 |
12 | Yu S, Manson J, White S, Bourne A, Waddy H, Davis M, Haan E(2003) X-linked myotubular myopathy in a family with three adult survivorsClin Genet64: 148152 |
13 | Florence JM, Pandya S, King WM, Robison JD, Baty J, Miller JP, Schierbecker J, Signore LC(1992) Intrarater reliability of manual muscle test (Medical Research Council Scale) grades in Duchenne’s muscular dystrophyPhysical Therapy72: 115126 |
14 | Berard C, Payan C, Hodgkinson I, Fermanian J(2005) MFM collaborative study group. A motor function measure for neuromuscular diseases. Construction and validation studyNeuromuscul Disord.15: 7463470 |
15 | The Leiden open Variation database (MTM1 gene) http://www.dmd.nl/nmdb/home.php?select_db=MTM1 |
16 | Maggi L, Scoto M, Cirak S, Robb SA, Klein A, Lillis S, Cullup T, Feng L, Manzur AY, Sewry CA, Abbs S, Jungbluth H, Muntoni F(2013) Congenital myopathies–clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United KingdomNeuromuscul Disord23: 3195205 |
17 | Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ(2011) Prevalence of congenital myopathies in a representative pediatric united states populationAnn neurol70: 662665 |
18 | Zanoteli E, Guimarães AS, Martins RJ, Yamashita HK, Toledo CS, Oliveira AS, Gabbai AA(2000) Temporomandibular joint involvement in a patient with centronuclear myopathyOral Surg Oral Med Oral Pathol Oral Radiol Endod90: 1118121 |
19 | Romero NB(2010) Centronuclear myopathies: A widening conceptNeuromuscul Disord20: 4223228 |
20 | Wilmshurst JM, Lillis S, Zhou H, Pillay K, Henderson H, Kress W, Müller CR, Ndondo A, Cloke V, Cullup T, Bertini E, Boennemann C, Straub V, Quinlivan R, Dowling JJ, Al-Sarraj S, Treves S, Abbs S, Manzur AY, Sewry CA, Muntoni F, Jungbluth H(2010) RYR1 mutations are a common cause of congenital myopathies with central nucleiAnn Neurol68: 5717726 |
21 | Majczenko K, Davidson AE, Camelo-Piragua S, Agrawal PB, Manfready RA, Li X, Joshi S, Xu J, Peng W, Beggs AH, Li JZ, Burmeister M, Dowling JJ(2012) Dominant mutation of CCDC78 in a unique congenital myopathy with prominent internal nuclei and atypical coresAm. J. Hum. Genet91: 365371 |
22 | Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC, Haliloglu G, Talim B, Yau KS, Allcock RJN, Laing NG, Perrella MA, Beggs AH(2014) SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathyAm J Hum Genet95: 218226 |
Figures and Tables
Fig.1
Patient 1 (18 y), patient 2 (14 y) and patient 3 (25 y). Note the identical morphological features with elongated face, wide-spaced eyes and the tent-shaped mouth in patients 1 and 2.
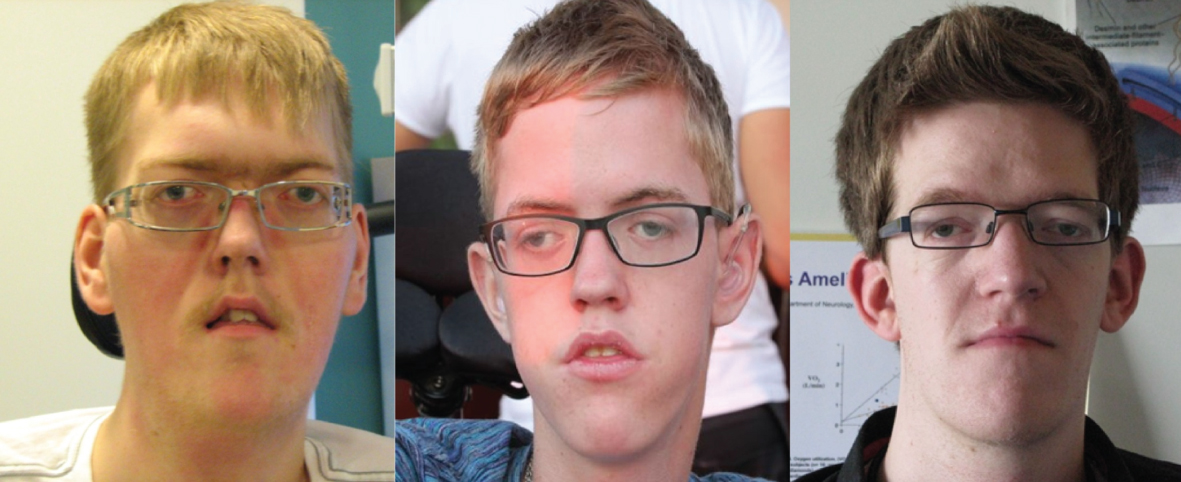
Fig.2
MMT scores, transformed to an 11-point rating scale (0–10), where 1 = 1, 2 = 2, 3 = 3–, 4 = 3. 5 = 3+, 6 = 4–, 7 = 4, 8 = 4+, 9 = 5–and 10 = 5. The vertical line illustrates the border between muscles that cannot/can overcome gravity. Note the stronger muscles in hands.
