
Possible Role of Activin in the Adiponectin Paradox-Induced Progress of Alzheimer’s Disease
Abstract
Accumulating evidence suggests that the adiponectin (APN) paradox might be involved in promoting aging-associated chronic diseases such as Alzheimer’s disease (AD). In human brain, APN regulation of the evolvability of amyloidogenic proteins (APs), including amyloid-β (Aβ) and tau, in developmental/reproductive stages, might be paradoxically manifest as APN stimulation of AD through antagonistic pleiotropy in aging. The unique mechanisms underlying APN activity remain unclear, a better understanding of which might provide clues for AD therapy. In this paper, we discuss the possible relevance of activin, a member of transforming growth factor β (TGFβ) superfamily of peptides, to antagonistic pleiotropy effects of APN. Notably, activin, a multiple regulator of cell proliferation and differentiation, as well as an endocrine modulator in reproduction and an organizer in early development, might promote aging-associated disorders, such as inflammation and cancer. Indeed, serum activin, but not serum TGFβ increases during aging. Also, activin/TGFβ signal through type II and type I receptors, both of which are transmembrane serine/threonine kinases, and the serine/threonine phosphorylation of APs, including Aβ42 serine 8 and αS serine 129, may confer pathological significance in neurodegenerative diseases. Moreover, activin expression is induced by APN in monocytes and hepatocytes, suggesting that activin might be situated downstream of the APN paradox. Finally, a meta-analysis of genome-wide association studies demonstrated that two SNPs relevant to the activin/TGFβ receptor signaling pathways conferred risk for major aging-associated disease. Collectively, activin might be involved in the APN paradox of AD and could be a significant therapeutic target.
INTRODUCTION
The physiological functions of amyloidogenic proteins (APs) relevant to neurodegenerative diseases, such as amyloid-β (Aβ) in Alzheimer’s disease (AD) and α-synuclein (αS) in Parkinson’s disease (PD), have remained unclear, despite decades of intense investigation. Given the similarity between yeast and human brain in terms of coping with diverse local stressors, we recently proposed that the concept of evolvability in yeast prion could be applied to the APs in neurodegenerative diseases (Fig. 1) [1, 2]. Evolvability should be evolutionarily beneficial because it might confer resistance against the forthcoming stressors in offspring’s brain. Evolvability, however, during young reproductive life may later manifest as neurodegeneration in aging parental brains (Fig. 1) [3]. This is based on antagonistic pleiotropy, a previously prominent concept regarding the mechanism of aging, suggesting that aging is due to the combined effect of multiple pleiotropic genes that each exerted a beneficial effect during an animal’s youth, but later lead to adverse effects in older age [4].
Fig. 1
Diagram showing that APN might stimulate both amyloidogenic evolvability and neurodegenerative disorders. Evolvability during developmental/reproductive stages and neurodegenerative diseases such as AD in post-reproductive senescence are driven by aggregated APs, and might exist in an antagonistic pleiotropy relationship. APN might be beneficial for amyloidogenic evolvability, while paradoxically detrimental (namely the APN paradox) in the form of neurodegenerative diseases through the antagonistic pleiotropy.
