
Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder without a cure. Most AD cases are sporadic where age represents the greatest risk factor. Lack of understanding of the disease mechanism hinders the development of efficacious therapeutic approaches. The loss of synapses in the affected brain regions correlates best with cognitive impairment in AD patients and has been considered as the early mechanism that precedes neuronal loss. Oxidative stress has been recognized as a contributing factor in aging and in the progression of multiple neurodegenerative diseases including AD. Increased production of reactive oxygen species (ROS) associated with age- and disease-dependent loss of mitochondrial function, altered metal homeostasis, and reduced antioxidant defense directly affect synaptic activity and neurotransmission in neurons leading to cognitive dysfunction. In addition, molecular targets affected by ROS include nuclear and mitochondrial DNA, lipids, proteins, calcium homeostasis, mitochondrial dynamics and function, cellular architecture, receptor trafficking and endocytosis, and energy homeostasis. Abnormal cellular metabolism in turn could affect the production and accumulation of amyloid-β (Aβ) and hyperphosphorylated Tau protein, which independently could exacerbate mitochondrial dysfunction and ROS production, thereby contributing to a vicious cycle. While mounting evidence implicates ROS in the AD etiology, clinical trials with antioxidant therapies have not produced consistent results. In this review, we will discuss the role of oxidative stress in synaptic dysfunction in AD, innovative therapeutic strategies evolved based on a better understanding of the complexity of molecular mechanisms of AD, and the dual role ROS play in health and disease.
MOLECULAR HALLMARKS OF ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) affects more than 5 million Americans, with numbers expected to grow as the population ages [1, 2]. Most AD cases are sporadic where the origin of the disease is not known but might be influenced by multiple factors including environmental exposure, genetic risk factors, mitochondrial haplotypes, age, and sex [2–4]. About 1% of cases are associated with familial mutations in the genes that encode either a transmembrane amyloid-β protein precursor (AβPP), or proteins presenilin 1 (PS1) and presenilin 2 (PS2), which are directly involved in the AβPP processing. While cleavage of AβPP at the plasma membrane by the α-secretase occurs without formation of pathologic amyloid-β (Aβ) peptides, cleavage with β- and γ-secretases leads to the release in the extracellular space of Aβ peptides with 40 or 42 residues where Aβ42 is more prone to aggregation and is the major component of extracellular amyloid plaques [5, 6]. Along with the formation of extracellular aggregates, Aβ peptides are present in neurons [5, 7]. Multiple studies conducted in vitro and in vivo using human tissue and transgenic mice demonstrated that intracellular Aβ accumulates prior to the development of extracellular plaques where it specifically affects synaptic function leading to a profound memory deficit [8–10]. The existence of intraneuronal Aβ could be explained by multiple mechanisms. Besides the plasma membrane, AβPP is present at several intracellular sites including the trans-Golgi network [11], endoplasmic reticulum (ER), and endosomal, lysosomal [12], and mitochondrial membranes [13] where Aβ could be generated via β- and γ-secretase cleavage. In addition, secreted Aβ peptides could be internalized via receptor-mediated or/and receptor-independent endocytosis [14–16]. Extensive studies also support the notion that soluble Aβ oligomers represent the most toxic species that affect multiple early molecular mechanisms leading to synaptic dysfunction in AD [15].
Intracellular neurofibrillary tangles (NFT) represent another hallmark of AD. Tau is a microtubule stabilizing protein. When it becomes hyperphosphorylated, it dislocates from the microtubules leading to their destabilization and a disruption of neuronal trafficking machinery [17]. Aβ-induced translocation of Tau to neuronal spines is associated with synaptic dysfunction early in AD pathogenesis [18]. The definitive diagnosis of AD can only be done by examining the postmortem brain tissue based on the presence of extracellular plaques formed by Aβ peptides, intracellular NFTs comprised of hyperphosphorylated Tau protein (pTau), Aβ deposits in blood vessels, neuronal and synaptic loss, and significant atrophy in selective brain regions involved in cognitive function (hippocampus, entorhinal and frontal cortices) [19]. The identification of familial AD mutations in APP, PS1, and PS2 genes gave rise to the amyloid cascade hypothesis that considered the formation of Aβ a culprit of the disease. While excessive production of Aβ peptides is observed early in patients that develop AD and is essential for AD pathology [20], it is not sufficient. Some aged individuals have significant Aβ load, but do not develop cognitive impairment [21, 22]. Recent studies conducted using positron-emission tomography (PET) and novel tracers that allow imaging of both amyloid and Tau distribution in the brain of living individuals suggest that there is a relationship between Tau protein deposition, Aβ plaques, and neurodegeneration [23]. Based on the pattern distribution and the manifestation of cognitive symptoms, it appears that the widespread presence of Aβ in the brain does not lead to the development of AD without Tau being present in the affected areas. These observations support the idea that the synergistic interaction between Aβ and Tau is essential to trigger neurodegeneration in AD [24, 25]. While this provides important insights into AD patient’s diagnostic and prognostic criteria, early molecular mechanisms leading to the accumulation of Aβ and pTau or driving factors that promote their spreading in the brain remain poorly understood hindering the development of efficacious therapeutic interventions [26–29].
THE ROLE OF OXIDATIVE STRESS IN ALZHEIMER’S DISEASE
In search for the underlying mechanisms of AD, the amyloid cascade hypothesis that dominated the field of AD research for the past decades has been challenged [30–32]. An alternative explanation of the disease mechanism has emerged from the observations linking mitochondrial dysfunction and increased production of reactive oxygen species (ROS) to the development of AD. The mitochondrial cascade hypothesis states that in sporadic, late-onset AD, loss of mitochondrial function associated with age affects the expression and processing of AβPP initiating Aβ accumulation [33]. Mitochondrial dysfunction has been well documented in AD [34, 35]. Abnormal mitochondrial axonal trafficking is already observed in embryonic neurons from multiple transgenic mouse models of familial AD with additional abnormalities in fission, fusion, and function detected prior to the development of amyloid plaques or memory impairment [36–50]. Brain glucose metabolism measured using fluorodeoxyglucose-positron emission tomography (FDG-PET) is reduced prior to the onset of disease in several groups of at-risk individuals including patients with mild cognitive impairment (MCI), a prodromal stage of AD, and in carriers of the apolipoprotein E epsilon-4 (ApoE4) allele, a strong genetic risk factor for late-onset AD. However, this hypometabolism does not correlate with an increase in brain Aβ deposition [51–53]. Furthermore, disruption in glucose metabolism associated with early mitochondrial dysfunction detected in multiple animal models and AD patients [38, 41, 43, 48, 54–60] may also be a direct determinant of oxidative stress and synaptic dysfunction that contribute to early disease mechanisms before any evidence of Aβ or Tau pathology [48, 61–63]. In the brain, the free energy necessary to drive most cellular reactions is primarily produced in mitochondria from the oxidation of glucose under aerobic conditions (Fig. 1). Oxidative stress, which is defined as ‘an imbalance in pro-oxidants and antioxidants with associated disruption of redox circuitry and macromolecular damage’ [64], is associated with increased production of ROS and reactive nitrogen species (RNS) including superoxide radical anion (O2–), hydrogen peroxide (H2O2), hydroxyl radical (HO–), nitric oxide (NO), and peroxynitrite (ONOO–). While there are multiple sources of ROS production in the cell including ER, peroxisomes, a family of NADH oxidases, and other enzymes such as monoamine oxidases [65, 66], mitochondria are the largest contributor to ROS production (Fig. 1) [67, 68]. During oxidative phosphorylation, H2O2 and O2– are produced as byproducts in mitochondria primarily by complexes I and III [69]. Under normal conditions, the antioxidant enzymes acting as free radical scavengers mediate levels of ROS. These include superoxide dismutases (SOD), glutathione peroxidase (GPX), glutaredoxins, thioredoxins, and catalase (Fig. 1). Additional mechanism of protection against oxidative stress involves the activation of nuclear factor erythroid-2-related factor 2 (Nrf2). Nrf2 is a transcription factor negatively regulated by its binding to the cytoplasmic repressor and stress sensor Kelch-like ECH associated protein 1 (KEAP1), which acts as a substrate adaptor to mediate ubiquitination and degradation of Nrf2 by the E3 ubiquitin ligase Cullin-3 [70]. In the presence of electrophiles and oxidants, KEAP1 releases Nrf2 with its subsequent translocation to the nucleus where it activates transcription of cytoprotective genes via promoter sequences containing conserved antioxidant response elements (AREs) [71, 72]. This increases levels of antioxidant enzymes and proteins such as glutathione-S-transferase, NAD(P)H: quinone oxidoreductase-1, SOD, GPX, heme oxygenase-1 (HO-1), glutamate cysteine ligase, thioredoxin, and catalase, and also promotes mitochondrial biogenesis ensuring a replacement of damaged organelles [73, 74]. However, there is conflicting evidence on whether Nrf2 is activated in AD. In one study, levels of Nrf2 expression were found to be decreased in AD patients despite the presence of oxidative stress [75]. Other studies reported an increase in the expression of the ARE-related genes in patients with MCI and AD [76, 77]. While the exact mechanism is presently unknown, these discrepancies could be associated with the variations in the levels of Nrf2 expression that could be influenced by aging and the diseasemechanisms [78].
Fig.1
ROS production in mitochondria during oxidative phosphorylation and antioxidant mechanisms. Complex I and complex III of the mitochondrial electron transport chain are the major sites of superoxide anion (O2–) production during aerobic respiration. O2– is converted to H2O2 by MnSOD or CuZnSOD in the intermembrane mitochondrial space. H2O2 is further reduced to water by detoxifying enzymes glutathione peroxidase (GPX) or catalase. GPX uses reduced glutathione (GSH) as the reductant, and the resulting oxidized glutathione reacts with another glutathione molecule to form glutathione disulfide (GSSG), which is restored to GSH by the enzyme glutathione reductase (GR). These reactions occur in mitochondrial matrix.
