
Comparative Analysis of Cortical Microinfarcts and Microbleeds using 3.0-Tesla Postmortem Magnetic Resonance Images and Histopathology
Abstract
Microvascular lesions including cortical microinfarctions (CMIs) and cerebral lobar microbleeds (CMBs) are usually caused by cerebral amyloid angiopathy (CAA) in the elderly and are correlated with cognitive decline. However, their radiological-histopathological coincidence has not been revealed systematically with widely used 3-Tesla (3T) magnetic resonance imaging (MRI). The purpose of the present study is to delineate the histopathological background corresponding to MR images of these lesions. We examined formalin-fixed 10-mm thick coronal brain blocks from 10 CAA patients (five were also diagnosed with Alzheimer’s disease, three with dementia with Lewy bodies, and two with CAA only) with dementia and six non CAA patients with neurodegenerative disease. Using 3T MRI, both 3D-fluid attenuated inversion recovery (FLAIR) and 3D-double inversion recovery (DIR) were examined to identify CMIs, and T2* and susceptibility-weighted images (SWI) were examined to identify CMBs. These blocks were subsequently examined histologically and immunohistochemically. In CAA patients, 48 CMIs and 6 lobar CMBs were invariably observed in close proximity to degenerated Aβ-positive blood vessels. Moreover, 16 CMIs (33%) of 48 were detected with postmortem MRI, but none were seen when the lesion size was smaller than 1 mm. In contrast, only 1 undeniable CMI was founded with MRI and histopathology in 6 non CAA patients. Small, cortical high-intensity lesions seen on 3D-FLAIR and 3D-DIR images likely represent CMIs, and low-intensity lesions in T2* and SWI correspond to CMBs with in vivo MRI. Furthermore, a close association between amyloid-laden vessels and these microvascular lesions indicated the contribution of CAA to their pathogenesis.
INTRODUCTION
With advancements in magnetic resonance imaging (MRI) technology, we are now able to visualize cerebral microbleeds (CMBs), which are considered to be small hemorrhages caused by cerebral small vessel diseases, including hypertensive small vessel disease and cerebral amyloid angiopathy (CAA). However, only a few studies have reported a direct comparison between CMBs seen in postmortem MRI and corresponding histopathological findings [1–4].
By contrast, the significance of cortical microinfarcts (CMIs) has been investigated pathologically because these lesions are thought to be too small to detect with MRI. The pathogenesis of CMIs has been attributed to CAA or alternatively, to hypertensive small vessel diseases. The frequency of CMIs is correlated with cognitive decline and is thought to be a strong predictor of dementia [5–10]. Unfortunately, CMIs remains invisible on conventional MRI [11, 12]. However, we first reported radiological detection of CMIs with high-resolution MRI and described a new protocol for enhanced detection with 3-Tesla (3T) MRI. This protocol is based on 3D imaging of either fluid attenuated inversion recovery (FLAIR) or double inversion recovery (DIR), the latter of which guarantees specific localization within the cerebral cortex [13]. Notably, we observed CMIs on both 3D-FLAIR and 3D-DIR images in nine out of 70 memory clinic patients. Of these, one familial Alzheimer’s disease (AD) patient with a PSEN1 mutation exhibited CMIs using this protocol, and an autopsy study of the brain of this patient’s mother demonstrated CMIs and severe CAA [14].
Our report has been confirmed by other researchers who compared hyperintense cortical microlesions observed on 7T in vivo MRI with the corresponding histopathologic findings [15–18]. Van Veluw et al., who reported detection of CMIs with 7T postmortem MRI, also performed 3T as well as 7T scans in vivo (27% of the CMIs detected on 7T were also detected on 3T), and suggested that CMIs may escape detection on regular clinical MRI [15]. However, our protocol for 3T MRI results in enhanced resolution with 3D imaging and can determine strict intracortical localization with DIR imaging. We consider that establishing a clinical protocol for 3T MRI is pivotal, because 7T MRI is not used for clinical investigations of a large number of patients. For this reason, we examined the histopathological counterparts of radiologically detected CMIs on 3T MRI by directly comparing the histopathological-radiological correlations in autopsied brains with CAA.
In the current study, we have hypothesized that cortical hyper-intense lesions detected by 3D-DIR imaging by 3 Tesla MRI histopathologically correspond to CMIs. For this purpose, we compared CMIs between CAA and non-CAA control brains, because the brains with CAA have been reported to have frequent CMIs, thereby facilitating their detection by histology and MRI.
MATERIALS AND METHODS
Conditions that may affect postmortem MR images
For optimization of the postmortem 3T MRI protocol (subjects C1 and C2 in Table 1), we examined 2 formalin-fixed 10-mm thick coronal brain blocks from 2 patients under variable conditions. We initially studied MR images of brain blocks in water at 0°C, 25°C, or approximately 37°C to investigate the effect of temperature on postmortem MRI. Second, we embedded the brain blocks in agar gel, so that the brain blocks were fixed in place to minimize motion artefacts. Third, we removed the pia mater of the brain, because the outer surface of the brain blocks exhibit artefactual hyperintensity in FLAIR or DIR images. Lastly, we washed the brain blocks with running water for 8 days because a previous paper reported deterioration of MR images in the presence of residual formalin [19, 20].
Table 1
Clinical features of autopsied brains and the number of CMIs and CMBs detected with postmortem MRI and histology.