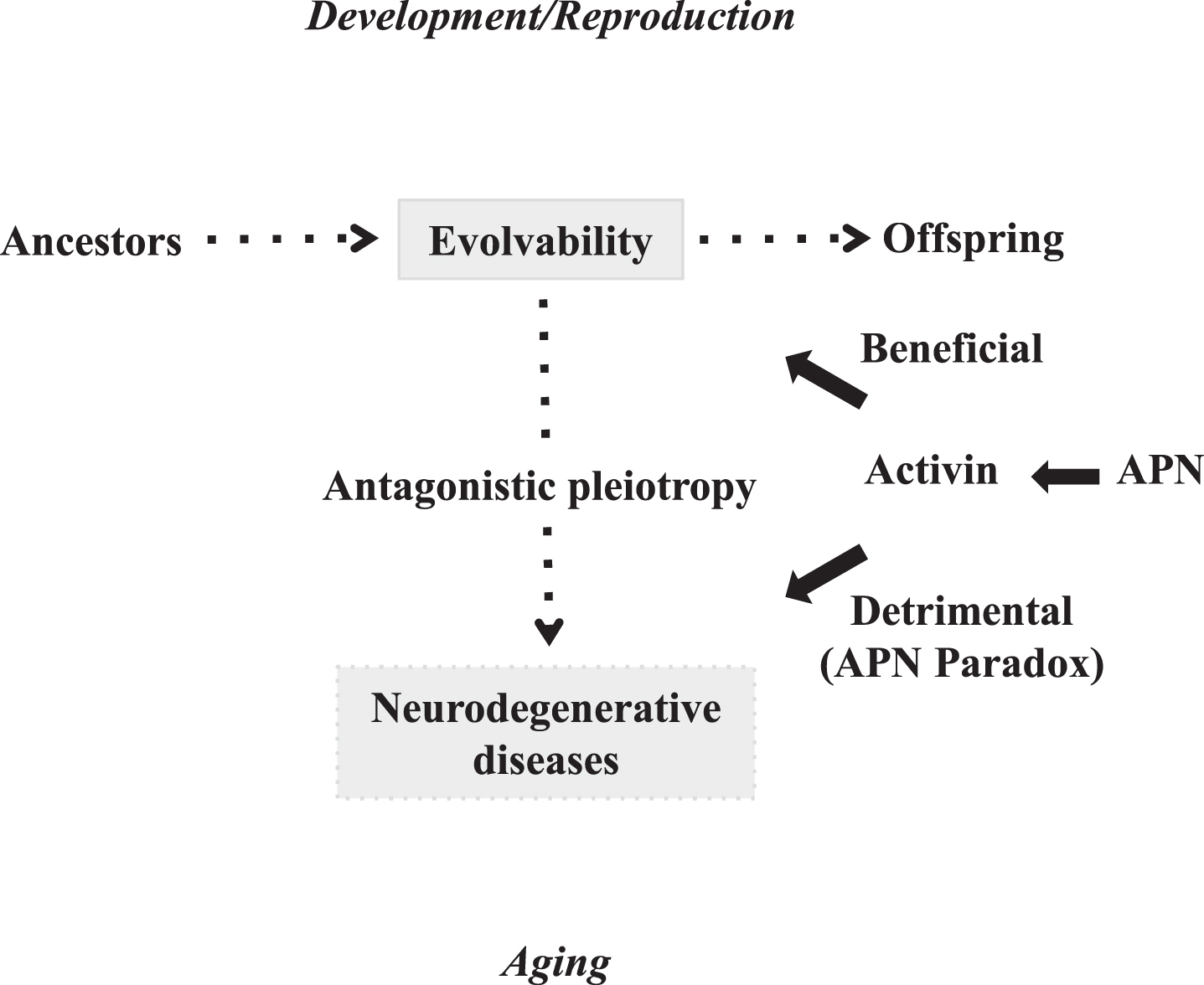
Regardless of the popularity of the antagonistic pleiotropy theory in aging research, the underlying molecular mechanisms of antagonistic pleiotropic action are poorly understood. In this context, the main objective of this paper is to discuss the role of the activin, a member of transforming growth factor β (TGFβ) family of peptides [5, 6] in the antagonistic pleiotropic actions in aging-associated neurodegenerative diseases such as AD. Activin was initially identified as a regulator of the endocrine system and embryonic development [5, 7], but subsequent accumulating data implicates activin as a pathologic factor in aging (Fig. 2). In particular, the activin/TGFβ family of peptides may promote inflammation [8], which is commonly associated with disease in aging. Furthermore, activin/TGFβ peptides may be involved in the serine/threonine phosphorylation of APs, conferring pathological significance in neurodegenerative diseases. Moreover, since serum activin levels increase during aging [9], activin might be important in chronic disorders. Recently, a number of studies showed that activin expression was induced by APN in various tissues [10, 11], suggesting that activin might be situated downstream of the APN paradox, leading to aging-associated disease including AD. Also consistent with these results, a recent genome-wide association study identified two SNPs relevant to activin/TGFβ receptor signaling pathways, that were linked with an increased risk for major human diseases, including AD, as well as human mortality [12]. Taken together, activin might play a central role in antagonistic pleiotropy during aging and might be an attractive therapeutic target of AD (Fig. 1).
Fig. 2
Multiple antagonistic pleiotropic effects of activin depending on life stage. a) Diagram showing the dual nature of activin activity depending on the life stage. Activin may be an important endocrine regulator in early development, cell growth, and differentiation in various biological systems and possibly in evolvability. In contrast, activin might be detrimental to the organism, such as neurodegenerative diseases in aging. Supporting this, activin may stimulate inflammation and increase risk for major human disease, including coronary heart disease, heart failure, stroke, diabetes and cancer and neurodegenerative diseases. Such dual actions of activin may be comparable to antagonistic pleiotropy. b) Schematic of the regulation of pituitary-gonadal (ovary and testis) axis by activin, inhibin and follistatin during reproduction. Secretion and production of FSH from anterior pituitary are stimulated by activin but are inhibited by inhibin and follistatin. c) Animal cap assay using Xenopus fertilized egg showed that embryonic development was promoted by activin. Scale bars = 100μm. Reprinted from Spicer et al. 2010 [56] with permission. d) Involvement of activin in inflammation of COPD (GOLD stage IV). Compared to the lung tissue of never-smokers (left), activin immunoreactivity in inflamed COPD lung tissue (middle and right), is significantly increased. The right image is a magnification of the square region of the middle figure. Scale bars = 100μm. Reprinted from Verhamme et al. [25] with permission. e) Activin immunoreactivity is stronger in esophageal cancer (down) compared to that in normal tissues (upper). Reprinted from Wang et al. 2015 [57] with permission.