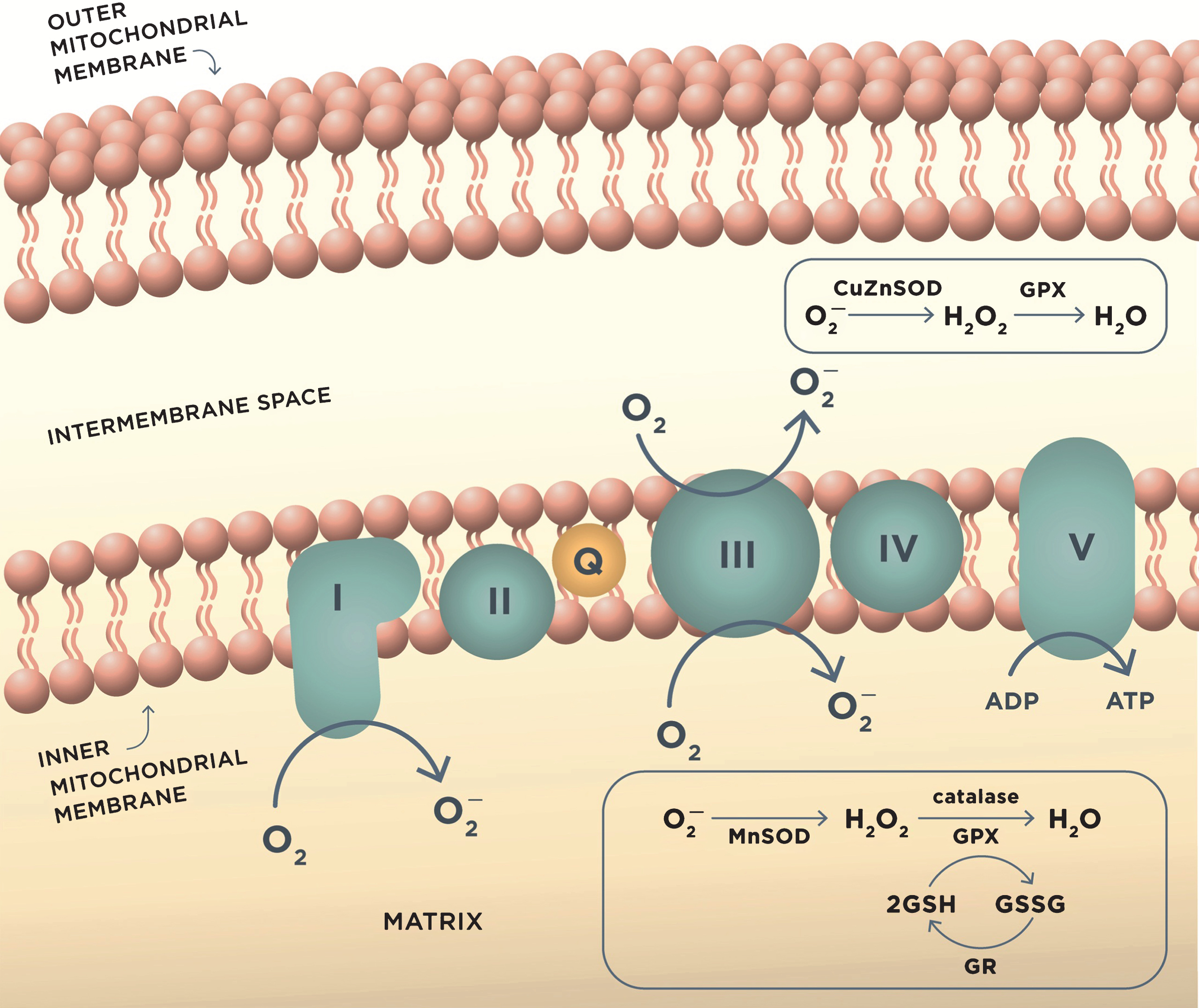
The balance between ROS production and the antioxidant defense is essential for normal cellular function. However, in AD, the activity of antioxidant enzymes is altered, thereby contributing to the unconstrained accumulation of oxidative damage [79]. When unbalanced, overproduction of ROS combined with the insufficient antioxidant defense leads to oxidative stress [80]. There is evidence that mitochondrial damage resulting in increased production of ROS contributes to the early stages of AD prior to the onset of clinical symptoms and the appearance of the Aβ pathology [80]. In support, markers of oxidative stress including high levels of oxidized proteins, glycosylated products, extensive lipid peroxidation, formation of alcohols, aldehydes, free carbonyls, ketones, cholestenone, and oxidative modifications in RNA and nuclear and mitochondrial DNA were found in postmortem brain tissue and in peripheral systems including cells and isolated mitochondria from people with preclinical or early stages of AD and ApoE4 carriers (Fig. 2) [58, 81–97]. Mitochondrial ROS could collapse mitochondrial membrane potential accelerating ROS production within the same organelle (Fig. 3). As a result, an increase in ROS production in a small subset of organelles could be sufficient to propagate ROS damage to other mitochondria eventually affecting the whole cell [66].
Fig.2
Molecular targets of ROS. While multiple sites in the cell can contribute to ROS production, uncontrolled ROS generation in mitochondria could impair a major source of energy in the cell resulting in detrimental consequences to the whole cellular environment. Intermediate levels of ROS can gradually affect multiple cellular functions including loss of synaptic activity, while critically damaged mitochondria can trigger a release of cytochrome c activating apoptosis.
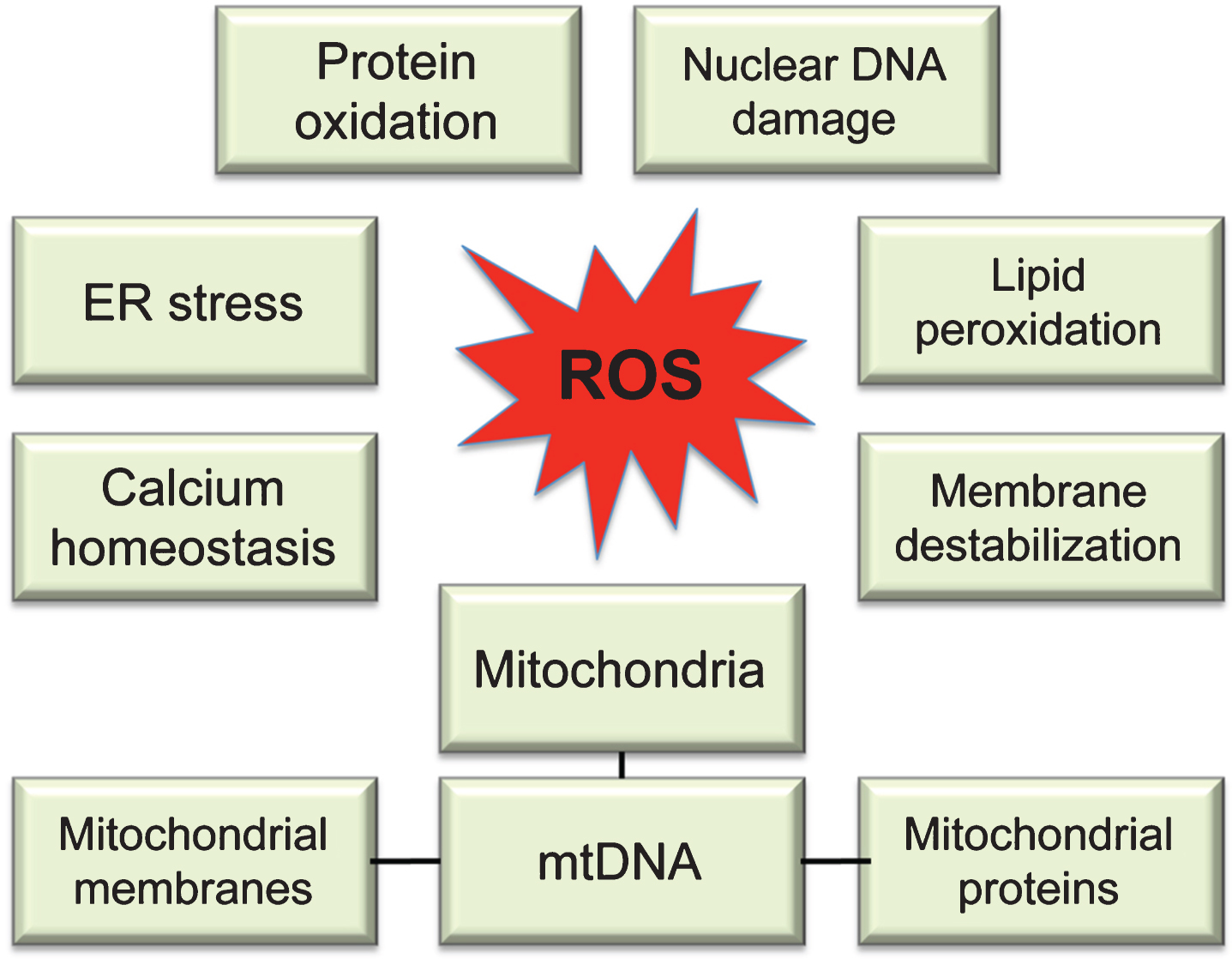
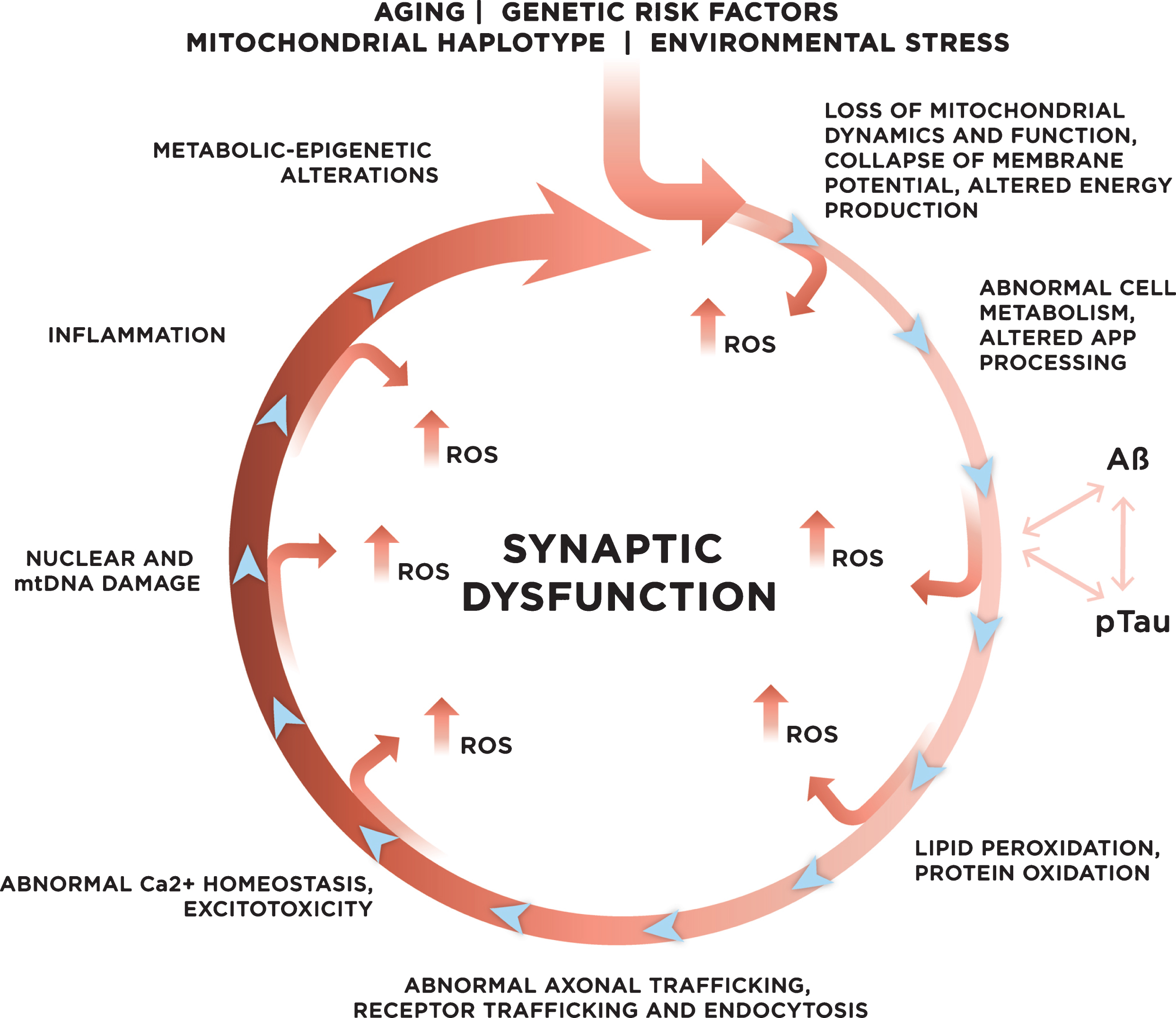
Fig.3
Genetic and environmental risk factors contribute to the development of late onset sporadic AD. With age, increased mitochondrial dysfunction and ROS production could initiate a vicious cycle where multiple systems and mechanisms affected by ROS exacerbate ROS production, accelerating cellular damage, and leading to synaptic dysfunction.
