Dysregulation of Autophagy, Mitophagy, and Apoptotic Genes in the Medial Temporal Lobe Cortex in an Ischemic Model of Alzheimer’s Disease
Abstract
Ischemic brain damage is a pathological incident that is often linked with medial temporal lobe cortex injury and finally its atrophy. Post-ischemic brain injury associates with poor prognosis since neurons of selectively vulnerable ischemic brain areas are disappearing by apoptotic program of neuronal death. Autophagy has been considered, after brain ischemia, as a guardian against neurodegeneration. Consequently, we have examined changes in autophagy (BECN 1), mitophagy (BNIP 3), and apoptotic (caspase 3) genes in the medial temporal lobe cortex with the use of quantitative reverse-transcriptase PCR following transient 10-min global brain ischemia in rats with survival 2, 7, and 30 days. The intense significant overexpression of BECN 1 gene was noted on the 2nd day, while on days 7–30 the expression of this gene was still upregulated. BNIP 3 gene was downregulated on the 2nd day, but on days 7–30 post-ischemia, there was a significant reverse tendency. Caspase 3 gene, associated with apoptotic neuronal death, was induced in the same way as BNIP 3 gene after brain ischemia. Thus, the demonstrated changes indicate that the considerable dysregulation of expression of BECN 1, BNIP 3, and caspase 3 genes may be connected with a response of neuronal cells in medial temporal lobe cortex to transient complete brain ischemia.
INTRODUCTION
Brain ischemia leads to chronic metabolic and structural changes within special brain regions: the hippocampus and temporal lobe [1–14]. Brain ischemia has been found as the most effective predictor for the later development of Alzheimer-type dementia [1–7]. Additionally, medial temporal lobe atrophy among ischemic brain survivors creates higher risks for memory dysfunction and the diagnosis of dementia [15]. Those patients with moderate to severe medial temporal lobe atrophy demonstrated poor performance on measures of learning, story recall, visual reproduction, visual spatial reasoning, and processing speed [15]. Post-ischemic dementia might be the result of direct influence of ischemia [12, 16–22] (also Pluta et al. unpublished results), ischemic white matter changes [23, 24], and Alzheimer-type neuropathology [12–14, 25–27], or combinations of these three [1].
Neuropathological changes associated with Alzheimer’s disease (AD) begin in the hippocampus including medial temporal lobe structures [28, 29]. Hippocampus damage in AD features neuron loss in the CA1 area and in medial temporal lobe early in the process. It has not been finally determined whether the primary event of pathology in AD starts in the hippocampus and proceeds to medial temporal lobe and other parts of brain or may be started in the medial temporal lobe itself [30]. There some studies underpinning the fact that memory impairment is directly correlated to the atrophy of medial temporal lobe during development of AD [31, 32]. Some other reports suggest that patients with ischemic brain injury and post-ischemic medial temporal lobe atrophy might have preclinical AD, which is clinically revealed by ischemic brain lesions [1, 33]. Currently, AD still lacks an etiology, early prediction, diagnosis, and effective treatment. AD can been called the “disease of theories”, e.g., amyloid hypothesis. While amyloid investigations have pivotal role in understanding some interactions and pathways, nothing is as powerful as an accurate animal model. Many transgenic models of AD have undoubtedly been of great benefit but the majority of mice genetically altered to overproduce amyloid-β peptide do not show significant tau protein pathology and have relatively little neurodegeneration [34, 35]. In summary, the results of the systematic review have revealed that mice bred to show increased levels of amyloid-β peptide do not perform significantly worse in cognitive tests than mice that do not have elevated amyloid-β peptide levels [36]. These results suggest that amyloid-β peptide may be considered as a biochemical “side effect” in the AD neuropathology [36]. Because of the large discrepancy in the behavioral findings observed across the AD mouse models, a question arises whether we are really any closer today to determining what these mechanisms actually are. In addition, another argument that complicates the use of transgenic models based exclusively on amyloid-β protein precursor and/or tau protein mutations is that other mechanisms may be also of importance. Is this simply because mice do not live long enough to trigger tau protein pathology and the associated neuronal death? Finally, the above models often fail to reproduce the selective vulnerability in the hippocampus CA1 area and it is impossible to follow how the neuropathology spreads into the other parts of brain. In using a non-genetic model to examine AD-like neuropathologies, we remove the variability that is associated with transgenic animals. For instance, transgene integration is apparently random. Also, experiments reveal that the genetic surrounding of the inserted transgenic construct is modulating the expression pattern of the transgene itself both quantitatively and qualitatively. To fully elucidate mechanisms of AD, it is essential that a good model is available [12, 37]. Ischemic brain injury with dementia and AD share apparently common features: protein aggregation [12, 16–19, 26, 27, 38, 39], specific vulnerability of certain classes of neurons [11–14], inflammation [13, 25, 40], long incubation period, and finally global brain atrophy [12–14] with dementia [4, 6, 7, 17]. It may suggest that these features reflect common neuropathological pathways in ischemic brain injury and AD and that the ischemic model is more faithfully recapitulating AD. In our experimental approach, we allow for normal aging and disease onset to occur instead of forcing the system to a disease state.
Based on known anatomical topography, we hypothesized that the local ischemic thickness of CA1 hippocampus field [12, 14] would be associated with medial temporal lobe damage and atrophy [8], reflecting an association between AD-related injury to hippocampal neurons and neurodegeneration in medial temporal lobe. To test this hypothesis, we decided to study disease progression from the hippocampus to the medial temporal lobe in the ischemic model of AD [12, 13, 37]. In this study, we present for the first time the time course of expression of autophagy, mitophagy, and apoptotic gene levels, all of which are implicated in AD, in the medial temporal lobe cortex subjected to transient complete brain ischemia.
MATERIALS AND METHODS
Brain ischemia, cortex sampling, and extraction of total cellular RNA
Female Wistar rats (n = 37, 2 month old, 160–180 g) were submitted to 10-min global brain ischemia due to cardiac arrest [37]. The rats were maintained in pairs per cage in a room temperature of 24±2°C, with 55±5% humidity, and with a 12 h light-dark cycle. All animals had free access to commercial laboratory chow and tap water ad libitum. All experimental procedures were performed during the light phase, under identical conditions. The rats used for experiments were treated in strict accordance with the NIH Guide for Care and Use of Laboratory Animals and European Communities Council Directive 142, as well as with the approval of the local Ethical Committee. After brain ischemia, the animals were allowed to survive 2 (n = 11), 7 (n = 10), and 30 days (n = 16). Sham-operated rats (n = 37) were exposed to the same procedures as ischemic animals but without induced cardiac arrest and thus served as controls.