![Multiple antagonistic pleiotropic effects of activin depending on life stage. a) Diagram showing the dual nature of activin activity depending on the life stage. Activin may be an important endocrine regulator in early development, cell growth, and differentiation in various biological systems and possibly in evolvability. In contrast, activin might be detrimental to the organism, such as neurodegenerative diseases in aging. Supporting this, activin may stimulate inflammation and increase risk for major human disease, including coronary heart disease, heart failure, stroke, diabetes and cancer and neurodegenerative diseases. Such dual actions of activin may be comparable to antagonistic pleiotropy. b) Schematic of the regulation of pituitary-gonadal (ovary and testis) axis by activin, inhibin and follistatin during reproduction. Secretion and production of FSH from anterior pituitary are stimulated by activin but are inhibited by inhibin and follistatin. c) Animal cap assay using Xenopus fertilized egg showed that embryonic development was promoted by activin. Scale bars = 100μm. Reprinted from Spicer et al. 2010 [56] with permission. d) Involvement of activin in inflammation of COPD (GOLD stage IV). Compared to the lung tissue of never-smokers (left), activin immunoreactivity in inflamed COPD lung tissue (middle and right), is significantly increased. The right image is a magnification of the square region of the middle figure. Scale bars = 100μm. Reprinted from Verhamme et al. [25] with permission. e) Activin immunoreactivity is stronger in esophageal cancer (down) compared to that in normal tissues (upper). Reprinted from Wang et al. 2015 [57] with permission.](https://content.iospress.com:443/media/jad/2021/81-2/jad-81-2-jad210206/jad-81-jad210206-g002.jpg)
DUAL ACTIONS OF ACTIVIN IN THE LIFE STAGES
Activin and its endogenous antagonist, inhibin, were initially purified from ovarian fluids as two closely related proteins that have opposing effects on the pituitary biosynthesis and secretion of follicle stimulating hormone (FSH) (Fig. 2) [13–15]. Activin is composed of a homodimer of beta subunits, while inhibin is a heterodimer of beta and alpha subunits [16]. Extensive studies characterizing the activin signaling pathway have revealed that, upon activin binding to the type II receptor, the type II receptor becomes phosphorylated, leading to subsequent phosphorylation and activation of type I receptor. The type I receptor then phosphorylates Smad2/3, which then binds to the common partner, Smad4, and migrates into the nucleus as a transcriptional regulator of target genes [17]. As mentioned, a number of biological functions are attributed to activin, including the regulation of the endocrine system, roles in embryonic development, and cell growth and differentiation in various biological systems [5, 7]. In the nervous system, activin promotes neural survival [18] and regulates neural differentiation [19]. In contrast to activin, the function of the inhibin receptor and its signal transduction are poorly understood, except for the finding that the proteoglycan, betaglycan, was shown to bind to inhibin to mediate the functional antagonism of activin signaling [20]. Similarly, follistatin, which was originally isolated from ovarian fluid as an inhibitor of pituitary FSH (Fig. 2) [21], was later revealed to be an activin-binding protein [22]. The activin/TGFβ protein superfamily has now expanded to include many other structurally related proteins such as TGFβ, anti-Mullerian hormone, bone morphogenetic protein, and growth differentiation factor [23].
Compared to the developmental and reproductive importance of activin signaling, the role of activin in molecular physiology of aging has largely been overlooked, and the molecule may, in fact, be detrimental during later life in mammals and humans (Fig. 2). For instance, activin may promote inflammation [24], a major underlying pathobiological factor for aging-associated conditions. Furthermore, a recent study has shown that activin is involved in various disorders of aging. First, activin mediates cigarette smoke-induced inflammation and chronic obstructive pulmonary disease (COPD) (Fig. 2) [25]. Also, muscle mass is negatively regulated by activin in primates, suggesting that activin might be involved in promoting sarcopenia [26]. Activin receptor is also activated in the skeleton, vasculature, heart, and kidney during chronic kidney disease [27]. Finally, serum activin levels are associated with hypertension in the elderly [28].
Although TGFβ is involved in the regulation of cell proliferation, differentiation, and immune functions in development/reproduction [29, 30], and is also involved in aging-associated diseases such as muscle atrophy, cancer, obesity, and AD in aging [31], few reports have addressed increased TGFβ expression in aging. Therefore, we mainly focus on activin in this context in the present paper.
POSSIBLE ROLE OF ACTIVIN IN THE REGULATION OF EVOLVABILITY AND NEURODEGENERATIVE DISEASES
Since activin might be centrally involved in the antagonistic pleiotropy during aging, it might also be involved in the regulation of antagonistic pleiotropy linking amyloidogenic evolvability with neurodegenerative diseases [3, 32].
Because protofibrillar APs may likely be associated with evolvability [2], it is possible that signaling of activin/TGFβ receptor pathways might be an important catalyst for oligomer formation of APs through their serine/threonine phosphorylation [17]. Activin is an important reproductive endocrine regulator and is expressed in a wide range of tissues, including seminal plasma, ovarian fluids, and brain [33]. Also, notably, semen also contains multiple types of amyloid fibrils, the biological roles of which are unknown, yet do not appear to be pathogenic [34]. Collectively, these findings would support the possibility of transgenerational transmission associated with AP evolvability, in which activin/ TGFβ receptor signaling might influence the oligomerization of APs.