Compelling data demonstrate that in addition to mitochondrial ROS production, abnormal homeostasis of bioactive metals including iron (Fe), copper (Cu), zinc (Zn), magnesium (Mg), manganese (Mn), and aluminum (Al) could be involved in free radical production and oxidative stress influencing Aβ and Tau aggregation [35, 98–100]. Increased levels of Fe, Cu, and Zn were detected using proton induced X-ray emission, immunohistochemistry, and synchrotron X-ray fluorescence in close proximity to the amyloid plaques in the brain tissue of AD patients and transgenic mouse models of AD [101–108]. The accumulation of these metals in the first place is thought to originate from the impaired neuronal metal homeostasis affected by aging, and exacerbated by amyloid and Tau pathologies in case of AD [109–111] There is a tight connection between protein misfolding, aggregation, and metal ion homeostasis. In particular, Zn directly affects AβPP processing by binding to the protein [112], and Al, Zn, Fe, and Cu directly bind Aβ promoting its aggregation [113–115]. Similarly, the redox metals could promote Tau phosphorylation, its release from the microtubules, and formation of NFTs [116, 117]. ROS production is facilitated by the redox-active metals including Cu, Fe, and Mn using catalytic reactions similar to the Fenton reaction where metals convert O2– and H2O2 to HO– species that are involved in lipid peroxidation [118]. Moreover, a direct binding of Aβ peptides to Cu or Fe has been shown to generate H2O2 [119]. Thus, the transition metals and Aβ could synergistically contribute to an increase in oxidative stress and extra-mitochondrial production of ROS. In agreement with a role of metal ions in pathology, restoration of metal dyshomeostasis after application of metal chelators [120–122] reduced levels of amyloid plaques and Aβ aggregation, and improved cognitive performance in humans and mouse models of AD [123].
Another source of ROS production directly mediated by Aβ involves microglia activated in the brain during an inflammatory response to the deposition of extracellular amyloid plaques [124]. Further, increased levels of Aβ could accelerate a production of ROS by directly binding to mitochondrial membranes, altering mitochondrial dynamics and function, ultimately leading to the abnormal energy metabolism and the loss of synaptic function [35, 37, 39, 46, 62, 125, 126]. Membrane-associated oxidative stress induced by Aβ peptides perturbs ceramide and cholesterol metabolism that, in turn, triggers a neurodegenerative cascade leading to additional Aβ accumulation, Tau phosphorylation, and clinical disease (Fig. 3) [127–135]. Furthermore, there is a direct link between altered membrane lipids and mitochondrial function, which is detrimental for brain bioenergetics [136, 137]. Strong data generated in animal models and humans suggest an intimate relationship between oxidative stress, Aβ accumulation, and abnormal Tau phosphorylation, where pTau specifically affects the activity of complex I synergistically contributing to the Aβ-mediated mitochondrial dysfunction and ROS production [138]. This could explain why accumulation of both Aβ and pTau may be required to initiate neurodegeneration in AD patients [23–25]. Moreover, emerging data suggest that mitochondria-mediated cellular bioenergetics could independently affect AβPP processing and Aβ production (Fig. 2) [139–145]. However, the details of causal relationship between oxidative stress, mitochondrial dysfunction, and Aβ and pTau accumulation in AD remain to be elucidated. Taken together, these data suggest that altered mitochondrial function, increased oxidative stress, exhausted antioxidant defense, production of Aβ and pTau, which furthermore affects mitochondrial function and ROS production, could represent a “vicious cycle” that with time exacerbates the disease process, eventually leading to neuronal death [47] (Fig. 3).
OXIDATIVE STRESS AND SYNAPTIC DYSFUNCTION IN ALZHEIMER’S DISEASE
Synapses are structurally specialized regions in neurons that propagate an electrical or chemical signal from one cell to another. During neurotransmission, signaling molecules such as glutamate, acetylcholine, dopamine, and others released from the active zones of a presynaptic neuron bind to and activate receptors on a postsynaptic neuron (Fig. 4) [146]. The strength of synaptic transmission depends on changes in neuronal activity where the dynamic nature of synaptic plasticity including long-term potentiation (LTP) and long-term depression (LTD) represents the fundamental mechanism of learning and memory [147, 148]. Neurons have a unique cellular architecture where formation or pruning and maintenance of dendritic spines are essential for neurotransmission and synaptic function (Fig. 4). Synaptic transmission critically relies on the fidelity of multiple cellular mechanisms including biosynthesis of neurotransmitters from amino acids to ensure their availability; the delivery of neurotransmitters to the sites of synapses requiring intact microtubule tracts and vesicle trafficking machinery; formation of synaptic vesicles that encapsulate neurotransmitters preparing for their release via exocytosis; binding of the neurotransmitter to the receptor on the postsynaptic neuron with subsequent activation of cellular response; and the removal of the neurotransmitter from the synaptic cleft after the release (Fig. 4) [149]. In addition, Ca2+ plays an essential role in mediating basal synaptic transmission, where an increase of its conductance through voltage gated Ca2+-channels clustered in the presynaptic membrane at the active zone triggers the release of synaptic vesicles [146]. Given the complexity of neurotransmission machinery, factors that affect any step of the process could have a detrimental effect on synaptic function in neurons and, ultimately, on cognitivefunction.
Fig.4
Structure of a synapse. Left: synapse between two neurons observed in the brain tissue of a wild type C57/Bl6 mouse using transmission electron microscopy (generated in Dr. Trushina laboratory [231]). An arrow indicates electron dense plasma membrane at the synapse. Presynaptic neurons contain a large number of synaptic vesicles (#). Both presynaptic and postsynaptic neurons have mitochondria at the site of synapse (*), which are delivered along the microtubule tracks (indicated with arrows). Scale bar, 0.5 μm. Right: a simplified cartoon of a synapse. Glutamate (blue spheres) released from the presynaptic neuron in a voltage dependent manner, activates the NMDA glutamate receptors present on pre- and postsynaptic neurons. These include AMPA (orange) and NMDA (green) receptors among others. Glutamate is cleared from the synaptic cleft primarily by the glial cells transporters (GLT-1). It is then recycled to neurons, repackaged into synaptic vesicles, and used in another synapse. An inadequate glutamate clearance could lead to the spillover and activation of extrasynaptic NMDA receptors. Memantine is believed to prevent this particular activation. Excessive entry of Ca2+ into presynaptic neuron (red spheres) could damage synaptic mitochondria leading to ROS production, altered synaptic transmission and neuronal dysfunction. This phenomenon is called excitotoxicity. Note that mitochondria are delivered to the site of synapse along the Tau-containing microtubule tracks. Destabilization of microtubules could affect mitochondrial localization and energy supply required for proper synaptic function.
![Structure of a synapse. Left: synapse between two neurons observed in the brain tissue of a wild type C57/Bl6 mouse using transmission electron microscopy (generated in Dr. Trushina laboratory [231]). An arrow indicates electron dense plasma membrane at the synapse. Presynaptic neurons contain a large number of synaptic vesicles (#). Both presynaptic and postsynaptic neurons have mitochondria at the site of synapse (*), which are delivered along the microtubule tracks (indicated with arrows). Scale bar, 0.5 μm. Right: a simplified cartoon of a synapse. Glutamate (blue spheres) released from the presynaptic neuron in a voltage dependent manner, activates the NMDA glutamate receptors present on pre- and postsynaptic neurons. These include AMPA (orange) and NMDA (green) receptors among others. Glutamate is cleared from the synaptic cleft primarily by the glial cells transporters (GLT-1). It is then recycled to neurons, repackaged into synaptic vesicles, and used in another synapse. An inadequate glutamate clearance could lead to the spillover and activation of extrasynaptic NMDA receptors. Memantine is believed to prevent this particular activation. Excessive entry of Ca2+ into presynaptic neuron (red spheres) could damage synaptic mitochondria leading to ROS production, altered synaptic transmission and neuronal dysfunction. This phenomenon is called excitotoxicity. Note that mitochondria are delivered to the site of synapse along the Tau-containing microtubule tracks. Destabilization of microtubules could affect mitochondrial localization and energy supply required for proper synaptic function.](https://content.iospress.com:443/media/jad/2017/57-4/jad-57-4-jad161088/jad-57-jad161088-g004.jpg)
AD is characterized by progressive memory impairment, which is associated with the inhibition of LTP and enhancement of LTD in the hippocampus [150]. Loss of synapses in the affected brain regions correlates best with cognitive impairment in AD patients and has been considered as the early mechanism that precedes neuronal loss [151–157]. Extensive studies conducted in vivo and in vitro support a direct relationship between oxidative stress and synaptic dysfunction in AD [39, 126, 158, 159]. In particular, it has been shown that independently and synergistically, ROS, Aβ, and pTau affect the activity of N-methyl-D-aspartate (NMDA) receptors. The NMDA receptors belong to the ionotropic family of glutamate receptors, which in coordination with α-amino-3-hydroxy-5-methyl-4-isoxa-zolepropionic acid (AMPA) receptors regulate the excitatory synaptic transmission and plasticity in the brain playing an essential role in learning and memory [160, 161]. Activation of NMDA receptors allows Ca2+ to enter the postsynaptic cells initiating a cascade of events that is critically involved in establishing LTP. The function of NMDA receptors declines with age, which could explain memory alterations associated with chronological aging. However, in AD, in addition to age-related changes, the expression of neurotoxic Aβ has been shown to reduce the amount of surface NMDA receptors in neurons and in brain tissue of AD mice [162], trigger NMDA-mediated Ca2+ influx inducing excitotoxicity and stress-related signaling pathways, exacerbating aging-related increase in oxidative stress, impaired energy metabolism, defective Ca2+ homeostasis, and altered regulation of transcription of genes important for neuronal development and plasticity [47, 163]. Memantine, the only FDA-approved drug for AD that is not an acetyl cholinesterase inhibitor, is a noncompetitive, low-affinity antagonist of NMDA receptors. Importantly, memantine has greater affinity to non-synaptic NMDA receptors, which are implicated in excitotoxicity associated with the glutamate spillover and have distinctly different composition of receptor subunits [164, 165]. In addition to the effect on NMDAR, soluble Aβ species have been shown to bind to AMPA receptors promoting their internalization via clathrin-mediated endocytosis after Ca2+-induced activation of calcineurin [166]. Altered internalization of AMPA receptors affects synaptic plasticity inducing synaptic dysfunction and loss of dendritic spines (Fig. 4).
Another type of synapses in the central nervous system utilizes γ-aminobutyric acid (GABA), which is a major neurotransmitter that induces inhibitory effect. In AD, levels of GABA are decreased with disease progression, and reduced levels of expression of GABAergic receptors has also been noted [167]. Degeneration of basal forebrain cholinergic cells that directly project to the cortex and hippocampus is well-documented in AD [168]. The cholinergic system is also implicated in cognitive functioning, especially in attention, memory, and emotion. Extensive data generated in human tissue and multiple animal models of AD demonstrated severe deficit in the activity of multiple acetylcholine synthesizing and degrading enzymes, acetylcholine transporters and receptors involved in synaptic signaling, along with reduction of presynaptic cholinergic markers. These investigations provided compelling evidence for the development of one of the few therapeutic approaches currently FDA-approved for AD, cholinesterase inhibitors. This approach allows increasing levels of acetylcholine at synapses by blocking the activity of acetylcholinesterase and butyrylcholinesterase enzymes, which are involved in acetylcholine hydrolysis [169].