| Subject | Neuropathologic diagnosis | Sex (F/M) | Age (years) | Number of CMIs | Number of CMBs | ||
| Histology | Both MRI and | MRI | Both MRI and | ||||
| Histology | Histology | ||||||
| C1 | DLB with CAA | F | 93 | 6 | 1 | 3 | 1 |
| C2 | DLB with CAA, Af | F | 86 | 12 | 4 | 0 | 0 |
| C3 | AD (B&B VI) with CAA | F | 83 | 3 | 2 | 1 | 1 |
| C4 | AD (B&B V) with CAA, cSS | F | 84 | 1 | 0 | 4 | 1 |
| C5 | AD (B&B VI) with CAA | F | 88 | 0 | 0 | 1 | 0 |
| C6 | AD (B&B V) with CAA | F | 82 | 2 | 2 | 3 | 1 |
| C7 | CAA | M | 85 | 9 | 4 | 1 | 0 |
| C8 | CAA | M | 98 | 3 | 1 | 3 | 0 |
| C9 | DLB with CAA, Af | M | 79 | 8 | 1 | 7 | 2 |
| C10 | AD (B&B V) with CAA, Af | M | 83 | 4 | 1 | 2 | 0 |
| Total | 48 | 16 | 22 | 6 | |||
| N1 | Amyotrophic lateral sclerosis | F | 72 | 0 | 0 | 0 | 0 |
| N2 | Multiple system atrophy | F | 78 | 0 | 0 | 2 | 1 |
| N3 | Amyotrophic lateral sclerosis with Af | M | 77 | 1 | 1* | 1 | 1 |
| N4 | Parkinson’s disease | M | 79 | 0 | 0 | 2 | 0 |
| N5 | Amyotrophic lateral sclerosis with Af | M | 77 | 0 | 0 | 0 | 0 |
| N6 | Limbic encephalitis | M | 62 | 0 | 0 | 2 | 0 |
| Total | 1 | 1 | 7 | 2 | |||
CMIs: cortical microinfarctions, CMBs: cerebral microbleeds, cSS: cortical superficial siderosis, CAA: cerebral amyloid angiopathy, DLB: Demetnia with Lewy bodies, AD: Alzheimer’s disease, B&B: Braak and Braak stage (Acta Neuropathol 1991; 239), Af: arterial fibrillations, C1: CAA subject 1, N1: non CAA subject 1, *: undeniable CMI.
Postmortem MRI protocol
The MRI studies were performed with a 3T MR unit (Achieva Philips Medical System, Best, the Netherlands) using an 8- or 32-channel phased-array head coil. We employed the previously described MR protocol with 3D-DIR and 3D-FLAIR images [13] to detect CMIs with 3T postmortem MRI. Axial DIR imaging sequences were performed using 2 different inversion pulses. The long inversion time and the short inversion time were defined as the intervals between the 180° inversion time and the 90° excitation pulse, respectively, which have been optimized for human brain imaging sequences and were provided by the vendor. Details of the 3D-DIR protocol were as follows: field of view, 250 mm; matrix, 256×256; section thickness, 1 mm; repetition time (ms)/echo time (ms), 5,500/260; long inversion time (ms)/short inversion time (ms), 2,550/450; number of signals acquired, 2.
3D-FLAIR imaging sequences were obtained in the sagittal direction, and then the axial and coronal images were reconstructed. The details of 3D-FLAIR were as follows: field of view, 260 mm; matrix, 352×352; section thickness, 1 mm; repetition time (ms)/echo time (ms), 6,000/400; inversion time, 2,000 ms; number of signals acquired, 2. Other sequences including susceptibility-weighted images (SWI) and T2*-weighted images (T2*) were performed to detect CMBs. The details of SWI were as follows: field of view, 230 mm; matrix, 512×512; section thickness, 1 mm; repetition time (ms)/echo time (ms), 22/11.5 (in-phase), 33 (shifted); number of signals acquired, 1; flip angle, 20°. And details of T2* were as follows: field of view, 230 mm; matrix, 512×512; section thickness, 3 mm; repetition time (ms)/echo time (ms), 475/16; number of signals acquired, 2; flip angle, 18°.
Comparison between postmortem MRI and histopathology
This study involves formalin-fixed 10-mm thick coronal brain blocks from 10 patients diagnosed with CAA (5 cases of AD with CAA, 3 of dementia with Lewy bodies with CAA, 2 of CAA only) and CMIs obtained from the Fukushimura brain bank in Fukushimura hospital (Medical Bioresource Database, B0028) and non CAA control same coronal brain blocks from 6 patients diagnosed with neurodegenerative disease (Amyotrophic lateral sclerosis, Multiple sclerosis, Parkinson’s disease, Limbic encephalitis) (Table 1). Five of 16 patients had atrial fibrillation (Af) in their clinical record. The average durations between death and formalin fixation ranged between 3–7 h. The target samples were 1 coronal block including gyri of parietal lobe per1 subject. Durations of formalin fixation of the CAA and the non CAA control brains were 30.2±15.0 months and 95.5±78.9 months, respectively. The postmortem diagnosis of AD was made according to stage V and VI of the classification of Braak and Braak [21], and those of dementia with Lewy bodies was made according to stage 4, 5, and 6 of the classification of Braak [22]. The severity of the CAA was evaluated according to the CERAD criteria [23]. The use of these materials for this study has been approved by both the medical ethics committees of Mie University Hospital and Fukushimura Hospital.
After postmortem MRI scanning, brain blocks were dehydrated and embedded in paraffin. Further, we cut brain blocks using a microtome to the depth of 3 mm from the surface, then serial 12 sections (7-μm thick) were cut. Standard histologic staining was performed on these serial sections, which included hematoxylin/eosin staining and Klüver-Barrera staining. Among these sections, we selected one representative section which contained largest number of CMIs. The number of CMIs was determined based on the presence of a sharply delineated area of ischemia with cellular death, tissue necrosis, and cavitation [7, 24]. Foci of mere astrogliosis and microgliosis with tissue rarefaction were excluded, because the diameter of these lesions was difficult to determine. According to the topographic distribution of CMIs in these sections, we searched for the corresponding CMI lesions among the sequential images of postmortem MRI. For detection of possible CMBs with MRI, we used T2*-weighted images or SWI. Then we examined these blocks histologically to verify microaneurysms or deposition of hemosiderin and hematoidin. The signals from blood vessels were markedly intensified on postmortem MRI T2* and SWI, possibly because blood flow was stopped. This observation is markedly different from that of conventional in vivo MRI, and therefore, we did not count low-intensity lesions in the sulcus and outer surface of the gyrus.
Sections with CMIs were further immunostained with anti-amyloid β (Aβ)40 and Aβ42 antibodies. For immunohistochemistry, rabbit polyclonal antibodies against Aβ40 and Aβ42 (44348A and 44–344, Invitrogen, Camarillo, CA, USA) were used. After incubation with the primary antibody, the sections were treated with a biotinylated secondary antibody. Immunolabeling with peroxidase-conjugated avidin was visualized with 3,3’-diaminobenzidine as a chromogen, and sections were counterstained lightly with Mayer’s hematoxylin.