In aging, serine/threonine phosphorylation of APs plays a crucial role in the pathogenesis of neurode-generation. For example, important for AD, Aβ42 phosphorylation at serine 8 exhibits increased agg-regative properties when compared to wild type Aβ42 [35]. Similarly, in PD and α-synucleinopathies, phosphorylation of αS at serine 129 increases αS aggregation [36], and serine 129 αS is a biomarker of Lewy bodies in PD [37]. Since the process of Lewy body formation, rather than simply αS fibrillization, might be the major driver of PD neurodegeneration [38], and a similar case might apply to AD as well, activin might play an important role in the progre-ssion of both AD and PD. Yet, presently, the kina-ses responsible for serine phosphorylation of APs in neurodegenerative conditions have not been clea-rly identified. Given that activin is pro-inflammatory [24], and that activin expression is increased in post-reproductive senescence [9], the activin receptor serine/threonine kinase is a strong candidate for the pathologic phosphorylation of APs. Despite possible involvement of activin in promoting inflammation in neurodegenerative diseases, including AD and PD, there are presently few reports describing a role for activin in inflammation in neurodegenerative diseases. Collectively, we predict that the serine/threonine phosphorylation of APs by activin/TGFβ receptor signaling might be critical for the regulation of both evolvability and neurodegenerative diseases.
POSSIBLE RELEVANCE OF ACTIVIN TO APN IN ANTAGONISTIC PLEIOTROPY
The antagonistic pleiotropic actions of activin in the life stages resemble that of APN. Indeed, the APN paradox has been well investigated in relation to aging-associated chronic diseases, such as chronic heart failure (CHF), chronic kidney disease (CKD), and COPD [39]. Despite beneficial properties for cardiovascular cells, APN is detrimental in these aging-associated diseases in which the increased APN in plasma is well correlated with the disease severity [40, 41].
Recent evidence suggests that the APN paradox might be involved in the progression of multiple aging-associated diseases, including AD and cancer [39, 41]. Regarding AD, a recent prospective cohort study showed that elevated serum APN levels were associated with the accumulation of amyloid deposits and the severity of cognitive deficits in elderly, suggesting that APN might promote Aβ amyloidosis in the elderly [42]. Similarly, increased serum APN was associated with cognitive decline in postmenopausal women [43, 44]. Furthermore, histopathological studies of the postmortem AD brains identified sequestration of APN by phospho-tau into the neurofibrillary tangle, suggesting that APN stimulates tau aggregation [45]. Thus, these results indicate that APN may promote AD through the antagonistic pleiotropy mechanism in aging, otherwise known as the APN paradox in AD (Fig. 1) [38]. Since AD and CHF may in some ways overlap mechanistically, the APN paradox in AD might be applicable to other diseases as well, including CHF and CKD.
In cancer, a recent prospective cohort study showed that significantly higher serum APN concentrations were observed in incident cancers, which also independently associates with cancer-related deaths in type 2 diabetes mellitus (T2DM) [46], suggesting that the APN paradox might be relevant for cancers comorbid with T2DM. Furthermore, although activin is tumor suppressive in various cancers, patients with high plasma activin levels had a significantly shorter survival period than those with low levels, such as in pancreatic cancer [47].
Certainly, it might be predicted that activin and APN might co-interact in the antagonistic pleiotropy mechanism. Along these lines, APN was shown to induce activin expression in various tissues, such as monocytes and hepatocytes [10, 11]. Certainly, pending further investigation, these results raise a possibility that activin might be the downstream effector of APN (Fig. 1).
GENOMIC WIDE ASSOCIATION STUDY (GWAS)
Recently, due to technological innovations such as the next generation sequencer, GWAS has become a powerful tool in aging science. In this regard, genetic observations strongly support the concept that activin might promote aging-associated chronic diseases. Notably, this concept was demonstrated in a combined meta-analysis of GWAS derived from the Atherosclerosis Risk in Communities study (N = 9,573), the Framingham Heart Study (N = 4,434), and the Health and Retirement Study (N = 9,679) [12]. Given the large combined sample, GWAS analysis was able to identify two SNPs (rs222826 and rs222827) situated in the 2q22 locus linked with risks for a wide variety of major human diseases and conditions, including neurodegenerative diseases, coronary heart disease, heart failure, stroke, diabetes and cancer, as well as mortality in an antagonistic fashion in different populations [12]. Interestingly, both of these SNPs appear to be relevant to activin/TGFβ receptor signaling pathways [12]. For instance, rs222826 is derived from the ZEB2 gene that functions as a transcriptional regulator interacting with activated SMADs in the TGFβ signaling pathway, while rs222827 corresponds to ACVR2A gene, namely activin receptor type-2A. Extensive work reveals that ZEB2 mutations cause severe developmental/genetic disorders [48], while the ACVR2A may contribute to pre-eclampsia in pregnancy [49]. This locus harbors an evolutionarily conserved gene-desert region with non-coding intergenic sequences likely involved in regulation of protein-coding flanking genes, ZEB2 and ACVR2A [12]. Combined with the dual physiological effects of activin as described earlier (Fig. 2), this further reinforces the notion that multiple actions of activin function through antagonistic pleiotropy.
ANTAGONISTIC PLEIOTROPY OF ACTIVIN AS A THERAPEUTIC TARGET
We recently proposed that the APN paradox might be a therapeutic target for AD. APN activity could be reduced by antisense oligonucleotide or anti-APN miRNA (Fig. 3) [50]. Yet, if APN resistance parallels insulin resistance in AD, this strategy might be ineffective. As described, activin might be the downstream effector of the APN paradox, and therefore, an attractive therapeutic target in AD.