Among numerous mechanisms that connect neurotoxic Aβ, Tau, oxidative stress, and synaptic dysfunction in AD are excitotoxicity, oxidation of proteins, and lipid peroxidation (Figs. 2, 3). Application of Systems Biology approaches including metabolomics and epigenetics to study early changes associated with AD progression in plasma, CSF, and brain tissue from individuals with different severity of AD and multiple animal models of AD confirmed that major alterations in metabolic networks identified early in disease are directly relevant to changes in neurotransmitter, lipid, and energy metabolism [48, 63, 170, 171]. Aβ-induced excitotoxicity associated with an excessive influx of calcium in postsynaptic neurons can lead to a cascade of events that increases ROS production, oxidative stress, Tau phosphorylation, and lipid peroxidation, ultimately leading to synaptic dysfunction (Fig. 3) [172–174]. Alteration of structure and fluidity of plasma membrane associated with lipid peroxidation could affect the organization and function of dendritic spines, signaling pathways, receptor trafficking, and localization [175]. Indeed, alterations in lipid trafficking and metabolism affect membrane fluidity and lipid homeostasis early in ApoE4 carriers [92, 137, 176, 177]. Moreover, lipid peroxidation of mitochondrial membranes could directly affect the dynamic and function of the organelle leading to reduced energy support at the sites of synapses, which is detrimental for brain bioenergetics [136, 137]. Altered mitochondrial fission, fusion, axonal motility, and function in turn could contribute to ROS production exacerbating synaptic function. As was mentioned earlier, Aβ-induced hyperphosphorylation of Tau destabilizes microtubule tracks, which alters axonal trafficking of mitochondria and their synaptic docking, and translocation of Tau to dendritic spines also may have a synergistic effect contributing to NMDA receptor destabilization, excitotoxicity, and increased oxidative stress with detrimental effect on synaptic function (Fig. 3). The role of protein oxidation in the mechanism of AD has been recently reviewed in [4].
THERAPEUTIC STRATEGIES FOR ALZHEIMER’S DISEASE
Currently approved treatments for AD are limited to three cholinesterase inhibitors, donepezil, rivastigmine, and galantamine, and a low affinity NMDA receptor antagonist, memantine. None of these approaches are disease modifying; they do not provide a “cure” but rather symptomatic treatment for some individuals [178]. Moreover, failure of the recent clinical trials focused on production or clearance of Aβ peptides emphasizes the urgency to consider alternative molecular mechanisms in order to design interventions that will delay or alleviate the development of AD [179]. While compelling evidence implicates oxidative stress in the early molecular mechanisms of AD [180], there is no FDA-approved antioxidant therapy for AD. Moreover, while antioxidant experimental therapeutics produced promising results in animal models of AD [181–183], clinical trials either failed or delivered inconclusive results [184]. For example, multiple trials assayed the effect of a strong antioxidant vitamin E (alpha tocopherol) on cognitive function in cognitively normal and generally healthy women 65 years or older [185], in cognitively normal women with preexisting cardiovascular disease or cardiovascular disease risk factors 40 years or older [186], in people with MCI [187], in patients with moderate to severe AD [188], and in individuals with mild to moderate AD [189]. Positive results where statistically significant changes in cognitive performance were achieved after vitamin E administration compared to placebo were found only in people with mild to moderate AD [189]. Importantly, there were no significant differences in the groups receiving memantine alone or memantine plus alpha tocopherol as a combination therapy. Moreover, meta-analysis of 19 randomized trials with vitamin E demonstrated its high toxicity and all-cause mortality at high doses [190]. Inconclusive results were also achieved in an open clinical trial where AD patients stably taking a cholinesterase inhibitor were supplemented with vitamin C and E over 1 year [191]. While oxidation of CSF lipids was significantly reduced after 1 year of the supplementation, the clinical course of AD did not differ between the vitamin-supplemented and the control group. Another failed trial involved the supplementation with vitamin E, C, and α-lipoic acid in patients with mild to moderate AD [192]. Despite a detection of reduced levels of markers of oxidative stress in CSF, a rapid cognitive decline observed in treated group raised significant safety concerns. Similar results were obtained in clinical trials with curcumin, a polyphenolic compound that has been demonstrated to have antioxidant and anti-inflammatory effects in preclinical studies [193]. Comprehensive update on the outcomes of the antioxidant treatments in recent clinical trials was provided in recent reviews [194, 195].
Multiple challenges associated with the design of clinical trials in elderly and the lack of a complete understanding of the molecular mechanism of antioxidant therapy may account for such diverse outcomes. First of all, there is no definitive test to diagnose AD in living individuals. The conclusive diagnosis of AD can only be done after the examination of postmortem brain tissue for the presence of amyloid plaques and NFTs. This introduces some ambiguity in the etiology behind cognitive impairment in the subjects recruited for clinical trials. Next, it is important at what stage of the disease the treatment is administered since some of the interactions may be efficacious only at the early stages. Furthermore, clinical trials in elderly are associated with the relatively small number of participants and short period of treatment, high frequency of death, inconsistent use of medication, and a lack of a follow-up data. However, there are clinical trials in progress that have greater number of participants and extended periods of treatment that may provide better results on the effect of the antioxidant therapy in AD [196, 197]. Along with the trials designed to test efficacy of a single compound found beneficial in preclinical trials, combination therapy for AD may hold a promise [198]. This approach includes treatment with multiple compounds with diverse properties that could improve several mechanisms and functions altered in AD without adverse side effects. In one of such trials, the administration of a nutraceutical formulation that included folate, alpha-tocopherol, B12, S-adenosyl methioinine, N-acetyl cysteine, and acetyl-L-carnitine to the AD patients over 1 year resulted in stabilization of cognitive function [199]. Similar antioxidant cocktails were shown beneficial in improving memory and cognitive performance in community-dwelling adults without dementia[200, 201].
In recent years, it has become apparent that strategies designed to target total ROS in the organism might not be productive since ROS have dual function. On one hand, increased ROS production contributes to age-related chronic conditions and neurodegeneration [47]. On the other, oxidant species, such as superoxide and hydrogen peroxide, can function as signaling molecules in a broad array of essential redox-dependent signaling pathways that are critical for the organismal survival including epidermal growth factor receptor signaling [202], inactivation of the tumor suppressor PTEN [203], circadian rhythms [204], the inflammatory response [205], and hormetic stress response [206–209]. Redox homeostasis with tight control over levels of ROS production is essential to protect cells from oxidative stress and, at the same time, to ensure presence of the important signaling molecules [210]. Thus, understanding how the dual role of ROS is maintained with age and in the context of different stages of the disease is important for the development of therapeutic approaches that target ROS production and clearance.
Based on the recognized contribution of mitochondria to cellular ROS, the development of novel antioxidants that directly target mitochondria represent a promising approach to mitigate local ROS production compared to the reduction of global levels of ROS. These compounds include coenzyme Q10, idebenone, creatine, MitoQ, MitoVitE, MitoTEMPOL, latrepirdine, methyleneblue, triterpenoids, series of Szeto-Schiller (SS) peptides, curcumin, Ginkgo biloba, and omega-3 polyunsaturated fatty acids. These mitochondria-targeted compounds have been extensively evaluated in multiple laboratories using various in vivo and in vitro models of AD where some of them including a peptide, 6’-dimethyltyrosine-Lys-Phe-NH2 (SS31), have been shown very efficacious in protecting against Aβ-induced oxidative stress, synaptic loss, mitochondrial dysfunction, and abnormal calcium homeostasis [62]. Some of these compounds demonstratedpromising results in clinical trials [211, 212]. Moreover, emerging data demonstrate that partial inhibition of OXPHOS with pharmacological inhibitors is beneficial in preventing obesity and type II diabetes, another risk factors contributing to AD [213–216], and promoting longevity in model organisms and in humans [217–220]. In particular, modulation of mitochondrial Complex I activity with small molecules was found efficacious in cognitive protection in multiple mouse models of AD [221] and in extending lifespan [222]. However, the details of molecular mechanism remain to be determined.
While supplementation with antioxidants so far appears to produce little modifying effect on AD development, non-pharmacological treatments and lifestyle interventions including exercise and caloric restriction have gained significant attention due to their overall positive effect on health and life span [223]. Specifically, grounded on a population-based perspective, the Alzheimer’s Association has identified regular physical exercise as one of the strategies to reduce the risk of cognitive decline and the development of dementia [224]. Indeed, regular physical activity was associated with reduced oxidative stress, increased antioxidant capacity, increased anti-inflammatory effects, reduced levels of ceramides that are elevated in AD, improved Aβ clearance associated with the upregulating Aβ transporters, and induced neurogenesis [223, 225, 226]. The molecular mechanisms implicated in the beneficial effect of exercise are not fully understood. One of the explanations is based on the concept of mitohormesis, which suggests that an exposure to low continuous or higher intermittent sub-lethal doses of exercise-associated stress could lead to a mitochondrial adaptation by inducing changes in gene expression through exercise-sensitive transcription factors such as PGC1α, mtTFA, NF-κB, HIF-1, and p53. Downstream effects result in increase in mitochondrial biogenesis and antioxidant response. Potential signaling factors that mediate this mitochondria-nuclear communication may include ROS, calcium, mitochondrial unfolded protein response, mitochondrial metabolites, and mitokines [66, 227]. In addition to exercise, modulation of diet, especially caloric restriction, has been shown not only to extend lifespan, but also to protect against cognitive decline [228, 229]. However, a recent study demonstrated that meals rich in saturated fat and foods with a high glycemic index have differential effect in adults with and without cognitive impairment [230]. In individuals without cognitive impairment, a consumption of high caloric food worsened cognitive performance, whereas consumption of high caloric food was beneficial in adults with cognitive impairment or the ApoE4 carriers. The authors also found that levels of Aβ in plasma were affected by meal type, suggesting a relationship between metabolic response and amyloid regulation. Therefore, a better understanding of the effect of diet modifications and exercise on metabolism, mitochondrial function and ROS production during different stages of disease progression is needed to develop safe and efficacious therapeutic strategies for AD.
CONCLUSIONS
Multiple lines of evidence provide strong support for the involvement of oxidative stress in the development of AD. At the same time, limited success of antioxidant therapies achieved to date emphasizes the need for better understanding of molecular mechanisms associated with different stages of AD development. Moreover, the dual role of ROS in essential neuroprotective cellular mechanisms versus detrimental effects of increased uncontrolled ROS production should be carefully considered while developing strategies to mitigate oxidative stress in neurodegenerative diseases.
ACKNOWLEDGMENTS
We thank Mr. Ivan Trushin for help with figures. This work was supported by grant from NIEHS R01ES020715 (to ET). Its content is solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1088r1).