RESULTS
Conditions that may affect postmortem MR images
The contrast between cerebral gray and white matter changed markedly depending on the temperature. In 0°C water, the signal intensity of both the gray and white matter was lowest, consequently showing no contrast between their borders. However, as the temperature of the brain blocks increased, the signal intensity of the gray matter increased markedly and that of the white matter increased moderately, allowing visualization of the border between the gray and white matter. Artefactual lines appeared in parallel to the outer surface of the brain in 60°C water, and thus, images were optimal in 37°C water. The resolution of MR images was not affected by the other procedures such as embedding the brain blocks in agar gel, removal of the pia mater, or washing out formalin from the brain.
Comparison between postmortem MRI and histopathology
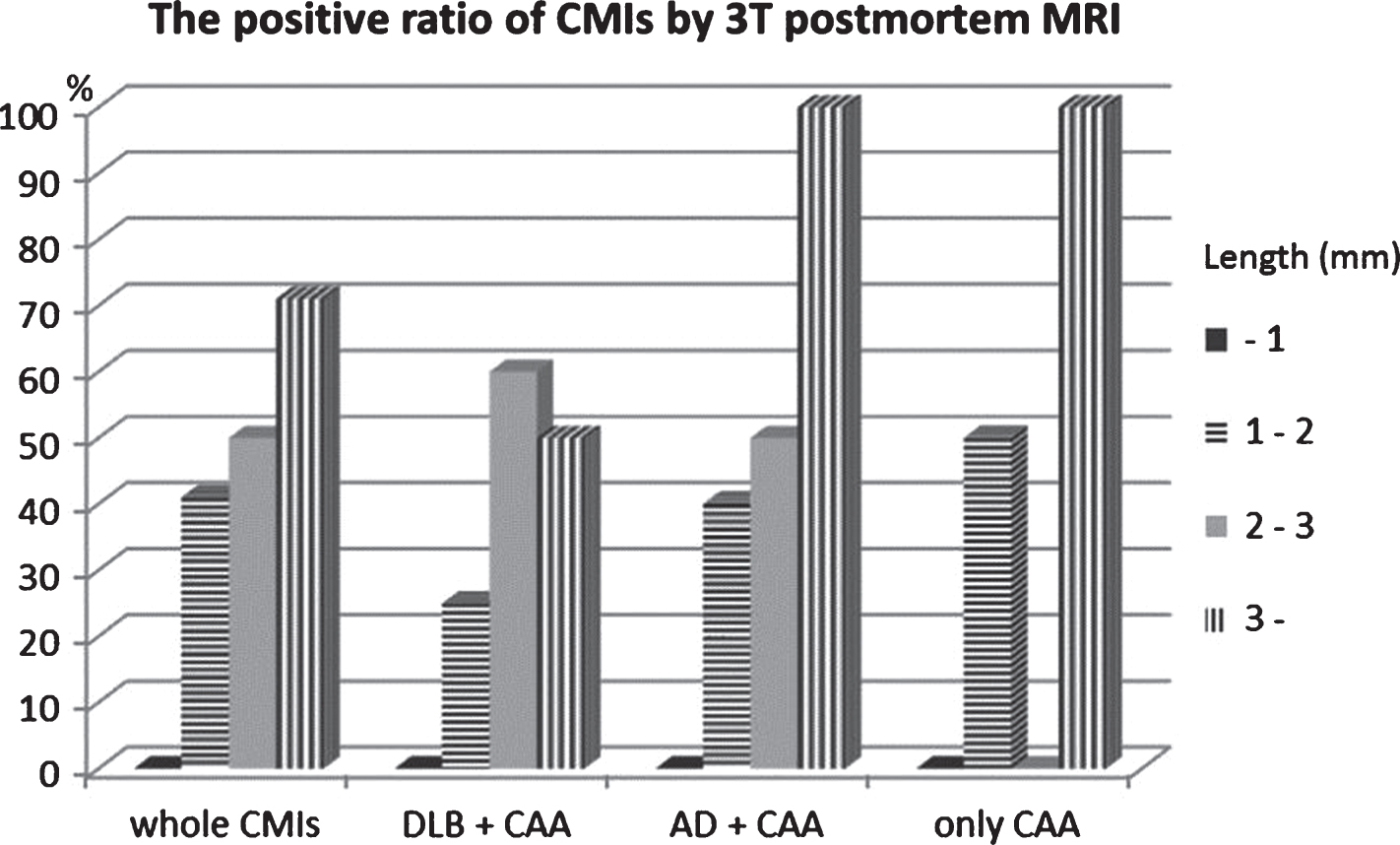
Using the present protocol, we found 48 CMIs and six lobar CMBs in 10 brain blocks (34.8±5.3 cm2) from 10 CAA subjects. Of the 48 CMIs, 16 (33%) were confirmed in FLAIR images and 8 (17%) were confirmed in DIR images to correspond to histological lesions. All eight CMIs observed in DIR images were also confirmed in FLAIR. In contrast, we found only one CMI in six brain blocks (26.3±4.2 cm2) from six non CAA control (Table 1). Some CMIs that were 2 mm or larger could be detected with 3T postmortem MRI, but lesions less than 1 mm (23/48, 48%) were not detectable (Fig. 1). Regarding underlying diseases with CAA, we observed no marked differences in the positive ratio of CMIs. On the contrary, we observed the CMIs on irregularly shaped cortices (18/48, 38%) in the superficial layer tended to be undetectable because of the partial volume effect.
Fig.1
Size distribution of the detection ratio of CMIs with postmortem 3T MRI. With only CAA or CAA complicated with Lewy body disease or Alzheimer’s disease, the positive ratio of CMIs is almost the same. CMIs of about 2 mm were detected with 3T postmortem MRI, but those less than 1 mm could not be detected. Between 1 and 2 mm, the environmental topography around CMIs in the cerebral gyrus determined whether or not they could be detected. CMIs that were located in complex cortical regions tended to be undetectable.