Fig. 3
Strategic therapies for neurodegeneration based on activin-mediated antagonistic pleiotropy. Given that the oligomerization of APs is regulated by phosphorylation through the activin receptor signaling pathway, which is stimulated by APN paradox, these steps might present attractive therapeutic targets for intervention. As such, in addition to suppression of APN expression by antisense strategies, such as antisense oligonucleotides and mi-RNA of APN, activin receptor signaling might be suppressed by various methods, including antisense oligonucleotides and mi-RNA of activin, and overexpression of inhibin and follistatin, and compounds of anti-Smads.
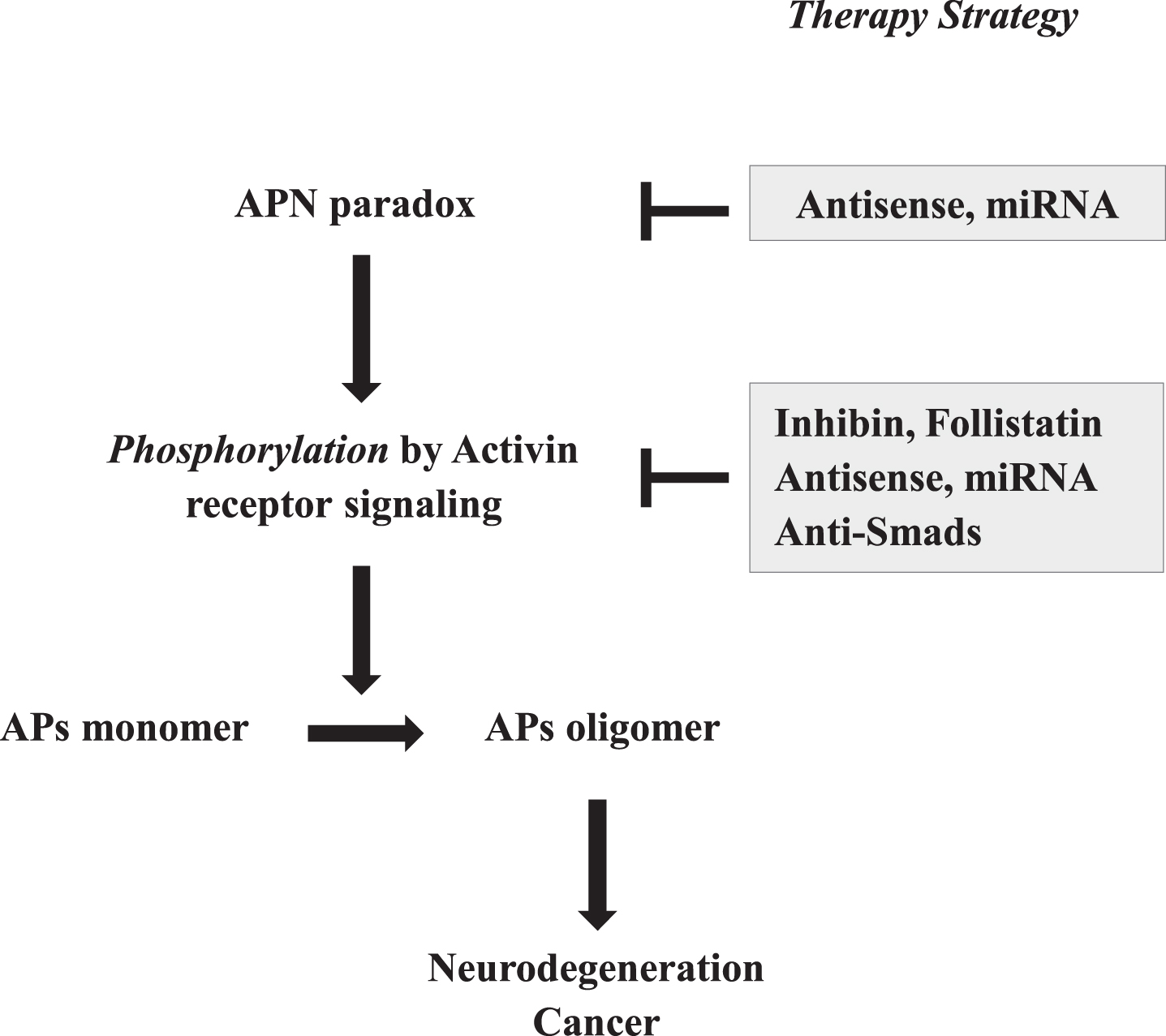
Since phosphorylation of APs by activin receptor signaling is critically involved in protofibril for-mation in neurodegenerative diseases of aging, suppressing activin expression might prove to be a more effective target compared to APN signaling inhibition (Fig. 3). To achieve this, suppressing activin expression might be effective, including reduction of activin mRNA by antisense and/or miRNA strategies. Next, endogenous activin inhibitors, including inhibin and follistatin, might also employed. To support this, peptides derived from follistatin were shown to inhibit muscle degeneration from myostatin, a member of TGFβ family, indicating the therapeutic potential of follistatin for the therapy of sarcopenia [51]. Lastly, suppressing activin receptor signaling by use of Smad-specific inhibitors might be effective, and it was previously described that the selective inhibition of Smad proteins would provide an important therapeutic benefit in TGFβ fibrotic diseases [52]. Conceivably, a combination of such strategies or with those against other related targets might be synergistically beneficial.