REFERENCES
[1] | Alzheimer’s Disease International, https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf. |
[2] | Kandimalla R , Thirumala V , Reddy PH ((2016) ) Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim Biophys Acta. doi: 10.1016/j.bbadis.2016.08.018 |
[3] | Wang Y , Brinton RD ((2016) ) Triad of risk for late onset Alzheimer’s: Mitochondrial haplotype, APOE genotype and chromosomal sex. Front Aging Neurosci 8: , 232. |
[4] | Tramutola A , Lanzillotta C , Perluigi M , Butterfield DA ((2016) ) Oxidative stress, protein modification and Alzheimer disease. Brain Res Bull. doi: 10.1016/j.brainresbull.2016.06.005 |
[5] | Selkoe DJ ((2008) ) Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer’s disease. Handb Clin Neurol 89: , 245–260. |
[6] | LaFerla FM , Green KN , Oddo S ((2007) ) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8: , 499–509. |
[7] | Selkoe DJ ((2008) ) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192: , 106–113. |
[8] | Gyure KA , Durham R , Stewart WF , Smialek JE , Troncoso JC ((2001) ) Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 125: , 489–492. |
[9] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[10] | Knobloch M , Konietzko U , Krebs DC , Nitsch RM ((2007) ) Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging 28: , 1297–1306. |
[11] | Xu H , Greengard P , Gandy S ((1995) ) Regulated formation of Golgi secretory vesicles containing Alzheimer beta-amyloid precursor protein. J Biol Chem 270: , 23243–23245. |
[12] | Kinoshita A , Fukumoto H , Shah T , Whelan CM , Irizarry MC , Hyman BT ((2003) ) Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci 116: , 3339–3346. |
[13] | Mizuguchi M , Ikeda K , Kim SU ((1992) ) Differential distribution of cellular forms of beta-amyloid precursor protein in murine glial cell cultures. Brain Res 584: , 219–225. |
[14] | Omtri RS , Davidson MW , Arumugam B , Poduslo JF , Kandimalla KK ((2012) ) Differences in the cellular uptake and intracellular itineraries of amyloid beta proteins 40 and 42: Ramifications for the Alzheimer’s drug discovery. Mol Pharm 9: , 1887–1897. |
[15] | Cleary JP , Walsh DM , Hofmeister JJ , Shankar GM , Kuskowski MA , Selkoe DJ , Ashe KH ((2005) ) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8: , 79–84. |
[16] | Lai AY , McLaurin J ((2010) ) Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int J Alzheimers Dis 2011: , 548380. |
[17] | Andreadis A , Brown WM , Kosik KS ((1992) ) Structure and novel exons of the human tau gene. Biochemistry 31: , 10626–10633. |
[18] | Miller EC , Teravskis PJ , Dummer BW , Zhao X , Huganir RL , Liao D ((2014) ) Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci 39: , 1214–1224. |
[19] | Selkoe DJ ((2001) ) Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis 3: , 75–80. |
[20] | Jack CR Jr , Holtzman DM ((2013) ) Biomarker modeling of Alzheimer’s disease. Neuron 80: , 1347–1358. |
[21] | Crystal H , Dickson D , Fuld P , Masur D , Scott R , Mehler M , Masdeu J , Kawas C , Aronson M , Wolfson L ((1988) ) Clinico-pathologic studies in dementia: Nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 38: , 1682–1687. |
[22] | Iacono D , Resnick SM , O’Brien R , Zonderman AB , An Y , Pletnikova O , Rudow G , Crain B , Troncoso JC ((2014) ) Mild cognitive impairment and asymptomatic Alzheimer disease subjects: Equivalent beta-amyloid and tau loads with divergent cognitive outcomes. J Neuropathol Exp Neurol 73: , 295–304. |
[23] | Sepulcre J , Schultz AP , Sabuncu M , Gomez-Isla T , Chhatwal J , Becker A , Sperling R , Johnson KA ((2016) ) In vivo tau, amyloid, and gray matter profiles in the aging brain. J Neurosci 36: , 7364–7374. |
[24] | Ossenkoppele R , Schonhaut DR , Scholl M , Lockhart SN , Ayakta N , Baker SL , O’Neil JP , Janabi M , Lazaris A , Cantwell A , Vogel J , Santos M , Miller ZA , Bettcher BM , Vossel KA , Kramer JH , Gorno-Tempini ML , Miller BL , Jagust WJ , Rabinovici GD ((2016) ) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139: , 1551–1567. |
[25] | Wang L , Benzinger TL , Su Y , Christensen J , Friedrichsen K , Aldea P , McConathy J , Cairns NJ , Fagan AM , Morris JC , Ances BM ((2016) ) Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between beta-amyloid and tauopathy. JAMA Neurol 73: , 1070–1077. |
[26] | Jack CR Jr , Knopman DS , Jagust WJ , Shaw LM , Aisen PS , Weiner MW , Petersen RC , Trojanowski JQ ((2010) ) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9: , 119–128. |
[27] | Holtzman DM , Morris JC , Goate AM ((2011) ) Alzheimer’s disease: The challenge of the second century. Sci Transl Med 3: , 77sr71. |
[28] | Bateman RJ , Xiong C , Benzinger TL , Fagan AM , Goate A , Fox NC , Marcus DS , Cairns NJ , Xie X , Blazey TM , Holtzman DM , Santacruz A , Buckles V , Oliver A , Moulder K , Aisen PS , Ghetti B , Klunk WE , McDade E , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Schofield PR , Sperling RA , Salloway S , Morris JC ((2012) ) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367: , 795–804. |
[29] | Clark CM , Pontecorvo MJ , Beach TG , Bedell BJ , Coleman RE , Doraiswamy PM , Fleisher AS , Reiman EM , Sabbagh MN , Sadowsky CH , Schneider JA , Arora A , Carpenter AP , Flitter ML , Joshi AD , Krautkramer MJ , Lu M , Mintun MA , Skovronsky DM ((2012) ) Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: A prospective cohort study. Lancet Neurol 11: , 669–678. |
[30] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[31] | Castello MA , Jeppson JD , Soriano S ((2014) ) Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurol 14: , 169. |
[32] | Castellani RJ , Smith MA ((2011) ) Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’. J Pathol 224: , 147–152. |
[33] | Swerdlow RH , Burns JM , Khan SM ((2014) ) The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta 1842: , 1219–1231. |
[34] | Beal MF ((1998) ) Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta 1366: , 211–223. |
[35] | Beal MF ((2005) ) Oxidative damage as an early marker of Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 26: , 585–586. |
[36] | Atamna H , Frey WH 2nd ((2007) ) Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 7: , 297–310. |
[37] | Baloyannis SJ , Costa V , Michmizos D ((2004) ) Mitochondrial alterations in Alzheimer’s disease. Am J Alzheimers Dis Other Demen 19: , 89–93. |
[38] | Beal MF ((2005) ) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58: , 495–505. |
[39] | Calkins MJ , Manczak M , Mao P , Shirendeb U , Reddy PH ((2011) ) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20: , 4515–4529. |
[40] | Cardoso SM , Pereira CF , Moreira PI , Arduino DM , Esteves AR , Oliveira CR ((2010) ) Mitochondrial control of autophagic lysosomal pathway in Alzheimer’s disease. Exp Neurol 223: , 294–298. |
[41] | Cardoso SM , Santana I , Swerdlow RH , Oliveira CR ((2004) ) Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem 89: , 1417–1426. |
[42] | Caspersen C , Wang N , Yao J , Sosunov A , Chen X , Lustbader JW , Xu HW , Stern D , McKhann G , Yan SD ((2005) ) Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 19: , 2040–2041. |
[43] | Du H , Guo L , Yan S , Sosunov AA , McKhann GM , Yan SS ((2010) ) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 107: , 18670–18675. |
[44] | Eckert A , Hauptmann S , Scherping I , Rhein V , Muller-Spahn F , Gotz J , Muller WE ((2008) ) Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis 5: , 157–159. |
[45] | Leuner K , Schutt T , Kurz C , Eckert SH , Schiller C , Occhipinti A , Mai S , Jendrach M , Eckert GP , Kruse SE , Palmiter RD , Brandt U , Drose S , Wittig I , Willem M , Haass C , Reichert AS , Muller WE ((2012) ) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16: , 1421–1433. |
[46] | Manczak M , Calkins MJ , Reddy PH ((2011) ) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum Mol Genet 20: , 2495–2509. |
[47] | Trushina E , McMurray CT ((2007) ) Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145: , 1233–1248. |
[48] | Trushina E , Nemutlu E , Zhang S , Christensen T , Camp J , Mesa J , Siddiqui A , Tamura Y , Sesaki H , Wengenack TM , Dzeja PP , Poduslo JF ((2012) ) Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One 7: , e32737. |
[49] | Zhu X , Perry G , Smith MA , Wang X ((2013) ) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 33: (Suppl 1), S253–S262. |
[50] | Kandimalla R , Reddy PH ((2016) ) Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim Biophys Acta 1862: , 814–828. |
[51] | Reiman EM , Chen K , Alexander GE , Caselli RJ , Bandy D , Osborne D , Saunders AM , Hardy J ((2004) ) Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A 101: , 284–289. |
[52] | Jagust WJ , Landau SM ((2012) ) Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci 32: , 18227–18233. |
[53] | Mosconi L , Pupi A , De Leon MJ ((2008) ) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147: , 180–195. |
[54] | Bubber P , Haroutunian V , Fisch G , Blass JP , Gibson GE ((2005) ) Mitochondrial abnormalities in Alzheimer brain: Mechanistic imlications. Ann Neurol 57: , 695–703. |
[55] | Blass JP ((2000) ) The mitochondrial spiral. An adequate cause of dementia in the Alzheimer’s syndrome. Ann N Y Acad Sci 924: , 170–183. |
[56] | Schmitt K , Grimm A , Kazmierczak A , Strosznajder JB , Gotz J , Eckert A ((2012) ) Insights into mitochondrial dysfunction: Aging, amyloid-beta, and tau-A deleterious trio. Antioxid Redox Signal 16: , 1456–1466. |
[57] | Reddy PH ((2011) ) Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res 1415: , 136–148. |
[58] | Sultana R , Mecocci P , Mangialasche F , Cecchetti R , Baglioni M , Butterfield DA ((2011) ) Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: Insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis 24: , 77–84. |
[59] | Caldwell CC , Yao J , Brinton RD ((2015) ) Targeting the prodromal stage of Alzheimer’s disease: Bioenergetic and mitochondrial opportunities. Neurotherapeutics 12: , 66–80. |
[60] | Yao J , Irwin RW , Zhao L , Nilsen J , Hamilton RT , Brinton RD ((2009) ) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106: , 14670–14675. |
[61] | Valla J , Yaari R , Wolf AB , Kusne Y , Beach TG , Roher AE , Corneveaux JJ , Huentelman MJ , Caselli RJ , Reiman EM ((2010) ) Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J Alzheimers Dis 22: , 307–313. |
[62] | Reddy PH , Tripathi R , Troung Q , Tirumala K , Reddy TP , Anekonda V , Shirendeb UP , Calkins MJ , Reddy AP , Mao P , Manczak M ((2012) ) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta 1822: , 639–649. |
[63] | Trushina E , Dutta T , Persson XM , Mielke MM , Petersen RC ((2013) ) Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS One 8: , e63644. |
[64] | Jones DP ((2006) ) Redefining oxidative stress. Antioxid Redox Signal 8: , 1865–1879. |