Prior to sampling, the brains were perfused with cold 0.9% NaCl via the left ventricle in order to flush blood vessels. After removing the brain from the skull, the brain was put on an ice chilled Petri dish. The ischemic and control pooled samples circa 1 mm3 volume of medial temporal lobe cortex left and right (all cortical layers) were taken with a narrow scalpel and were immediately placed in RNALater solution (Life Technologies, USA) [20–22].
Total cellular RNA isolation was performed according to the method described by Chomczynski and Sacchi [41]. The RNA quality and quantity was assessed using the NanoDrop 2000 spectrophotometer (Thermo Scientific, USA) [20–22]. Obtained RNA was stored in 80% ethanol at –20°C for further analysis [20–22].
The cDNA synthesis
The cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit, according to manufacturer’s instructions (Applied Biosystems, USA). Each reactive mixture contained the following set of reagents: 1×RT buffer, 20 U RNase inhibitor, 50 U reverse transcriptase (Multiscribe Reverse Transcriptase), 1×RT Random Primers, 4 mM of each deoxynucleotide: dATP, dGTP, dTTP and dCTP plus examined 1μg RNA in DNase-, RNase- and protease-free water (Sigma-Aldrich, USA) to complete the volume required for reaction [20]. Afterwards, the reactive components were thoroughly mixed and centrifuged to fuse them well. The cDNA was synthesized on Veriti Dx (Applied Biosystems, USA) under the following conditions: stage I: 25°C, 10 min, stage II: 37°C, 120 min, stage III: 85°C, 5 min, stage IV: 4°C.
The RT-qPCR protocol
The cDNA, which was obtained by reverse transcription (RT) procedure, was amplified by real-time gene expression analysis (qPCR) on 7900HT Real-Time Fast System (Applied Biosystems, USA), using the manufacturer’s SDS software [20]. Reaction components included: 1.25μl mixture of probe and oligonucleotide starters specific for genes examined, 12.5μl buffer TaqMan Universal PCR Master Mix, 1μl cDNA, DNase-, RNase- and protease-free water (Sigma Aldrich, USA) to complete the required reactive volume. The reaction was performed on an optic 25μl-well reaction plate, using probe sets of TaqMan Gene Expression Assays (Applied Biosystems, USA) with FAM-NFQ markers and oligonucleotide starters for rat genes: autophagy (BECN 1), mitophagy (BNIP 3) and caspase 3 and the housekeeping gene: Rpl13a was used as an internal control gene [42]. Amplification protocol included the following cycles: initial denaturation: 95°C, 10 min and 40 cycles, each composed of two temperatures: 95°C, 15 s and 60°C, 1 min. The number of copies of DNA molecules was monitored and calculated on 7900HT Real-Time Fast System (Applied Biosystems, USA) in each amplification cycle. To calculate the number of examined DNA molecules present in the mixture at the onset of reaction, the number of PCR cycles after which the level of fluorescence exceeded the defined threshold cycle (CT) RQ Study Software (Applied Biosystems, USA) was used. The CT value for each sample of endogenous control gene (Rpl13a) was used to normalize the level of the examined gene expression. The relative level of gene expression was calculated according to the formula [43]: 1) Calculating CT differences between the examined gene and the reference gene are presented below: for the examined gene after ischemia of medial temporal lobe cortex: ΔCT ischemic sample = CT target gene from ischemic sample–CT reference gene, ischemic sample and for the calibrator gene (control sample): ΔCT calibrator = CT target gene from control sample –CT reference gene, control sample. 2) Normalizing ΔCT of the ischemic sample to the ΔCT of the calibrator ΔΔCT =ΔCT ischemic sample–ΔCT calibrator. 3) Relative expression (RQ) of rat genes was calculated by the formula: RQ = 2–ΔΔCT. The RQ defines the expression of the examined gene in the medial temporal lobe cortex taken from an ischemic rat with reference to the gene expression in the same structure of a control rat. Finally, the RQs were analyzed after their logarithmic conversion into logarithm of RQ (LogRQ) [20]. Thus, the obtained results were more legible. LogRQ = 0 means that gene expression in the calibrated sample and the ischemic one are the same. LogRQ <0 points to decreased gene expression in the ischemic sample, whereas LogRQ >0 points to increased gene expression in the ischemic sample compared to the calibrated one.
Statistical analyses
The results were statistically analyzed by SPSS v. 17 software by means of the non-parametric Kruskal-Wallis test and Spearman rho correlation analysis. Data were presented as mean±SEM. The level of statistical significance was set at p≤0.05.
RESULTS
Changes in expression of BECN 1, BNIP 3, and caspase 3 genes in rats 2 days following brain ischemia
BECN 1 gene expression increased to a maximum of 1.029–fold change but both BNIP 3 and caspase 3 genes decreased to a minimum of –1,887 and –0.523 – fold change, respectively. The mean expression levels of genes were as follows: BECN 1 0.434±0.115, BNIP 3 –0.877±0.204, and caspase 3 –0.160±0.110.
Changes in expression of BECN 1, BNIP 3, and caspase 3 genes in rats 7 days following brain ischemia
BECN 1 gene expression decreased to a minimum of 0.550 – fold change, but both BNIP 3 and caspase 3 genes increased to a maximum of 3.668 and 0.471 – fold change, respectively. The mean expression levels of genes were as follows: BECN 1 0.161±0.066, BNIP 3 1.487±0.547, and caspase 3 0.196±0.053.
Changes in expression of BECN 1, BNIP 3, and caspase 3 genes in rats 30 days following brain ischemia
Both BECN 1 and BNIP 3 genes expression decreased to a minimum of 0.490 and 0.437 – fold change, respectively. Caspase 3 gene increased to a maximum of 0.536 – fold change. The mean expression levels of genes were as follows: BECN 1 0.129±0.048, BNIP 3 0.176±0.037, and caspase 3 0.258±0.040.
