
A Genetic Variant of the Sortilin 1 Gene is Associated with Reduced Risk of Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder represented by the accumulation of intracellular tau protein and extracellular deposits of amyloid-β (Aβ) in the brain. The gene sortilin 1 (SORT1) has previously been associated with cardiovascular disease in gene association studies. It has also been proposed to be involved in AD pathogenesis through facilitating Aβ clearance by binding apoE/Aβ complexes prior to cellular uptake. However, the neuropathological role of SORT1 in AD is not fully understood. To evaluate the associations between gene variants of SORT1 and risk of AD, we performed genetic analyses in a Swedish case-control cohort. Ten single nucleotide polymorphisms (SNPs), covering the whole SORT1 gene, were selected and genotyped in 620 AD patients and 1107 controls. The SNP rs17646665, located in a non-coding region of the SORT1 gene, remained significantly associated with decreased risk of AD after multiple testing (pc = 0.0061). In addition, other SNPs were found to be nominally associated with risk of AD, as well as altered cognitive function and the CSF biomarker Aβ42, but these associations did not survive correction for multiple testing. The fact that SORT1 has been strongly associated with risk of cardiovascular disease is intriguing as cardiovascular disease is also regarded as a risk factor for AD. Finally, increased knowledge about SORT1 function has a potential to increase our understanding of APOE, the strongest risk factor for AD.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder and the most predominant form of dementia in the elderly population, with a prevalence of 10% in adults older than 65 years of age [1]. The disease is characterized by a gradual synaptic and neuronal loss that causes cognitive impairment with gradual progression into severe dementia with a terminal outcome. The histopathological hallmarks of AD include the accumulation of intracellular neurofibrillary tangles composed of hyperphosphorylated protein tau (P-tau) and extracellular deposits of amyloid-β (Aβ) in the brain of AD patients [1]. Today, a genetic background can partly explain the incidence of early-onset AD, also called familial AD (fAD), where the genes involved in Aβ production and processing contain mutations, namely amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) [2].
For late-onset, sporadic AD (sAD), which is the predominant form of AD, the APOE ɛ4 allele has been shown to be the major genetic risk factor [3, 4]. ApoE is mainly produced by astrocytes and microglia in the central nervous system [5] where it delivers peptides to neurons for synaptic maintenance [6]. With regard to AD, the role of apoE is not fully understood. Although some evidence shows that apoE can bind to Aβ and facilitate cellular uptake of the peptide from the extracellular space into endosomes for catabolism [7, 8], the underlying molecular mechanisms are still unclear.
Sortilin 1 (referred to as sortilin here), is a transmembrane sorting receptor encoded by the SORT1 gene. Sortilin is expressed both in neurons and non-neuronal cells and was first discovered in a screen for endocytic receptors [9]. In the brain, high levels of sortilin expression and immunoreactivity have been found in neuronal cell bodies and dendrites of allocortical areas such as the piriform cortex and hippocampus [10]. Sortilin was initially recognized as an intracellular transporter located in the trans-Golgi network [11], showing high similarities to the sorting receptor VPS10P in Saccharomyces cerevisiae [12]. The VPS10P domain of sortilin encompasses the extracellular part of the protein [13] and is shared among receptors in the VPS10P domain receptor family [14]. Sortilin is involved in complex trafficking patterns in which it can relocate from the cell surface and bind unrelated ligands, mediating transport to various cellular compartments including endosomes and lysosomes [15].
With regard to AD, sortilin has been proposed to be involved in the pathogenesis through several, sometimes contradictory, mechanisms. In cell studies, sortilin facilitates retrograde trafficking of the AβPP-cleaving enzyme beta secretase-1 (BACE-1) to Golgi and increases the cleavage of AβPP, stimulating Aβ production [16, 17]. Also, oligomerized Aβ has been shown to act as a ligand for sortilin, inducing endocytosis of Aβ and apoptosis [18]. Additionally, other experiments have revealed that sortilin also binds to and targets AβPP for lysosomal degradation, as well as promotes α-secretase cleavage of AβPP [19]. Studies in rodents demonstrate that sortilin is capable of binding extracellular apoE/Aβ complexes delivering them to lysosomes for degradation. Hence, rodents lacking the sortilin gene displayed increased apoE levels and Aβ plaque burden [20]. Furthermore, a significant increase in sortilin protein levels has been observed in postmortem brains has of patients with AD[21].
Single nucleotide polymorphisms (SNPs) within and in the vicinity of the SORT1 gene have previously been associated with cardiovascular diseases (CVD) [22–27] and serum LDL levels [25, 28–33]. Also, altered SORT1 mRNA expression [34] and increased serum levels of sortilin has been found in patients suffering from depression, where two SNPs within SORT1 were significantly associated with increased serum levels of the protein [35].
Gene association studies have highlighted sortilin-related receptor 1 (SORL1), another member of the VPS10P domain receptor family, as a candidate gene for AD [36]. According to AlzGene [37], a database collecting published genetic association data in AD patients, no studies so far have shown any significant associations between gene variants of SORT1 and the risk of developing AD. However, since preclinical findings indicate promisingfunctional roles for sortilin in AD pathogenesis, we were intrigued to test whether SORT1 was associated with AD in a clinical AD material. In the present study we report a case-control associationstudy of SORT1 in a Swedish cohort of AD patients and controls of Caucasian descent. The genetic variation in SORT1 was tested against diagnosis, mini-mental state examination (MMSE)and characteristic cerebrospinal fluid (CSF) bio-markers for AD, i.e., total tau (T-tau), P-tau, andAβ42.
MATERIALS AND METHODS
Study cohort
The Swedish case-control study population consisted of 620 AD patients and 1,107 controls of Caucasian origin. AD patients were recruited from Piteå (n = 182) and Malmö (n = 438), whereas control patients were recruited from Malmö (n = 433) and Gothenburg (Mölndal) (n = 674). Demographics for the participants are found in Table 1. All diagnoses were set according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders (NINCDS-ADRDA) criteria [38] following detailed clinical investigation including medical history, physical, neurological and psychiatric examination, screening laboratory tests, ECG, chest X-rays, EEG, and computerized tomography (CT) of the brain. No patient had a family history raising suspicion of familial AD, but PSEN1, PSEN2, and APP genes were not sequenced. Controls had no dementia and showed no signs of other psychiatric or neurological disorders including stroke, malignant disorders or infectious diseases. Controls with a Mini-Mental State Examination (MMSE) score lower than 28 were excluded from the study to reduce the risk of including controls with potential of developing AD. MMSE score data were available for 568 AD patients and 837 controls and were tested according to Folstein et al. [39]. The APOE ɛ4 carrier status is presented in Table 1. Quantifications of AD-related CSF biomarkers (T-tau, P-tau, and Aβ42) were available for a subset of the AD patients (T-tau n = 493, P-tau n = 266, and Aβ42 n = 481). The diagnosis was set without prior knowledge of genetic background or biochemical analysis and vice versa.