[65] | Hauptmann N , Grimsby J , Shih JC , Cadenas E ((1996) ) The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch Biochem Biophys 335: , 295–304. |
[66] | Di Meo S , Reed TT , Venditti P , Victor VM ((2016) ) Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev 2016: , 1245049. |
[67] | Ray PD , Huang BW , Tsuji Y ((2012) ) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24: , 981–990. |
[68] | Holmstrom KM , Finkel T ((2014) ) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15: , 411–421. |
[69] | Quinlan CL , Perevoshchikova IV , Hey-Mogensen M , Orr AL , Brand MD ((2013) ) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol 1: , 304–312. |
[70] | Joshi G , Johnson JA ((2012) ) The Nrf2-ARE pathway: A valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat CNS Drug Discov 7: , 218–229. |
[71] | Kensler TW , Wakabayashi N , Biswal S ((2007) ) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: , 89–116. |
[72] | Denzer I , Munch G , Friedland K ((2016) ) Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol Res 103: , 80–94. |
[73] | Patel M ((2016) ) Targeting oxidative stress in central nervous system disorders. Trends Pharmacol Sci 37: , 768–778. |
[74] | Bhat AH , Dar KB , Anees S , Zargar MA , Masood A , Sofi MA , Ganie SA ((2015) ) Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed Pharmacother 74: , 101–110. |
[75] | Ramsey CP , Glass CA , Montgomery MB , Lindl KA , Ritson GP , Chia LA , Hamilton RL , Chu CT , Jordan-Sciutto KL ((2007) ) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66: , 75–85. |
[76] | Tanji K , Maruyama A , Odagiri S , Mori F , Itoh K , Kakita A , Takahashi H , Wakabayashi K ((2013) ) Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J Neuropathol Exp Neurol 72: , 18–28. |
[77] | Schipper HM , Bennett DA , Liberman A , Bienias JL , Schneider JA , Kelly J , Arvanitakis Z ((2006) ) Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging 27: , 252–261. |
[78] | Kanninen K , Malm TM , Jyrkkanen HK , Goldsteins G , Keksa-Goldsteine V , Tanila H , Yamamoto M , Yla-Herttuala S , Levonen AL , Koistinaho J ((2008) ) Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci 39: , 302–313. |
[79] | Kim TS , Pae CU , Yoon SJ , Jang WY , Lee NJ , Kim JJ , Lee SJ , Lee C , Paik IH , Lee CU ((2006) ) Decreased plasma antioxidants in patients with Alzheimer’s disease. Int J Geriatr Psychiatry 21: , 344–348. |
[80] | Uttara B , Singh AV , Zamboni P , Mahajan RT ((2009) ) Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7: , 65–74. |
[81] | Smith MA , Rudnicka-Nawrot M , Richey PL , Praprotnik D , Mulvihill P , Miller CA , Sayre LM , Perry G ((1995) ) Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. J Neurochem 64: , 2660–2666. |
[82] | Butterfield DA , Di Domenico F , Barone E ((2014) ) Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim Biophys Acta 1842: , 1693–1706. |
[83] | Kosenko EA , Solomadin IN , Tikhonova LA , Reddy VP , Aliev G , Kaminsky YG ((2014) ) Pathogenesis of Alzheimer disease: Role of oxidative stress, amyloid-beta peptides, systemic ammonia and erythrocyte energy metabolism. CNS Neurol Disord Drug Targets 13: , 112–119. |
[84] | Castellani RJ , Moreira PI , Perry G , Zhu X ((2012) ) The role of iron as a mediator of oxidative stress in Alzheimer disease. Biofactors 38: , 133–138. |
[85] | Zhu X , Raina AK , Lee HG , Chao M , Nunomura A , Tabaton M , Petersen RB , Perry G , Smith MA ((2003) ) Oxidative stress and neuronal adaptation in Alzheimer disease: The role of SAPK pathways. Antioxid Redox Signal 5: , 571–576. |
[86] | Markesbery WR ((1999) ) The role of oxidative stress in Alzheimer disease. Arch Neurol 56: , 1449–1452. |
[87] | Pratico D ((2008) ) Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol Sci 29: , 609–615. |
[88] | Schippling S , Kontush A , Arlt S , Buhmann C , Sturenburg HJ , Mann U , Muller-Thomsen T , Beisiegel U ((2000) ) Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic Biol Med 28: , 351–360. |
[89] | Smith MA , Kutty RK , Richey PL , Yan SD , Stern D , Chader GJ , Wiggert B , Petersen RB , Perry G ((1994) ) Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol 145: , 42–47. |
[90] | Ramassamy C , Krzywkowski P , Averill D , Lussier-Cacan S , Theroux L , Christen Y , Davignon J , Poirier J ((2001) ) Impact of apoE deficiency on oxidative insults and antioxidant levels in the brain. Brain Res Mol Brain Res 86: , 76–83. |
[91] | Picklo MJ , Montine TJ , Amarnath V , Neely MD ((2002) ) Carbonyl toxicology and Alzheimer’s disease. Toxicol Appl Pharmacol 184: , 187–197. |
[92] | Bassett CN , Montine TJ ((2003) ) Lipoproteins and lipid peroxidation in Alzheimer’s disease. J Nutr Health Aging 7: , 24–29. |
[93] | Mecocci P , Beal MF , Cecchetti R , Polidori MC , Cherubini A , Chionne F , Avellini L , Romano G , Senin U ((1997) ) Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Mol Chem Neuropathol 31: , 53–64. |
[94] | Lovell MA , Markesbery WR ((2007) ) Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 35: , 7497–7504. |
[95] | Lovell MA , Markesbery WR ((2008) ) Oxidatively modified RNA in mild cognitive impairment. Neurobiol Dis 29: , 169–175. |
[96] | Bradley-Whitman MA , Timmons MD , Beckett TL , Murphy MP , Lynn BC , Lovell MA ((2014) ) Nucleic acid oxidation: An early feature of Alzheimer’s disease. J Neurochem 128: , 294–304. |
[97] | Migliore L , Fontana I , Trippi F , Colognato R , Coppede F , Tognoni G , Nucciarone B , Siciliano G ((2005) ) Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging 26: , 567–573. |
[98] | Greenough MA , Camakaris J , Bush AI ((2013) ) Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 62: , 540–555. |
[99] | Wang P , Wang ZY ((2016) ) Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev. doi: 10.1016/j.arr.2016.10.003 |
[100] | Liu G , Huang W , Moir RD , Vanderburg CR , Lai B , Peng Z , Tanzi RE , Rogers JT , Huang X ((2006) ) Metal exposure and Alzheimer’s pathogenesis. J Struct Biol 155: , 45–51. |
[101] | Danscher G , Jensen KB , Frederickson CJ , Kemp K , Andreasen A , Juhl S , Stoltenberg M , Ravid R ((1997) ) Increased amount of zinc in the hippocampus and amygdala of Alzheimer’s diseased brains: A proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J Neurosci Methods 76: , 53–59. |
[102] | Friedlich AL , Lee JY , van Groen T , Cherny RA , Volitakis I , Cole TB , Palmiter RD , Koh JY , Bush AI ((2004) ) Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer’s disease. J Neurosci 24: , 3453–3459. |
[103] | Lee JY , Cole TB , Palmiter RD , Suh SW , Koh JY ((2002) ) Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A 99: , 7705–7710. |
[104] | Lee JY , Mook-Jung I , Koh JY ((1999) ) Histochemically reactive zinc in plaques of the Swedish mutant beta-amyloid precursor protein transgenic mice. J Neurosci 19: , RC10. |
[105] | Lovell MA , Robertson JD , Teesdale WJ , Campbell JL , Markesbery WR ((1998) ) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158: , 47–52. |
[106] | Miller LM , Wang Q , Telivala TP , Smith RJ , Lanzirotti A , Miklossy J ((2006) ) Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J Struct Biol 155: , 30–37. |
[107] | Stoltenberg M , Bush AI , Bach G , Smidt K , Larsen A , Rungby J , Lund S , Doering P , Danscher G ((2007) ) Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150: , 357–369. |
[108] | Suh SW , Jensen KB , Jensen MS , Silva DS , Kesslak PJ , Danscher G , Frederickson CJ ((2000) ) Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res 852: , 274–278. |
[109] | Boom A , Pochet R , Authelet M , Pradier L , Borghgraef P , Van Leuven F , Heizmann CW , Brion JP ((2004) ) Astrocytic calcium/zinc binding protein S100A6 over expression in Alzheimer’s disease and in PS1/APP transgenic mice models. Biochim Biophys Acta 1742: , 161–168. |
[110] | Heizmann CW , Cox JA ((1998) ) New perspectives on S100 proteins: A multi-functional Ca(2+)-, Zn(2+)- and Cu(2+)-binding protein family. Biometals 11: , 383–397. |
[111] | Barnham KJ , Bush AI ((2014) ) Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev 43: , 6727–6749. |
[112] | Lammich S , Kojro E , Postina R , Gilbert S , Pfeiffer R , Jasionowski M , Haass C , Fahrenholz F ((1999) ) Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A 96: , 3922–3927. |
[113] | Bush AI , Pettingell WH , Multhaup G , d Paradis M , Vonsattel JP , Gusella JF , Beyreuther K , Masters CL , Tanzi RE ((1994) ) Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265: , 1464–1467. |
[114] | Tougu V , Karafin A , Palumaa P ((2008) ) Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J Neurochem 104: , 1249–1259. |
[115] | Mantyh PW , Ghilardi JR , Rogers S , DeMaster E , Allen CJ , Stimson ER , Maggio JE ((1993) ) Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem 61: , 1171–1174. |
[116] | Sayre LM , Perry G , Harris PL , Liu Y , Schubert KA , Smith MA ((2000) ) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J Neurochem 74: , 270–279. |
[117] | Boom A , Authelet M , Dedecker R , Frederick C , Van Heurck R , Daubie V , Leroy K , Pochet R , Brion JP ((2009) ) Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn2+ in human-tau transfected cells. Biochim Biophys Acta 1793: , 1058–1067. |
[118] | Stohs SJ , Bagchi D ((1995) ) Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18: , 321–336. |
[119] | Huang X , Atwood CS , Hartshorn MA , Multhaup G , Goldstein LE , Scarpa RC , Cuajungco MP , Gray DN , Lim J , Moir RD , Tanzi RE , Bush AI ((1999) ) The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38: , 7609–7616. |
[120] | Adlard PA , Cherny RA , Finkelstein DI , Gautier E , Robb E , Cortes M , Volitakis I , Liu X , Smith JP , Perez K , Laughton K , Li QX , Charman SA , Nicolazzo JA , Wilkins S , Deleva K , Lynch T , Kok G , Ritchie CW , Tanzi RE , Cappai R , Masters CL , Barnham KJ , Bush AI ((2008) ) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59: , 43–55. |
[121] | Cherny RA , Atwood CS , Xilinas ME , Gray DN , Jones WD , McLean CA , Barnham KJ , Volitakis I , Fraser FW , Kim Y , Huang X , Goldstein LE , Moir RD , Lim JT , Beyreuther K , Zheng H , Tanzi RE , Masters CL , Bush AI ((2001) ) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30: , 665–676. |
[122] | Opazo C , Barria MI , Ruiz FH , Inestrosa NC ((2003) ) Copper reduction by copper binding proteins and its relation to neurodegenerative diseases. Biometals 16: , 91–98. |
[123] | Cristovao JS , Santos R , Gomes CM ((2016) ) Metals and Neuronal Metal Binding Proteins Implicated in Alzheimer’s Disease. Oxid Med Cell Longev 2016: , 9812178. |
[124] | Nakajima K , Kohsaka S ((2001) ) Microglia: Activation and their significance in the central nervous system. J Biochem 130: , 169–175. |