Summary of the mean levels of genes expression in medial temporal lobe cortex in rats 2, 7, and 30 days following brain ischemia
Figures 1–3 show changes in the mean expression levels of genes with statistical significance: BECN 1 (Fig. 1), BNIP 3 (Fig. 2), and caspase 3 (Fig. 3) after 10-min complete brain ischemia in rats with survival 2, 7, and 30 days. Significant correlations between the expression of genes in the medial temporal lobe cortex 2, 7 and 30 days after global brain ischemia was evident for BNIP 3 and caspase 3 (Spearman rho correlation test p≤0.01).
DISCUSSION
In the present study, we provide evidence for the first time for the activation of an autophagic BECN 1 gene in the medial temporal lobe cortex during 30 days after brain ischemia. The overexpression of the BECN 1 gene was accompanied by an early (during 2 days) downregulation of both BNIP 3 and caspase 3 genes. Next, during 7–30 days expression of both genes was significantly upregulated. It has been suggested that ischemia induces classical apoptotic neuronal death in the cortex [44–46] but our data have indicated that the situation is more complex and that this form of neuronal death in temporal cortex is to some extent delayed for 7 days by protective overexpression of BECN 1 gene.
Overexpression of BECN 1 gene in the medial temporal lobe cortex was noted in all studied times after brain ischemia and suggest a possible efficient role of autophagy-mediated neuronal protection [47, 48]. Some studies indicate that the autophagy has an important role in amyloid protein precursor processing as well as in the autophagic clearance of aggregation-prone proteins in neurodegeneration [48–50]. We have postulated that an efficient autophagic response in the temporal cortex might protect cortical neurons from acute and/or fast cell death triggered by ischemia [44–46], which is not observed in the hippocampus (Pluta et al., unpublished results).
In this study, we have noted that BNIP 3 gene is significantly downregulated 2 days after ischemia, which is not in accordance with data from the focal brain ischemia [51] and the ischemic hippocampus (Pluta et al., unpublished results). Next during 7–30 days post-ischemia, this gene expression was significantly upregulated. In this context, it is important to stress that there is an association between the increased activity of BNIP 3 gene and different mitochondrial neurotoxic proteins such as AIF, Endo G, or PARP 1 [52–55], which are involved in neuronal death. Brain ischemia appears to cause redistribution of BNIP 3, AIF, Endo G, and PARP 1 from the mitochondria to the neuronal nucleus [52, 53, 55–59]. It has been shown that BNIP 3 induces cell death mostly via mitochondrial dysfunction, in that homodimeric BNIP 3 inserts into mitochondrial outer membrane to increase its permeability leading to a release of different cytotoxic proteins such as Endo G, AIF, and others [60]. In addition, it has been shown that BNIP 3 interacts with LC3 to target damaged mitochondria to autophagosomes, initiating the process of mitophagy in the ischemic neurons [61]. Moreover, double immunostaining for BNIP 3 and caspase 3 indicates that most ischemic neurons were stained for both proteins [51, 62]. However, some BNIP 3 positive neurons did not stain for caspase 3 [51, 62]. These observations open the possibility that the ischemia-regulated pro-apoptotic protein BNIP 3 enters the nucleus and could interact with other proteins involved in DNA structure, transcription or mRNA splicing after brain ischemia. The precise mechanism by which BNIP 3, AIF, Endo G, and PARP 1 execute cortical neurons death is not fully understood, however, it likely involves both nuclear-independent and nuclear-dependent processes. Thus, our data suggest a novel insight into BNIP 3 gene in regulating neuronal damage and death in the ischemic medial temporal lobe cortex.
We have recently noted that caspase 3 gene is significantly downregulated 2 days after medial temporal lobe ischemia. These data are contradictory to caspase 3 overexpression in the hippocampus during 2 days after ischemia (Pluta et al., unpublished results). In the hippocampus, at that time caspase 3 appears to be a key trigger for apoptotic neuronal death. Seven and 30 days after ischemia, caspase 3 was significantly upregulated in the temporal cortex. Then, it is possible that the above mediated process in the medial temporal lobe cortex is delayed by 7 days as compared to the same process in the ischemic hippocampus (Pluta et al., unpublished results). On the other hand, it can suggest the mechanism of slow spreading neuronal pathology from the hippocampus into the medial temporal lobe cortex in ischemic brain with Alzheimer-phenotype and dementia [63]. Caspase can use some substrates such as PARP 1, an inhibitor of caspase-activated DNAse, amyloid-β protein precursor, presenilins [62], and tau protein [64–67]. Activated caspase 3 cleaves nuclear DNA repair enzyme, such as PARP 1, which leads to nuclear DNA damage and finally apoptosis [44–46]. Moreover, caspase 3 could cleave the GGA3 protein and thus increase BACE 1 activity [68, 69]. Importantly, as amyloid-β protein precursor has been elevated long-term after brain ischemia [10, 12, 14, 23, 24, 70, 71] and has been shown a substrate for caspase 3 [62] and caspase 3 can enhance BACE 1 activity [68, 69], this pathway may also have influence on the development of neuronal changes in the medial temporal lobe cortex. Recent studies have indicated that increased level of activated caspase significantly correlates to elevated levels of truncated tau protein and formation of neurofibrillary tangles [72, 73]. In addition, cognitive decline was significantly negatively associated with increased levels of caspase activity and tau protein truncated by caspase 3 [67].
Presented data suggest a possible efficient role of autophagy gene in slowing cortical neuronal damage and death mediated by ischemic injury [47]. Our data may suggest that brain ischemia induced neuronal death in the medial temporal lobe cortex with delay by 7 days as compared to hippocampus (Pluta et al., unpublished results), by caspase 3 and BNIP 3 gene activation. This study suggests that BNIP 3 gene regulates ischemia-induced caspase-independent cortical neuronal death probably through influence on Endo G, AIF, and PARP 1 in the neuronal cell nucleus [52, 74]. Mitochondrial BNIP 3 gene is probably an upstream signal of these proteins. The levels of anti- and pro-apoptotic proteins were studied before now in our model what recently is in accordance with gene induction [75]. For already examined proteins such as beclin 1 and BNIP 3 in the brain ischemia by other scientists [52, 53, 61, 76, 77], we have added now new data related to their genes. Caspase-dependent and caspase-independent neuronal death can occur coincidently within ischemic neurons of the medial temporal lobe cortex, leading to cell death of mixed neurochemical features [59, 62].