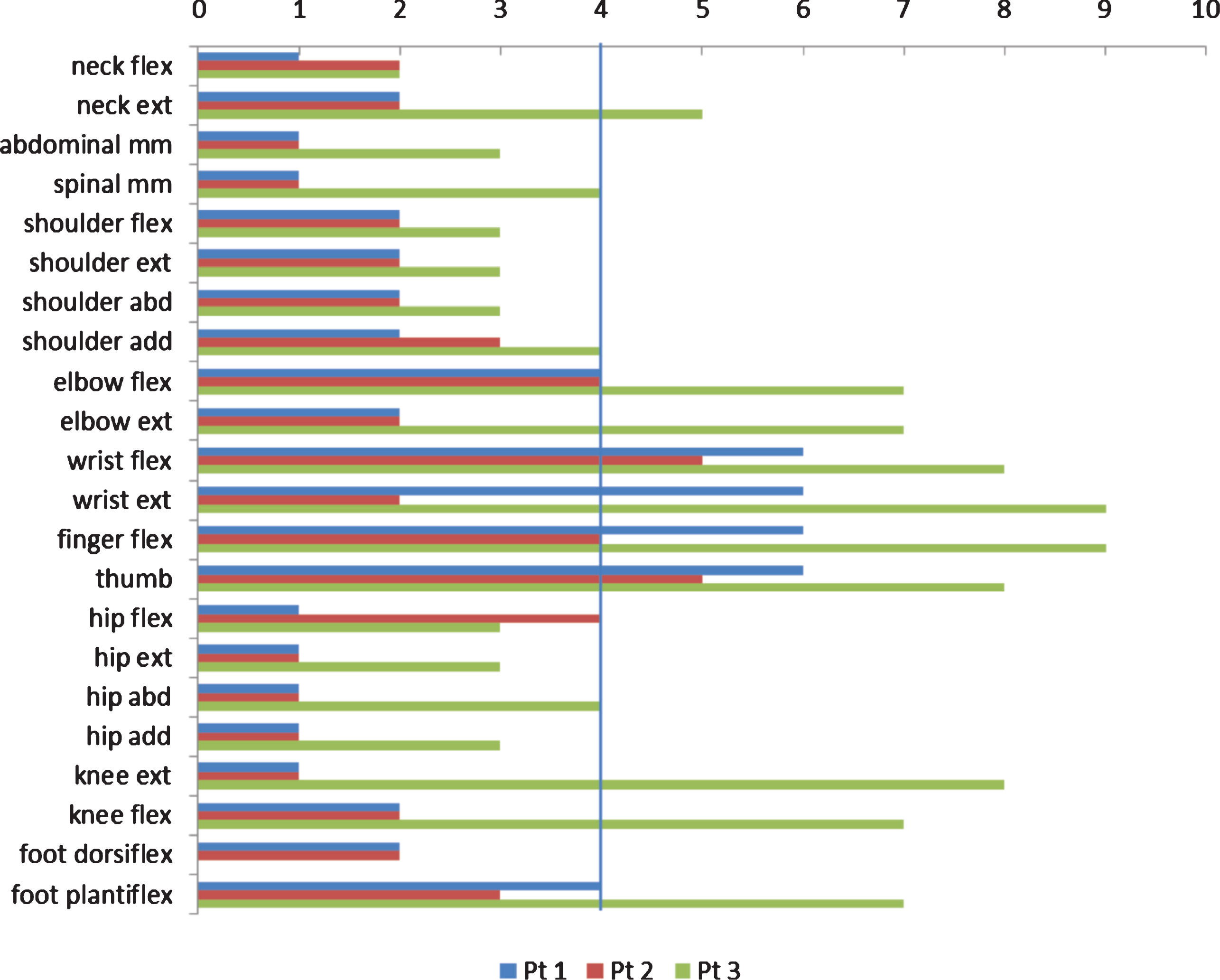
Table 1
Clinical and genetic findings in 11 patients. *Patients 7 and 8 are mother and daughter. **Patients 10 and 11 are sisters; b = needs banisters, c = compensatory movements. Y = year, mo = months. S = stable, P = progressive, A = ambulant, NA = non-ambulant
| No | Gene | Gene mutations | Inheritance | Sex | Age years | Age (months) independent ambulation | Course | Current function |
| 1 | MTM1 | c.674T>C, exon 8, (earlier reported as c.728T>C) | X - mother carrier | M | 19 | 18 m | P | NA (since 9 y old) |
| 2 | MTM1 | c.1353+1G>A, exon 12, (earlier reported as G(1407+1)A) | X - | M | 14 | Never | S | NA |
| 3 | MTM1 | c.1037G>C, p.W346S, exon 10 | X - | M | 25 | 18 m | S | A, stairs (+b) |
| 4 | DNM2 | c.1393C>T, (p.R465W), exon 11 | AD - sporadic | M | 25 | 18 m | S | A, stairs (+b), run (+c) |
| 5 | DNM2 | c.1102G>A, (p.E368K), exon 8 | AD - sporadic | F | 20 | 14 m | S | A, stairs (+b), run (+c) |
| 6 | DNM2 | c.1565G>A, (p.R522H), exon 14 | AD -sporadic | F | 52 | 12 m | S | A, stairs (+b), run (+c) |
| 7 | DNM2 | c.1393C>T, (p.R465W), exon 11 | AD -familiar | F * | 70 | unknown | S till 4th decade | Walks with support |
| 8 | DNM2 | c.1393C>T –(p.R465W), exon 11 | AD (mother = no 7) | F * | 42 | 9 m | S | A, stairs, run (+c) |
| 9 | DNM2 | c.1852G>A, (p.Ala618Thr), exon 16 | AD - sporadic | M | 19 | 17 m | P | NA (since 14 y old) |
| 10 | RYR1 | c.325C>T (p.Arg109Trp), exon 4+c.7308_7309delTG+c.1453A>G (p.Met485Val), exon 14 | AR - parents carriers | F** | 28 | 15 m | S | A, walks indoors |