Genotyping and DNA preparation
The sequence of SORT1 (Gene ID: 6272) was obtained from the UCSC Genome Browser, assembly GRCh38/hg38 [40] and the HapMap project [41]. The software Haploview 4.2 was used to create a linkage disequilibrium (LD) block and to identify genetic variation (tag SNPs) in the gene. The TAGGER function in Haploview was used to select tag SNPs with the criteria of a minor allele frequency (MAF) ≥0.05 and a pairwise tagging was set to r2 = 0.8.
A regional linkage disequilibrium (LD) plot was created for the SNP rs17646665 using SNAP software [42], to localize SNPs in the vicinity of rs17646665 which are in LD. The plot was created using the parameters: 1000 Genomes Pilot 1 dataset, CEU population panel, r2 threshold set to 0.8 with a distance limit of 500.
In total, nine tag SNPs were chosen to fully cover the SORT1 gene, forming one LD block (Fig. 1). In addition, the SNP rs646776 was also included in the study since it has been shown to regulate the expression of SORT1 and has previously been associated with LDL serum levels as well as cardiovascular implications [28, 43].
Genomic DNA was extracted from whole blood using GenoPrep™ DNA Blood and DNA MagAttract kit (Qiagen, Germany), together with theGenoMTM-48 Robotic Workstation (GenoVision, Norway) and stored in –20°C until it was used for analysis. Samples were sent to LGC Genomics [44] for sequencing using a KASP™ genotyping assay.
Cerebrospinal fluid analysis
Studies have shown that AD patients have decreased levels of Aβ42 and increased levels of T-tau and P-tau in CSF compared with controls [45]. Levels of T-tau, P-tau, and Aβ42 were therefore measured and used as biochemical markers in this study [46, 47]. CSF samples were obtained from AD patients by lumbar puncture in the L3/L4 or L4/L5 interspace as previously described [48]. T-tau and P-tau were determined using a sandwich ELISA (Innotest™ hTAU-Ag, Fujirebio, Ghent, Belgium) [46] and Aβ42 levels were also measured in a sandwich ELISA (Innotest β-amyloid (1-42), Fujirebio, Ghent, Belgium) specifically constructed for Aβ42 as described previously [47].
Statistical analyses
Controls and AD patients were compared using t-test for continuous parameters (MMSE, age, and CSF biomarkers) whereas Pearson chi-square test was used when categorical parameters (APOE ɛ4 carrier status and sex) were analyzed. All SNPs were tested for deviation from Hardy-Weinberg equilibrium (HWE). SNP association to diagnosis was performed using logistic regression with an additive genetic model with sex and number of APOE ɛ4 alleles as covariates. Single marker associations to continuous parameters (MMSE and CSF biomarkers) were only performed in the AD patient group and analyzed using linear regression with sex, age, and number of APOE ɛ4 alleles as covariates.P-values <0.05 were considered statistically significant. A permutation test (set to 10 000 permutations) was performed for all SNPs to correct for multiple testing. Haplotype analyses were performed using a window size of 2-3. The statistical analyses in this study were carried out using IBM SPSS Statistics version 20, (New York, NY, USA) and Plink v1.07 [49].
Ethics
The clinical evaluation of all study participants was set without any prior knowledge of genetic background or any other biochemical analysis results, and vice versa. Participants (or their close relatives) gave their consent for participating in the study as well as for future results being published, which was directed in accordance with the provisions of the Helsinki Declaration. This study was approved by the ethical committees in Lund, Gothenburg and Umeå.
RESULTS
In this study, a total of 1727 samples were analyzed, including 620 AD patients and 1107 controls of Swedish, Caucasian descent (Table 1). When background parameters were compared, APOE ɛ4 allele frequencies, sex, age, and MMSE were shown to differ between AD cases and controls. In the case of APOE ɛ4 and MMSE, our results are expected and in line with prior knowledge that AD patients carry the APOE ɛ4 allele more frequently and show lower MMSE scores. Regarding distribution differences in sex, a portion of the controls were recruited from a longitudinal population study of female participants which explains the skewed ratio.
We selected nine SNPs to cover the full SORT1 gene, and also included a SNP located downstream of SORT1 (Table 2 and Fig. 1), which has shown a strong association with LDL serum levels [25, 28, 30, 31, 33]. The genotyping of SORT1 generated an average call rate of 98.4% for all selected SNPs in this study. Genotype frequencies were in agreement with HWE for both AD patients and controls. The MAFs ranged from 0.063 to 0.47 which was in the same range as the published HapMap data for the same SNPs in other Caucasian cohorts [41].
Next, we used the genotyping data to investigate possible associations between SORT1 and the risk of AD (Table 3). When only the SNPs in SORT1 were used in the calculations for association to AD, i.e., without any covariates in the analysis, the SNPs rs17646665 (p = 0.0034) and rs72646553 (p = 0.014) were both nominally associated with a reduced risk for AD. When the identified covariates for this population (sex and number of APOE ɛ4 alleles) were included in the analysis, rs72646553 was no longer significantly associated with diagnosis (padj = 0.052) (Table 3). However, rs17646665 remained strongly significantly (padj = 0.00063) associated with risk of AD (Table 3). The association also survived a multiple testing correction (pc = 0.0061).
Since the patients originated from different regions in Sweden, there is a risk that the association would be the result of regional genetic differences. To evaluate this possibility, we performed an analysis of rs17646665 in the Malmö/Lund population only where both controls and AD patients were available. This analysis showed the same association with risk of AD (ORadj (95% CI) = 0.5 (0.3–0.8); padj = 0.0015) as our analysis of the whole population. This indicates that the association was not due to regional genetic differences in the populations studied. A haplotype analysis was also performed although the significant associations found did not add anything beyond the association of the SNP associated with the disease, rs17646665.
To further investigate genes that could be related to our gene variant of SORT1, we created a regional LD plot from available CEU population panel data for rs17646665 to localize SNPs in its vicinity, which are in LD (Fig. 2). According to the regional LD plot, it is clear that no other SNP within the SORT1 gene are in strong LD with rs17646665. In total, six SNPs were identified (r2 >0.8) and localized in or near the genes synaptophysin-like 2 (SYPL2; rs2272272), adhesion molecule with Ig-like domain 1 (AMIGO1; rs17575427), 5’ of G protein-coupled receptor 61 (GPR61; rs552101), between AMIGO1 and GPR61 (rs56018934), guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 3 (GNAI3; rs1279195) and glutathione S-transferase mu 4 (GSTM4; rs650985). This indicates that these SNPs are also likely to be associated with risk ofAD.
Finally, all SNPs in this study were also analyzed in relation to MMSE scores and levels of CSF biomarkers T-tau, P-tau, and Aβ42 in the AD patients. For MMSE, the minor allele of rs7536292 (adjusted slope (βadj) = 0.85, padj = 0.044) was nominally significantly associated with a higher MMSE score, although the results did not survive a multiple testing correction (pc = 0.26).
In the case of Aβ42, the two SNPs rs646776 (βadj = 26.60, padj = 0.031) and rs72646553 (βadj = –33.07, padj = 0.010) were both nominally associated with altered levels of this biomarker. The minor allele of rs646776 was associated with higher levels of Aβ42, whereas rs72646553 showed a reverse trend, with a minor allele associated with lower Aβ42 levels. These associations were however not significant after a correction (rs646776 pc = 0.20; rs72646553 pc = 0.082). Regarding T-tau and P-tau, no SNPs were found to be associated with levels of the two biomarkers. A haplotype analysis was also performed, but no significant associations were found.