ACKNOWLEDGMENTS
The authors acknowledge support by the Polish National Science Centre (DEC-2013/09/B/NZ7/01345-RP) and by the Mossakowski Medical Research Centre, Polish Academy of Sciences, Poland (T3-RP). Also, this study was supported by a grant from Medical University of Lublin, Poland (DS 475/15-SJC and DS 222/14-JK). The research was developed using the equipment purchased within the Project “The equipment of innovative laboratories doing research on new medicines used in the therapy of civilization and neoplastic diseases” within the Operation Program Development of Eastern Poland 2007–2013, Priority Axis I Modern Economy, Operations I.3 Innovation Promotion (JK, SJC).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0387r1).
REFERENCES
[1] | Leys D , Henon H , Mackowiak-Cordoliani MA , Pasquier F ((2005) ) Poststroke dementia. Lancet Neurol 4: , 752–759. |
[2] | De la Torre JC ((2006) ) How do heart disease and stroke become risk factors for Alzheimer’s disease? Neurol Res 28: , 637–644. |
[3] | Pinkston JB , Alekseeva N , Gonzalez Toledo E ((2009) ) Stroke and dementia. Neurol Res 31: , 824–831. |
[4] | Barra de la Tremblaye P , Plamondon H ((2011) ) Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav Brain Res 219: , 227–233. |
[5] | Kiryk A , Pluta R , Figiel I , Mikosz M , Ułamek M , Niewiadomska G , Jabłoński M , Kaczmarek L ((2011) ) Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 219: , 1–7. |
[6] | Li J , Wang YJ , Zhang M , Fang CQ , Zhou HD ((2011) ) Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci 89: , 86–92. |
[7] | Cohan CH , Neumann JT , Dave KR , Alekseyenko A , Binkert M , Stransky K , Lin HW , Barnes CA , Wright CB , Perez-Pinzon MA ((2015) ) Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS One 10: , e0124918. |
[8] | Pluta R ((1985) ) Influence of prostacyclin on early morphological changes in the rabbit brain after complete 20–min ischemia. J Neurol Sci 70: , 305–316. |
[9] | De la Torre JC ((2008) ) Pathophysiology of neuronal energy crisis in Alzheimer’s disease. Neurodegener Dis 5: , 126–132. |
[10] | Pluta R , Januszewski S , Jabłoński M , Ułamek M ((2010) ) Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. (Suppl). Acta Neurochir 106: , 37–41. |
[11] | Pluta R , Jabłoński M , Czuczwar SJ ((2012) ) Postischemic dementia with Alzheimer phenotype: Selectively vulnerable versus resistant areas of the brain and neurodegeneration versus β-amyloid peptide. Folia Neuropathol 50: , 101–109. |
[12] | Pluta R ((2000) ) The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann N Y Acad Sci 903: , 324–334. |
[13] | Pluta R ((2002) ) Astroglial expression of the beta-amyloid in ischemia-reperfusion brain injury. Ann N Y Acad Sci 977: , 102–108. |
[14] | Pluta R , Ułamek M , Jabłoński M ((2009) ) Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec 292: , 1863–1881. |
[15] | Jokinen H , Kalska H , Ylikoski R , Hietanen M , Mäntylä R , Pohjasvaara T , Kaste M , Erkinjuntti T ((2004) ) Medial temporal lobe atrophy and memory deficits in elderly stroke patients. Eur J Neurol 11: , 825–832. |
[16] | Pluta R , Kida E , Lossinsky AS , Golabek AA , Mossakowski MJ , Wisniewski HM ((1994) ) Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res 649: , 323–328. |