Finally, the timing of administration of such treatments in the course of neurodegeneration is extremely important. Recently, consensus has been favoring early treatment, even pre-symptomatic therapy, as a paradigm for neurodegenerative disease modification [53]. Although it is predicted that suppression of activin receptor signaling by disease-modifying treatment may lead to the reduced evolvability, it remains unknown how reduced evolvability might in turn affect offspring’s brain. Therefore, such treatments should be avoided during reproductive life, until the unexpected side effects are clarified.
CONCLUSION
Although more than 60 years have passed since G.C. Williams described the concept of antagonistic pleiotropy [4], little progress has been made in terms of the underlying molecular mechanisms [54]. Distinct from evolvability, which may presumably be a primitive phenomenon in evolution, neurodegenerative disorders generated by antagonistic pleiotropy in aging may be a relatively recent phenomenon in human brain because of our extended postmenopausal lifespan. Indeed, the length of the post-reproductive senescence depends on environmental conditions, such as the presence of predators and available nutrition [3, 55]. In modern times, only humans have neither predators nor lack of nutrition through development of plant and livestock farming. Because of this context, animal models might not sufficiently reproduce the human experience for the proper analysis of antagonistic pleiotropy.
On the other hand, aging science reaps the benefit of technological innovation, and GWAS findings support a role of activin/TGFβ in antagonistic pleiotropy in aging. Thus, it is predicted that activin might be involved in linking evolvability with neurodegenerative disorders. Furthermore, it is tempting to speculate that other morphogenic factors such as retinoic acid may be active in modulating activin-related antagonistic pleiotropy. Naturally, inhibin and follistatin, endogenous inhibitors of activin activity, may negatively regulate antagonistic pleiotropy related to activin, suggesting that these molecules might be therapeutically important for aging disorders. In summary, a better understanding of the role of the activin signaling pathway in antagonistic pleiotropy which links development with aging may lead to more effective therapies for aging-associated conditions such as neurodegenerative disease.
ACKNOWLEDGMENTS
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (University of Chicago).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0206r1).
REFERENCES
[1] | Wickner RB ((2016) ) Yeast and fungal prions. Cold Spring Harb Perspect Biol 8: , a023531. |
[2] | Hashimoto M , Ho G , Sugama S , Takamatasu Y , Shimizu Y , Takenouchi T , Waragai M , Masliah E ((2018) ) Evolvability of amyloidogenic proteins in human brain. J Alzheimers Dis 62: , 73–83. |
[3] | Hashimoto M , Ho G , Takamatsu Y , Shimizu Y , Sugama S , Takenouchi T , Waragai M , Masliah E ((2018) ) Evolvability and neurodegenerative disease: antagonistic pleiotropy phenomena derived from amyloid aggregates. J Parkinsons Dis 8: , 405–408. |
[4] | Williams GC ((1957) ) Pleiotropy, natural selection, and the evolution of senescence. Evolution 11: , 398–411. |
[5] | Ueno N , Nishimatsu S , Murakami K ((1990) ) Activin as a cell differentiation factor. Prog Growth Factor Res 2: , 113–124. |
[6] | Robertson DM ((1991) ) Transforming growth factor beta/inhibin family. Baillieres Clin Endocrinol Metab 5: , 615–634. |
[7] | Asashima M , Nakano H , Shimada K , Kinoshita K , Ishii K , Shibai H , Ueno N ((1990) ) Mesodermal induction in early amphibian embryos by activin A (erythroid differentiation factor). Roux Arch Dev Biol 198: , 342–357. |
[8] | Wijayarathna R , de Kretser DM ((2016) ) Activins in reproductive biology and beyond. Hum Reprod Update 22: , 330–335. |
[9] | Baccarelli A , Morpurgo PS , Corsi A , Vaghi I , Fanelli M , Cremonesi G , Vaninetti S , Beck-Peccoz P , Spada A ((2001) ) Activin A serum levels and aging of the pituitary-gonadal axis: a cross-sectional study in middle-aged and elderly healthy subjects. Exp Gerontol 36: , 1403–1412. |
[10] | Weigert J , Neumeier M , Wanninger J , Schober F , Sporrer D , Weber M , Schramm A , Wurm S , Stögbauer F , Filarsky M , Schäffler A , Aslanidis C , Schölmerich J , Buechler C ((2009) ) Adiponectin upregulates monocytic activin A but systemic levels are not altered in obesity or type 2 diabetes. Cytokine 45: , 86–91. |
[11] | Wanninger J , Neumeier M , Hellerbrand C , Schacherer D , Bauer S , Weiss TS , Huber H , Schäffler A , Aslanidis C , Schölmerich J , Buechler C ((2011) ) Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochim Biophys Acta 1811: , 626–633. |