DISCUSSION
The main objective of this study was to investigate possible associations between variants of the SORT1 gene and the risk of AD. In addition, in the AD cohort, the SNPs were also tested for associations to the AD CSF biomarker levels, as well as MMSE scores, with the latter serving as a measure for cognitive function. To this end, SNPs selected in this study consisted of both new tag SNPs as well as SNPs previously investigated for association to AD or other diseases (Table 2). Our results showed that the minor allele of rs17646665 was strongly associated (padj = 0.00063; pc = 0.0061) with a reduced risk of AD. The association thus remained significant after adjusting for relevant covariates (sex and number of APOE ɛ4 alleles) and correcting for multiple testing. To evaluate this association further, future replications in Swedish and other populations are warranted.
As the SNP rs17646665 has not previously been associated with any disease, we were interested in studying it further. According to the regional LD plot created for rs17646665 (Fig. 2), it is clear that no other SNPs within the SORT1 gene are in LD with rs17646665. The six SNPs in LD with rs17646665 were localized in or near the genes SYPL2 (rs2272272), AMIGO1 (rs17575427), 5’ of GPR61 (rs552101), between AMIGO1 and GPR61 (rs56018934), GNAI3 (rs1279195) and GSTM4 (rs650985). This indicates that variants in these genes have the potential to be involved in the pathogenesis of AD. Interestingly, AMIGO1 has been shown to be involved in both neuronal development and survival, with a potential correlation to Aβ42 [50, 51]. In addition, analysis of rs17646665 in Genotype-Tissue Expression (GTEx) databases at the GTEx Portal [52] revealed that this SNP is associated with reduced SYPL2 expression in many different tissues. Moreover, SYPL2 (also known as mitsugumin 29) expression has been identified in astrocytes around Aβ plaques in AD brains [53].
For AD patients, genetic variations in SORT1 were also investigated with regard to characteristic AD CSF biomarkers, namely T-tau, P-tau and Aβ42, as well as MMSE scores. While rs17646665, which was associated with risk of AD, showed no significant association to any of the CSF biomarkers, rs646776 and rs72646553 were nominally associated with CSF levels of Aβ42. The two SNPs had different effects on Aβ42 levels, with the minor allele of rs646776 being associated with increased CSF Aβ42 levels, whilst the minor allele of rs72646553 caused an opposing outcome. This is in line with the LD pattern between the two SNPs.
The SNP rs646776 is located in a non-coding DNA region in-between the genes cadherin EGF LAG seven-pass G-type receptor 2 (CELSR2) and proline and serine rich coiled-coil 1 (PSRC1), where the minor allele has been associated with increase in expression of SORT1. This is in agreement with the hypothesis that increased sortilin levels are protective against Aβ42 accumulation by facilitating uptake and degradation of apoE/Aβ complexes [20], as increased CSF levels of Aβ42 correspond to lower Aβ42 aggregation in the brain [47].
The SNP rs72646553 is localized in the first exon of SORT1 where the genetic variation generates a synonymous mutation. Data from the GTEx portal indicate that the minor allele is associated with a slight increase in expression of SORT1 that could affect Aβ42 metabolism. However, in the present study, rs72646553 was both nominally associated with decreased Aβ42 CSF levels, indicative of AD, but it was also nominally associated with reduced risk of AD. It is therefore difficult to draw any conclusions from this finding. Although the results were nominally significant, neither of the SNPs associated with Aβ42 levels survived correction for multiple testing.
For MMSE, the minor C allele of another SNP, rs7536292, was nominally associated with higher MMSE scores, but this association did not remain significant after multiple testing correction. None of the SNPs were associated with T-tau or P-tau, neither before nor after correction. This would be logical as unaltered P-tau levels has been reported in Sort1(-/-) mice crossed with an AD model strain [54].
When comparing with previous studies, all SNPs have previously been analyzed in different AD populations, in relation to sAD [55–59], fAD [36], or dementia [60]. In the latter study, Reynolds et al. found rs17585335 to be nominally associated with risk of dementia, but the authors concluded that the association was not convincing enough [60]. Zeng et al. investigated a possible association of SORT1 and AD in a Han Chinese population where none of the tested SNPs showed any significant result [56]. Indeed, two of the SNPs in the study (rs646776 and rs464218) were also included in our study, confirming no associations with AD. On the other hand, the SNP rs17646665 that our study highlighted as strongly associated with a lower risk of AD, was not included in theirs and this SNP shows a very low LD to the tested SNPs in the Han Chinese study, which might explain the negative outcome. Additionally, we assume that a genetic variation may exist between cohorts from different regions, i.e., when comparing Han Chinese and European Caucasians. Moreover, rs17646665 has previously been included in a large scale study where several members of the VPS10P domain receptors family were tested for association with AD [55], but no associations were found for rs17646665 in this study or in previous GWAS analyses [57–59]. Due to differences in reported studies and our material, it is possible that the association in our cohort could be restricted to a Swedish/Scandinavian population.
CVD and serum lipid levels are two of the major risk factors for developing AD [61], and as previously described, SORT1 has been shown to be associated with both serum lipid levels and CVD. In other words, SORT1 might play an important role in explaining the underlying relation between CVD, serum lipid levels, and AD. In the present study, we have indeed found an association between SORT1 and the risk of AD. To expand our knowledge in this field, we need to further investigate how SORT1 is linked to the disease by exploring underlying mechanisms. We suggest that future research will explore measurements of AD CSF biomarkers in combination with biomarkers for CVD and serum lipid levels, since this combined may generate useful information and knowledge regarding the link between CVD and AD, which would be beneficial for future drug development. We also suggest that pharmacological tools that modulate the uptake and clearance of apoE-Aβ-sortilin complexes could serve as future therapeutics for AD.
ACKNOWLEDGMENTS
Funding for this study was provided by the Swedish Dementia Association (CHA, PK and AW), the Foundation Professor Bror Gadelius minnesfond (CHA), the Nilsson-Ehle Donations (PK), the Kungl. Vetenskaps- och Vitterhets- Samhället in Gothenburg (PK), the Foundation Sigurd och Elsa Goljes Minne (PK), the Swedish Alzheimer Foundation (PK and AW), The Swedish Society of Medicine (PK and AW), the Torsten Söderberg’s Foundation (PK), Region Västra Götaland (PK), Stiftelsen Längmanska kulturfonden (PK), Magnus Bergvalls Stiftelse (PK), Tore Nilsons Stiftelse (PK), Gunvor & Josef Anérs stiftelse (PK), Marcus Borgströms stiftelse (PK), K. och O.F. Hedströms minnesfond (PK), Wilhelm och Martina Lundgrens stiftelser (PK), Åhlén-stiftelsen (PK), O. E. och Edla Johanssons vetenskapliga stiftelse (PK), Åke Wibergs stiftelse (PK), the Knut and Alice Wallenberg Foundation (HZ), the Swedish Research Council (OH, KB, HZ, IS and AW), ERC (OH and HZ), American Alzheimer Association (KB, IS), Frimurarestiftelsen (AW, HZ), Stiftelsen Psykiatriska forskningsfonden (AW), the Gothenburg Medical Society (AW), Swedish Brain Power (AW) and Sahlgrenska University Hospital (AW). The research leading to these results has received funding to PK from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement n° 608743. We would also like to thank Rakesh Banote, Anna Zettergren, and Mona Seibt Palmér for help with handling of samples and Bryn Farnsworth, Tad Heppner, and Jenny Landin for critical reading of the manuscript.