[17] | Kida E , Pluta R , Lossinsky AS , Gołąbek A , Choi–Miura NH , Wiśniewski HM , Mossakowski MJ ((1995) ) Complete cerebral ischemia with short–term survival in rat induced by cardiac arrest. II. Extracellular and intracellular accumulation of apolipoproteins E and J in the brain. Brain Res 674: , 341–346. |
[18] | Wen Y , Yang SH , Liu R , Perez EJ , Brun-Ziukemagel AM , Koulen P , Simpkins JW ((2007) ) Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta 1772: , 473–483. |
[19] | Pluta R , Furmaga-Jabłońska W , Maciejewski R , Ułamek-Kozioł M , Jabłoński M ((2013) ) Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in soradic Alzheimer’s disease. Mol Neurobiol 47: , 425–434. |
[20] | Kocki J , Ułamek-Kozioł M , Bogucka-Kocka A , Januszewski S , Jabłoński M , Gil-Kulik P , Brzozowska J , Petniak A , Furmaga-Jabłońska W , Bogucki J , Czuczwar SJ , Pluta R ((2015) ) Dysregulation of amyloid-β protein precursor, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J Alzheimers Dis 47: , 1047–1056. |
[21] | Pluta R , Kocki J , Ułamek-Kozioł M , Bogucka-Kocka A , Gil-Kulik P , Januszewski S , Jabłoński M , Petniak A , Brzozowska J , Bogucki J , Furmaga-Jabłońska W , Czuczwar SJ ((2016) ) Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol Rep 68: , 155–161. |
[22] | Pluta R , Kocki J , Ułamek-Kozioł M , Petniak A , Gil-Kulik P , Januszewski S , Bogucki J , Jabłoński M , Brzozowska J , Furmaga-Jabłońska M , Bogucka-Kocka A , Czuczwar SJ ((2016) ) Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J Alzheimers Dis 51: , 1023–1031. |
[23] | Pluta R , Ułamek M , Januszewski S ((2006) ) Micro-blood-brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. (Suppl). Acta Neurochir 96: , 267–271. |
[24] | Pluta R , Januszewski S , Ułamek M ((2008) ) Ischemic blood-brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. (Suppl). Acta Neurochir 102: , 353–356. |
[25] | Sekeljic V , Bataveljic D , Stamenkovic S , Ułamek M , Jabłoński M , Radenovic L , Pluta R , Andjus PR ((2012) ) Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct Funct 217: , 411–420. |
[26] | Pluta R , Jabłoński M , Ułamek-Kozioł M , Kocki J , Brzozowska J , Januszewski S , Furmaga-Jabłońska W , Bogucka-Kocka A , Maciejewski R , Czuczwar SJ ((2013) ) Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol 48: , 500–515. |
[27] | Ułamek-Kozioł M , Pluta R , Bogucka-Kocka A , Januszewski S , Kocki J , Czuczwar SJ ((2016) ) Brain ischemia with Alzheimer phenotype dysregulates Alzheimer’s disease-related proteins. Pharmacol Rep 68: , 582–591. |
[28] | Lee DY , Fletcher E , Carmichael OT , Singh B , Mungas D , Reed B , Martinez O , Buonocore MH , Persianinova M , De Carli C ((2012) ) Sub-regional hippocampal injury is associated with fornix degeneration in Alzheimer’s disease. Front Aging Neurosci 4: , 1. |
[29] | Louwersheimer E , Keulen MA , Steenwijk MD , Wattjes MP , Jiskoot LC , Vrenken H , Teunissen CE , van Berckel BN , van der Flier WM , Scheltens P , van Swieten JC , Pijnenburg YA ((2016) ) Heterogeneous language profiles in patients with primary progressive aphasia due to Alzheimer’s disease. J Alzheimers Dis 51: , 581–590. |
[30] | Magnin B , Mesrob L , Kinkingnéhun S , Pélégrini-Issac M , Colliot O , Sarazin M , Dubois B , Lehéricy S , Benali H ((2009) ) Support vector machine-based classification of Alzheimer’s disease from whole-brain anatomical MRI. Neuroradiology 51: , 73–83. |
[31] | Petersen RC , Jack CR JR , Xu YC , Waring SC , O’Brien PC , Smith GE , Ivnik RJ , Tangalos EG , Boeve BF , Kokmen E ((2000) ) Memory and MRI-based hippocampal volumes in aging and AD. Neurology 54: , 581–587. |