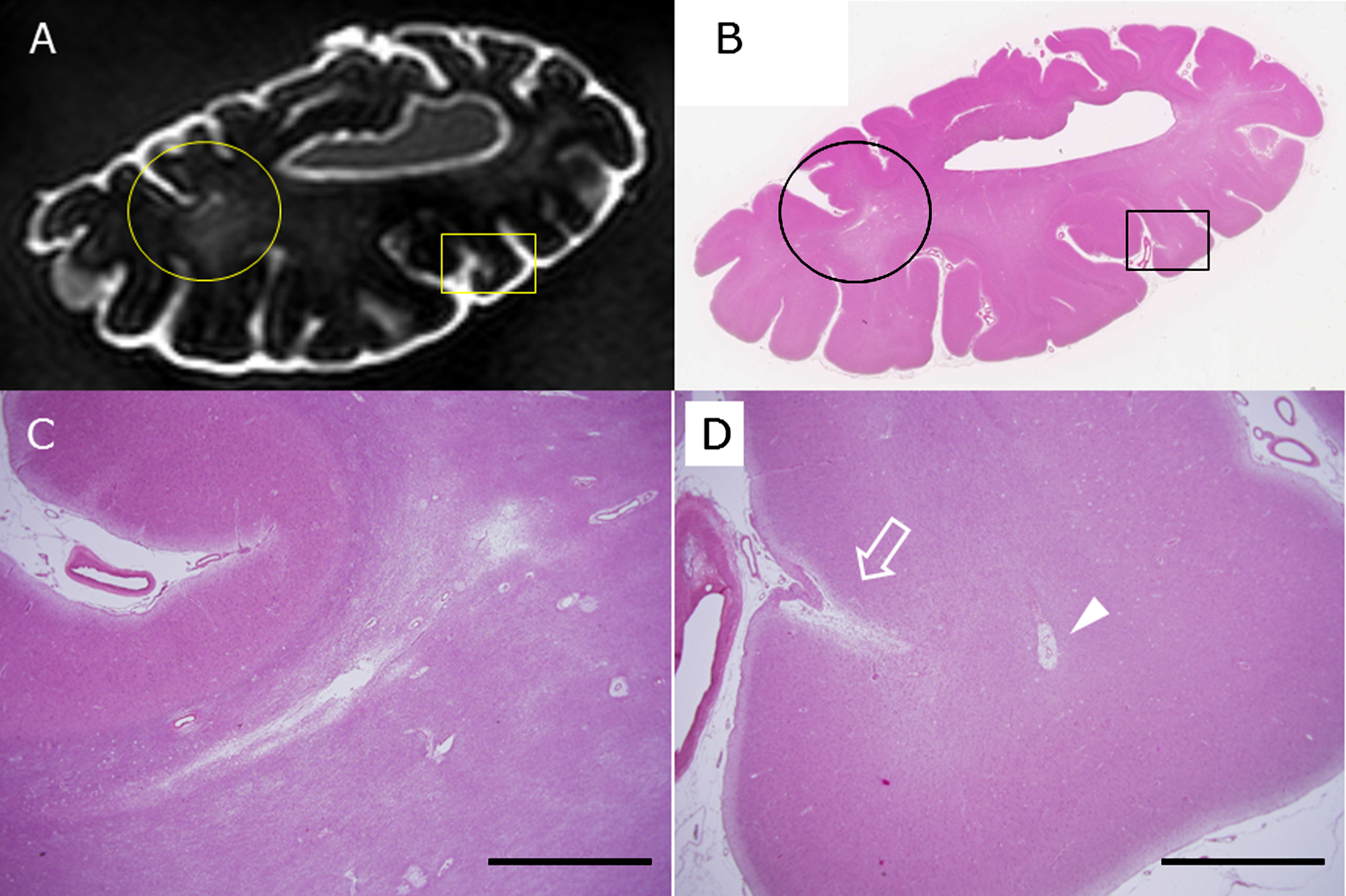
On the other hand, we found 29 lesions by SWI or T2* weighted image of 3T postmortem MRI which were judged to be CMBs, but we could verify only 8 CMBs histopathologically (Fig. 2). CMBs coexisted with CMIs in the same block frequently, but not in the same sites, and, often distributed in the other gyri. Of the 8 CMBs, a microhematoma around small vessels corresponded to 1 CMB, and clusters of hemosiderin-laden macrophages corresponded to 7 CMBs. Figure 3 shows the coincidence of radiological and histological findings of a CMB and demonstrated correspondence between MRI findings and clusters of perivascular hemosiderin-laden macrophages (Fig. 3).
Fig.2
Lesions with a small signal loss (the center of the red circle) represent CMBs in the cerebral cortex of subject C4 and were detected with postmortem 3T MRI (SWI). Other lesions with a small signal loss in sulci are leptomeningeal arterioles (A). This formalin-fixed brain block was embedded in paraffin, sliced thin, and stained with hematoxylin and eosin (HE) as in B. C (the black rectangle shown in B) and D (high-power field) are photomicrographs that show the histopathologic lesion of a CMB. The lesion has degenerated vessel walls and deposition of hematoidin (dark yellow) surrounded by hemosiderin (dark brown). Scale bars in C: 2 mm, D: 200 μm.