Authors’ disclosures are available online (http://j-alz.com/manuscript-disclosures/16-0319r2).
REFERENCES
[1] | Blennow K , de Leon MJ , Zetterberg H ((2006) ) Alzheimer’s disease. Lancet 368: , 387–403. |
[2] | Tanzi RE , Bertram L ((2005) ) Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 120: , 545–555. |
[3] | Saunders AM , Strittmatter WJ , Schmechel D , George-Hyslop PH , Pericak-Vance MA , Joo SH , Rosi BL , Gusella JF , Crapper-MacLachlan DR , Alberts MJ , Hulette C , Crain B , Goldgaber D , Roses AD ((1993) ) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43: , 1467–1472. |
[4] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[5] | Pitas RE , Boyles JK , Lee SH , Foss D , Mahley RW ((1987) ) Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta 917: , 148–161. |
[6] | Mauch DH , Nagler K , Schumacher S , Goritz C , Muller EC , Otto A , Pfrieger FW ((2001) ) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294: , 1354–1357. |
[7] | Koistinaho M , Lin S , Wu X , Esterman M , Koger D , Hanson J , Higgs R , Liu F , Malkani S , Bales KR , Paul SM ((2004) ) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10: , 719–726. |
[8] | Jiang Q , Lee CY , Mandrekar S , Wilkinson B , Cramer P , Zelcer N , Mann K , Lamb B , Willson TM , Collins JL , Richardson JC , Smith JD , Comery TA , Riddell D , Holtzman DM , Tontonoz P , Landreth GE ((2008) ) ApoE promotes the proteolytic degradation of Abeta. Neuron 58: , 681–693. |
[9] | Petersen CM , Nielsen MS , Nykjaer A , Jacobsen L , Tommerup N , Rasmussen HH , Roigaard H , Gliemann J , Madsen P , Moestrup SK ((1997) ) Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J Biol Chem 272: , 3599–3605. |
[10] | Sarret P , Krzywkowski P , Segal L , Nielsen MS , Petersen CM , Mazella J , Stroh T , Beaudet A ((2003) ) Distribution of NTS3 receptor/sortilin mRNA and protein in the rat central nervous system. J Comp Neurol 461: , 483–505. |
[11] | Morinville A , Martin S , Lavallee M , Vincent JP , Beaudet A , Mazella J ((2004) ) Internalization and trafficking of neurotensin via NTS3 receptors in HT29 cells. Int J Biochem Cell Biol 36: , 2153–2168. |
[12] | Marcusson EG , Horazdovsky BF , Cereghino JL , Gharakhanian E , Emr SD ((1994) ) The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 77: , 579–586. |
[13] | Quistgaard EM , Madsen P , Groftehauge MK , Nissen P , Petersen CM , Thirup SS ((2009) ) Ligands bind to Sortilin in the tunnel of a ten-bladed beta-propeller domain. Nat Struct Mol Biol 16: , 96–98. |
[14] | Willnow TE , Petersen CM , Nykjaer A ((2008) ) VPS10P-domain receptors - regulators of neuronal viability and function. Nat Rev Neurosci 9: , 899–909. |
[15] | Nykjaer A , Willnow TE ((2012) ). Sortilin: A receptor to regulate neuronal viability and function. Trends Neurosci 35: , 261–270. |
[16] | Finan GM , Okada H , Kim TW ((2011) ) BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J Biol Chem 286: , 12602–12616. |
[17] | Tan J , Evin G ((2012) ) Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J Neurochem 120: , 869–880. |
[18] | Takamura A , Sato Y , Watabe D , Okamoto Y , Nakata T , Kawarabayashi T , Oddo S , Laferla FM , Shoji M , Matsubara E ((2012) ) Sortilin is required for toxic action of Abeta oligomers (AbetaOs): Extracellular AbetaOs trigger apoptosis, and intraneuronal AbetaOs impair degradation pathways. Life Sci 91: , 1177–1186. |
[19] | Gustafsen C , Glerup S , Pallesen LT , Olsen D , Andersen OM , Nykjaer A , Madsen P , Petersen CM ((2013) ) Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. J Neurosci 33: , 64–71. |
[20] | Carlo AS , Gustafsen C , Mastrobuoni G , Nielsen MS , Burgert T , Hartl D , Rohe M , Nykjaer A , Herz J , Heeren J , Kempa S , Petersen CM , Willnow TE ((2013) ) The pro-neurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-beta peptide in the brain. J Neurosci 33: , 358–370. |
[21] | Saadipour K , Yang M , Lim Y , Georgiou K , Sun Y , Keating D , Liu J , Wang YR , Gai WP , Zhong JH , Wang YJ , Zhou XF ((2013) ) Amyloid beta(1)(-)(4)(2) (Abeta(4)(2)) up-regulates the expression of sortilin via the p75(NTR)/RhoA signaling pathway. J Neurochem 127: , 152–162. |
[22] | Takeuchi F , Isono M , Katsuya T , Yokota M , Yamamoto K , Nabika T , Shimokawa K , Nakashima E , Sugiyama T , Rakugi H , Yamaguchi S , Ogihara T , Yamori Y , Kato N ((2012) ) Association of genetic variants influencing lipid levels with coronary artery disease in Japanese individuals. PLoS One 7: , e46385. |
[23] | Jones GT , Bown MJ , Gretarsdottir S , Romaine SP , Helgadottir A , Yu G , Tromp G , Norman PE , Jin C , Baas AF , Blankensteijn JD , Kullo IJ , Phillips LV , Williams MJ , Topless R , Merriman TR , Vasudevan TM , Lewis DR , Blair RD , Hill AA , Sayers RD , Powell JT , Deloukas P , Thorleifsson G , Matthiasson SE , Thorsteinsdottir U , Golledge J , Ariens RA , Johnson A , Sohrabi S , Scott DJ , Carey DJ , Erdman R , Elmore JR , Kuivaniemi H , Samani NJ , Stefansson K , van Rij AM ((2013) ) A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum Mol Genet 22: , 2941–2947. |
[24] | Lee JY , Lee BS , Shin DJ , Woo Park K , Shin YA , Joong Kim K , Heo L , Young Lee J , Kyoung Kim Y , Jin Kim Y , Bum Hong C , Lee SH , Yoon D , Jung Ku H , Oh IY , Kim BJ , Lee J , Park SJ , Kim J , Kawk HK , Lee JE , Park HK , Lee JE , Nam HY , Park HY , Shin C , Yokota M , Asano H , Nakatochi M , Matsubara T , Kitajima H , Yamamoto K , Kim HL , Han BG , Cho MC , Jang Y , Kim HS , Euy Park J , Lee JY ((2013) ) A genome-wide association study of a coronary artery disease risk variant. J Hum Genet 58: , 120–126. |
[25] | Arvind P , Nair J , Jambunathan S , Kakkar VV , Shanker J ((2014) ) CELSR2-PSRC1-SORT1 gene expression and association with coronary artery disease and plasma lipid levels in an Asian Indian cohort. J Cardiol 64: , 339–346. |