[32] | Jauhiainen AM , Pihlajamäki M , Tervo S , Niskanen E , Tanila H , Hänninen T , Vanninen RL , Soininen H ((2009) ) Discriminating accuracy of medial temporal lobe volumetry and fMRI in mild cognitive impairment. Hippocampus 19: , 166–175. |
[33] | Pasquier F , Leys D ((1997) ) Why are stroke patients prone to develop dementia? J Neurol 244: , 135–142. |
[34] | Koistinaho M , Kettunen MI , Goldsteins G , Keinanen R , Salminen A , Ort M , Bures J , Liu D , Kauppinen RA , Higgins LS , Koistinaho J ((2002) ) Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: Role of inflammation. Proc Natl Acad Sci U S A 99: , 1610–1615. |
[35] | Spires TL , Hyman BT ((2005) ) Transgenic models of Alzheimer’s disease: Learning from animals. NeuroRx 2: , 423–437. |
[36] | Foley AM , Ammar ZM , Lee RH , Mitchell CS ((2015) ) Systematic review of the relationship between amyloid-β levels and measures of transgenic mouse cognitive deficit in Alzheimer’s disease. J Alzheimers Dis 44: , 787–795. |
[37] | Pluta R , Lossinsky AS , Mossakowski MJ , Faso L , Wisniewski HM ((1991) ) Reassessment of a new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol 83: , 1–11. |
[38] | Qi J , Wu H , Yang Y , Wand D , Chen Y , Gu Y , Liu T ((2007) ) Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J Alzheimers Dis 12: , 335–341. |
[39] | Yang SH , Simpkins JW ((2007) ) Ischemia-reperfusion promotes tau and beta-amyloid pathology and a progressive cognitive impairment. In: Ischemia-reperfusion pathways in Alzheimer’s disease, Pluta R, ed. Nova Science Publishers, Inc., New York, pp. 113–138. |
[40] | Pluta R ((2002) ) Glial expression of the beta-amyloid peptide in cardiac arrest. J Neurol Sci 203-204: , 277–280. |
[41] | Chomczynski P , Sacchi N ((1987) ) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 62: , 156–159. |
[42] | Yao L , Chen X , Tian Y , Lu X , Zhang P , Shi Q , Zhang J , Liu Y ((2012) ) Selection of housekeeping genes for normalization of RT-PCR in hypoxic neural stem cells of rat in vitro. Mol Biol Rep 39: , 569–576. |
[43] | Livak KJ , Schmittgen TD ((2001) ) Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods 25: , 402–408. |
[44] | Popa-Wagner A , Badan I , Walker L , Groppa S , Patrana N , Kessler C ((2007) ) Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathpl 113: , 277–293. |
[45] | Buga AM , Sascau M , Pisoschi C , Herndon JG , Kessler C , Popa-Wagner A. ((2008) ) The genomic response of the ipsilateral and contralateral cortex to stroke in aged rats. J Cell Mol Med 12: , 2731–2753. |
[46] | Broughton BRS , Reutens DC , Sobey ChG ((2009) ) Apoptotic mechanisms after cerebral ischemia. Stroke 40: , e331–e339. |
[47] | Garcia-Arencibia M , Hochfeld WE , Toh PPC , Rubinsztein DC ((2010) ) Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol 21: , 691–698. |
[48] | Yang DS , Stavrides P , Mohan PS , Kaushik S , Kumar A , Ohno M , Schmidt SD , Wesson D , Bandyopadhyay U , Jiang Y , Pawlik M , Peterhoff CM , Yang AJ , Wilson DA , St George-Hyslop P , Westaway D , Mathews PM , Levy E , Cuervo AM , Nixon RA ((2011) ) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 134: , 258–277. |
[49] | Lucin KM , O’Brien CE , Bieri G , Czirr E , Mosher KI , Abbey RJ , Mastroeni DF , Rogers J , Spencer B , Masliah E , Wyss-Coray T ((2013) ) Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79: , 873–886. |