| 11 | RYR1 | c.325C>T (p.Arg109Trp), exon 4+c.7308_7309delTG+c.1453A>G (p.Met485Val), exon 14 | AR - parents carriers | F ** | 34 | 22 m | P | NA (since 13 y) |
Table 2
Medical history and clinical findings in patients with MTM1 mutations. *FVC (Forced vital capacity) percentage of predicted value
| Patient 1 | Patient 2 | Patient 3 | |
| Birth height/weight | 54 cm/3.9 kg | 57 cm/3.2 kg | 54 cm/3.1 kg |
| Histology | central nuclei + FTD (12 mo) | central nuclei (at 2.6 y) | i.a. (one week), myopathic (12 mo) central nuclei (7 y; 25 y) |
| Independent walking (age) | 18 months (Lost at 9 y) | Non-ambulant | 18 months |
| Height in the first years of life | >75th percentile | 75th percentile | >75th percentile |
| Ptosis | + | + | + |
| Ophthalmoplegia | +diagnosed at 7 y | +diagnosed at 11 y | +diagnosed at 5 y |
| Myopia | + | + | + |
| High-arched palate | + | +(extreme) | + |
| Surgery ankles (TA) | 9 y | 9 y (+knees) | 15 y (+pes cavus) |
| Scoliosis | 14 y | 10 y | – |
| Retentio testis | – | 5 y | 3 y |
| Feeding –G tube | 15 y | always | – |
| Respiratory function (FVC% ) | 19% - used NIV from 6 y | IV from 3 y | 35% - used NIV from 9 y |
| MRC % total/proximal/ distal | 26% /14% /45% | 24% / 19% /32% | 57% /36% /80% |
| MFM % total | 31/100 | 27/100 | 89/100 |
| Axial/proximal | 36/100 | 33/100 | 92/100 |
| distal | 81/100 | 66/100 | 100/100 |
| Joint contractures | Neck, shoulder, elbow, wrist, hips, knees, jaws | Neck, shoulder, elbow, wrist, hips, knees, jaws | Feet, jaws |