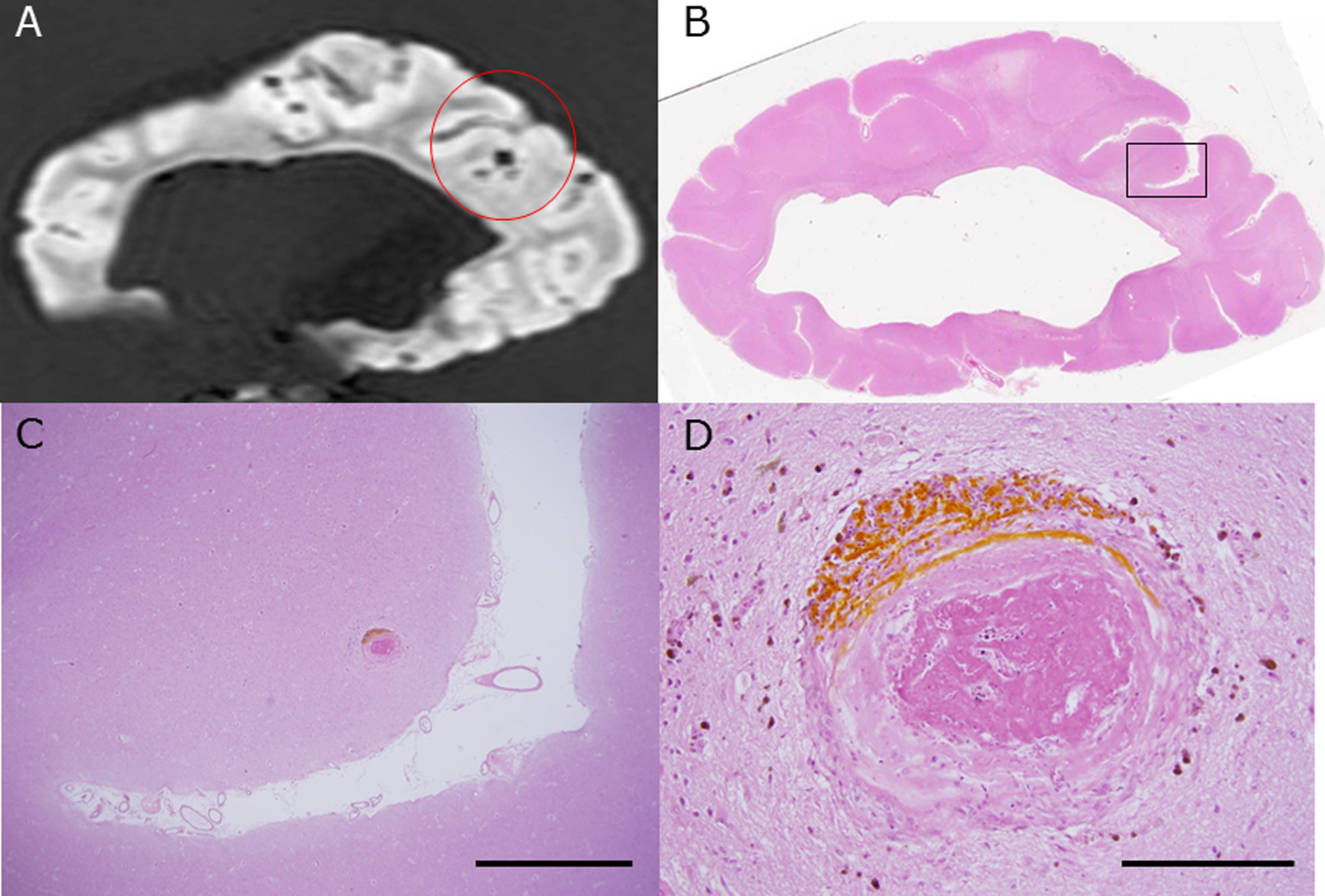
Fig.3
A CMB detected with SWI with postmortem 3T MRI (A) is compared with HE staining of the brain block (B) of subject C1. The rectangle in B is enlarged in C and shows accumulation of hemosiderin-laden macrophages around the small vessels (Inset). Scale bars in C: 200 μm (inset: 20 μm).
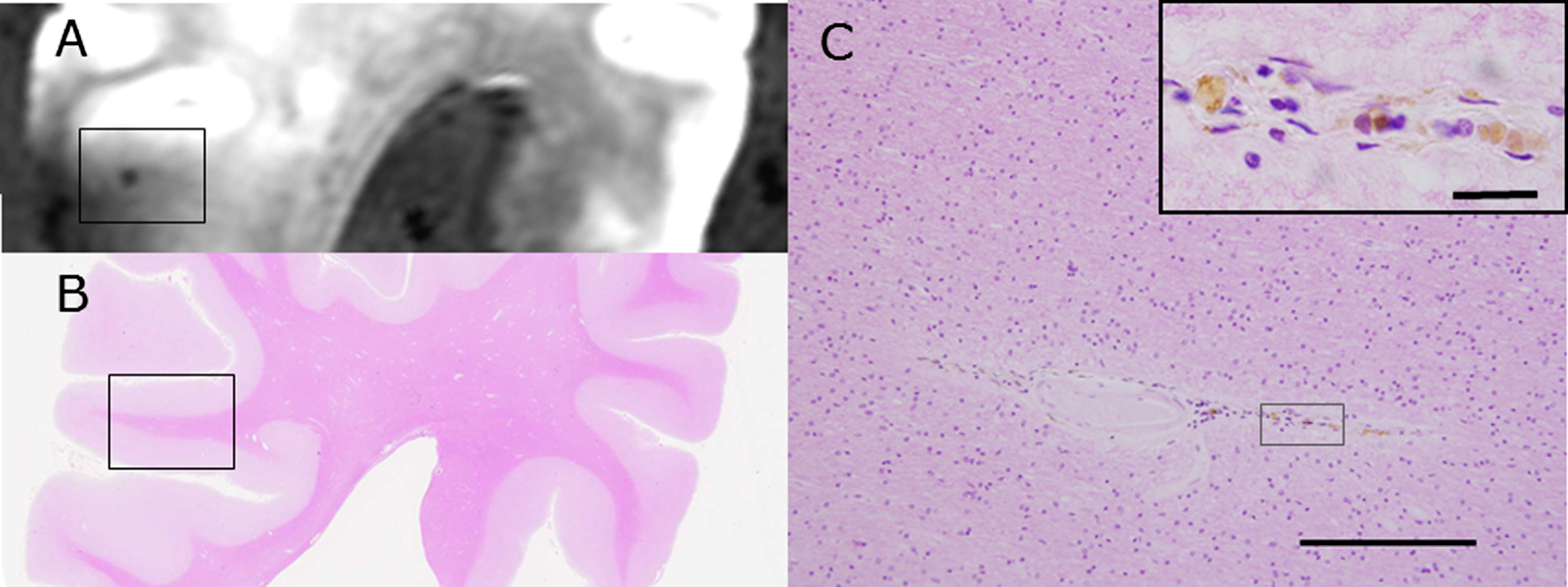
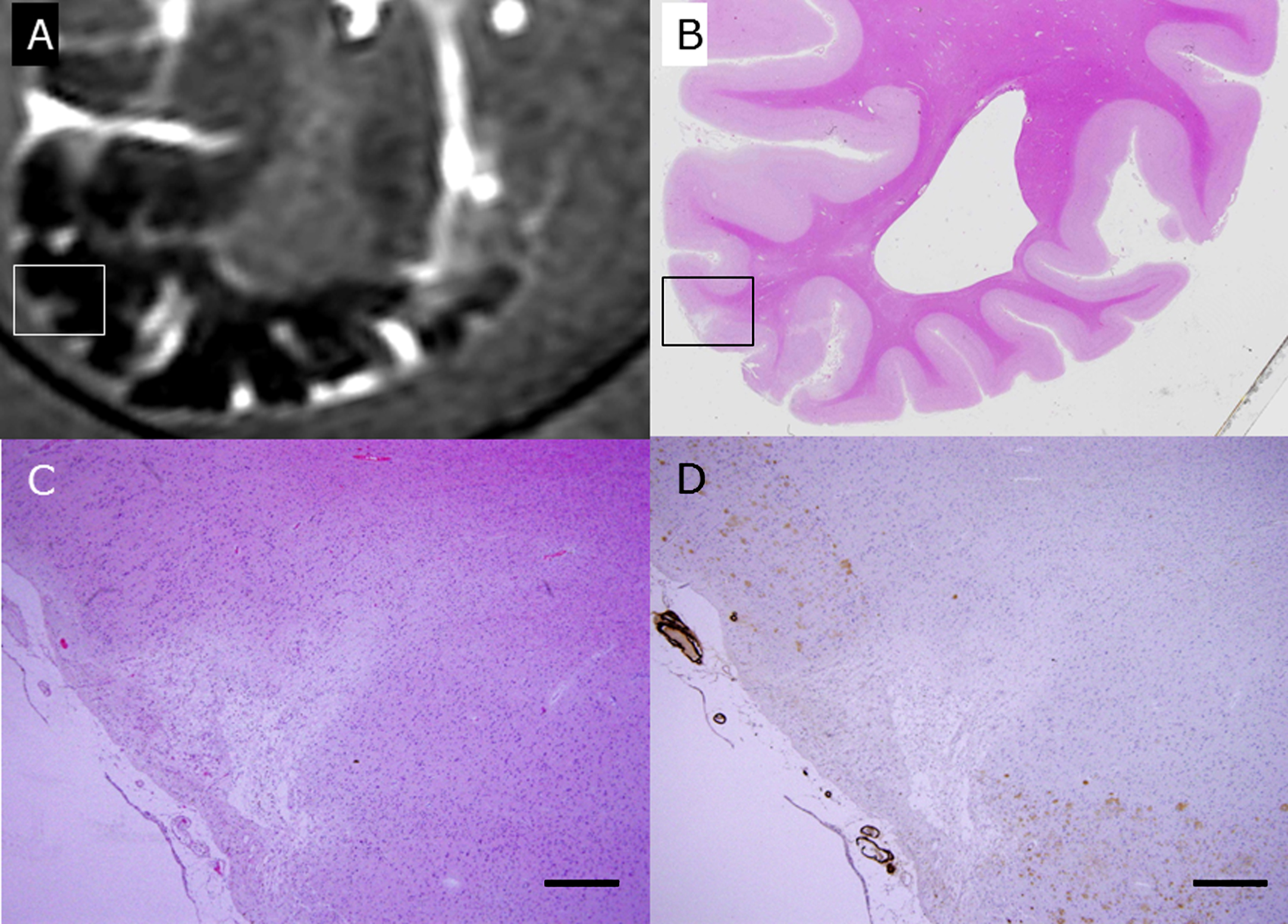
Figure 4 shows the coincidence of radiological and histological finding of a CMI, which was apposed to degenerated Aβ-positive arterioles (Fig. 4). With immunohistochemistry for Aβ42, senile plaques disappeared in the CMIs and their surrounding areas. Degenerated Aβ-positive arterioles adjacent to CMIs often showed double-barrel changes and exhibited both Aβ40 and Aβ42 positivity, with more prominent positivity for Aβ40. In 10 CAA cases, 33 of 48 CMIs (68.8%) were opposed to Aβ-positive arterioles, and 15 of 48 CMIs (31.3%) were apposed to those with double-barrel changes. On the contrary, in 6 non CAA control cases, there was only one CMI which show no proximity to amyloid angiopathy (0%).
Fig.4
The rectangle in A is a wedge-shaped CMI detected by postmortem 3T MRI FLAIR imaging in the brain of subject C1 (A). This block was subsequently fixed, sliced, and stained with HE (B, C). The rectangle in B is enlarged in C and D. With immunohistochemistry for Aβ (D), senile plaques (brown) are lost in the surrounding CMI, and degenerated Aβ-positive vessels are apposed to the CMI. Scale bars in C and D: 500 μm.
Cholesterol crystal embolus, which indicates artery to artery embolus, was not observed in our all materials. Using 3T postmortem MRI, a small subcortical infarction was detected below cerebral cortices harboring CMIs. This small subcortical infarction was crescent-shaped, was located just beneath the cortico-medullary junction, and seemed to be distributed in the terminal zones of the subcortical branch of the meningeal artery (Fig. 5). We also observed an enlarged perivascular space, which appeared as high-intensity spots, similar to CMIs. However, the enlarged perivascular space showed a different distribution from CMIs (arrow in Fig. 5D) in terms of frequent localization just beneath the cortico-medullary junction (arrowhead in Fig. 5D).
Fig.5