[50] | Tan CC , Yu JT , Tan MS , Jiang T , Zhu XCh , Tan L ((2014) ) Autophagy in aging and neurodegenerative diseases: Implications for pathogenesis and therapy. Neurobiol Aging 35: , 941–957. |
[51] | Althaus J , Bernaudin M , Petit E , Toutain J , Touzani O , Rami A ((2006) ) Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1α) protein following focal cerebral ischemia in rats. Neurochem Int 48: , 687–695. |
[52] | Schmidt-Kastner R , Aguirre Chen C , Kietzmann T , Saul I , Busto R , Ginsberg MD ((2004) ) Nuclear localization of the hypoxia-regulated pro-apoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res 1001: , 133–142. |
[53] | Zhang Z , Yang X , Zhang S , Ma X , Kong J ((2007) ) BNIP3 upregulation and EndoG translocation in delayed neuronal death in stroke and in hypoxia. Stroke 38: , 1606–1613. |
[54] | Zhu C , Wang X , Huang Z , Qiu L , Xu F , Vahsen N , Nilsson M , Eriksson PS , Hagberg H , Culmsee C , Plesnila N , Kroemer G , Blomgren K ((2007) ) Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia ischemia. Cell Death Differ 14: , 775–784. |
[55] | Lu P , Kamboj A , Gibson SB , Anderson ChM ((2014) ) Poly(ADP-Ribose) polymerase-1 causes mitochondrial damage and neuron death mediated by Bnip3. J Neurosci 34: , 15975–15987. |
[56] | Plesnila N , Zhu C , Culmsee C , Gröger M , Moskowitz MA , Blomgren K ((2004) ) Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab 24: , 458–466. |
[57] | Culmsee C , Zhu C , Landshamer S , Becattini B , Wagner E , Pellecchia M , Blomgren K , Plesnila N ((2005) ) Apoptosis inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci 25: , 10262–10272. |
[58] | Lee BI , Lee DJ , Cho KJ , Kim GW ((2005) ) Early nuclear translocation of endonuclease G and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Neurosci Lett 386: , 23–27. |
[59] | Zhao ST , Chen M , Li SJ , Zhang MH , Li BX , Das M , Bean JC , Kong JM , Zhu XH , Gao TM ((2009) ) Mitochondrial BNIP3 upregulation precedes endonuclease G translocation in hippocampal neuronal death following oxygen-glucose deprivation. BMC Neurosci 10: , 113. |
[60] | Nakamura Y , Kitamura N , Shinogi D , Yoshida M , Goda O , Murai R , Kamino H , Arakawa H ((2012) ) BNIP3 and NIX mediate Mieap-induced accumulation of lysosomal proteins within mitochondria. PLoS One 7: , e30767. |
[61] | Shi RY , Zhu SH , Li V , Gibson SB , Xu XS , Kong JM ((2014) ) BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther 20: , 1045–1055. |
[62] | Rami A ((2003) ) Ischemic neuronal death in the rat hippocampus: The calpain-calpastatin-caspase hypothesis. Neurobiol Dis 13: , 75–88. |
[63] | Buga AM , Scholz CJ , Kumar S , Herndon JG , Alexandru D , Cojocaru GR , Dandekar T , Popa-Wagner A ((2012) ) Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PLoS One 7: , e50985. |
[64] | Quintanilla RA , Dolan PJ , Jin YN , Johnson GV ((2012) ) Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol Aging 33: , 619e25–619e35. |
[65] | Jarero-Basulto JJ , Luna-Munoz J , Mena R , Kristofikova Z , Ripova D , Perry G , Binder LI , Garcia-Sierra F ((2013) ) Proteolytic cleavage of polymeric tau protein by caspase-3: Implications for Alzheimer’s disease. J Neuropathol Exp Neurol 72: , 1145–116. |
[66] | McIlwain DR , Berger T , Mak TW ((2013) ) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5: , a008656. |