[12] | Kulminski AM , He L , Culminskaya I , Loika Y , Kernogitski Y , Arbeev KG , Loiko E , Arbeeva L , Bagley O , Duan M , Yashkin A , Fang F , Kovtun M , Ukraintseva SV , Wu D , Yashin AI ((2016) ) Pleiotropic associations of allelic variants in a 2q22 region with risks of major human diseases and mortality. PLoS Genet 12: , e1006314. |
[13] | Chari S , Hopkinson CR , Daume E , Sturm G ((1979) ) Purification of “inhibin” from human ovarian follicular fluid. Acta Endocrinol (Copenh) 90: , 157–166. |
[14] | Ling N , Ying SY , Ueno N , Shimasaki S , Esch F , Hotta M , Guillemin R ((1986) ) Pituitary FSH is released by a heterodimer of the beta-subunits from the two forms of inhibin. Nature 321: , 779–782. |
[15] | Vale W , Rivier J , Vaughan J , McClintock R , Corrigan A , Woo W , Karr D , Spiess J ((1986) ) Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature 321: , 776–779. |
[16] | Bilezikjian LM , Blount AL , Donaldson CJ , Vale WW ((2006) ) Pituitary actions of ligands of the TGF-beta family: activins and inhibins. Reproduction 132: , 207–215. |
[17] | Gaddy-Kurten D , Tsuchida K , Vale W ((1995) ) Activins and the receptor serine kinase superfamily. Recent Prog Horm Res 50: , 109–129. |
[18] | Schubert D , Kimura H , LaCorbiere M , Vaughan J , Karr D , Fischer WH ((1990) ) Activin is a nerve cell survival molecule. Nature 344: , 868–870. |
[19] | Hashimoto M , Nakamura T , Inoue S , Kondo T , Yamada R , Eto Y , Sugino H , Muramatsu M ((1992) ) Follistatin is a developmentally regulated cytokine in neural differentiation. J Biol Chem 267: , 7203–7206. |
[20] | Lewis KA , Gray PC , Blount AL , MacConell LA , Wiater E , Bilezikjian LM , Vale W ((2000) ) Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 404: , 411–414. |
[21] | Esch FS , Shimasaki S , Mercado M , Cooksey K , Ling N , Ying S , Ueno N , Guillemin R ((1987) ) Structural characterization of follistatin: a novel follicle-stimulating hormone release-inhibiting polypeptide from the gonad. Mol Endocrinol 1: , 849–855. |
[22] | Nakamura T , Takio K , Eto Y , Shibai H , Titani K , Sugino H ((1990) ) Activin-binding protein from rat ovary is follistatin. Science 247: , 836–838. |
[23] | Wu MY , Hill CS ((2009) ) TGF-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 16: , 329–343. |
[24] | Jones KL , Mansell A , Patella S , Scott BJ , Hedger MP , de Kretser DM , Phillips DJ ((2007) ) Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A 104: , 16239–16244. |
[25] | Verhamme FM , Bracke KR , Amatngalim GD , Verleden GM , Van Pottelberge GR , Hiemstra PS , Joos GF , Brusselle GG ((2014) ) Role of activin-A in cigarette smoke-induced inflammation and COPD. Eur Respir J 43: , 1028–1041. |
[26] | Latres E , Mastaitis J , Fury W , Miloscio L , Trejos J , Pangilinan J , Okamoto H , Cavino K , Na E , Papatheodorou A , Willer T , Bai Y , Hae Kim J , Rafique A , Jaspers S , Stitt T , Murphy AJ , Yancopoulos GD , Gromada J ((2017) ) Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun 8: , 15153. |
[27] | Williams MJ , Sugatani T , Agapova OA , Fang Y , Gaut JP , Faugere MC , Malluche HH , Hruska KA ((2018) ) The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease. Kidney Int 93: , 147–158. |
[28] | Tsai YL , Chang CC , Liu LK , Huang PH , Chen LK , Lin SJ ((2018) ) The association between serum activin A levels and hypertension in the elderly: a cross-sectional analysis from I-Lan Longitudinal Aging Study. Am J Hypertens 31: , 369–374. |
[29] | Morikawa M , Derynck R , Miyazono K ((2016) ) TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol 8: , A021873. |
[30] | Tominaga K , Suzuki HI ((2019) ) TGF-β signaling in cellular senescence and aging-related pathology. Int J Mol Sci 20: , 5002. |
[31] | Papageorgis P ((2017) ) Complex interplay between aging and cancer: role of TGF-β signaling. Crit Rev Oncog 22: , 313–321. |
[32] | Wei J , Takamatsu Y , Wada R , Fujita M , Ho G , Masliah E , Hashimoto M ((2021) ) Therapeutic potential of αS evolvability for neuropathic Gaucher disease. Biomolecules 11: , 1453–1459. |
[33] | Luisi S , Florio P , Reis FM , Petraglia F ((2001) ) Expression and secretion of activin A: possible physiological and clinical implications. Eur J Endocrinol 145: , 225–236. |
[34] | Roan NR , Sandi-Monroy N , Kohgadai N , Usmani SM , Hamil KG , Neidleman J , Montano M , Ständker L , Röcker A , Cavrois M , Rosen J , Marson K , Smith JF , Pilcher CD , Gagsteiger F , Sakk O , O’Rand M , Lishko PV , Kirchhoff F , Münch J , Greene WC ((2017) ) Semen amyloids participate in spermatozoa selection and clearance. Elife 6: , e24888. |