[26] | Angelakopoulou A , Shah T , Sofat R , Shah S , Berry DJ , Cooper J , Palmen J , Tzoulaki I , Wong A , Jefferis BJ , Maniatis N , Drenos F , Gigante B , Hardy R , Laxton RC , Leander K , Motterle A , Simpson IA , Smeeth L , Thomson A , Verzilli C , Kuh D , Ireland H , Deanfield J , Caulfield M , Wallace C , Samani N , Munroe PB , Lathrop M , Fowkes FG , Marmot M , Whincup PH , Whittaker JC , de Faire U , Kivimaki M , Kumari M , Hypponen E , Power C , Humphries SE , Talmud PJ , Price J , Morris RW , Ye S , Casas JP , Hingorani AD ((2012) ) Comparative analysis of genome-wide association studies signals for lipids, diabetes, and coronary heart disease: Cardiovascular Biomarker Genetics Collaboration. Eur Heart J 33: , 393–407. |
[27] | Qi L , Ma J , Qi Q , Hartiala J , Allayee H , Campos H ((2011) ) Genetic risk score and risk of myocardial infarction in Hispanics. Circulation 123: , 374–380. |
[28] | Kathiresan S , Melander O , Guiducci C , Surti A , Burtt NP , Rieder MJ , Cooper GM , Roos C , Voight BF , Havulinna AS , Wahlstrand B , Hedner T , Corella D , Tai ES , Ordovas JM , Berglund G , Vartiainen E , Jousilahti P , Hedblad B , Taskinen MR , Newton-Cheh C , Salomaa V , Peltonen L , Groop L , Altshuler DM , Orho-Melander M ((2008) ) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40: , 189–197. |
[29] | Willer CJ , Sanna S , Jackson AU , Scuteri A , Bonnycastle LL , Clarke R , Heath SC , Timpson NJ , Najjar SS , Stringham HM , Strait J , Duren WL , Maschio A , Busonero F , Mulas A , Albai G , Swift AJ , Morken MA , Narisu N , Bennett D , Parish S , Shen H , Galan P , Meneton P , Hercberg S , Zelenika D , Chen WM , Li Y , Scott LJ , Scheet PA , Sundvall J , Watanabe RM , Nagaraja R , Ebrahim S , Lawlor DA , Ben-Shlomo Y , Davey-Smith G , Shuldiner AR , Collins R , Bergman RN , Uda M , Tuomilehto J , Cao A , Collins FS , Lakatta E , Lathrop GM , Boehnke M , Schlessinger D , Mohlke KL , Abecasis GR ((2008) ) Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 40: , 161–169. |
[30] | Shirts BH , Hasstedt SJ , Hopkins PN , Hunt SC ((2011) ) Evaluation of the gene-age interactions in HDL cholesterol, LDL cholesterol, and triglyceride levels: The impact of the SORT1 polymorphism on LDL cholesterol levels is age dependent. Atherosclerosis 217: , 139–141. |
[31] | Keebler ME , Sanders CL , Surti A , Guiducci C , Burtt NP , Kathiresan S ((2009) ) Association of blood lipids with common DNA sequence variants at 19 genetic loci in the multiethnic United States National Health and Nutrition Examination Survey III. Circ Cardiovasc Genet 2: , 238–243. |
[32] | Gupta R , Ejebe K , Butler J , Lettre G , Lyon H , Guiducci C , Wilks R , Bennett F , Forrester T , Tayo B , Musunuru K , Hirschhorn J , Kathiresan S , Cooper RS , McKenzie CA ((2010) ) Association of common DNA sequence variants at 33 genetic loci with blood lipids in individuals of African ancestry from Jamaica. Hum Genet 128: , 557–561. |
[33] | Walia GK , Gupta V , Aggarwal A , Asghar M , Dudbridge F , Timpson N , Singh NS , Kumar MR , Kinra S , Prabhakaran D , Reddy KS , Chandak GR , Smith GD , Ebrahim S ((2014) ) Association of common genetic variants with lipid traits in the Indian population. PLoS One 9: , e101688. |
[34] | Belzeaux R , Formisano-Treziny C , Loundou A , Boyer L , Gabert J , Samuelian JC , Feron F , Naudin J , Ibrahim EC ((2010) ) Clinical variations modulate patterns of gene expression and define blood biomarkers in major depression. J Psychiatr Res 44: , 1205–1213. |
[35] | Buttenschon HN , Demontis D , Kaas M , Elfving B , Molgaard S , Gustafsen C , Kaerlev L , Petersen CM , Borglum AD , Mors O , Glerup S ((2015) ) Increased serum levels of sortilin are associated with depression and correlated with BDNF and VEGF. Transl Psychiatry 5: , e677. |
[36] | Rogaeva E , Meng Y , Lee JH , Gu Y , Kawarai T , Zou F , Katayama T , Baldwin CT , Cheng R , Hasegawa H , Chen F , Shibata N , Lunetta KL , Pardossi-Piquard R , Bohm C , Wakutani Y , Cupples LA , Cuenco KT , Green RC , Pinessi L , Rainero I , Sorbi S , Bruni A , Duara R , Friedland RP , Inzelberg R , Hampe W , Bujo H , Song YQ , Andersen OM , Willnow TE , Graff-Radford N , Petersen RC , Dickson D , Der SD , Fraser PE , Schmitt-Ulms G , Younkin S , Mayeux R , Farrer LA , St George-Hyslop P ((2007) ) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39: , 168–177. |
[37] | AlzGene database, http://www.alzgene.org/. |
[38] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[39] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[40] | USCS Genome Browser, http://genome.ucsc.edu/ |
[41] | |
[42] | SNAP Software, https://www.broadinstitute.org/mpg/snap/ldplot.php |
[43] | Musunuru K , Strong A , Frank-Kamenetsky M , Lee NE , Ahfeldt T , Sachs KV , Li X , Li H , Kuperwasser N , Ruda VM , Pirruccello JP , Muchmore B , Prokunina-Olsson L , Hall JL , Schadt EE , Morales CR , Lund-Katz S , Phillips MC , Wong J , Cantley W , Racie T , Ejebe KG , Orho-Melander M , Melander O , Koteliansky V , Fitzgerald K , Krauss RM , Cowan CA , Kathiresan S , Rader DJ ((2010) ) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466: , 714–719. |
[44] | LGC Genomics, http://www.lgcgroup.com/ |
[45] | Blennow K , Vanmechelen E , Hampel H ((2001) ) CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol Neurobiol 24: , 87–97. |
[46] | Blennow K , Wallin A , Agren H , Spenger C , Siegfried J , Vanmechelen E ((1995) ) Tau protein in cerebrospinal fluid: A biochemical marker for axonal degeneration in Alzheimer disease?. Mol Chem Neuropathol 26: , 231–245. |
[47] | Andreasen N , Hesse C , Davidsson P , Minthon L , Wallin A , Winblad B , Vanderstichele H , Vanmechelen E , Blennow K ((1999) ) Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: Differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol 56: , 673–680. |
[48] | Andreasen N , Minthon L , Vanmechelen E , Vanderstichele H , Davidsson P , Winblad B , Blennow K ((1999) ) Cerebrospinal fluid tau and Abeta42 as predictors of development of Alzheimer’s disease in patients with mild cognitive impairment. Neurosci Lett 273: , 5–8. |
[49] | PLINK Software, http://pngu.mgh.harvard.edu/ purcell/plink/. |