The circle and rectangle in A and B indicate a small subcortical infarction and a CMI, respectively. The circle is enlarged in C, and the rectangle is in D. The arrow indicates a CMI, and the arrowhead shows enlarged perivascular space (D) in the brain of subject C2 as detected by postmortem 3T MRI (A) and by HE staining of a section from the same block (B-D). Scale bars in C and D: 2 mm.
DISCUSSION
This study revealed radiological and histological coincidence of CMIs with 3T postmortem MRI, which strongly supports the idea that small cortical high-intensity lesions detected by our in vivo MRI protocol represent CMIs. Furthermore, closeapposition of Aβ-laden degenerated arterioles to CMIs may indicate that CAA is responsible for the majority of CMIs. Such close contact between CAA and CMIs has been reported in our previous study [7]. However, these results do not exclude a possible involvement of hypertensive vasculopathy, because the present data did not include patients with cerebrovascular diseases. In recent years, CMIs have been suggested to be caused by heterogeneous types of pathogenesis including hypertensive small vessel disease [25, 26]. Of note, CMIs should be differentiated from small cortical infarctions caused bymicroembolism from ruptured plaques or cardiac sources [27], although further studies will be required to precisely discern these two entities.
The novelty of this paper is the demonstration of the radiological and histological coincidence of CMIs using 3T MRI and histological backgrounds of such CMIs. The present protocol is exactly same as that reported by Ii et al. and has been optimized for sensitivity with widely used 3T MR units [13]. With 3D FLAIR imaging, judging whether lesions are juxta-cortical, mixed white and gray matter, or intracortical is sometimes difficult because the exact anatomic border between the cerebral cortex and white matter appears ambiguous. DIR imaging was commercially installed in the 3T Philips MR machine recently and shows superior delineation of gray matter. However, pulsation artifacts of blood vessels or cerebrospinal fluid may be more pronounced on DIR than on FLAIR [28]. With in vivo MRI, DIR imaging has been used to detect cortical lesions of various pathologies such as multiple sclerosis and small cortical infarctions [13, 27, 29, 30], because DIR provides intracortical localization and sensitive detection. In the current postmortem MRI experiment, however, 3D-FLAIR appeared to be superior to DIR for detection of CMIs, probably because artefacts frequently occur in the interface with surrounding water or air bubbles. It is also novel finding in this study.