[125] | Gibson GE , Karuppagounder SS , Shi Q ((2008) ) Oxidant-induced changes in mitochondria and calcium dynamics in the pathophysiology of Alzheimer’s disease. Ann N Y Acad Sci 1147: , 221–232. |
[126] | Manczak M , Anekonda TS , Henson E , Park BS , Quinn J , Reddy PH ((2006) ) Mitochondria are a direct site of Abeta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: , 1437–1449. |
[127] | Mattson MP , Cutler RG , Jo DG ((2005) ) Alzheimer peptides perturb lipid-regulating enzymes. Nat Cell Biol 7: , 1045–1047. |
[128] | Lee JT , Xu J , Lee JM , Ku G , Han X , Yang DI , Chen S , Hsu CY ((2004) ) Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol 164: , 123–131. |
[129] | Grimm MO , Grimm HS , Patzold AJ , Zinser EG , Halonen R , Duering M , Tschape JA , De Strooper B , Muller U , Shen J , Hartmann T ((2005) ) Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol 7: , 1118–1123. |
[130] | Cutler RG , Kelly J , Storie K , Pedersen WA , Tammara A , Hatanpaa K , Troncoso JC , Mattson MP ((2004) ) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101: , 2070–2075. |
[131] | Chalfant CE , Kishikawa K , Mumby MC , Kamibayashi C , Bielawska A , Hannun YA ((1999) ) Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem 274: , 20313–20317. |
[132] | Haughey NJ , Bandaru VV , Bae M , Mattson MP ((2010) ) Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim Biophys Acta 1801: , 878–886. |
[133] | Blass JP , Baker AC , Ko L , Black RS ((1990) ) Induction of Alzheimer antigens by an uncoupler of oxidative phosphorylation. Arch Neurol 47: , 864–869. |
[134] | Szabados T , Dul C , Majtenyi K , Hargitai J , Penzes Z , Urbanics R ((2004) ) A chronic Alzheimer’s model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behav Brain Res 154: , 31–40. |
[135] | Ichimura H , Parthasarathi K , Quadri S , Issekutz AC , Bhattacharya J ((2003) ) Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest 111: , 691–699. |
[136] | Colell A , Fernandez A , Fernandez-Checa JC ((2009) ) Mitochondria, cholesterol and amyloid beta peptide: A dangerous trio in Alzheimer disease. J Bioenerg Biomembr 41: , 417–423. |
[137] | Rosales-Corral SA , Lopez-Armas G , Cruz-Ramos J , Melnikov VG , Tan DX , Manchester LC , Munoz R , Reiter RJ ((2012) ) Alterations in lipid levels of mitochondrial membranes induced by amyloid-beta: A protective role of melatonin. Int J Alzheimers Dis 2012: , 459806. |
[138] | Mondragon-Rodriguez S , Perry G , Zhu X , Moreira PI , Acevedo-Aquino MC , Williams S ((2013) ) Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid Med Cell Longev 2013: , 940603. |
[139] | Vlassenko AG , Vaishnavi SN , Couture L , Sacco D , Shannon BJ , Mach RH , Morris JC , Raichle ME , Mintun MA ((2010) ) Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc Natl Acad Sci U S A 107: , 17763–17767. |
[140] | Onyango IG , Ahn JY , Tuttle JB , Bennett JP Jr , Swerdlow RH ((2010) ) Nerve growth factor attenuates oxidant-induced beta-amyloid neurotoxicity in sporadic Alzheimer’s disease cybrids. J Neurochem 114: , 1605–1618. |
[141] | Khan SM , Cassarino DS , Abramova NN , Keeney PM , Borland MK , Trimmer PA , Krebs CT , Bennett JC , Parks JK , Swerdlow RH , Parker WD Jr , Bennett JP Jr ((2000) ) Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol 48: , 148–155. |
[142] | Gasparini L , Racchi M , Benussi L , Curti D , Binetti G , Bianchetti A , Trabucchi M , Govoni S ((1997) ) Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett 231: , 113–117. |
[143] | Gabuzda D , Busciglio J , Chen LB , Matsudaira P , Yankner BA ((1994) ) Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem 269: , 13623–13628. |
[144] | Webster MT , Pearce BR , Bowen DM , Francis PT ((1998) ) The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm (Vienna) 105: , 839–853. |
[145] | Henriques AG , Domingues SC , Fardilha M , da Cruz e Silva EF , da Cruz e Silva OA ((2005) ) Sodium azide and 2-deoxy-D-glucose-induced cellular stress affects phosphorylation-dependent AbetaPP processing. J Alzheimers Dis 7: , 201–212; discussion 255-262. |
[146] | Szule JA , Jung JH , McMahan UJ ((2015) ) The structure and function of ‘active zone material’ at synapses. Philos Trans R Soc Lond B Biol Sci 370: , 1672. |
[147] | Malenka RC , Bear MF ((2004) ) LTP and LTD: An embarrassment of riches. Neuron 44: , 5–21. |
[148] | Neves G , Cooke SF , Bliss TV ((2008) ) Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat Rev Neurosci 9: , 65–75. |
[149] | Mattson MP , Magnus T ((2006) ) Ageing and neuronal vulnerability. Nat Rev Neurosci 7: , 278–294. |
[150] | Jang SS , Chung HJ ((2016) ) Emerging link between Alzheimer’s disease and homeostatic synaptic plasticity. Neural Plas 2016: , 7969272. |
[151] | DeKosky ST , Scheff SW ((1990) ) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27: , 457–464. |
[152] | Hamos JE , DeGennaro LJ , Drachman DA ((1989) ) Synaptic loss in Alzheimer’s disease and other dementias. Neurology 39: , 355–361. |
[153] | Robinson JL , Molina-Porcel L , Corrada MM , Raible K , Lee EB , Lee VM , Kawas CH , Trojanowski JQ ((2014) ) Perforant path synaptic loss correlates with cognitive impairment and Alzheimer’s disease in the oldest-old. Brain 137: , 2578–2587. |
[154] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[155] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT ((2011) ) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: , a006189. |
[156] | Cooper LN , Bear MF ((2012) ) The BCM theory of synapse modification at 30: Interaction of theory with experiment. Nat Rev Neurosci 13: , 798–810. |
[157] | Spires-Jones TL , Hyman BT ((2014) ) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82: , 756–771. |
[158] | Kamat PK , Kalani A , Rai S , Swarnkar S , Tota S , Nath C , Tyagi N ((2016) ) Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: Understanding the therapeutics strategies. Mol Neurobiol 53: , 648–661. |
[159] | Reddy PH , Beal MF ((2008) ) Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14: , 45–53. |
[160] | Newcomer JW , Farber NB , Olney JW ((2000) ) NMDA receptor function, memory, and brain aging. Dialogues Clin Neurosci 2: , 219–232. |
[161] | Frankland PW , Bontempi B ((2005) ) The organization of recent and remote memories. Nat Rev Neurosci 6: , 119–130. |
[162] | Snyder EM , Nong Y , Almeida CG , Paul S , Moran T , Choi EY , Nairn AC , Salter MW , Lombroso PJ , Gouras GK , Greengard P ((2005) ) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8: , 1051–1058. |
[163] | Bezprozvanny I , Mattson MP ((2008) ) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31: , 454–463. |
[164] | Parsons MP , Raymond LA ((2014) ) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82: , 279–293. |
[165] | Lipton SA ((2006) ) Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat Rev Drug Discov 5: , 160–170. |
[166] | Hsieh H , Boehm J , Sato C , Iwatsubo T , Tomita T , Sisodia S , Malinow R ((2006) ) AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52: , 831–843. |
[167] | Li Y , Sun H , Chen Z , Xu H , Bu G , Zheng H ((2016) ) Implications of GABAergic neurotransmission in Alzheimer’s disease. Front Aging Neurosci 8: , 31. |
[168] | Schliebs R , Arendt T ((2011) ) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221: , 555–563. |
[169] | Campos C , Rocha NB , Vieira RT , Rocha SA , Telles-Correia D , Paes F , Yuan T , Nardi AE , Arias-Carrion O , Machado S , Caixeta L ((2016) ) Treatment of cognitive deficits in Alzheimer’s disease: A psychopharmacological review. Psychiatr Danub 28: , 2–12. |
[170] | Ellis B , Hye A , Snowden SG ((2015) ) Metabolic modifications in human biofluids suggest the involvement of sphingolipid, antioxidant, and glutamate metabolism in Alzheimer’s disease pathogenesis. J Alzheimers Dis 46: , 313–327. |
[171] | Trushina E , Mielke MM ((2013) ) Recent advances in the application of metabolomics to Alzheimer’s Disease. Biochim Biophys Acta 1842: , 1232–1239. |
[172] | De Felice FG , Velasco PT , Lambert MP , Viola K , Fernandez SJ , Ferreira ST , Klein WL ((2007) ) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282: , 11590–11601. |
[173] | Rai S , Kamat PK , Nath C , Shukla R ((2013) ) A study on neuroinflammation and NMDA receptor function in STZ (ICV) induced memory impaired rats. J Neuroimmunol 254: , 1–9. |
[174] | Kamat PK , Rai S , Swarnkar S , Shukla R , Ali S , Najmi AK , Nath C ((2013) ) Okadaic acid-induced Tau phosphorylation in rat brain: Role of NMDA recetor. Neuroscience 238: , 97–113. |
[175] | Morris G , Walder K , Puri BK , Berk M , Maes M ((2016) ) The deleterious effects of oxidative and nitrosative stress on palmitoylation, membrane lipid rafts and lipid-based cellular signalling: New drug targets in neuroimmune disorders. Mol Neurobiol 53: , 4638–4658. |
[176] | Han X ((2010) ) Multi-dimensional mass spectrometry-based shotgun lipidomics and the altered lipids at the mild cognitive impairment stage of Alzheimer’s disease. Biochim Biophys Acta 1801: , 774–783. |
[177] | Han X , Rozen S , Boyle SH , Hellegers C , Cheng H , Burke JR , Welsh-Bohmer KA , Doraiswamy PM , Kaddurah-Daouk R ((2011) ) Metabolomics in early Alzheimer’s disease: Identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One 6: , e21643. |
[178] | Schneider LS , Sano M ((2009) ) Current Alzheimer’s disease clinical trials: Methods and placebo outcomes. Alzheimers Dement 5: , 388–397. |
[179] | Cummings J , Aisen PS , DuBois B , Frolich L , Jack CR Jr , Jones RW , Morris JC , Raskin J , Dowsett SA , Scheltens P ((2016) ) Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res Ther 8: , 39. |
[180] | Padurariu M , Ciobica A , Lefter R , Serban IL , Stefanescu C , Chirita R ((2013) ) The oxidative stress hypothesis in Alzheimer’s disease. Psychiatr Danub 25: , 401–409. |
[181] | Sancheti H , Kanamori K , Patil I , Diaz Brinton R , Ross BD , Cadenas E ((2014) ) Reversal of metabolic deficits by lipoic acid in a triple transgenic mouse model of Alzheimer’s disease: A 13C NMR study. J Cereb Blood Flow Metab 34: , 288–296. |
[182] | Quinn JF , Bussiere JR , Hammond RS , Montine TJ , Henson E , Jones RE , Stackman RW Jr ((2007) ) Chronic dietary alpha-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol Aging 28: , 213–225. |
[183] | Rajasekar N , Dwivedi S , Tota SK , Kamat PK , Hanif K , Nath C , Shukla R ((2013) ) Neuroprotective effect of curcumin on okadaic acid induced memory impairment in mice. Eur J Pharmacol 715: , 381–394. |
[184] | Zhou X , Li Y , Shi X , Ma C ((2016) ) An overview on therapeutics attenuating amyloid beta level in Alzheimer’s disease: Targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am J Transl Res 8: , 246–269. |
[185] | Kang JH , Cook N , Manson J , Buring JE , Grodstein F ((2006) ) A randomized trial of vitamin E supplementation and cognitive function in women. Arch Intern Med 166: , 2462–2468. |