[50] | Piras S , Furfaro AL , Piccini A , Passalacqua M , Borghi R , Carminati E , Parodi A , Colombo L , Salmona M , Pronzato MA , Marinari UM , Tabaton M , Nitti M ((2014) ) Monomeric Abeta1-42 and RAGE: Key players in neuronal differentiation. Neurobiol Aging 35: , 1301–1308. |
[51] | Chen Y , Hor HH , Tang BL ((2012) ) AMIGO is expressed in multiple brain cell types and may regulate dendritic growth and neuronal survival. J Cell Physiol 227: , 2217–2229. |
[52] | ((2013) ) The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: , 580–585. |
[53] | Satoh K , Akatsu H , Yamamoto T , Kosaka K , Yokota H , Yamada T ((2012) ) Mitsugumin 29 is transcriptionally induced in senile plaque-associated astrocytes. Brain Res 1441: , 9–16. |
[54] | Capsoni S , Amato G , Vignone D , Criscuolo C , Nykjaer A , Cattaneo A ((2013) ) Dissecting the role of sortilin receptor signaling in neurodegeneration induced by NGF deprivation. Biochem Biophys Res Commun 431: , 579–585. |
[55] | Reitz C , Tosto G , Vardarajan B , Rogaeva E , Ghani M , Rogers RS , Conrad C , Haines JL , Pericak-Vance MA , Fallin MD , Foroud T , Farrer LA , Schellenberg GD , George-Hyslop PS , Mayeux R , Alzheimer’s Disease Genetics C ((2013) ) Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP). Transl Psychiatry 3: , e256. |
[56] | Zeng F , Deng YP , Yi X , Cao HY , Zou HQ , Wang X , Liang CR , Wang YR , Zhang LL , Gao CY , Xu ZQ , Lian Y , Wang L , Zhou XF , Zhou HD , Wang YJ ((2013) ) No association of SORT1 gene polymorphism with sporadic Alzheimer’s disease in the Chinese Han population. Neuroreport 24: , 464–468. |
[57] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thorton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin CF , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau MT , Choi SH , Reitz C , Pasquier F , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Moron FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fievet N , Huentelman MW , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuiness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossu P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Deniz Naranjo MC , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F , Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannefelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O’Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley TH Jr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RF , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JS , Boerwinkle E , Riemenschneider M , Boada M , Hiltuenen M , Martin ER , Schmidt R , Rujescu D , Wang LS , Dartigues JF , Mayeux R , Tzourio C , Hofman A , Nothen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[58] | Naj AC , Jun G , Beecham GW , Wang LS , Vardarajan BN , Buros J , Gallins PJ , Buxbaum JD , Jarvik GP , Crane PK , Larson EB , Bird TD , Boeve BF , Graff-Radford NR , De Jager PL , Evans D , Schneider JA , Carrasquillo MM , Ertekin-Taner N , Younkin SG , Cruchaga C , Kauwe JS , Nowotny P , Kramer P , Hardy J , Huentelman MJ , Myers AJ , Barmada MM , Demirci FY , Baldwin CT , Green RC , Rogaeva E , St George-Hyslop P , Arnold SE , Barber R , Beach T , Bigio EH , Bowen JD , Boxer A , Burke JR , Cairns NJ , Carlson CS , Carney RM , Carroll SL , Chui HC , Clark DG , Corneveaux J , Cotman CW , Cummings JL , DeCarli C , DeKosky ST , Diaz-Arrastia R , Dick M , Dickson DW , Ellis WG , Faber KM , Fallon KB , Farlow MR , Ferris S , Frosch MP , Galasko DR , Ganguli M , Gearing M , Geschwind DH , Ghetti B , Gilbert JR , Gilman S , Giordani B , Glass JD , Growdon JH , Hamilton RL , Harrell LE , Head E , Honig LS , Hulette CM , Hyman BT , Jicha GA , Jin LW , Johnson N , Karlawish J , Karydas A , Kaye JA , Kim R , Koo EH , Kowall NW , Lah JJ , Levey AI , Lieberman AP , Lopez OL , Mack WJ , Marson DC , Martiniuk F , Mash DC , Masliah E , McCormick WC , McCurry SM , McDavid AN , McKee AC , Mesulam M , Miller BL , Miller CA , Miller JW , Parisi JE , Perl DP , Peskind E , Petersen RC , Poon WW , Quinn JF , Rajbhandary RA , Raskind M , Reisberg B , Ringman JM , Roberson ED , Rosenberg RN , Sano M , Schneider LS , Seeley W , Shelanski ML , Slifer MA , Smith CD , Sonnen JA , Spina S , Stern RA , Tanzi RE , Trojanowski JQ , Troncoso JC , Van Deerlin VM , Vinters HV , Vonsattel JP , Weintraub S , Welsh-Bohmer KA , Williamson J , Woltjer RL , Cantwell LB , Dombroski BA , Beekly D , Lunetta KL , Martin ER , Kamboh MI , Saykin AJ , Reiman EM , Bennett DA , Morris JC , Montine TJ , Goate AM , Blacker D , Tsuang DW , Hakonarson H , Kukull WA , Foroud TM , Haines JL , Mayeux R , Pericak-Vance MA , Farrer LA , Schellenberg GD ((2011) ) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43: , 436–441. |
[59] | Beecham GW , Hamilton K , Naj AC , Martin ER , Huentelman M , Myers AJ , Corneveaux JJ , Hardy J , Vonsattel JP , Younkin SG , Bennett DA , De Jager PL , Larson EB , Crane PK , Kamboh MI , Kofler JK , Mash DC , Duque L , Gilbert JR , Gwirtsman H , Buxbaum JD , Kramer P , Dickson DW , Farrer LA , Frosch MP , Ghetti B , Haines JL , Hyman BT , Kukull WA , Mayeux RP , Pericak-Vance MA , Schneider JA , Trojanowski JQ , Reiman EM , Schellenberg GD , Montine TJ ((2014) ) Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet 10: , e1004606. |
[60] | Reynolds CA , Hong MG , Eriksson UK , Blennow K , Wiklund F , Johansson B , Malmberg B , Berg S , Alexeyenko A , Gronberg H , Gatz M , Pedersen NL , Prince JA ((2010) ) Analysis of lipid pathway genes indicates association of sequence variation near SREBF1/TOM1L2/ATPAF2 with dementia risk. Hum Mol Genet 19: , 2068–2078. |
[61] | Stampfer MJ ((2006) ) Cardiovascular disease and Alzheimer’s disease: Common links. J Intern Med 260: , 211–223. |
Figures and Tables
Fig.1
Linkage disequilibrium (LD) plot for the ten selected SNPs within and in the vicinity of the SORT1 gene. The correlation between SNPs is shown by their pairwise r2 values indicated by numbers and greyscale. The plot was generated using Haploview software from the genotyping data in the cohort.