7T MRI is a powerful tool in neuroscience for investigation of brain structures and the function of healthy subjects. Using their 3T MRI protocol, van Veluw et al. reported lower detection of three CMIs that were visible with 2D-FLAIR and T1-weighted imaging compared to detection of 15 CMIs with 7T MRI [15]. However, the availability of 7T MRI for clinical use is limited because of the high expense and possible unwanted effects on the disease condition. Moreover, application of FLAIR images is limited in 7T MRI units because of low signal-to-noise ratios, especially in the temporal lobes [31]. Therefore, our original protocol with 3D-FLAIR and 3D-DIR in 3T MRI may be a powerful tool in clinical research that can be used with a large number of patients. Indeed, studies investigating the clinical significance of CMIs using 3T MRI have already been launched with a large number of patients [8].
CMIs larger than 2 mm are detectable on 3T MRI, whereas those with sizes over 1 mm can be detected with 7T MRI [15]. Previous neuropathological studies defined microinfarcts as lesions that are ‘not visible with the naked eye’ or ‘only visible upon light microscopy’, and included the smallest range of 50 to 100 μm in diameter [32]. Westover et al. [33] indicated that a count of one or two microinfarcts in routine postmortem examination implies a maximum likelihood of 552 or 1,104 microinfarcts, respectively, throughout the brain. Therefore, detection of CMIs on 3T or 7T MRI represents only a small portion of the total CMI load. In view of these findings, the difference between 3T and 7T MRI is likely the visibility and detection ratio of CMIs. Therefore, 3T MRI combined with 3D imaging is appropriate for clinical studies, and 7T MRI will be most useful for investigation of the pathophysiology of CMIs.
Our study has some limitations. One is the relatively small number of subjects. This drawback is due to the limited number of patients with severe CAA with CMIs. Clearly, a study that involves a larger number of patients is warranted. The second is some ambiguity to rule out the microembolic mechanism in their pathogenesis. However, all subjects had no stroke episode, nor larger embolic lesions in the vicinity of CMIs. Moreover, all CMIs were adjacent to Aβ positive subpial arterioles, thereby indicating preferential involvement of CAA as their mechanism.
In current histopathologic observation of CMBs, seven of eight sites of CMBs had deposits of hemosiderin-laden macrophages. Fisher [34] proposed classification and pathogenesis of CMBs, and suggested the possibility that angiophagy of emboli by endothelial cells may be a mechanism of embolus extravasation and recanalization of microvessels [35]. We were unable to investigate this mechanism of CMBs in our current autopsy samples. Further studies are required to definitely determine the etiology of CMBs using postmortem 3T MRI.
Conclusions
Direct comparison between 3T MRI findings and histopathology revealed coincident findings for CMIs and CMBs. The current protocol involving a postmortem MRI scan become a useful method for future studies to investigate the pathogenesis of CMIs and CMBs. Moreover, similar small cortical high-intensity lesions detected with in vivo MRI represent CMIs in the histopathology of patients with dementia.
ACKNOWLEDGMENTS
The content of this manuscript has not been published or submitted for publication elsewhere. We gratefully acknowledge Kaori Kawamura fortechnical help.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1242r2).
REFERENCES
[1] | Fazekas F , Kleinert R , Roob G , Kleinert G , Kapeller P , Schmidt R , Hartung HP ((1999) ) Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: Evidence of microangiopathy-related microbleeds. Am J Neuroradiol 20: , 637–642. |
[2] | Tanaka A , Ueno Y , Nakayama Y , Takano K , Takebayashi S ((1999) ) Small chronic hemorrhage and ischemic lesions in association with spontaneous intracerebral hematomas. Stroke 30: , 1637–1642. |
[3] | Schrag M , McAuley G , Pomakian J , Jiffry A , Tung S , Mueller C , Vinters HV , Haacke EM , Holshouser B , Kido D , Kirsch WM ((2010) ) Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: A postmortem MRI study. Acta Neuropathol 119: , 291–302. |
[4] | De Reuck J , Auger F , Cordonnier C , Deramecourt V , Durieux N , Pasquier F , Bordet R , Maurage CA , Leys D ((2011) ) Comparison of 7.0-T T2*-magnetic resonance imaging of cerebral bleeds in post-mortem brain sections of Alzheimer patients with their neuropathological correlates. Cerebrovasc Dis 31: , 511–517. |
[5] | Kövari E , Gold G , Herrmann FR , Canuto A , Hof PR , Bouras C , Giannakopoulos P ((2007) ) Cortical microinfarcts and demyelination affect cognition in cases at high risk for dementia. Neurology 68: , 927–931. |
[6] | Arvanitakis Z , Leurgans SE , Barnes LL , Bennett DA , Schneider JA ((2011) ) Microinfarct pathology, dementia, and cognitive systems. Stroke 42: , 722–727. |
[7] | Okamoto Y , Yamamoto T , Kalaria RN , Senzaki H , Maki T , Hase Y , Kitamura A , Washida K , Yamada M , Ito H , Tomimoto H , Takahashi R , Ihara M ((2012) ) Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123: , 381–394. |
[8] | Ueda Y , Satoh M , Tabei K , Kida H , Ii Y , Asahi M , Maeda M , Sakuma H , Tomimoto H ((2016) ) Neuropsychological features of microbleeds and cortical microinfarct detected by high resolution magnetic resonance imaging. J Alzheimers Dis 53: , 315–325. |
[9] | Wang Z , van Veluw SJ , Wong A , Liu W , Shi L , Yang J , Xiong Y , Lau A , Biessels GJ , Mok VCT ((2016) ) Risk factors and cognitive relevance of cortical cerebral microinfarcts in patients with ischemic stroke or transient ischemic attack. Stroke 47: , 2450–2455. |