[186] | Kang JH , Cook NR , Manson JE , Buring JE , Albert CM , Grodstein F ((2009) ) Vitamin E, vitamin C, beta carotene, and cognitive function among women with or at risk of cardiovascular disease: The Women’s Antioxidant and Cardiovascular Study. Circulation 119: , 2772–2780. |
[187] | Petersen RC , Thomas RG , Grundman M , Bennett D , Doody R , Ferris S , Galasko D , Jin S , Kaye J , Levey A , Pfeiffer E , Sano M , van Dyck CH , Thal LJ , Alzheimer’s Disease Cooperative Study Group ((2005) ) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352: , 2379–2388. |
[188] | Sano M , Ernesto C , Thomas RG , Klauber MR , Schafer K , Grundman M , Woodbury P , Growdon J , Cotman CW , Pfeiffer E , Schneider LS , Thal LJ ((1997) ) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med 336: , 1216–1222. |
[189] | Dysken MW , Sano M , Asthana S , Vertrees JE , Pallaki M , Llorente M , Love S , Schellenberg GD , McCarten JR , Malphurs J , Prieto S , Chen P , Loreck DJ , Trapp G , Bakshi RS , Mintzer JE , Heidebrink JL , Vidal-Cardona A , Arroyo LM , Cruz AR , Zachariah S , Kowall NW , Chopra MP , Craft S , Thielke S , Turvey CL , Woodman C , Monnell KA , Gordon K , Tomaska J , Segal Y , Peduzzi PN , Guarino PD ((2014) ) Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 311: , 33–44. |
[190] | Miller ER 3rd , Pastor-Barriuso R , Dalal D , Riemersma RA , Appel LJ , Guallar E ((2005) ) Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 142: , 37–46. |
[191] | Arlt S , Muller-Thomsen T , Beisiegel U , Kontush A ((2012) ) Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem Res 37: , 2706–2714. |
[192] | Galasko DR , Peskind E , Clark CM , Quinn JF , Ringman JM , Jicha GA , Cotman C , Cottrell B , Montine TJ , Thomas RG , Aisen P , Alzheimer’s Disease Cooperative Study ((2012) ) Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 69: , 836–841. |
[193] | Ringman JM , Frautschy SA , Teng E , Begum AN , Bardens J , Beigi M , Gylys KH , Badmaev V , Heath DD , Apostolova LG , Porter V , Vanek Z , Marshall GA , Hellemann G , Sugar C , Masterman DL , Montine TJ , Cummings JL , Cole GM ((2012) ) Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res Ther 4: , 43. |
[194] | Wojtunik-Kulesza KA , Oniszczuk A , Oniszczuk T , Waksmundzka-Hajnos M ((2016) ) The influence of common free radicals and antioxidants on development of Alzheimer’s Disease. Biomed Pharmacother 78: , 39–49. |
[195] | Mecocci P , Polidori MC ((2012) ) Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta 1822: , 631–638. |
[196] | Tolonen M , Halme M , Sarna S ((1985) ) Vitamin E and selenium supplementation in geriatric patients: A double-blind preliminary clinical trial. Biol Trace Elem Res 7: , 161–168. |
[197] | Kryscio RJ , Abner EL , Schmitt FA , Goodman PJ , Mendiondo M , Caban-Holt A , Dennis BC , Mathews M , Klein EA , Crowley JJ , Investigators SELECT ((2013) ) A randomized controlled Alzheimer’s disease prevention trial’s evolution into an exposure trial: The PREADViSE Trial. J Nutr Health Aging 17: , 72–75. |
[198] | Dias KS , Viegas C Jr ((2014) ) Multi-target directed drugs: A modern approach for design of new drugs for the treatment of Alzheimer’s disease. Curr Neuropharmacol 12: , 239–255. |
[199] | Remington R , Bechtel C , Larsen D , Samar A , Page R , Morrell C , Shea TB ((2016) ) Maintenance of cognitive performance and mood for individuals with Alzheimer’s disease following consumption of a nutraceutical formulation: A one-year, open-label study. J Alzheimers Dis 51: , 991–995. |
[200] | Chan A , Remington R , Kotyla E , Lepore A , Zemianek J , Shea TB ((2010) ) A vitamin/nutriceutical formulation improves memory and cognitive performance in community-dwelling adults without dementia. J Nutr Health Aging 14: , 224–230. |
[201] | Banerjee P , Maity S , Das T , Mazumder S ((2011) ) A double-blind randomized placebo-controlled clinical study to evaluate the efficacy and safety of a polyherbal formulation in geriatric age group: A phase IV clinical report. J Ethnopharmacol 134: , 429–433. |
[202] | Meng TC , Fukada T , Tonks NK ((2002) ) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell 9: , 387–399. |
[203] | Kwon J , Lee SR , Yang KS , Ahn Y , Kim YJ , Stadtman ER , Rhee SG ((2004) ) Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A 101: , 16419–16424. |
[204] | Edgar RS , Green EW , Zhao Y , van Ooijen G , Olmedo M , Qin X , Xu Y , Pan M , Valekunja UK , Feeney KA , Maywood ES , Hastings MH , Baliga NS , Merrow M , Millar AJ , Johnson CH , Kyriacou CP , O’Neill JS , Reddy AB ((2012) ) Peroxiredoxins are conserved markers of circadian rhythms. Nature 485: , 459–464. |
[205] | Ha EM , Oh CT , Bae YS , Lee WJ ((2005) ) A direct role for dual oxidase in Drosophila gut immunity. Science 310: , 847–850. |
[206] | Mathers J , Fraser JA , McMahon M , Saunders RD , Hayes JD , McLellan LI ((2004) ) Antioxidant and cytoprotective responses to redox stress. Biochem Soc Symp 157–176. |
[207] | Mattson MP ((2008) ) Hormesis defined. Ageing Res Rev 7: , 1–7. |
[208] | Ristow M ((2014) ) Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat Med 20: , 709–711. |
[209] | Schmeisser S , Priebe S , Groth M , Monajembashi S , Hemmerich P , Guthke R , Platzer M , Ristow M ((2013) ) Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol Metab 2: , 92–102. |
[210] | Droge W ((2002) ) Free radicals in the physiological control of cell function. Physiol Rev 82: , 47–95. |
[211] | Kumar A , Singh A ((2015) ) A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front Pharmaco 6: , 206. |
[212] | Feniouk BA , Skulachev VP ((2016) ) Cellular and molecular mechanisms of action of mitochondria-targeted antioxidants. Curr Aging Sci 9: , doi: 10.2174-1874609809666160921113706 |
[213] | Pospisilik JA , Knauf C , Joza N , Benit P , Orthofer M , Cani PD , Ebersberger I , Nakashima T , Sarao R , Neely G , Esterbauer H , Kozlov A , Kahn CR , Kroemer G , Rustin P , Burcelin R , Penninger JM ((2007) ) Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131: , 476–491. |
[214] | Wredenberg A , Freyer C , Sandstrom ME , Katz A , Wibom R , Westerblad H , Larsson NG ((2006) ) Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun 350: , 202–207. |
[215] | Vernochet C , Mourier A , Bezy O , Macotela Y , Boucher J , Rardin MJ , An D , Lee KY , Ilkayeva OR , Zingaretti CM , Emanuelli B , Smyth G , Cinti S , Newgard CB , Gibson BW , Larsson NG , Kahn CR ((2012) ) Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab 16: , 765–776. |
[216] | Quintens R , Singh S , Lemaire K , De Bock K , Granvik M , Schraenen A , Vroegrijk IO , Costa V , Van Noten P , Lambrechts D , Lehnert S , Van Lommel L , Thorrez L , De Faudeur G , Romijn JA , Shelton JM , Scorrano L , Lijnen HR , Voshol PJ , Carmeliet P , Mammen PP , Schuit F ((2013) ) Mice deficient in the respiratory chain gene Cox6a2 are protected against high-fat diet-induced obesity and insulin resistance. PLoS One 8: , e56719. |
[217] | Raule N , Sevini F , Li S , Barbieri A , Tallaro F , Lomartire L , Vianello D , Montesanto A , Moilanen JS , Bezrukov V , Blanche H , Hervonen A , Christensen K , Deiana L , Gonos ES , Kirkwood TB , Kristensen P , Leon A , Pelicci PG , Poulain M , Rea IM , Remacle J , Robine JM , Schreiber S , Sikora E , Eline Slagboom P , Spazzafumo L , Antonietta Stazi M , Toussaint O , Vaupel JW , Rose G , Majamaa K , Perola M , Johnson TE , Bolund L , Yang H , Passarino G , Franceschi C ((2014) ) The co-occurrence of mtDNA mutations on different oxidative phosphorylation subunits, not detected by haplogroup analysis, affects human longevity and is population specific. Aging Cell 13: , 401–407. |
[218] | Copeland JM , Cho J , Lo T Jr , Hur JH , Bahadorani S , Arabyan T , Rabie J , Soh J , Walker DW ((2009) ) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol 19: , 1591–1598. |
[219] | Lee SS , Lee RY , Fraser AG , Kamath RS , Ahringer J , Ruvkun G ((2003) ) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33: , 40–48. |
[220] | Liu X , Jiang N , Hughes B , Bigras E , Shoubridge E , Hekimi S ((2005) ) Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 19: , 2424–2434. |
[221] | Zhang L , Zhang S , Maezawa I , Trushin S , Minhas P , Pinto M , Jin LW , Prasain K , Nguyen TD , Yamazaki Y , Kanekiyo T , Bu G , Gateno B , Chang KO , Nath KA , Nemutlu E , Dzeja P , Pang YP , Hua DH , Trushina E ((2015) ) Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. E Bio Medicine 2: , 294–305. |
[222] | Baumgart M , Priebe S , Groth M , Hartmann N , Menzel U , Pandolfini L , Koch P , Felder M , Ristow M , Englert C , Guthke R , Platzer M , Cellerino A ((2016) ) Longitudinal RNA-Seq analysis of vertebrate aging identifies mitochondrial complex i as a small-molecule-sensitive modifier of lifespan. Cell Syst 2: , 122–132. |
[223] | Mendiola-Precoma J , Berumen LC , Padilla K , Garcia-Alcocer G ((2016) ) Therapies for prevention and treatment of Alzheimer’s disease. Biomed Res Int 2016: , 2589276. |
[224] | Baumgart M , Snyder HM , Carrillo MC , Fazio S , Kim H , Johns H ((2015) ) Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement 11: , 718–726. |
[225] | Bertram S , Brixius K , Brinkmann C ((2016) ) Exercise for the diabetic brain: Howhysical training may help prevent dementia and Alzheimer’s disease in T2DM patients. Endocrine 53: , 350–363. |
[226] | Baker LD , Frank LL , Foster-Schubert K , Green PS , Wilkinson CW , McTiernan A , Cholerton BA , Plymate SR , Fishel MA , Watson GS , Duncan GE , Mehta PD , Craft S ((2010) ) Aerobic exercise improves cognition for older adults with glucose intolerance, a risk factor for Alzheimer’s disease. J Alzheimers Dis 22: , 569–579. |
[227] | Merry TL , Ristow M ((2016) ) Mitohormesis in exercise training. Free Radic Biol Med 98: , 123–130. |
[228] | Ntsapi C , Loos B ((2016) ) Caloric restriction and the precision-control of autophagy: A strategy for delaying neurodegenerative disease progression. Exp Gerontol 83: , 97–111. |
[229] | Van Cauwenberghe C , Vandendriessche C , Libert C , Vandenbroucke RE ((2016) ) Caloric restriction: Beneficial effects on brain aging and Alzheimer’s disease. Mamm Genome 27: , 300–319. |
[230] | Hanson AJ , Bayer JL , Baker LD , Cholerton B , VanFossen B , Trittschuh E , Rissman RA , Donohue MC , Moghadam SH , Plymate SR , Craft S ((2015) ) Differential effects of meal challenges on cognition, metabolism, and biomarkers for apolipoprotein E ɛ4 carriers and adults with mild cognitive impairment. J Alzheimers Dis 48: , 205–218. |
[231] | Zhang L , Trushin S , Christensen TA , Bachmeier BV , Gateno B , Schroeder A , Yao J , Itoh K , Sesaki H , Poon WW , Gylys KH , Patterson ER , Parisi JE , Diaz Brinton R , Salisbury JL , Trushina E ((2016) ) Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci Rep 6: , 18725. |