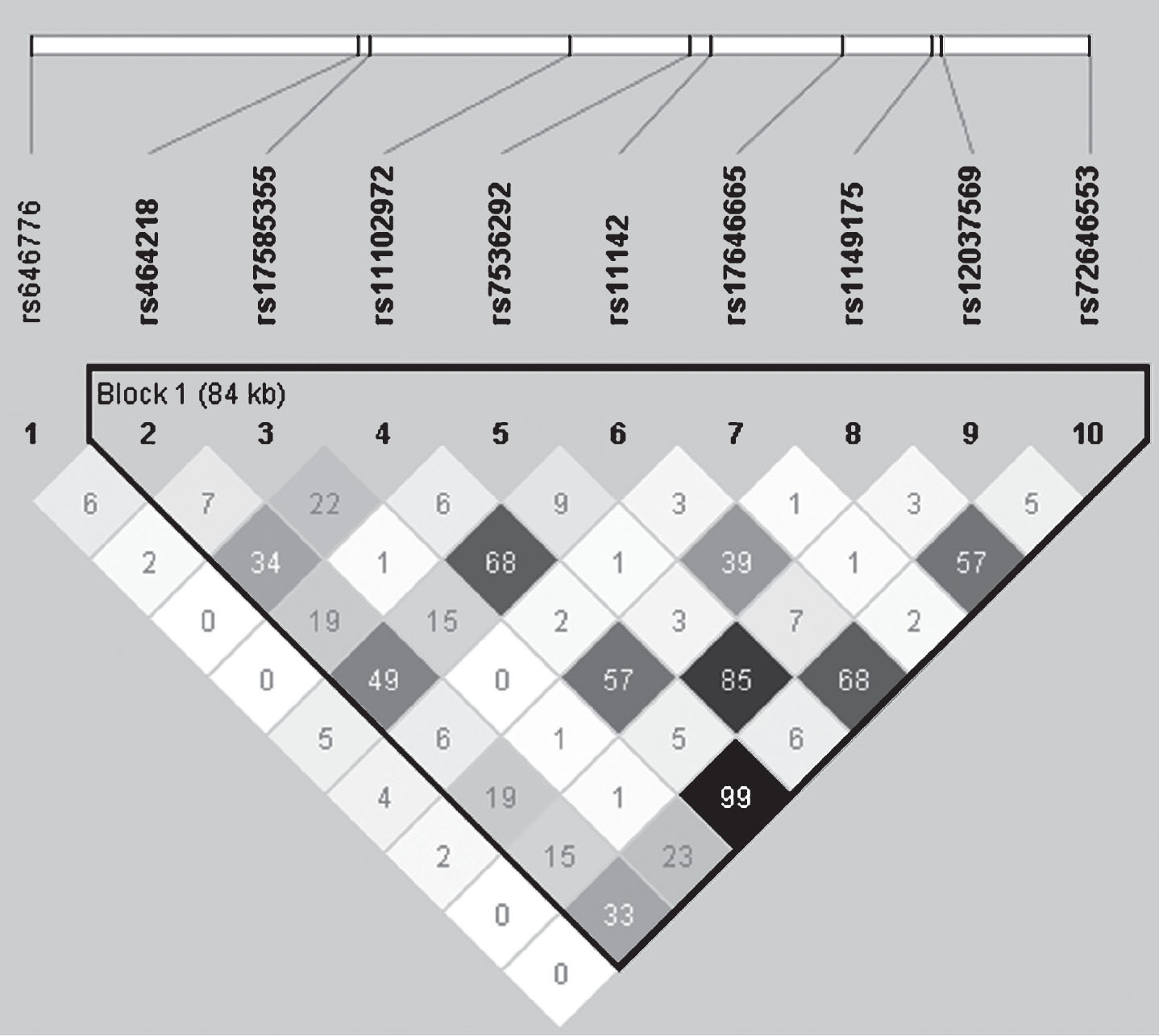
Fig.2
The regional LD plot was created from a published CEU population panel for rs17646665, located in SORT1 (marked by the arrow) with 500 kb flanking genomic regions on each side. The r2 threshold was set to 0.8 and six SNPs were identified with r2 >0.8 for this region, all of which were located in other genes than SORT1. The SNPs are indicated by the numbers over the boxes (1: SYPL2 (rs2272272), 2: AMIGO1 (rs17575427), 3: 5’ of GPR61 (rs552101), 4: between AMIGO1 and GPR61 (rs56018934), 5: GNAI3 (rs1279195) and 6: GSTM4 (rs650985)).