[67] | Means JC , Venkatesan A , Gerdes B , Fan JY , Bjes ES , Price JL ((2015) ) Drosophila spaghetti and double time link the circadian clock and light to caspases, apoptosis and tauopathy. PLoS Genet 7: , e1005171. |
[68] | Tesco G , Koh YH , Kang EL , Cameron AN , Das S , Sena-Esteves M , Hiltunen M , Yang SH , Zhong Z , Shen Y , Simpkins JW , Tanzi RE ((2007) ) Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 54: , 721–737. |
[69] | Sarajarvi T , Haapasalo A , Viswanathan J , Mäkinen P , Laitinen M , Soininen H , Hiltunen M ((2009) ) Down-regulation of seladin-1 increases BACE1 levels and activity through enhance GGA3 depletion during apoptosis. J Biol Chem 284: , 34433–34443. |
[70] | Pluta R ((2003) ) Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival. (Suppl). Acta Neurochir 86: , 117–122. |
[71] | Jabłoński M , Maciejewski R , Januszewski S , Ułamek M , Pluta R ((2011) ) One year follow up in ischemic brain injury and the role of Alzheimer factors. (Suppl 1), S113-S. Physiol Res 60: , 119. |
[72] | De Calignon A , Fox LM , Pitstick R , Carlson GA , Bacskai BJ , Spires-Jones TL , Hyman BT ((2010) ) Caspase activation precedes and leads to tangles. Nature 464: , 1201–1204. |
[73] | Hanger DP , Wray S ((2010) ) Tau cleavage and tau aggregation in neurodegenerative disease. Biochem Soc Trans 38: , 1016–1020. |
[74] | Walls KC , Ghosh AP , Ballestas ME , Klocke BJ , Roth KA ((2009) ) Bcl-2/Adenovirus E1B 19 kDa interacting protein-3 (BNIP3) regulates hypoxia-induced neural precursor cell death. J Neuropathol Exp Neurol 68: , 1326–1338. |
[75] | Krajewski S , Mai JK , Krajewska M , Sikorska M , Mossakowski MJ , Reed JC ((1995) ) Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci 15: , 6364–6376. |
[76] | Zheng YQ , Liu JX , Li XZ , Xu L , Xu YG ((2009) ) RNA interference-mediated downregulation of Beclin 1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin 30: , 919–927. |
[77] | Xingyong C , Xicui S , Huanxing S , Jingsong O , Yi H , Xu Z , Ruxun H , Hong P ((2013) ) Upregulation of myeloid cell leukemia-1 potentially modulates beclin-1-dependent autophagy in ischemic stroke in rats. BMC Neurosci 14: , 56. |
Figures and Tables
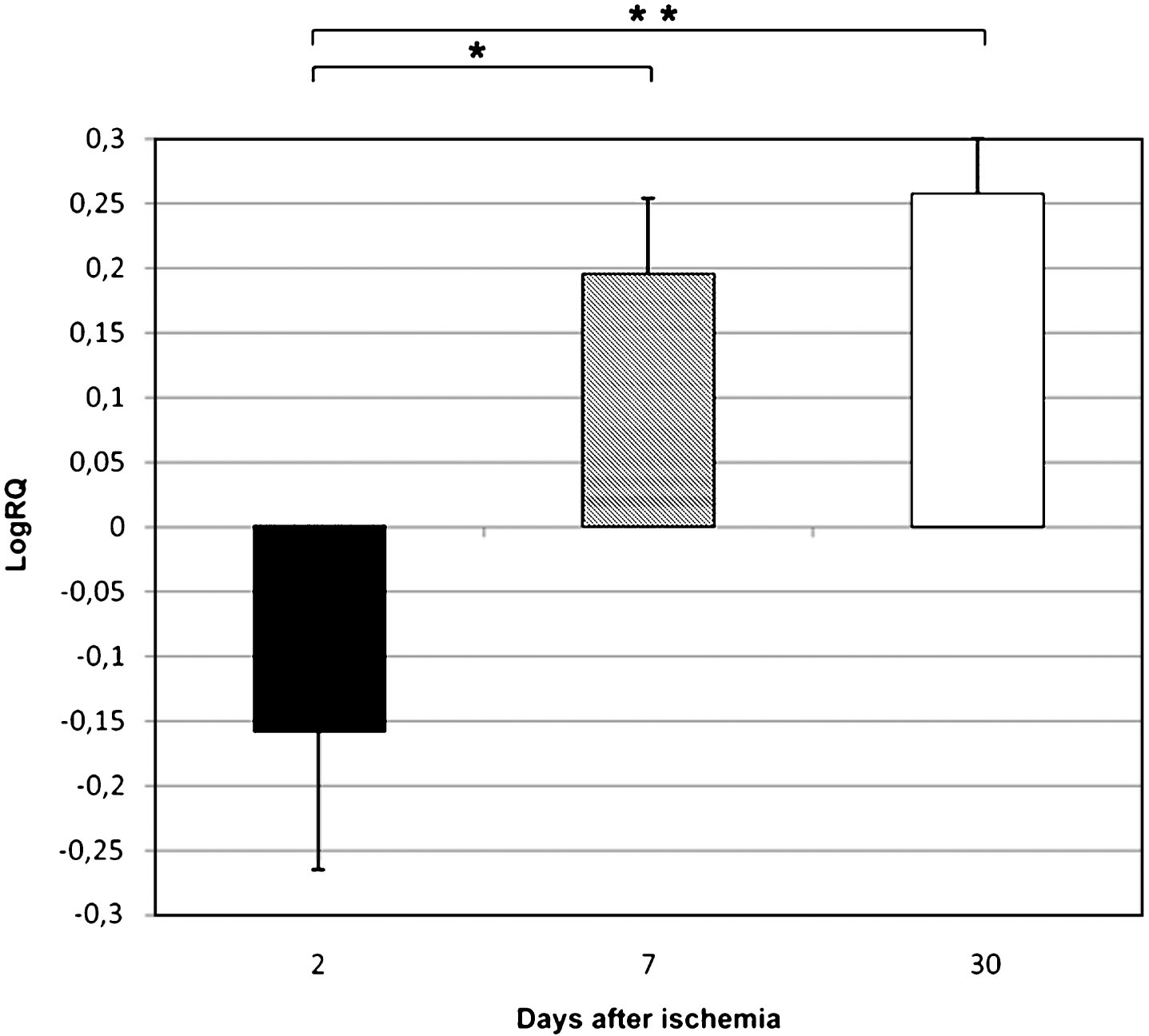
Fig.1
The mean expression levels of BECN 1 gene in the medial temporal lobe cortex in rats 2, 7, and 30 days after 10-min of global brain ischemia. Marked SEM–standard error of the mean. Indicated statistically significant difference in levels of gene expression between 2 and 30 days after 10-min of global brain ischemia (Kruskal-Wallis test). *p≤0.05.
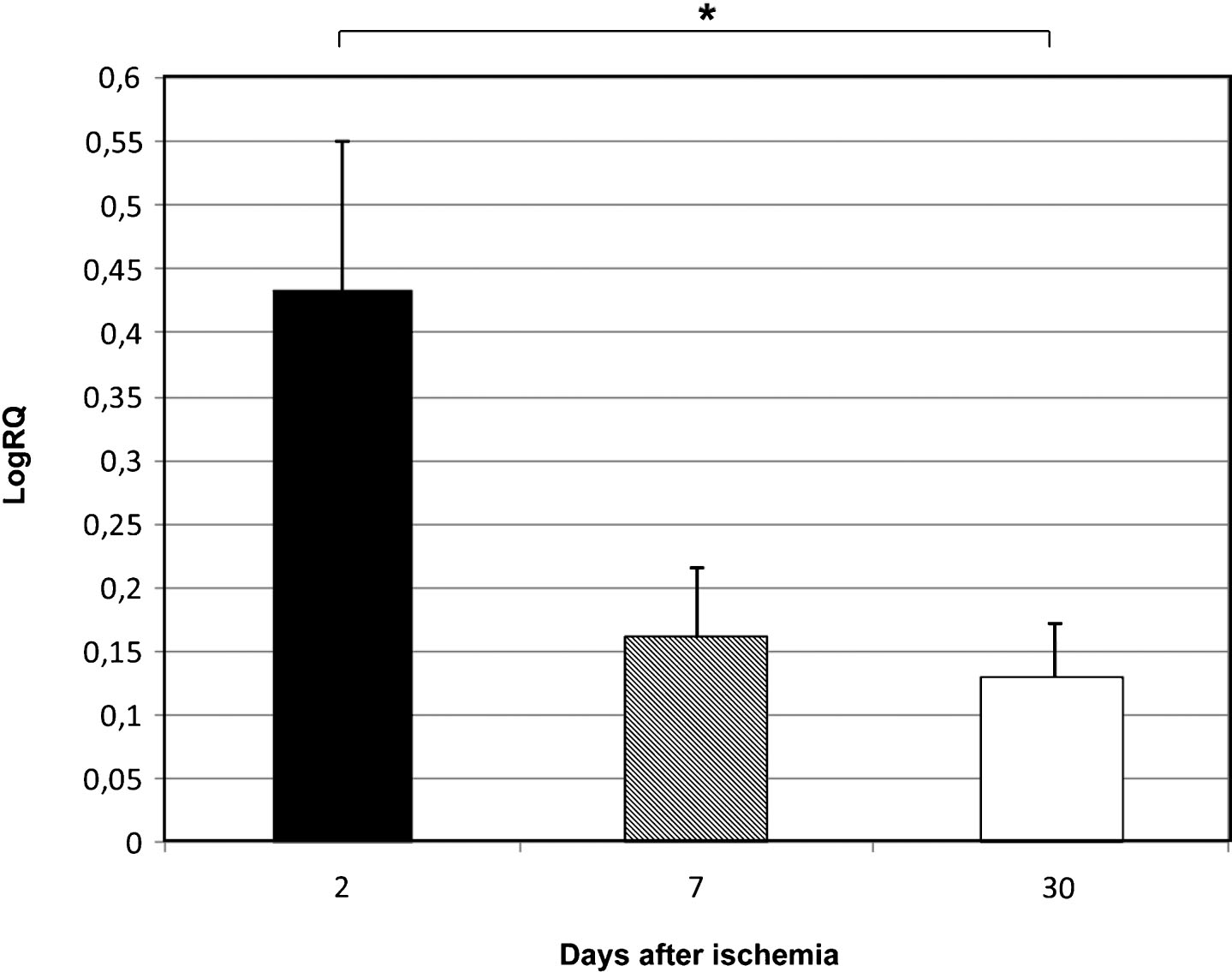
Fig.2
The mean expression levels of BNIP 3 gene in the medial temporal lobe cortex in rats 2, 7, and 30 days after 10-min of global brain ischemia. Marked SEM–standard error of the mean. Indicated statistically significant differences in levels of gene expression between 2 and 7 and between 2 and 30 days after 10-min of global brain ischemia (Kruskal-Wallis test). *p≤0.05, **p≤0.01.
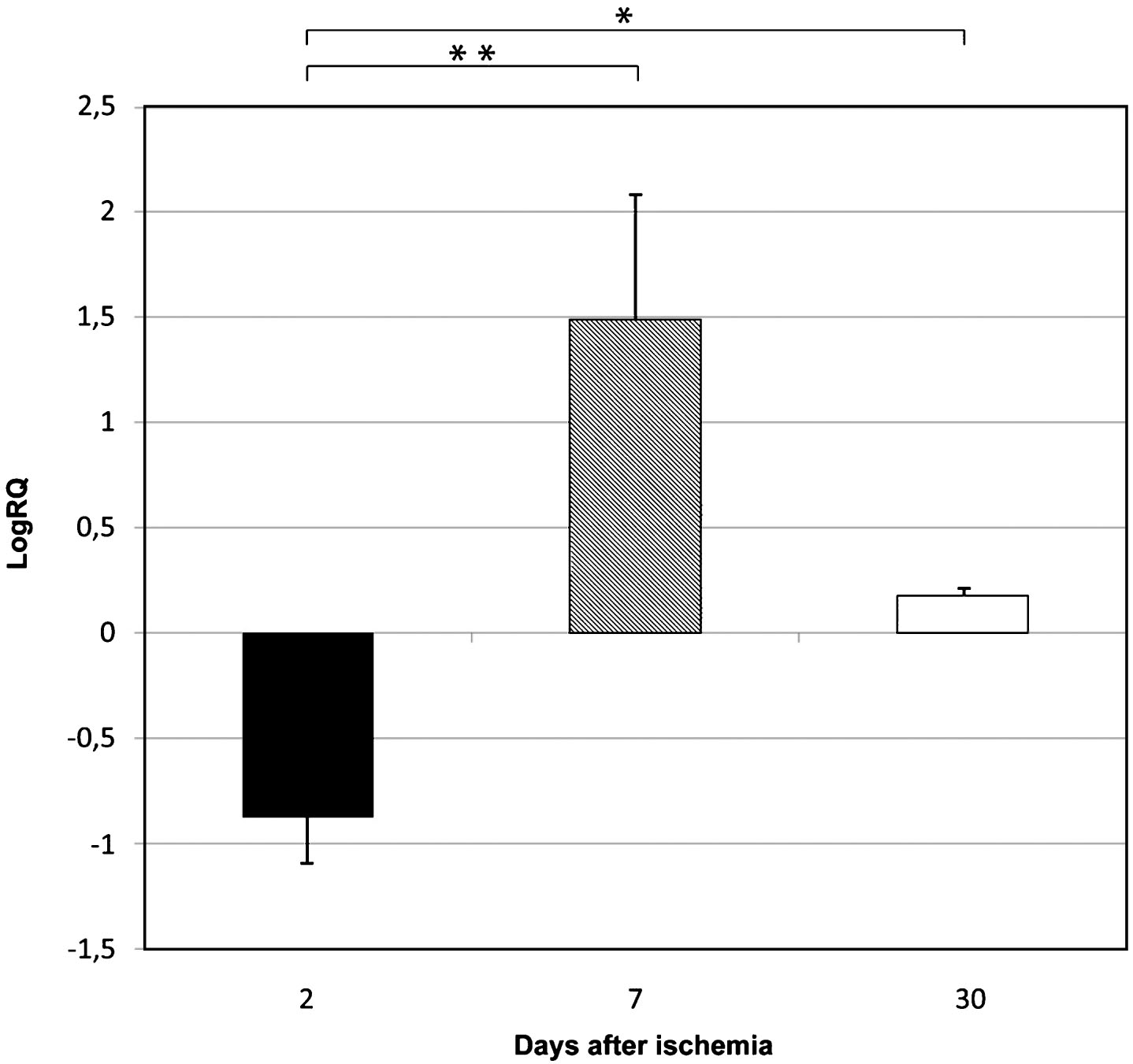
Fig.3
The mean expression levels of caspase 3 gene in the medial temporal lobe cortex in rats 2, 7, and 30 days after 10-min of global brain ischemia. Marked SEM–standard error of the mean. Indicated statistically significant differences in levels of gene expression between 2 and 7 and between 2 and 30 days after 10-min of global brain ischemia (Kruskal-Wallis test). *p≤0.05, **p≤0.01.