[35] | Jamasbi E , Separovic F , Hossain MA , Ciccotosto GD ((2017) ) Phosphorylation of a full length amyloid-beta peptide modulates its amyloid aggregation, cell binding and neurotoxic properties. Mol Biosyst 13: , 1545–1551. |
[36] | Fujiwara H , Hasegawa M , Dohmae N , Kawashima A , Masliah E , Goldberg MS , Shen J , Takio K , Iwatsubo T ((2002) ) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4: , 160–164. |
[37] | Fares MB , Jagannath S , Lashuel HA ((2021) ) Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci 22: , 111–131. |
[38] | Mahul-Mellier AL , Burtscher J , Maharjan N , Weerens L , Croisier M , Kuttler F , Leleu M , Knott GW , Lashuel HA ((2021) ) The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A 117: , 4971–4982. |
[39] | Waragai M , Ho G , Takamatsu Y , Shimizu Y , Sugino H , Sugama S , Takenouchi T , Masliah E , Hashimoto M ((2018) ) Dual-therapy strategy for modification of adiponectin receptor signaling in aging-associated chronic diseases. Drug Discov Today 23: , 1305–1311. |
[40] | Woodward L , Akoumianakis I , Antoniades C ((2017) ) Unravelling the adiponectin paradox: novel roles of adiponectin in the regulation of cardiovascular disease. Br J Pharmacol 174: , 4007–4020. |
[41] | Waragai M , Ho G , Takamatsu Y , Wada R , Sugama S , Takenouchi T , Masliah E , Hashimoto M ((2020) ) Adiponectin paradox in Alzheimer’s disease; relevance to amyloidogenic evolvability? Front Endocrinol (Lausanne) 11: , 108. |
[42] | Wennberg AM , Gustafson D , Hagen CE , Roberts RO , Knopman D , Jack C , Petersen RC , Mielke MM ((2016) ) Serum adiponectin levels, neuroimaging, and cognition in the Mayo Clinic Study of Aging. J Alzheimers Dis 53: , 573–581. |
[43] | van Himbergen TM , Beiser AS , Ai M , Seshadri S , Otokozawa S , Au R , Thongtang N , Wolf PA , Schaefer EJ ((2012) ) Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and alzheimer disease: results from the Framingham Heart Study. Arch Neurol 69: , 594–600. |
[44] | De Franciscis P , Barbieri M , Leo S , Dalise AM , Sardu C , Marfella R , Colacurci N , Paolisso G , Rizzo MR ((2017) ) Serum adiponectin levels are associated with worse cognitive function in postmenopausal women. PLoS One 12: , e0186205. |
[45] | Waragai M , Adame A , Trinh I , Sekiyama K , Takamatsu Y , Une K , Masliah E , Hashimoto M ((2016) ) Possible involvement of adiponectin, the anti-diabetes molecule, in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 52: , 1453–1459. |
[46] | Lee CH , Lui DTW , Cheung CYY , Fong CHY , Yuen MMA , Chow WS , Woo YC , Xu A , Lam KSL ((2020) ) Higher circulating adiponectin concentrations predict incident cancer in type 2 diabetes - the adiponectin paradox. J Clin Endocrinol Metab 105: , dgaa075. |
[47] | Loumaye A , de Barsy M , Nachit M , Lause P , van Maanen A , Trefois P , Gruson D , Thissen JP ((2017) ) Circulating activin A predicts survival in cancer patients. J Cachexia Sarcopenia Muscle 8: , 768–777. |
[48] | Saunders CJ , Zhao W , Ardinger HH ((2009) ) Comprehensive ZEB2 gene analysis for Mowat-Wilson syndrome in a North American cohort: a suggested approach to molecular diagnostics. Am J Med Genet A 149a: , 2527–2531. |
[49] | Yong HE , Melton PE , Johnson MP , Freed KA , Kalionis B , Murthi P , Brennecke SP , Keogh RJ , Moses EK ((2015) ) Genome-wide transcriptome directed pathway analysis of maternal pre-eclampsia susceptibility genes. PLoS One 10: , e0128230. |
[50] | Waragai M , Ho G , Takamatsu Y , Wada R , Sugama S , Takenouchi T , Masliah E , Hashimoto M ((2020) ) Adiponectin paradox as a therapeutic target in Alzheimer’s disease. J Alzheimers Dis 76: , 1249–1253. |
[51] | Tsuchida K ((2008) ) Myostatin inhibition by a follistatin-derived peptide ameliorates the pathophysiology of muscular dystrophy model mice. Acta Myol 27: , 14–18. |
[52] | Laping NJ ((1999) ) Therapeutic uses of smad protein inhibitors: Selective inhibition of specific TGF-beta activities. IDrugs 2: , 907–914. |
[53] | Sperling R , Mormino E , Johnson K ((2014) ) The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 84: , 608–622. |
[54] | Kirkwood TB ((2005) ) Understanding the odd science of aging. Cell 120: , 437–447. |
[55] | Croft DP , Brent LJ , Franks DW , Cant MA ((2015) ) The evolution of prolonged life after reproduction. Trends Ecol Evol 30: , 407–416. |
[56] | Spicer E , Suckert C , Al-Attar H , Marsden M ((2010) ) Integrin alpha5beta1 function is regulated by XGIPC/kermit2 mediated endocytosis during Xenopus laevis gastrulation. PLoS One 5: , e10665. |
[57] | Wang Z , Zhang N , Song R , Fan R , Yang L , Wu L ((2015) ) Activin A expression in esophageal carcinoma and its association with tumor aggressiveness and differentiation. Oncol Lett 10: , 143–148. |