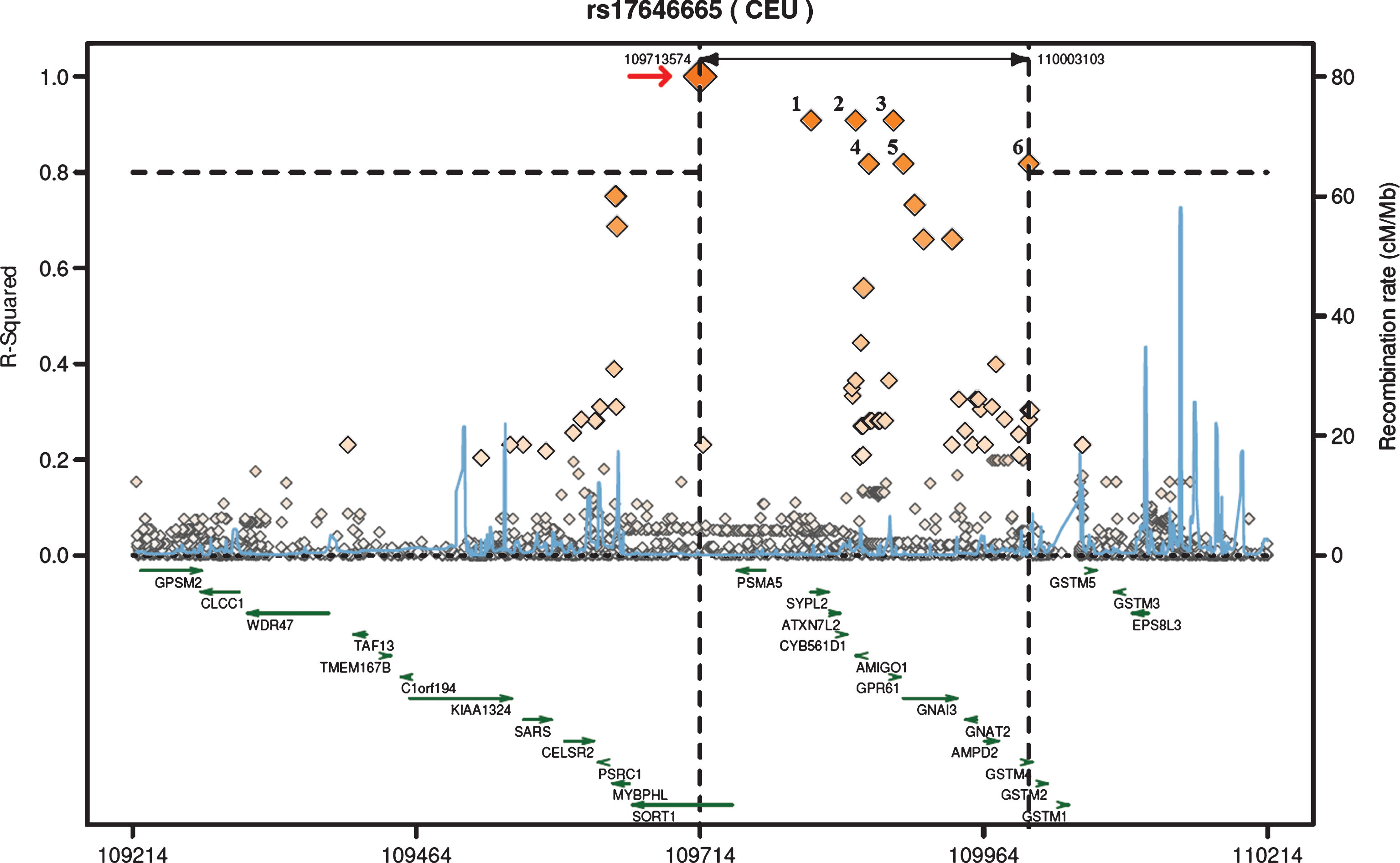
Table 1
Demographics for Alzheimer’s disease (AD) cases and controls
| Parameter | AD | Control | p | |
| Number of subjects | n | 620 | 1107 | |
| Age1 | years | 75.4 ± 7.3 | 71.8 ± 5.6 | <0.001 |
| Sex | male | 243 (39.5) | 372 (33.7) | <0.05 |
| female | 372 (60.5) | 733 (66.3) | ||
| MMSE2 | 20 ± 5.6 | 29 ± 0.8 | <0.001 | |
| Number of | 0 | 186 (30.3) | 774 (70.2) | |
| APOE ɛ4 | 1 | 317 (51.6) | 298 (27.0) | <0.001 |
| alleles | 2 | 111 (18.1) | 31 (2.8) |
Data are presented as absolute numbers with percentages in parentheses or as mean ± SD. P-values for categorical variables were calculated using chi-square statistical test and p-values for continuous variables were calculated using t-test. 1nAD = 614; nControl = 1104. 2nAD = 568; nControl = 837.
Table 2
Summary of SORT1 SNPs studied
| SNP | Genomic position | Alleles | MAF (%) | SNP location |
| SORT1 | Chromosome 1 | minor:major | ||
| rs646776 | 109275908 | C:T | 23.6 | Near gene 3’ |
| rs464218 | 109313684 | G:A | 46.5 | 3’UTR |
| rs17585355 | 109315193 | C:A | 6.3 | Intron |
| rs11102972 | 109338099 | C:T | 23.0 | Intron |
| rs7536292 | 109352071 | C:T | 17.4 | Intron |
| rs11142 | 109354481 | A:G | 30.2 | Coding syn. |
| rs17646665 | 109369429 | G:A | 6.9 | Intron |
| rs1149175 | 109379755 | A:G | 14.5 | Intron |
| rs12037569 | 109381055 | T:G | 15.2 | Intron |
| rs72646553 | 109397881 | C:G | 22.4 | Coding syn. |
Presented are single-nucleotide polymorphisms (SNPs) numbered according to the gene location. Genome positions were obtained from the NCBI genome database. MAF, minor allele frequency, presented as percentages for each SNP. UTR, untranslated region. Coding syn., Coding synonymous.
Table 3
SORT1 single marker (SNP) frequencies and associations with risk of Alzheimer’s disease
| SNP | Genotype | AD (%) | Control (%) | ORadj (95% CI) | padj |
| rs646776 | CC | 27 (4.4) | 70 (6.4) | ||
| CT | 221 (36.2) | 388 (35.5) | 1.0 (0.8–1.2) | 0.81 | |
| TT | 362 (59.3) | 636 (58.1) | |||
| rs464218 | GG | 124 (20.3) | 254 (23.1) | ||
| GA | 308 (50.5) | 527 (47.9) | 1.0 (0.8–1.2) | 0.88 | |
| AA | 178 (29.2) | 319 (29.0) | |||
| rs17585355 | CC | 2 (0.3) | 3 (0.3) | ||
| CA | 64 (10.4) | 142 (12.9) | 0.8 (0.6–1.2) | 0.27 | |
| AA | 552 (89.3) | 952 (86.8) | |||
| rs11102972 | CC | 33 (5.5) | 59 (5.4) | ||
| CT | 192 (31.7) | 405 (37.2) | 0.9 (0.7–1.1) | 0.15 | |
| TT | 380 (62.8) | 626 (57.4) | |||
| rs7536292 | CC | 23 (3.8) | 30 (2.7) | ||
| CT | 177 (29.0) | 311 (28.3) | 1.2 (0.9–1.4) | 0.17 | |
| TT | 410 (67.2) | 757 (68.9) | |||
| rs11142 | AA | 50 (8.3) | 99 (9.3) | ||
| AG | 239 (39.8) | 466 (44.0) | 0.9 (0.7–1.1) | 0.17 | |
| GG | 311 (51.8) | 494 (46.6) | |||
| rs17646665 | GG | 0 (0.0) | 7 (0.6) | ||
| GA | 64 (10.5) | 159 (14.5) | 0.6 (0.4–0.8) | 0.00063 | |
| AA | 547 (89.5) | 930 (84.9) | |||
| rs1149175 | AA | 17 (2.8) | 25 (2.3) | ||
| AG | 133 (21.6) | 279 (25.4) | 0.9 (0.7–1.1) | 0.35 | |
| GG | 466 (75.6) | 796 (72.4) | |||
| rs12037569 | TT | 14 (2.3) | 24 (2.2) | ||
| TG | 158 (25.9) | 285 (26.0) | 1.0 (0.8–1.3) | 0.75 | |
| GG | 437 (71.8) | 786 (71.8) | |||
| rs72646553 | CC | 33 (5.6) | 58 (5.4) | ||
| CG | 169 (28.7) | 398 (36.7) | 0.8 (0.7–1.0) | 0.05 | |
| GG | 386 (65.6) | 628 (57.9) |
Genotype data are presented as absolute numbers (percentages). OR, odds ratio per minor allele; CI, confidence interval; ORadj, adjusted odds ratio; padj, adjusted p-value using covariates. P-values were calculated using a logistic regression model adjusted for sex and number of APOE ɛ4 alleles. Bold numbers indicate p-values <0.05.