[10] | Hiral S , Sikking E , Shaik MA , Chan QL , van Veluw SJ , Vrooman H , Cheng CY , Sabanayagam C , Cheung CY , Wong TY , Venketasubramanian N , Biessels GJ , Chen C , Ikram MK ((2016) ) Cortical cerebral microinfarcts on 3T MRI: A novel marker of cerebrovascular disease. Neurology 87: , 1583–1590. |
[11] | Gouw AA , Seewann A , van der Flier WM , Barkhof F , Rozemuller AM , Scheltens P , Geurts JJG ((2011) ) Heterogeneity of small vessel disease: A systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 82: , 126–135. |
[12] | Smith EE , Schneider JA , Wardlaw JM , Greenberg SM ((2012) ) Cerebral microinfarcts: The invisible lesions. Lancet Neurol 11: , 272–282. |
[13] | Ii Y , Maeda M , Kida H , Matsuo K , Shindo A , Taniguchi A , Tomimoto H ((2013) ) In vivo detection of cortical microinfarcts on ultrahigh-field MRI. J Neuroimaging 23: , 28–32. |
[14] | Niwa A , Matsuo K , Shindo A , Yata K , Shiraishi T , Tomimoto H ((2013) ) Clinical and neuropathological findings in a patient with familial Alzheimer disease showing a mutation in the PSEN1 gene. Neuropathology 33: , 199–203. |
[15] | van Veluw SJ , Zwanenburg JJ , Engelen-Lee JY , Spliet WGM , Hendrikse J , Luijten PR , Biessels GJ ((2013) ) In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J Cereb Blood Flow Metab 33: , 322–329. |
[16] | De Reuck J , Deramecourt V , Auger F , Durieux N , Cordonnier C , Devos D , Defebvre L , Moreau C , Caparros-Lefebvre D , Bordet R , Maurage CA , Pasquier F , Leys D ((2014) ) Post-mortem 7.0-tesla magnetic resonance study of cortical microinfarcts in neurodegenerative diseases and vascular dementia with neuropathological correlates. J Neurol Sci 346: , 85–89. |
[17] | van Veluw SJ , Zwanenburg JJM , Rozemuller AJM , Luijten PR , Spliet WGM , Biessels GJ ((2015) ) The spectrum of MR detectable cortical microinfarcts: A classification study with 7-tesla postmortem MRI and histopathology. J Cereb Blood Flow Metab 35: , 676–683. |
[18] | De Reuck JL , Auger F , Durieux N , Cordonnier C , Deramecourt V , Pasquier F , Maurage CA , Leys D , Bordet R ((2016) ) The topography of cortical microinfarcts in neurodegenerative diseases and in vascular dementia: A postmortem 7.0-Tesla magnetic resonance imaging study. Eur Neurol 76: , 57–61. |
[19] | Thelwall PE , Shepherd TM , Stanisz GJ , Blackband SJ ((2006) ) Effects of temperature and aldehyde fixation on tissue water diffusion properties, studied in an erythrocyte ghost tissue model. Magn Reson Med 56: , 282–289. |
[20] | Shepherd TM , Thelwall PE , Stanisz GJ , Blackband SJ ((2009) ) Aldehyde fixative solutions alter the water relaxation and diffusion properties of nervous tissue. Magn Reson Med 62: , 26–34. |
[21] | Braak H , Braak E ((1991) ) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[22] | Braak H , Del Tredici K , Rüb U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[23] | Ellis RJ , Olichney JM , Thal LJ , Mirra SS , Morris JC , Beekly D , Heyman A ((1996) ) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, part XV. Neurology 46: , 1592–1596. |
[24] | Okamoto Y , Ihara M , Fujita Y , Ito H , Takahashi R , Tomimoto H ((2009) ) Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport 20: , 990–996. |
[25] | Kövari E , Herrmann FR , Hof PR , Bouras C ((2013) ) The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol 39: , 498–509. |
[26] | Zheng L , Vinters HV , Mack WJ , Zarow C , Ellis WG , Chui HC ((2013) ) Cerebral atherosclerosis is associated with cystic infarcts and microinfarcts but not Alzheimer pathologic changes. Stroke 44: , 2835–2841. |
[27] | Landi D , Maggio P , Lupoi D , Palazzo P , Altamura C , Falato E , Altavilla R , Vollaro S , Coniglio AD , Tibuzzi F , Passarelli F , Silvestrini M , Pasqualetti P , vernieri F ((2015) ) Cortical ischemic lesion burden measured by DIR is related to carotid artery disease severity. Cerebrovasc Dis 39: , 23–30. |
[28] | Turetschek K , Wunderbaldinger P , Bankier AA , Zontsich T , Graf O , Mallek R , Hittmair K ((1998) ) Double inversion recovery imaging of the brain: Initial exerience and comparison with fluid attenuated inversion recovery imaging. Magn Reson Imaging 16: , 127–135. |
[29] | Geurts JJG , Pouwels PJW , Uitdehaag BMJ , Polman CH , Barkhof F , Castelijns JA ((2005) ) Intracortical lesions in multiple sclerosis: Improved detection with 3D double inversion-recovery MR imaging. Radiology 236: , 254–260. |
[30] | Wattjes MP , Lutterbey GG , Gieseke J , Träber F , Kloz L , Schmidt S , Schild HH ((2007) ) Double invasion recovery brain imaging at 3T: Diagnostic value in the detection of multiple sclerosis lesions. Am J Neuroradiol 28: , 54–59. |
[31] | Visser F , Zwanenburg JJM , Hoogduin JM , Luijten PR ((2010) ) High-resolution magnetization-prepared 3D-FLAIR imaging at 7.0 tesla. Magn Reson Med 64: , 194–202. |
[32] | Brundel M , de Bresser J , van Dillen JJ , Kappelle LJ , Biessels GJ ((2012) ) Cerebral microinfarcts: A systematic review of neuropathological studies. J Cereb Blood Flow Metab 32: , 425–436. |
[33] | Westover MB , Bianchi MT , Yang C , Schneider JA, Greenberg SM ((2013) ) Estimating cerebral microinfarct burden from autopsy samples. Neurology 80: , 1365–1369. |
[34] | Fisher M ((2014) ) Cerebral microbleeds: Where are we now? Neurology 83: , 1304–1305. |
[35] | Grutzendler J , Murikinati S , Hiner B , Ji L , Lam CK , Yoo T , Gupta S , Hafler BP , Adelman RA , Yuan P , Rodriguez G ((2014) ) Angiophagy prevents early embolus washout but recanalizes microvessels through embolus extravasation. Sci Transl Med 226: , 226ra31. |

